

Abstract
We evaluated the promoter methylation levels of the APC, MGMT, hMLH1, RASSF1A and CDKN2A genes in 107 colorectal cancer (CRC) samples and 80 healthy adjacent tissues. We searched for correlation with both physical and pathological features, polymorphisms of folate metabolism pathway genes (MTHFR, MTRR, MTR, RFC1, TYMS, and DNMT3B), and data on circulating folate, vitamin B12 and homocysteine, which were available in a subgroup of the CRC patients. An increased number of methylated samples were found in CRC respect to adjacent healthy tissues, with the exception of APC, which was also frequently methylated in healthy colonic mucosa. Statistically significant associations were found between RASSF1A promoter methylation and tumor stage, and between hMLH1 promoter methylation and tumor location. Increasing age positively correlated with both hMLH1 and MGMT methylation levels in CRC tissues, and with APC methylation levels in the adjacent healthy mucosa. Concerning gender, females showed higher hMLH1 promoter methylation levels with respect to males. In CRC samples, the MTR 2756AG genotype correlated with higher methylation levels of RASSF1A, and the TYMS 1494 6bp ins/del polymorphism correlated with the methylation levels of both APC and hMLH1. In adjacent healthy tissues, MTR 2756AG and TYMS 1494 6bp del/del genotypes correlated with APC and MGMT promoter methylation, respectively. Low folate levels were associated with hMLH1 hypermethylation. Present results support the hypothesis that DNA methylation in CRC depends from both physiological and environmental factors, with one-carbon metabolism largely involved in this process.
Keywords: Colorectal Cancer, epigenetics, DNA methylation, folate metabolism, polymorphisms, APC, MGMT, CDKN2A, hMLH1, RASSF1A
Introduction
Colorectal cancer (CRC) represents a serious health concern, with over one-million new cases diagnosed worldwide every year.1 The disease occurs sporadically in most of the cases (75–80%) as a result of a multi-step process leading to the accumulation of genetic and epigenetic alterations in colon mucosa cells, primarily affecting oncogenes, tumor suppressor genes and DNA repair genes.2 DNA methylation involves the covalent addition of a methyl group to the 5′ position on cytosine residues, usually in CpG dinucleotides, and is one of the most studied epigenetic marks in CRC.3 Methylation of CpG islands (domains unusually enriched with CpG dinucleotides) in the promoter region of a gene is commonly associated with gene silencing as it inhibits the access of the transcriptional machinery to the promoter, while promoter demethylation is a condition enabling gene expression.3
The genes frequently methylated in CRC tissues, as well as the factors that can contribute to the methylation levels of those genes, including aging, gender, dietary habits, life-styles, environmental agents, and medications, have been recently reviewed.2-5 The ever-growing number of genes that show impaired methylation in CRC emphasizes the crucial role of epigenetic alterations for future diagnosis, prognosis and choice of therapeutic strategies, and active research is currently ongoing to develop rapid, cost effective and reproducible tools for the detection of epigenetic marks.3,4
DNA methylation is largely dependent on folate bioavailability, and impairments within the folate (one-carbon) metabolic pathway can be of relevance for cancer development.5 There is consensus in the literature indicating that individuals who habitually consume the greatest quantities of folate, or who have the highest concentrations of blood folate, are at decreased risk for the development of CRC.6,7 However, folate derivatives are cofactors in nucleotide synthesis and high levels of the vitamin could therefore promote the proliferation of rapidly dividing cells.6,7 Indeed, the current opinion is that higher folate intake is protective against CRC development in nearly all circumstances except for those individuals who consume an excessive amount of the vitamin and have existing neoplastic lesions.6,7
Given its pivotal role in DNA methylation processes, one-carbon metabolism has been largely investigated as a potential modulator of DNA methylation in CRC. Indeed, researchers started addressing the relationship between promoter methylation of CRC-related genes and folate intake at the end of the last century.8 Those studies were followed by several papers aimed at addressing the possible contribution of polymorphisms of genes involved in folate metabolism as modulators of DNA methylation changes in CRC.9-12
In the present study we assessed the methylation levels in the promoters of five key CRC genes (APC, MGMT, hMLH1, RASSF1A, and CDKN2A/p16) in both surgically resected cancer tissues and healthy adjacent mucosa of a group of diagnostically confirmed sporadic CRC individuals. We then searched for correlation between the methylation levels of each gene and age, gender, tumor size, cancer stage, and nine functional polymorphisms of six genes (MTHFR, MTR, MTRR, RFC1 [SLC19A1], TYMS, and DNMT3B) involved in one-carbon metabolism. Moreover, in a subgroup of the patients, data on circulating levels of folate, homocysteine, and vitamin B12 were available, and we searched for correlation between those biomarkers and the methylation profiles of the genes under investigation.
Results
Comparison between tumor and healthy tissues in CRC patients
Table 1 shows the demographic and clinical characteristics of the 107 CRC patients recruited for the present study, and the number of methylated samples for each of the studied genes in the CRC tissue. For 80 out of 107 CRC patients (34 females, 46 males) both tumor and healthy adjacent tissue specimens, located near the cancerous lesion (about 20 cm distance), were collected to analyze the methylation levels in the promoters of the chosen genes: APC, MGMT, hMLH1, RASSF1A, and CDKN2A/p16 (Table 2).
Table 1. Demographic characteristics of the study population.
| Total subjects | Age (Mean ± SD) | Gender | Stage (TNM) | Tumour size | Locationa | Methylated genesb |
|---|---|---|---|---|---|---|
| 107 | 71,07 ± 12,98 | M: 61 | Adenoma: 11 | T1: 3 | C: 73 | APC: 53 (49.5%) |
| F: 46 | I: 17 | T2: 19 | S: 12 | MGMT: 45 (42.0%) | ||
| II: 32 | T3: 66 | SR: 17 | CDKN2A: 31 (29.0%) | |||
| III: 33 | T4: 8 | R: 3 | hMLH1: 18 (16.8%) | |||
| IV: 14 | M: 1 | RASSF1A: 18 (16.8%) |
a Location: C = Colon, S: Sigma, SR = Sigma-Rectum, R = Rectum, M = Mixed b Methylated genes: number (percent) of methylated samples in CRC tissues
Table 2. Methylation data observed in CRC and adjacent healthy tissues in terms of numbers of methylated samples and percentage for each of the studied genes.
| Total patients = 80 (Females = 34; Males = 46) |
Cancer tissue N° (%) |
Adjacent healthy tissue N° (%) |
|---|---|---|
| APC | 39 (48.7) | 37 (46.2) |
| MGMT | 36 (45.0) | 14 (17.5)a |
| CDKN2A | 24 (30.0) | 5 (6.25)a |
| hMLH1 | 13 (16.2) | 1 (1.25)a |
| RASSF1A | 10 (12.5) | 1 (1.25)a |
a Significant difference CRC vs. Adjacent healthy tissue (P < 0.01, Chi square or Fisher Exact Test).
Table 2 shows the methylation data observed in CRC and adjacent healthy tissues in terms of numbers of methylated samples for each of the studied genes. For all of them, with the exception of APC, we observed a statistically significant increased number of methylated samples in CRC vs. adjacent healthy tissues (Chi square or Fisher exact test P < 0.01). By contrast, APC showed a similar frequency of methylated samples in both CRC and healthy tissues.
For each of the studied subjects and for each of the analyzed genes the percentage of promoter methylation observed in both CRC and healthy specimens is shown (Fig. 1). APC and MGMT resulted frequently methylated in healthy tissues; however, promoter methylation levels were relatively low and not higher than 10% (Fig. 1A and B). Similar results were obtained also for the other three genes, which, however, resulted methylated only in very few healthy tissue samples (Figs. 1C–E). Interestingly, only for CDKN2A, one subject showed almost 40% promoter methylation also in the normal mucosa. That patient showed 100% promoter methylation in the cancerous tissue and experienced CRC recurrence (Fig. 1C). No correlation between the methylation levels in CRC and healthy tissues was observed (linear regression P > 0.05) for each of the studied genes.

Figure 1.APC (A), MGMT (B), CDKN2A (C), hMLH1 (D) and RASSF1A (E) methylation in tumor and adjacent healthy tissue for each patient.
Correlation between methylation levels and both clinico-pathological and physical features
For each of the studied genes we analyzed the correlation between promoter methylation and both physical and clinico-pathological features of the CRC patients such as age, gender, TNM stage, tumor size and tumor location in all the 107 CRC specimens and also assessed the contribution of age and gender with respect to gene promoter methylation in the 80 healthy tissues analyzed. This last analysis was restricted to APC and MGMT genes, since they were those most frequently methylated in healthy tissues (Table 2).
A statistically significant association between RASSF1A promoter methylation and stage was found (P = 0.01), with stages I and III showing higher methylation than adenomas and/or other stages (Fig. 2A). A statistically significant association between hMLH1 promoter methylation and tumor location was observed (P = 0.04). Particularly, after stratification of the samples into three groups (right colon, left colon, and sigma/rectum) higher hMLH1methylation was observed in the right colon with respect to cancers of the sigma/rectum (Fig. 2B).
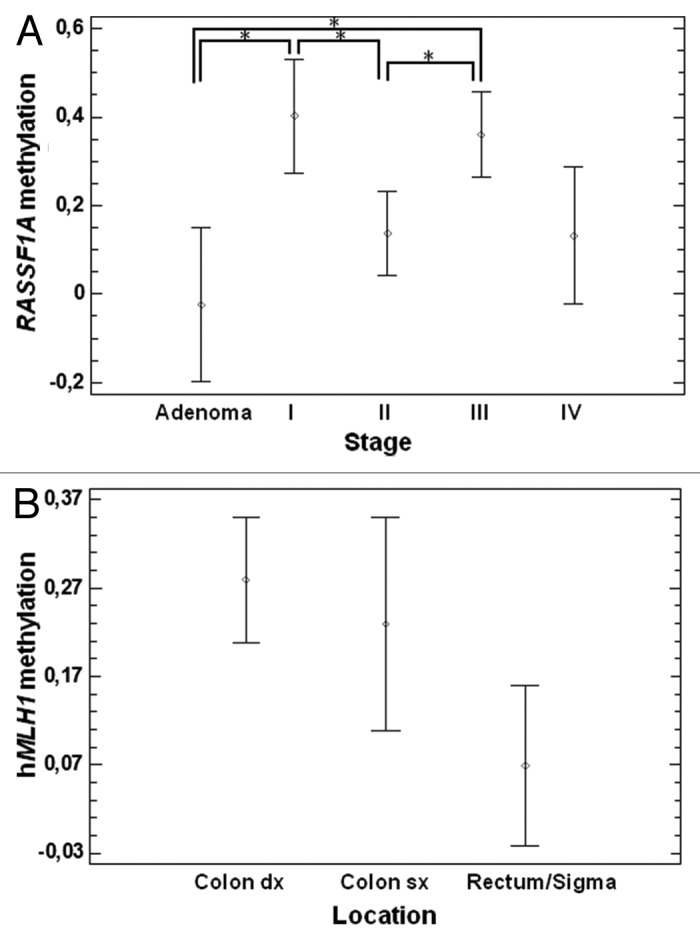
Figure 2. Correlation between gene promoter methylation, tumor stage and location. (A) A correlation between stages and RASSF1A promoter methylation was found, and particularly stage I and III showed hypermethylation with respect to adenomas and/or other stages (P = 0.01). (B) Correlation between location and hMLH1 promoter methylation. We observed a significant difference between right colon and sigma/rectum (P = 0.04).
By correlating promoter methylation status with gender, a statistically significant association between hMLH1 promoter methylation in CRC tissues and gender was found. Particularly, females showed higher methylation levels with respect to males (P = 0.03) (Fig. 3). No statistically significant association between the methylation levels of other genes and gender was found in either CRC or adjacent healthy tissues (data not shown). A statistically significant association between MGMT and hMLH1 promoter methylation and age (P = 0.002 and P = 0.0006, respectively) has been observed in CRC tissues. Particularly, an increase of methylation levels of these two genes with aging was noticed (Fig. 4A and B). Moreover, a significant correlation between age and APC promoter methylation in adjacent healthy tissues was observed (P = 0.01, Fig. 4C).
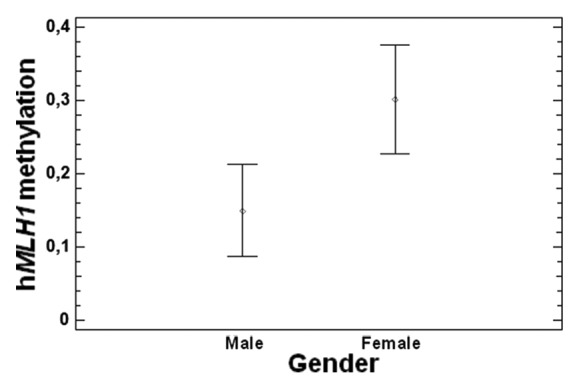
Figure 3. Correlation between gender and hMLH1 promoter methylation (P = 0.03).
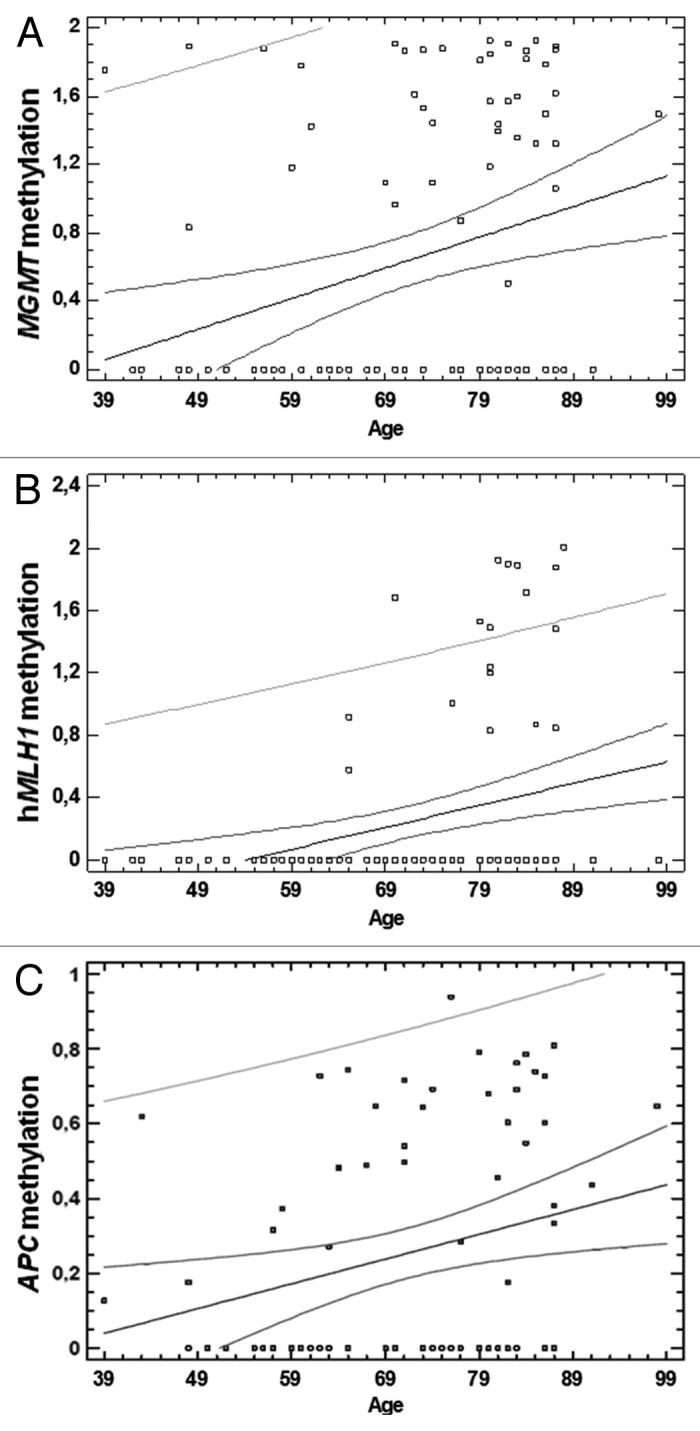
Figure 4. Correlation between age and gene promoter methylation. (A) Correlation between age and MGMT promoter methylation in CRC tissues (P = 0.002). (B) Correlation between age and hMLH1 promoter methylation in CRC tissues (P = 0.0006). (C) Correlation between age and APC promoter methylation in adjacent healthy tissues (P = 0.01).
Polymorphisms in one-carbon metabolism genes and gene promoter methylation
Allele and genotype frequencies of each of the studied polymorphisms (MTHFR 677C > T, MTHFR 1298A > C, MTRR 66A > G, MTR 2756A > G, RFC1 80G > A, TYMS 28 bp repeats, TYMS 1494 6 bp ins/del, DNMT3B -149C > T, and DNMT3B -579G > T) are shown in Table 3, and their correlation with CpG island methylation of APC, MGMT, hMLH1, RASSF1A and CDKN2A gene promoters was tested in CRC samples, as well as in the adjacent healthy tissues (only for APC and MGMT promoters). A statistically significant correlation was found between the MTR 2756A > G polymorphism and: (1) RASSF1A in CRC samples (P = 0.02, Fig. 5A), (2) APC in the healthy mucosa samples (P = 0.03, Figure 5B). Particularly, the AG genotype correlates with increased RASSF1A and APC promoter methylation with respect to the AA one (Fig. 5). Moreover, we observed a statistically significant correlation between the TYMS 1494 6bp ins/del polymorphism and both APC and hMLH1 promoter methylation levels in CRC tissues (P = 0.02 and 0.03, respectively, Fig. 6A and B), and MGMT promoter methylation in the healthy tissue (P = 0.02, Fig. 6C). In CRC specimens the presence of the 6bp del/del genotype correlates with decreased promoter methylation levels, while in the normal mucosa with increased methylation.
Table 3. Distribution of genotypes.
| Polymorphisms | Total | Genotype frequencies | ||
|---|---|---|---|---|
| RFC1 80G > A | 93 | GG (32.3%) | GA (50.5%) | AA (17.2%) |
| MTHFR 677C > T | 93 | CC (34.4%) | CT (47.3%) | TT (18.3%) |
| MTHFR 1298 A > C | 94 | AA (55.3%) | AC (36.2%) | CC (8.5%) |
| MTRR 66A > G | 94 | AA (41.5%) | AG (39.4%) | GG (19.1%) |
| MTR 2756A > G | 94 | AA (72.3%) | AG (27.7%) | GG (0.0%) |
| TYMS 1494 6bp ins/del | 92 | 6+6+ (34.8%) | 6+6- (51.1%) | 6-6- (14.1%) |
| TYMS 28 bp repeats | 92 | 3R3R (33%) | 3R2R (46%) | 2R2R (21%)* |
| DNMT3B -149C > T | 92 | CC (41.3%) | CT (44.6%) | TT (12.0%) |
| DNMT3B -579G > T | 92 | GG (45.7%) | GT (44.6%) | TT (9.7%) |
For four patients the genotypes were 2R4R and for two other patients 3R4R and 3R5R, respectively.
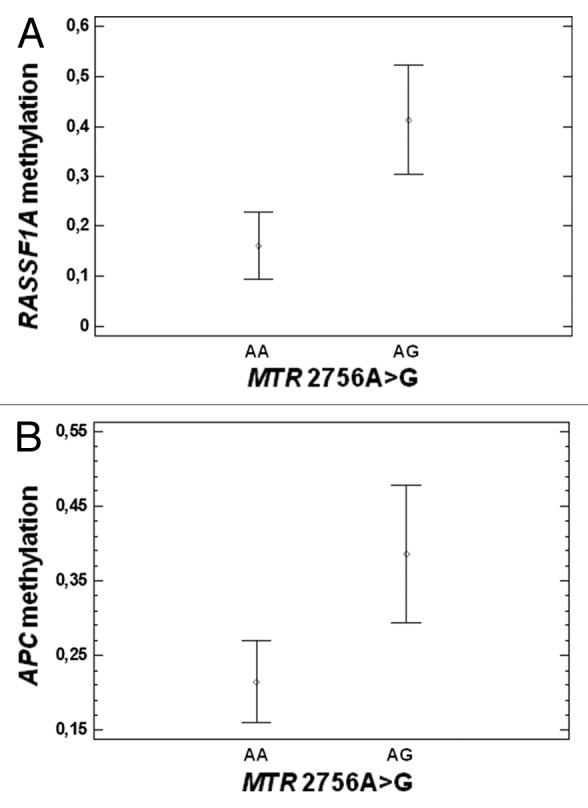
Figure 5. Correlation between the MTR 2756A > G polymorphism and gene promoter methylation. (A) Correlation with RASSF1A promoter methylation in CRC tissues (P = 0.02). (B) Correlation with APC promoter methylation in healthy tissues (P = 0.03).
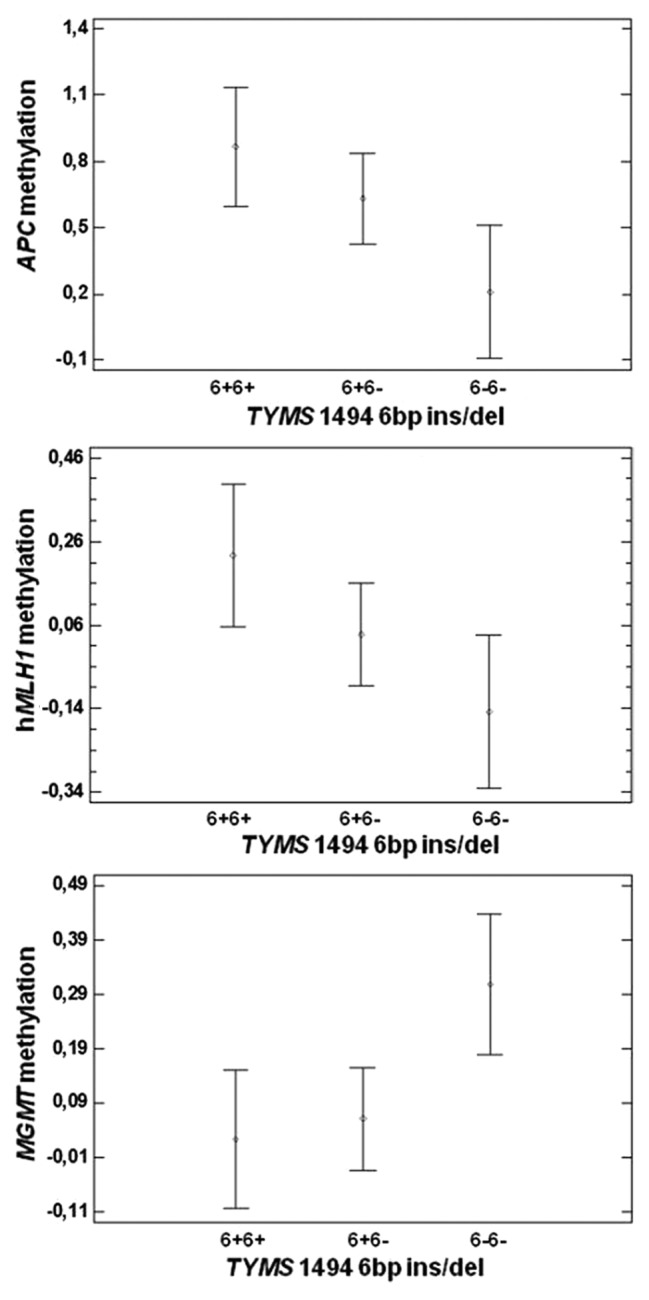
Figure 6. (A) Correlation between TYMS 1494 6bp ins/del polymorphism and APC promoter methylation in CRC tissues (P = 0.02). (B) Correlation between TYMS 1494 6bp ins/del polymorphism and hMLH1 promoter methylation in CRC tissues (P = 0.03). (C) Correlation between TYMS 1494 6bp ins/del polymorphism and MGMT promoter methylation in healthy tissues (P = 0.02).
Folate and homocysteine values in CRC patients and their correlation with gene promoter methylation
Folates, homocysteine, and vitamin B12 values have been measured in blood at colonoscopy. For some patients these biomarkers were not available, due to technical or other reasons as explained in details in the materials and methods section. Overall, we collected those values for about 40 CRC patients (Table 4). We observed a correlation (P = 0.05) between plasma folate levels and hMLH1 promoter methylation, showing individuals with folate levels below the normal range higher methylation (Fig. 7).
Table 4. Folate, homocysteine and vitamin B12 values in CRC patients (mean and standard deviation).
| Folate (ng/ml) | Homocysteine (µmol/l) | Vitamin B12 | |||||||
|---|---|---|---|---|---|---|---|---|---|
| Total | 4.6–18.7 ng/ml a |
< 4.6 ng/mlb |
Total | 4.3–11.1 µmol/l a |
> 11.1 µmol/lc |
Total | 191–663 pg/ml a |
> 663 pg/mlc |
|
| N° of patients | 36 | 23 | 13 | 37 | 22 | 15 | 39 | 35 | 4 |
| Mean + SD | 5.5 ± 1.7 | 6.5 ± 1.1 | 3.6 ± 0.6 | 10.8 ± 4.3 | 8.0 ± 1.9 | 14.3 ± 3.2 | 447 ± 152 | 414 ± 119 | 738 ± 226 |
a Normal range, b below the normal range, c above the normal range.
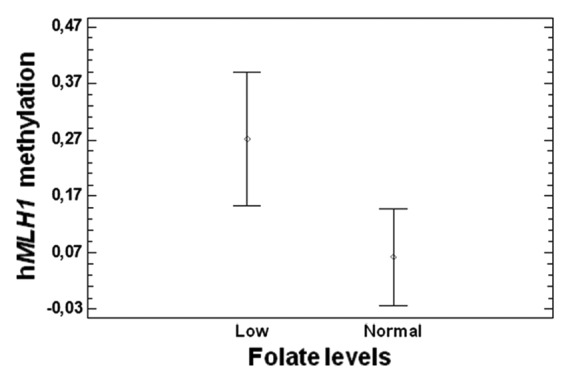
Figure 7. Correlation between plasma folate levels and hMLH1 promoter methylation (P = 0.05).
Immunohistochemical analysis for MLH1 protein and correlation with the BRAF V600E mutation
Immunohistochemical analysis was performed to evaluate the correlation between hMLH1 promoter methylation and MLH1 protein expression (Fig. 8). A total of 30 samples was assessed, 15 of them showing hMLH1 promoter methylation and the other 15 with 0% promoter methylation. 80% of the subjects showing hMLH1 promoter methylation were negative for MLH1 immunostaining. Moreover, those few subjects (20%) showing hMLH1 promoter methylation and positive MLH1 immunostaining had very low percentages of gene promoter methylation (Table 5). By contrast, 93% of the subjects showing 0% promoter methylation were positive for MLH1 immunostaining (Table 5). Present results indicate a very good correlation between hMLH1 promoter methylation and protein expression (P < 0.001). In addition, 10 out of the 15 samples with hMLH1 promoter methylation were carriers of the BRAF V600E mutation, most of them showing also lack of MLH1 protein and an elevated number of methylated genes among the five under investigation in the present study (Table 5). The BRAF V600E mutation was not found among the 15 individuals showing 0% hMLH1 promoter methylation (Table 5).
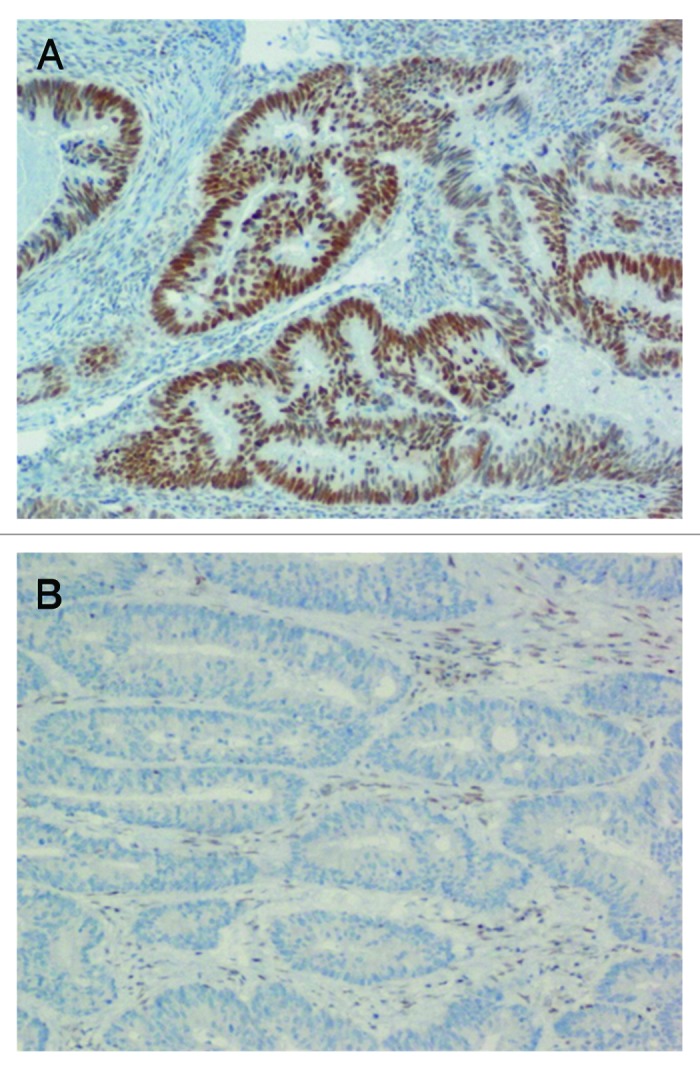
Figure 8. Immunohistochemical analysis showing the correlation between hMLH1 promoter methylation and MLH1 protein expression. (A) Unmethylated sample showing positive MLH1 nuclear staining (nuclei of brown color). (B) Methylated sample showing lack of MLH1 nuclear staining (absence of brown color in the nuclei).
Table 5. Immunohistochemical analysis and BRAF V600E mutation in individuals with different percentage of hMLH1 methylation.
| Gender | Age | hMLH1 methylation % |
MLH1 IHCa | N° of methylated genesb |
BRAF mutationc |
|---|---|---|---|---|---|
| F | 80 | 30.0 | - | 4 | V600E |
| F | 71 | 0 | + | 0 | |
| F | 79 | 0 | + | 0 | |
| M | 87 | 0 | + | 1 | |
| M | 81 | 83.5 | - | 4 | V600E |
| F | 64 | 0 | + | 1 | |
| F | 82 | 0 | + | 2 | |
| F | 82 | 78.4 | - | 5 | V600E |
| F | 62 | 0 | + | 0 | |
| M | 81 | 0 | + | 2 | |
| F | 84 | 51.1 | - | 2 | V600E |
| F | 87 | 6.0 | - | 3 | V600E |
| F | 85 | 6.4 | + | 2 | |
| F | 70 | 47.3 | - | 4 | V600E |
| F | 84 | 0 | + | 1 | |
| M | 91 | 0 | - | 1 | |
| M | 87 | 29.3 | - | 4 | |
| F | 79 | 32.9 | - | 2 | |
| M | 80 | 0 | - | 1 | |
| F | 83 | 76.5 | - | 4 | V600E |
| F | 65 | 7.3 | - | 1 | |
| M | 88 | 100 | - | 4 | V600E |
| F | 87 | 75 | - | 5 | V600E |
| F | 77 | 0 | + | 2 | |
| F | 80 | 14.8 | + | 3 | |
| M | 81 | 0 | + | 0 | |
| F | 79 | 0 | + | 1 | |
| M | 80 | 16.4 | + | 3 | V600E |
| F | 83 | 0 | + | 0 | |
| M | 76 | 0 | + | 0 |
a IHC = immunohistochemical analysis (+ = positive sample, - = negative sample), b Number of methylated genes among the five investigated in the present study, c Carriers of the V600E mutation are indicated, wild type subjects are left blank.
Discussion
It is now largely accepted that cancer is a multi-step process resulting from the accumulation of both genetic and epigenetic alterations of the genome.13 Gene mutations and epigenetic modifications have been initially viewed as two separate mechanisms participating in carcinogenesis. However, recent evidence points to a crosstalk between these two mechanisms in cancer formation, suggesting that gene mutations have the potential of disrupting several epigenetic patterns and that epigenetic modifications can drive genome instability and mutagenesis.14,15 For example, the epigenetic inactivation of DNA repair genes, such as hMLH1, MGMT, and others is often associated with genome instability and increased frequency of point mutations in cancer-related genes.14 Physiological aging and gender differences have been often associated with changes of epigenetic patterns, that can also be induced by exposure to environmental or lifestyle factors.2-5 Among dietary habits linked to changes of DNA methylation, particular attention has been dedicated to the folate metabolic pathway, given its pivotal role in providing one-carbon moieties for DNA methylation reactions.5
To further address this issue, here we evaluated the methylation profiles of five CRC-related genes, namely APC, MGMT, hMLH1, RASSF1A, and CDKN2A by means of MS-HRM technique in cancer tissues of 107 CRC patients as well as in the healthy adjacent mucosa of 80 of them. We then searched for correlation of the promoter methylation profiles of the selected genes with both physiological and pathological characteristics of the patients, and with a panel of biomarkers of one-carbon metabolism.
Our analysis revealed that APC was the most frequently methylated gene in both CRC (49.5%) and healthy mucosa tissues (46.2%), although the level of promoter methylation in healthy tissues was relatively low and not higher than 10%. No correlation between the methylation levels in CRC and healthy tissues was observed, as indicated by several subjects showing low levels of methylation in the healthy tissue and no methylation in the CRC one, or high levels of methylation in the CRC tissue but no or very low promoter methylation in the adjacent healthy mucosa. The tumor suppressor gene APC is one of the key components of the Wnt pathway, germline mutations in APC are associated with hereditary familial adenomatous polyposis (FAP) or attenuated FAP, and somatic mutations are common in sporadic CRC.16 APC hypermethylation is also frequent in sporadic CRC and may cause transcriptional silencing occurring early during colon neoplasia progression, but the reported methylation status of the APC promoter varies greatly among studies performed in different populations.16-19 Moreover, hypermethylation of the APC promoter has been shown to be relatively common in the normal colonic mucosa.19 Present results are in agreement with recent reports in Swedish and Vietnamese CRC patients, where methylation of the APC gene was detected with similar frequencies between the cancerous and normal tissues.19 We also observed that APC methylation in normal colonic mucosa increased significantly with age. This is in agreement with some previous reports of an age-related methylation of tumor suppressor genes in normal colorectal mucosa, that might represent physiological changes and/or constitute pre-neoplastic lesions.20,21
The MGMT gene is involved in DNA repair processes (mismatch repair) and in our cohort resulted hypermethylated in more than 40% of CRC tissues and in 17.5% of normal colonic mucosa. Again, we observed higher methylation levels in the affected tissue than in the adjacent healthy mucosa. Several previous reports of the literature suggest that MGMT hypermethylation results in gene silencing and increased rate of mutations in normal colonic mucosa that might represent an initiating step in the development of mismatch repair-deficient CRC.22-25 We also observed an age-related increase in MGMT methylation in CRC tissues in agreement with previous reports by Tserga et al.26 that showed a correlation between age and MGMT promoter methylation in breast cancer specimens, and Menigatti et al.21 that showed an age related increase of MGMT methylation in healthy colonic mucosa samples.
CDKN2A was methylated in 29% of CRC samples and both RASSF1A and hMLH1 in 16.8% of them; by contrast, they resulted methylated only in a few healthy mucosa tissues. Also the reported methylation frequencies of those genes varied within studies. For example, concerning the DNA repair hMLH1 gene, Huang et al.27 observed hMLH1 methylation in 20% of the CRC tissues analyzed, and others reported that it was methylated only in 1.8% of 112 adenomas.24 Arai et al.28 observed that the proportion of gastric and colorectal carcinomas with hMLH1 hypermethylation increases with age, reaching 25–30% of all carcinomas in elderly subjects, and those cancers are usually characterized by microsatellite instability and favorable prognosis. We also observed a significant increased hMLH1 promoter methylation with aging, as well as gender differences with females showing higher hMLH1 methylation than males. Those data are in agreement with results by Menigatti et al.21 that showed increased hMLH1 methylation with both increasing age and female gender in the healthy colonic mucosa. Ramírez et al.29 observed that methylation of the DNA repair genes hMLH1 and MGMT in normal mucosa correlated significantly with microsatellite instability and k-ras activation in the neighboring cancerous mucosa tissue, suggesting that epigenetic alterations in the mucosa surrounding cancerous neoplastic lesions might be due to a “field effect” occurring in early stages of carcinogenesis and working as a substrate for the subsequent accumulation of genetic alterations.
We also observed a strong correlation between hMLH1 promoter methylation and MLH1 protein levels evaluated by means of immunohistochemical analysis. Moreover, almost 70% of the samples showing hMLH1 promoter methylation were also carriers of the BRAF V600E mutation. Promoter hypermethylation of the mismatch repair gene hMLH1 is associated with microsatellite instability (MSI) and BRAF mutations in CRC.30 Indeed, hMLH1 methylation, MSI, and the BRAF V600E mutation, are often observed in CIMP high tumors, a specific subgroup of CRC denoted as the “CpG island methylator phenotype” as it displays extensive levels of methylated genes.30 Therefore, it is reasonable to speculate that factors linked to hMLH1 methylation are also linked to the CIMP high status. In this regard, we observed increased hMLH1 methylation in females than males, as well as increased hMLH1 methylation with age and in right colon tumors than in those of the sigma/rectum. Present data are in agreement with several previous reports suggesting that CIMP high tumors are associated with older age, female gender, proximal tumor location, microsatellite instability, BRAF mutation, and hMLH1 methylation.28,30,31 However, the panel of biomarkers used to evaluate the CIMP high status is not yet standardized,30 we had no opportunity to include all the most commonly studied CIMP biomarkers in the present investigation, and we only had a limited number of subjects with both hMLH1 methylation and BRAF mutation. Therefore, present factors linked to hMLH1 methylation are only indicative of a possible link to the CIMP high condition.
Also, CDKN2A methylation has been largely studied in CRC tissues and adjacent healthy mucosa.29 A recent large-scale study revealed that CDKN2A methylation in the normal mucosa can range from 0 to > 90% in some cases.32 Indeed, we observed a patient with more than 40% CDKN2A methylation in the normal mucosa. Similarly to the present study, CDKN2A methylation was observed in 24.8% of CRC specimens of Spanish CRC patients.33 Despite some reports suggesting that CDKN2A methylation might be associated with good prognosis,33 recent large-scale studies and a literature meta-analysis revealed that it is a marker of poor prognosis in CRC and other surgically treated cancers.32,34
RASSF1A methylation levels have been also largely investigated in CRC tissues. Nillson et al.35 reported it to be methylated in 14% of Swedish CRC tissues and 0% of the adjacent healthy mucosa, values that are really similar to the present 16.8% in CRC tissues and 1.25% in adjacent mucosa. Gene promoter methylation was also associated with poor prognosis in a 20 y follow up study of those patients.35 Also in this case, RASSF1A methylation levels varied with studies ranging from 10–20% of CRC subjects up to 40–50%, with some authors observing RASSF1A methylation in early stages of CRC, and others reporting it more frequently methylated in later stages.35-37 Some correlations with tumor stage were also observed in the present study.
The correlation between polymorphisms of genes involved in folate metabolism and DNA methylation has been investigated by several authors, and we recently reviewed the literature in the field.5
A very interesting finding of the present study was the association between the MTR 2756A > G polymorphism and promoter methylation levels of RASSF1A in CRC tissues and APC in healthy mucosa. The possible contribution of the MTR 2756A > G polymorphism to CRC risk has been largely investigated, often with conflicting results among studies, and a recent meta-analysis suggests that it could be associated with increased CRC risk in alcohol consumers.38 Methionine synthase is the enzyme that catalyzes the transmethylation of hcy to methionine, which is then used to form S-adenosylmethionine (SAM), the major intracellular methylating agent, and alcohol consumption reduces MTR activity and the production of SAM.39 Noteworthy, 89% of our CRC patients reported to be daily alcohol consumers, usually wine twice a day. Association of the MTR 2756A > G polymorphism with CRC risk has been also reported in smokers,38 but in this case only a few of our patients (7.5%) were smokers or ex-smokers from less than 10 y.
Another interesting finding was a correlation between the TYMS 1494 6bp ins/del polymorphism and both APC and hMLH1 promoter methylation in CRC tissues, and with MGMT methylation in the healthy mucosa. Thymidylate synthase is required for the conversion of deoxyuridine monophosphate (dUMP) to deoxythymine monophosphate (dTMP) in the de novo synthesis of pyrimidines, and the studied polymorphism is believed to impair TYMS mRNA expression or stability, ultimately leading to reduced protein levels in del/del homozygous individuals.40 Reduced TYMS levels in del/del individuals might impair the one-carbon metabolic pathway, particularly in rapidly dividing cells such as cancerous ones,41 thereby resulting in impaired methylation of certain genes, as we observed. Differences between cancer tissue and normal mucosa cells might reflect the different rate of cellular division in the two tissues and the consequent different need of TYMS activity and DNA precursors. In addition, genetic association studies and their meta-analysis suggest a contribution of TYMS polymorphisms to CRC risk.42,43
Data on folate, vitamin B12 and homocysteine, were available only for a small subgroup of the patients, however we observed a significant correlation between folate levels and hMLH1 promoter methylation in CRC tissues. Complex interactions among folate, hcy, vitamin B12, other B group vitamins and polymorphisms of one-carbon metabolic genes are known to affect DNA methylation in CRC,5 that we could not evaluate due to the scarce availability of biochemical data.
The method used in the present study does not allow to get information on the methylation status of each single CpG within the region under investigation, but provides an average methylation value of all the CpG sites in the fragment analyzed.44 Our recent investigation of the methylation levels of APC, MGMT, hMLH1, and CDKN2A promoter regions by means of pyrosequencing revealed that all the CpG sites analyzed tended to be methylated and no difference in mean methylation levels among different sites was observed for APC, hMLH1, and CDKN2A promoters,45 while a few CpG sites in the promoter of the MGMT gene tended to be less methylated (average 35–40% methylation) than the others (average 60–70% methylation).45
Conclusion
In summary, we screened a large cohort of CRC and healthy adjacent tissues by means of MS-HRM, a rapid and cost-effective technique for DNA methylation analyses. Our screening confirmed several previously reported observations in CRC, such as the high frequency of APC promoter methylation in both CRC tissues and healthy mucosa, and the correlation between hMLH1 promoter methylation with both age, gender, lack of MLH1 protein in the nuclei, and BRAF mutations among others. We also noticed several interesting correlations between markers of one-carbon metabolism and gene promoter methylation in CRC, further supporting both the contribution of this pathway to CRC pathogenesis and the evidence that DNA methylation in colonic mucosa cells is a multifactorial trait depending from both physiological and environmental factors.
Materials and Methods
Study population
DNA was obtained from both surgically resected tumor tissues of 107 patients (Table 1) and the adjacent normal tissue (at 20 cm of distance), available from 80 of them (Table 2). CRC diagnosis was performed by Medical Doctors at Department of Surgery, Medical, Molecular and Critical Area Pathology, University of Pisa, that provided tissue specimens. Staging was assessed after pathological examination of specimens based on TNM classification (Table 1). Family history of CRC was ascertained and all the subjects included in the present study had no family history of the disease. The study was approved by the ethical committee of the Pisa University Hospital. The individuals gave their written informed consent.
Extraction of genomic DNA
Genomic DNA was extracted using QIAmp DNA blood Mini Kit (Qiagen, Milan, Italy) according to the manufacturer’s instruction. The extracted DNA was quantified using a Nano Drop ND 2000c spectrophotometer (NanoDrop Thermo scientific).
Bisulfite modification
200 ng of DNA from each sample were treated with sodium bisulfite using the EpiTectH Bisulfite Kit (Qiagen) according to the manufacturer’s protocol. Sodium bisulfite treatment converts all unmethylated cytosines into uracil, while methylated cytosines are left unchanged.
Methylation sensitive-high resolution melting (MS-HRM) analysis
Promoter methylation was assessed by means of methylation sensitive-high resolution melting (MS-HRM) analysis in a CFX96 Real-Time PCR detection system (Bio-Rad). For the MS-HRM analysis we developed in-house protocols according to literature recommendations, using methylation independent primers (MIP).46,47 All analyses were run according to the following conditions: 1 cycle of 95 °C for 12 min, 60 cycles of 95 °C for 30s, Ta for 30s and 72 °C for 15s; followed by an HRM step of 95 °C for 10s and 50 °C for 1 min, 65 °C for 15s, and continuous acquisition to 95 °C at one acquisition per 0.2 °C. PCR was performed in a final volume of 25 ml, containing 12,5 ml of master mix (Qiagen), 10 pmol of each primer and 1 ml (almost 10 ng) of bisulfite modified DNA template. Each reaction was performed in duplicate. We analyzed 10% of the samples independently on separate occasions to verify the inter-assay variability and we observed a good reproducibility. Table 6 shows the conditions (primers, annealing temperature, CpG sites, and amplicon length) used for each gene. Fully methylated and unmethylated DNA (EpiTectH methylated and unmethylated human control DNA, bisulfite converted, Qiagen) were mixed to obtain the following ratios of methylation: 0%, 12,5%, 25%, 50%, 75%, 100%. Standard curves with known methylation ratios were included in each assay and were used to deduce the methylation ratio of each tumor and normal sample (Fig. 9). Validation of the MS-HRM assays was performed by means of pyrosequencing, as detailed elsewhere.44 In order to obtain single methylation percentage values from MS-HRM assays, rather than a range, we applied an interpolation method recently developed and described by us, that allowed to obtain precise HRM methylation values comparable to those obtained by pyrosequencing.44
Table 6. Primers and annealing temperature (Ta) used during MS-HRM analysis, as well as amplicon length and number of CpG sites for each of the studied genes.
| Gene | Primer sequences: 5′-3′ | Ta | CpG sites | Amplicon lenght |
|---|---|---|---|---|
| APC | F: CGGGGTTTTGTGTTTTATTG R: TCCAACGAATTACACAACTAC |
56 °C | 4 | 71 bp |
| MGMT | F: GCGTTTCGGATATGTTGGGATAAGT R: AACGACCCAAACACTCACCAAA |
58 °C | 12 | 110 bp |
| hMLH1 | F: GGTTATAAGAGTAGGGTTAA R: ATACCAATCAAATTTCTC |
56 °C | 5 | 81 bp |
| RASSF1A | F: TCGGGTTTTATAGTTTTTGTATTTAGGTTTT R: CCTCCCCCAAAATCCAAACTAA |
60 °C | 7 | 87 bp |
| CDKN2A | F: CGGAGGAAGAAAGAGGAGGGGT R: CGCTACCTACTCTCCCCCTCT |
62 °C | 7 | 93 bp |

Figure 9. Melting curves for each of the studied genes. (A) APC. (B) MGMT. (C) CDKN2A. (D) RASSF1A. (E) hMLH1. Each curve shows the standards (0%, 12.5%, 25%, 50%, 75% and 100% methylation) and a sample in duplicate (indicated with an arrow).
Genotyping
Genotyping for SLC19A1 (RFC1) 80A > G (rs1051266), MTHFR 677C > T (rs1801133), MTHFR 1298A > C (rs1801131), MTRR 66A > G (rs1801394), MTR 2756A > G (rs1805087), TYMS 28bp repeats (rs34743033), TYMS 1494 6bp ins/del (rs34489327), DNMT3B -149C > T (rs2424913), and DNMT3B -579G > T (rs1569686) were performed according to PCR-RFLP methods previously described by us.48,49 They are all functional polymorphisms, commonly studied in CRC genetic association studies, and with reported minor allele frequencies ranging from 15% to 49% in healthy Caucasians.48-50 Internal quality control samples with confirmed genotypes were added to each PCR-RFLP reaction. Digestion fragments were visualized after electrophoresis on a 3% agarose gel stained with ethidium bromide. Genotyping was possible only on 92–94 of the total CRC samples (Table 3) due to DNA run-out during MS-HRM analyses.
Biochemical analyses
Peripheral blood samples from CRC patients had been collected before surgery. Plasma was immediately separated and stored in freezer at -80 °C. All the analyses were performed with standard protocols at the diagnostic laboratory of the Pisa University Hospital, as detailed elsewhere.49 Those data are available for a subgroup of the patients (n = 39 for vitamin B12, n = 37 for hcy, and n = 36 for folates) because of blood drawings for biochemical markers was not possible for all of them or for technical problems during analyses, and also because we excluded individuals taking vitamin supplements, metformin, or other drugs known to interfere with those biomarkers.
Immunohistochemical analysis
5-μm sections from formalin-fixed, paraffin-embedded tissue were stained using the avidin-biotin complex method of Ventana Medical System (Ultraview DAB detection kit, Ventana Medical System) and using the BenchMark XT (Ventana Medical System) automated immunohistochemical stain. Staining was performed using antibodies to MLH1 (clone M1, Ventana Medical System). Nuclear immunostaining of normal colonic mucosal epithelial cells, lymphocytes and stromal cells served as internal positive controls. Positive nuclear staining of more than 10% of tumor cells was considered positive for MLH1 protein expression (Fig. 8). Loss of expression was recorded when all malignant cells showed absent nuclear staining or when less than 10% of tumor cells showed positive nuclear staining (Fig. 8). A total of 30 samples was assessed, 15 showing hMLH1 methylation and 15 showing 0% hMLH1 promoter methylation (Table 5).
Microdissection and DNA extraction for the evaluation of BRAF mutation
Serial 5-μm sections were taken from the above described formalin fixed paraffin embedded tissues. The last section was stained with hematoxilin-eosin; the tumor area was marked and the percentage of tumor cells was estimated by a pathologist. The tumor tissue was manually microdissected from one to three unstained sections previously submitted to xylene deparaffination and was lysed overnight at 56 °C in 180 μl of ATL buffer and 20 μl of proteinase K. DNA was purified using the spin column procedure (QIAamp DNA Mini Kit, QIAGEN) and finally reconstituted in 40 μl of AE buffer. DNA content was measured with a Nanodrop-1000 spectrophotometer (Thermo Fisher Scientific Inc.) and was kept at 4 °C before use.
Detection of BRAF V600E mutation by Real Time PCR
Five microliters of genomic DNA concentrated 10 ng/μl was tested for BRAF V600E mutation using the CE-marked Easy® BRAF kit (Diatech Pharmacogenetics), a real-time PCR based assay that uses allele-specific primers and probes in association with a mutant enrichment technique, following the manufacturer’s instructions. The kit enables the detection of a low percentage of mutant in a background of wild-type genomic DNA and the oligo mix allows the co-amplification of BRAF mutated target sequence and an endogenous control gene. The latter is used for the assessment of the quality and quantity of DNA in the sample. All reactions was performed on Rotor-GeneTM 6000 instrument (Corbett Research) as follows: hold at 95 °C for 2 min; denaturation at 95 °C (10 s) and annealing at 58 °C (60 s) for 40 cycles. The mutation status of sample was determined by considering the ΔCt values in according to the manufacturer’s recommendations. If the ΔCt value was less than 10, the sample was considered as V600E positive, otherwise as negative (Table 5).
Statistical analyses
Differences in the number of methylated samples in CRC vs. healthy adjacent tissues have been evaluated by means of chi square analyses or Fisher exact test. Linear regression analysis was performed to search for correlation between age and methylation data, as well as to search for correlation between methylation levels in CRC samples and methylation levels in the adjacent healthy mucosa. The effect of gender on mean methylation levels was assessed by means of analysis of variance (ANOVA), including age at sampling, the number of methylated genes and the levels of methylation of each of the other genes as covariates. Multifactorial analysis of variance (MANOVA), including age at sampling, gender, the number of methylated genes for each sample (ranging from 0 to 5), and the levels of methylation of each of the other four genes as covariates, was used to correlate methylation data with tumor size, staging and with each of the studied polymorphisms (SNPs). By means of MANOVA we evaluated simultaneously the effect of the nine SNPs on the methylation levels of a certain gene. In such a way the number of tests performed has been significantly reduced, and the analysis allowed correcting the effect of each given SNP for the presence of all the other ones that were tested simultaneously. Since methylation data were not normally distributed, natural logarithm transformation of all values was done before analysis. Folate, vitamin B12, and hcy levels were available only for about 40 out of 107 individuals. For each of the studied markers the patients were stratified into two groups (i.e., low folates [< 4.6 ng/ml] and normal folates [4.6–18.7 ng/ml], normal vitamin B12 [191–663 pg/ml] and high vitamin B12 [> 663 pg/ml], and normal hcy [4.3–11.1 µmol/l] and high hcy [> 11.1 µmol/l] [Table 4]) and mean methylation differences among the two groups have been evaluated for each of the studied genes by means of MANOVA, as explained above. Analyses were performed with the STATGRAPHICS 5.1 Plus software package for Windows.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
The study was funded by Istituto Toscano Tumori (ITT) (Prot.AOOGRT/325424/Q.80.110 16/12/2009) “Correlation among epigenetic, environmental and genetic factors in colorectal carcinoma.” FC, FM and AF have been supported by ITT fellowships. We acknowledge Fondazione Bracco for the donation of the C1000TM thermal cycler coupled with the CFX96TM Real-Time PCR Detection System (Bio-Rad).
References
- 1.Harrison S, Benziger H. The molecular biology of colorectal carcinoma and its implications: a review. Surgeon. 2011;9:200–10. doi: 10.1016/j.surge.2011.01.011. [DOI] [PubMed] [Google Scholar]
- 2.Migliore L, Migheli F, Spisni R, Coppedè F. Genetics, cytogenetics, and epigenetics of colorectal cancer. J Biomed Biotechnol 2011; 2011:792362. [DOI] [PMC free article] [PubMed]
- 3.Lao VV, Grady WM. Epigenetics and colorectal cancer. Nat Rev Gastroenterol Hepatol. 2011;8:686–700. doi: 10.1038/nrgastro.2011.173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Heyn H, Esteller M. DNA methylation profiling in the clinic: applications and challenges. Nat Rev Genet. 2012;13:679–92. doi: 10.1038/nrg3270. [DOI] [PubMed] [Google Scholar]
- 5.Coppedè F. Epigenetic biomarkers of colorectal cancer: Focus on DNA methylation. Cancer Lett. 2014;342:238–47. doi: 10.1016/j.canlet.2011.12.030. [DOI] [PubMed] [Google Scholar]
- 6.Duthie SJ. Folate and cancer: how DNA damage, repair and methylation impact on colon carcinogenesis. J Inherit Metab Dis. 2011;34:101–9. doi: 10.1007/s10545-010-9128-0. [DOI] [PubMed] [Google Scholar]
- 7.Mason JB. Unraveling the complex relationship between folate and cancer risk. Biofactors. 2011;37:253–60. doi: 10.1002/biof.174. [DOI] [PubMed] [Google Scholar]
- 8.Cravo M, Fidalgo P, Pereira AD, Gouveia-Oliveira A, Chaves P, Selhub J, Mason JB, Mira FC, Leitao CN. DNA methylation as an intermediate biomarker in colorectal cancer: modulation by folic acid supplementation. Eur J Cancer Prev. 1994;3:473–9. doi: 10.1097/00008469-199411000-00004. [DOI] [PubMed] [Google Scholar]
- 9.Mokarram P, Naghibalhossaini F, Saberi Firoozi M, Hosseini SV, Izadpanah A, Salahi H, Malek-Hosseini SA, Talei A, Mojallal M. Methylenetetrahydrofolate reductase C677T genotype affects promoter methylation of tumor-specific genes in sporadic colorectal cancer through an interaction with folate/vitamin B12 status. World J Gastroenterol. 2008;14:3662–71. doi: 10.3748/wjg.14.3662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.de Vogel S, Wouters KA, Gottschalk RW, van Schooten FJ, de Goeij AF, de Bruïne AP, Goldbohm RA, van den Brandt PA, Weijenberg MP, van Engeland M. Genetic variants of methyl metabolizing enzymes and epigenetic regulators: associations with promoter CpG island hypermethylation in colorectal cancer. Cancer Epidemiol Biomarkers Prev. 2009;18:3086–96. doi: 10.1158/1055-9965.EPI-09-0289. [DOI] [PubMed] [Google Scholar]
- 11.Kim JW, Park HM, Choi YK, Chong SY, Oh D, Kim NK. Polymorphisms in genes involved in folate metabolism and plasma DNA methylation in colorectal cancer patients. Oncol Rep. 2011;25:167–72. [PubMed] [Google Scholar]
- 12.Wettergren Y, Odin E, Carlsson G, Gustavsson B. MTHFR, MTR, and MTRR polymorphisms in relation to p16INK4A hypermethylation in mucosa of patients with colorectal cancer. Mol Med. 2010;16:425–32. doi: 10.2119/molmed.2009.00156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dawson MA, Kouzarides T. Cancer epigenetics: from mechanism to therapy. Cell. 2012;150:12–27. doi: 10.1016/j.cell.2012.06.013. [DOI] [PubMed] [Google Scholar]
- 14.Toyota M, Suzuki H. Epigenetic drivers of genetic alterations. Adv Genet. 2010;70:309–23. doi: 10.1016/B978-0-12-380866-0.60011-3. [DOI] [PubMed] [Google Scholar]
- 15.You JS, Jones PA. Cancer genetics and epigenetics: two sides of the same coin? Cancer Cell. 2012;22:9–20. doi: 10.1016/j.ccr.2012.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Esteller M, Sparks A, Toyota M, Sanchez-Cespedes M, Capella G, Peinado MA, Gonzalez S, Tarafa G, Sidransky D, Meltzer SJ, et al. Analysis of adenomatous polyposis coli promoter hypermethylation in human cancer. Cancer Res. 2000;60:4366–71. [PubMed] [Google Scholar]
- 17.Lee S, Hwang KS, Lee HJ, Kim JS, Kang GH. Aberrant CpG island hypermethylation of multiple genes in colorectal neoplasia. Lab Invest. 2004;84:884–93. doi: 10.1038/labinvest.3700108. [DOI] [PubMed] [Google Scholar]
- 18.Chen SP, Chiu SC, Wu CC, Lin SZ, Kang JC, Chen YL, Lin PC, Pang CY, Harn HJ. The association of methylation in the promoter of APC and MGMT and the prognosis of Taiwanese CRC patients. Genet Test Mol Biomarkers. 2009;13:67–71. doi: 10.1089/gtmb.2008.0045. [DOI] [PubMed] [Google Scholar]
- 19.Dimberg J, Hong TT, Skarstedt M, Löfgren S, Zar N, Matussek A. Analysis of APC and IGFBP7 promoter gene methylation in Swedish and Vietnamese colorectal cancer patients. Oncol Lett. 2013;5:25–30. doi: 10.3892/ol.2012.967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Krakowczyk L, Strzelczyk JK, Adamek B, Zalewska-Ziob M, Arendt J, Półtorak S, Maciejewski B, Wiczkowski A. Methylation of the MGMT and p16 genes in sporadic colorectal carcinoma and corresponding normal colonic mucosa. Med Sci Monit. 2008;14:BR219–25. [PubMed] [Google Scholar]
- 21.Menigatti M, Truninger K, Gebbers JO, Marbet U, Marra G, Schär P. Normal colorectal mucosa exhibits sex- and segment-specific susceptibility to DNA methylation at the hMLH1 and MGMT promoters. Oncogene. 2009;28:899–909. doi: 10.1038/onc.2008.444. [DOI] [PubMed] [Google Scholar]
- 22.Abouzeid HE, Kassem AM, Abdel Wahab AH, El-mezayen HA, Sharad H, Abdel Rahman S. Promoter hypermethylation of RASSF1A, MGMT, and HIC-1 genes in benign and malignant colorectal tumors. Tumour Biol. 2011;32:845–52. doi: 10.1007/s13277-011-0156-7. [DOI] [PubMed] [Google Scholar]
- 23.Svrcek M, Buhard O, Colas C, Coulet F, Dumont S, Massaoudi I, Lamri A, Hamelin R, Cosnes J, Oliveira C, et al. Methylation tolerance due to an O6-methylguanine DNA methyltransferase (MGMT) field defect in the colonic mucosa: an initiating step in the development of mismatch repair-deficient colorectal cancers. Gut. 2010;59:1516–26. doi: 10.1136/gut.2009.194787. [DOI] [PubMed] [Google Scholar]
- 24.Lee KH, Lee JS, Nam JH, Choi C, Lee MC, Park CS, Juhng SW, Lee JH. Promoter methylation status of hMLH1, hMSH2, and MGMT genes in colorectal cancer associated with adenoma-carcinoma sequence. Langenbecks Arch Surg. 2011;396:1017–26. doi: 10.1007/s00423-011-0812-9. [DOI] [PubMed] [Google Scholar]
- 25.Farzanehfar M, Vossoughinia H, Jabini R, Tavassoli A, Saadatnia H, Khorashad AK, Ahadi M, Afzalaghaee M, Ghayoor Karimiani E, Mirzaei F, et al. Evaluation of methylation of MGMT (O⁶-methylguanine-DNA methyltransferase) gene promoter in sporadic colorectal cancer. DNA Cell Biol. 2013;32:371–7. doi: 10.1089/dna.2012.1949. [DOI] [PubMed] [Google Scholar]
- 26.Tserga A, Michalopoulos NV, Levidou G, Korkolopoulou P, Zografos G, Patsouris E, Saetta AA. Association of aberrant DNA methylation with clinicopathological features in breast cancer. Oncol Rep. 2012;27:1630–8. doi: 10.3892/or.2011.1576. [DOI] [PubMed] [Google Scholar]
- 27.Huang Q, Huang JF, Zhang B, Baum L, Fu WL. Methylation variable position profiles of hMLH1 promoter CpG islands in human sporadic colorectal carcinoma. Diagn Mol Pathol. 2012;21:24–33. doi: 10.1097/PDM.0b013e318230effd. [DOI] [PubMed] [Google Scholar]
- 28.Arai T, Kasahara I, Sawabe M, Honma N, Aida J, Tabubo K. Role of methylation of the hMLH1 gene promoter in the development of gastric and colorectal carcinoma in the elderly. Geriatr Gerontol Int. 2010;10(Suppl 1):S207–12. doi: 10.1111/j.1447-0594.2010.00590.x. [DOI] [PubMed] [Google Scholar]
- 29.Ramírez N, Bandrés E, Navarro A, Pons A, Jansa S, Moreno I, Martínez-Rodenas F, Zárate R, Bitarte N, Monzó M, et al. Epigenetic events in normal colonic mucosa surrounding colorectal cancer lesions. Eur J Cancer. 2008;44:2689–95. doi: 10.1016/j.ejca.2008.09.004. [DOI] [PubMed] [Google Scholar]
- 30.de Vogel S, Weijenberg MP, Herman JG, Wouters KAD, de Goeij AFPM, van den Brandt PA, de Bruïne AP, van Engeland M. MGMT and MLH1 promoter methylation versus APC, KRAS and BRAF gene mutations in colorectal cancer: indications for distinct pathways and sequence of events. Ann Oncol. 2009;20:1216–22. doi: 10.1093/annonc/mdn782. [DOI] [PubMed] [Google Scholar]
- 31.Barault L, Charon-Barra C, Jooste V, de la Vega MF, Martin L, Roignot P, Rat P, Bouvier AM, Laurent-Puig P, Faivre J, et al. Hypermethylator phenotype in sporadic colon cancer: study on a population-based series of 582 cases. Cancer Res. 2008;68:8541–6. doi: 10.1158/0008-5472.CAN-08-1171. [DOI] [PubMed] [Google Scholar]
- 32.Bihl MP, Foerster A, Lugli A, Zlobec I. Characterization of CDKN2A(p16) methylation and impact in colorectal cancer: systematic analysis using pyrosequencing. J Transl Med. 2012;10:173. doi: 10.1186/1479-5876-10-173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Veganzones-de-Castro S, Rafael-Fernández S, Vidaurreta-Lázaro M, de-la-Orden V, Mediero-Valeros B, Fernández C, Maestro-de las Casas ML. p16 gene methylation in colorectal cancer patients with long-term follow-up. Rev Esp Enferm Dig. 2012;104:111–7. doi: 10.4321/s1130-01082012000300002. [DOI] [PubMed] [Google Scholar]
- 34.Xing XB, Cai WB, Luo L, Liu LS, Shi HJ, Chen MH. The Prognostic Value of p16 Hypermethylation in Cancer: A Meta-Analysis. PLoS One. 2013;8:e66587. doi: 10.1371/journal.pone.0066587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Nilsson TK, Löf-Öhlin ZM, Sun XF. DNA methylation of the p14ARF, RASSF1A and APC1A genes as an independent prognostic factor in colorectal cancer patients. Int J Oncol. 2013;42:127–33. doi: 10.3892/ijo.2012.1682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sinha R, Hussain S, Mehrotra R, Kumar RS, Kumar K, Pande P, Doval DC, Basir SF, Bharadwaj M. Kras gene mutation and RASSF1A, FHIT and MGMT gene promoter hypermethylation: indicators of tumor staging and metastasis in adenocarcinomatous sporadic colorectal cancer in Indian population. PLoS One. 2013;8:e60142. doi: 10.1371/journal.pone.0060142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Fernandes MS, Carneiro F, Oliveira C, Seruca R. Colorectal cancer and RASSF family--a special emphasis on RASSF1A. Int J Cancer. 2013;132:251–8. doi: 10.1002/ijc.27696. [DOI] [PubMed] [Google Scholar]
- 38.Ding W, Zhou DL, Jiang X, Lu LS. Methionine synthase A2756G polymorphism and risk of colorectal adenoma and cancer: evidence based on 27 studies. PLoS One. 2013;8:e60508. doi: 10.1371/journal.pone.0060508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Halsted CH, Villanueva JA, Devlin AM, Chandler CJ. Metabolic interactions of alcohol and folate. J Nutr. 2002;132(Suppl):2367S–72S. doi: 10.1093/jn/132.8.2367S. [DOI] [PubMed] [Google Scholar]
- 40.Ulrich CM, Bigler J, Bostick R, Fosdick L, Potter JD. Thymidylate synthase promoter polymorphism, interaction with folate intake, and risk of colorectal adenomas. Cancer Res. 2002;62:3361–4. [PubMed] [Google Scholar]
- 41.Skibola CF, Forrest MS, Coppedé F, Agana L, Hubbard A, Smith MT, Bracci PM, Holly EA. Polymorphisms and haplotypes in folate-metabolizing genes and risk of non-Hodgkin lymphoma. Blood. 2004;104:2155–62. doi: 10.1182/blood-2004-02-0557. [DOI] [PubMed] [Google Scholar]
- 42.Gao CM, Ding JH, Li SP, Liu YT, Cao HX, Wu JZ, Tajima K. Polymorphisms in the thymidylate synthase gene and risk of colorectal cancer. Asian Pac J Cancer Prev. 2012;13:4087–91. doi: 10.7314/apjcp.2012.13.8.4087. [DOI] [PubMed] [Google Scholar]
- 43.Lu M, Sun L, Yang J, Li YY. 3R variant of thymidylate synthase 5′-untranslated enhanced region contributes to colorectal cancer risk: a meta-analysis. Asian Pac J Cancer Prev. 2012;13:2605–10. doi: 10.7314/apjcp.2012.13.6.2605. [DOI] [PubMed] [Google Scholar]
- 44.Migheli F, Stoccoro A, Coppedè F, Wan Omar WA, Failli A, Consolini R, Seccia M, Spisni R, Miccoli P, Mathers JC, et al. Comparison study of MS-HRM and pyrosequencing techniques for quantification of APC and CDKN2A gene methylation. PLoS One. 2013;8:e52501. doi: 10.1371/journal.pone.0052501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Failli A, Legitimo A, Migheli F, Coppedè F, Mathers JC, Spisni R, Miccoli P, Migliore L, Consolini R. Efficacy and Feasibility of the Epithelial Cell Adhesion Molecule (EpCAM) Immunomagnetic Cell Sorter for Studies of DNA Methylation in Colorectal Cancer. Int J Mol Sci. 2013;15:44–57. doi: 10.3390/ijms15010044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Huang KT, Dobrovic A, Yan M, Karim RZ, Lee CS, Lakhani SR, Fox SB. DNA methylation profiling of phyllodes and fibroadenoma tumours of the breast. Breast Cancer Res Treat. 2010;124:555–65. doi: 10.1007/s10549-010-0970-4. [DOI] [PubMed] [Google Scholar]
- 47.Wojdacz TK, Dobrovic A. Methylation-sensitive high resolution melting (MS-HRM): a new approach for sensitive and high-throughput assessment of methylation. Nucleic Acids Res. 2007;35:e41. doi: 10.1093/nar/gkm013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Coppedè F, Migheli F, Bargagna S, Siciliano G, Antonucci I, Stuppia L, Palka G, Migliore L. Association of maternal polymorphisms in folate metabolizing genes with chromosome damage and risk of Down syndrome offspring. Neurosci Lett. 2009;449:15–9. doi: 10.1016/j.neulet.2008.10.074. [DOI] [PubMed] [Google Scholar]
- 49.Coppedè F, Bosco P, Tannorella P, Romano C, Antonucci I, Stuppia L, Romano C, Migliore L. DNMT3B promoter polymorphisms and maternal risk of birth of a child with Down syndrome. Hum Reprod. 2013;28:545–50. doi: 10.1093/humrep/des376. [DOI] [PubMed] [Google Scholar]
- 50.Coppedè F, Tannorella P, Pezzini I, Migheli F, Ricci G, Caldarazzo lenco E, Piaceri I, Polini A, Nacmias B, Monzani F, et al. Folate, homocysteine, vitamin B12, and polymorphisms of genes participating in one-carbon metabolism in late-onset Alzheimer’s disease patients and healthy controls. Antioxid Redox Signal. 2012;17:195–204. doi: 10.1089/ars.2011.4368. [DOI] [PubMed] [Google Scholar]