

Abstract
The Slit family of secreted proteins and their transmembrane receptor, Robo, were originally identified in the nervous system where they function as axon guidance cues and branching factors during development. Since their discovery, a great number of additional roles have been attributed to Slit/Robo signaling, including regulating the critical processes of cell proliferation and cell motility in a variety of cell and tissue types. These processes are often deregulated during cancer progression, allowing tumor cells to bypass safeguarding mechanisms in the cell and the environment in order to grow and escape to new tissues. In the last decade, it has been shown that the expression of Slit and Robo is altered in a wide variety of cancer types, identifying them as potential therapeutic targets. Further, studies have demonstrated dual roles for Slits and Robos in cancer, acting as both oncogenes and tumor suppressors. This bifunctionality is also observed in their roles as axon guidance cues in the developing nervous system, where they both attract and repel neuronal migration. The fact that this signaling axis can have opposite functions depending on the cellular circumstance make its actions challenging to define. Here, we summarize our current understanding of the dual roles that Slit/Robo signaling play in development, epithelial tumor progression and tumor angiogenesis.
Keywords: Slit, Robo, Cancer, Tumor, Tumor Angiogenesis, Axon Guidance Molecules
Introduction
The existence of axon guidance molecules was postulated by Ramon y Cajal in the late 1800s, but 100 years elapsed before their molecular identification. As their name suggests, these cues act to instruct the migration of neurons and their axons in the developing nervous system, establishing the initial pattern of axonal projections that is subsequently refined by activity-dependent mechanisms. Axon guidance cues are secreted by both target and non-target cells, and they are bi-functional, acting as both attractants and repellents when patterning the nervous system. Cell surface receptors are responsible for directing the response of a cell to these cues. These receptors translate directional information provided by the cues to the cytoskeleton, generating the movement and, in the case of axons, directional outgrowth required for pathfinding.
In addition to their role as axon guidance cues, these molecules also function outside the nervous system to regulate the development of other organs, including the immune and vascular systems, epithelial organs and glands. In these contexts, “axon guidance” cues do not simply function as instructional signals, but rather act more broadly to control the growth, branching, adhesion and position of cells in complex tissues. Like many molecules that play key roles in development, “axon guidance” cues are often deregulated in disease processes, especially cancer, with many of these cues acting to either promote or suppress tumor growth and progression.
Among the many families of “axon guidance” molecules, Slits, signaling through their Roundabout (Robo) receptors, constitute a relatively small group of factors. They were originally identified as chemorepellents that play a crucial role in preventing developing commissural neurons from inappropriately recrossing the midline. Once believed to have only restricted functions in the developing nervous system as guidance cues and branching factors (Brose et al., 1999; Wang et al., 1999), members of this family are now identified as key regulators of many cellular processes in multiple tissue types, including the mammary gland, heart, lung and kidney (Greenberg et al., 2004; Hinck, 2004; Medioni et al., 2010; Piper et al., 2000). In addition, they have also been implicated in multiple human pathologies including cancer and inflammation (Legg et al., 2008; London and Li, 2011; London et al., 2010; Wu et al., 2001).
As we learn more about the mechanisms of cancer progression, it is becoming clear that tumor cells hijack normal cellular processes to survive and metastasize to secondary tissues. Specifically, a cell’s proliferative, adhesive and migratory properties are often altered in the process of tumor cell transformation, allowing rapid proliferation and tumor growth, detachment from the surrounding tissue, and invasion into the vasculature leading to metastasis. Over the last decade, many studies have implicated Slit/Robo signaling in the regulation of cell proliferation, cell adhesion, and cell migration, raising the possibility that this pathway represents a key target for alteration in cells undergoing tumor cell transformation. In fact, Slit and Robo expression levels are altered in a majority of human cancers. However, recent work suggests that the role of Slit/Robo in tumor progression is anything but simple. Emerging evidence postulates that Slits and Robos function both as oncogenes and tumor suppressors, often in the same tissue. In this review, we summarize how Slit/Robo signaling confers both tumor suppressive and oncogenic effects on the progression of various types of cancers, focusing most of our discussion on vertebrate systems, though invertebrate studies will be touched upon when relevant.
The Slit/Robo Signaling Interaction is Well Characterized
Slit is a Large Secreted Factor
Slits are secreted extracellular matrix proteins expressed in many cell types and tissues. While flies and worms express only one Slit molecule, mice and humans express three (Slit1, Slit2, and Slit3) that share a high degree of structural conservation (Dickson and Gilestro, 2006). Slits are large multi-domain proteins with a unique tandem of four leucine-rich repeats (LRRs, D1–D4), each connected via disulfide bonds near the N terminus. These LRRs are followed by seven to nine epidermal growth factor (EGF)-like domains, and a laminin G-like domain capped by a C-terminal cysteine-rich module. Structural studies have revealed that the LRR domains each contain a conserved motif that creates a concave shape that might be important for modulating the interaction of Slits with their cognate receptors (Howitt et al., 2004; Morlot et al., 2007a; Morlot et al., 2007b). Further structural studies have shown that Slits undergo post-translational modifications (Figure 1A). Slit proteins are proteolytically cleaved within the fifth EGF region to release an N-terminal fragment that binds Robo receptors and mediates all assayed cell guidance functions of Slit/Robo signaling (Nguyen Ba-Charvet et al., 2001).
Figure 1. Structural Representation of Slits, Robos and Their Interaction.
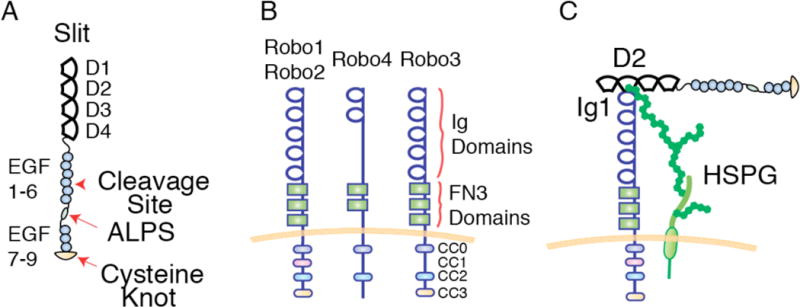
(A) At their N-terminus, vertebrate and invertebrate Slits consist of four Leucine-Rich Repeats (LRRs), termed D1 – D4. These LRRs are followed by seven-nine epidermal growth factor (EGF)-like domains, a laminin G-like domain (ALPS), and a C-terminal cysteine-rich knot. Slits are proteolytically cleaved between two EGF-like domains. (B) Vertebrates have four Robos (Robo1–4), while fly, chick and zebrafish have three (Robo1–3). Robo1, 2 and 3 contain five immunoglobulin (Ig) domains and 3 fibronectin type 3 (FN3) domains. Robo4 contains only two Ig domains and two FN3 domains. Zebrafish Robo4 is unique in that it contains three Ig domains instead of two. In their cytoplasmic tail, Robos contain between two and four conserved proline-rich domains (CC0-CC3). (C) The Slit/Robo signaling pair can be stabilized via heparan sulfate glycosaminoglycans (GAGs) that are present either in the extracellular matrix or attached to membrane-associated proteins such as the heparan sulfate proteoglycan (HSPG) syndecan.
The most commonly studied member of the Slit family of proteins is Slit2. It is known to regulate many aspects of tissue morphogenesis and cell function, including cell migration, proliferation, adhesion and death. While the function of Slit1 remains largely unknown, recent studies are beginning to ascribe functions to Slit3 in embryonic angiogenesis. Studies have shown that Slit3 may function as an angiogenic factor involved in regulating endothelial cell proliferation and motility, in addition to regulating formation of vascular networks (Zhang et al., 2009). Thus, while our current knowledge confirms that the Slit family of axon guidance molecules plays important roles in many aspects of development, it is also clear that there are likely many unknown functions yet to be discovered.
Robo is a Highly Conserved Transmembrane Receptor
The Robo family of receptors is highly conserved, though the number of Robo genes differs between invertebrates and vertebrates. Whereas worms have only one Robo (Sax-3) receptor, flies, chick and zebrafish have three (Robo1–3), and mammals have four (Robo1/Dutt1, Robo2, Robo3/Rig-1, and Robo4/Magic Roundabout) (Figure 1B) (Challa et al., 2001; Hohenester, 2008; Huminiecki et al., 2002; Lee et al., 2001). While Robo1–3 contain a high degree of structural and functional similarity, Robo4 appears to function distinctly from the other Robo family members and, therefore, will be discussed in a separate section.
Robo1–3 are single-pass transmembrane receptors that belong to the immunoglobulin (Ig) superfamily of cell adhesion molecules (CAMs) and are conserved between vertebrates and invertebrates (Hohenester, 2008). The ectodomain of mammalian Robo receptors contains five Ig domains followed by three fibronectin type 3 (FN3) repeats (Figure 1B). The Robo intracellular domain has no inherent catalytic activity, but confers a downstream signal by recruiting various factors to conserved proline-rich domains, referred to as CC0-CC3, and a number of phosphorylatable sites present within the intracellular domain. It was recently demonstrated that the different functions of Robo receptors in neuronal development are due to both gene expression levels and to specific characteristics of the different Robo receptors themselves (Spitzweck et al., 2010). For example, by engineering the expression of each of the three Robo receptors from each Robo gene locus, the authors found that lateral positioning of longitudinal axon pathways depends on gene expression, not on the type of Robo expressed, whereas commissure formation depends on the expression of specific combinations of Robos. This suggests that the receptors have structural differences that confer specific signaling responses, such as prevention of midline crossing by Robo1, and promotion of midline crossing by Robo2 (Spitzweck et al., 2010).
It has long been believed that the different signaling responses induced by each receptor in response to Slit binding are primarily due to variations in the number and combinations of CC motifs in their intracellular domains. However, recent studies reaveal that both intracellular and extracellular domains dictate the resulting signaling response induced by Slit binding. In Drosophila neuronal development, Robo2 has both lateral positioning and midline crossing activities. In a 2010 study, Evans and Bashaw demonstrate that Ig1 and Ig3 are required for lateral positioning, whereas promidline crossing seems to be dictated by Ig2. The authors performed studies in which the cytoplasmic domains of Robo2 and Robo3 were replaced with that of Robo1 and found that stimulation with Slit confers wild-type activities, strongly suggesting that the specificity lies in the ectodomains. However, complete loss of the cytoplasmic domains ablate all receptor activity, indicating that these are also required for signaling. Furthermore, the authors demonstrate that the differences in signaling response are not simply due to differences in Slit2 binding to the ectodomain, but rather due to differences in multimerization and receptor/ligand stoichiometry dictated by the Ig domains (Evans and Bashaw, 2010). These results suggest that, while the cytoplasmic domain is important for downstream signaling, the specificity of the signaling response might be determined by the ectodomains. As Slits and Robos are known to regulate multiple types of signaling responses, including cell motility and cell proliferation, and to play critical roles in the development of many vital organs, including the kidney and breast, it will be interesting to learn if the Ig domains of Robo ectodomains play an equally important role in conferring Slit signaling in diverse systems.
Cleavage of Robo Reveals Additional Regulatory Complexities
Current efforts examining post-translational modifications of Robo suggest that regulation of this receptor, and consequently the signaling pathways it mediates, is more complex than initially proposed. This has been demonstrated using the Alexander hepatoma cell line, PLC/PRF/5, in which the intracellular domain of Robo is successively cleaved by metalloproteases and γ-secretases, yielding two distinct intracellular Robo1 fragments (Robo1-CTF1 and Robo1-CTF2) (Seki et al., 2010). The identification of several nuclear localization signals (NLSs) within these intracellular fragments of Robo1 suggests a potential transcriptional role for the receptor, at least in cancer cells. Biochemical fractionation of PLC/PRF/5 cells treated with the proteasome inhibitor MG-132 show Robo1-CTF2 exclusively located in the nucleus, whereas Robo1-CTF1 is found in each of the membrane, cytosolic and nuclear fractions (Seki et al., 2010). This suggests that the successive cleavage of the Robo1-CTF is critical for proper localization within the cell and may play a regulatory role. However, removal of all three potential NLSs does not abolish nuclear localization, suggesting that perhaps nuclear localization of Robo1-CTF relies on other currently unidentified effector molecules (Seki et al., 2010). It is clear that further studies of NLS-containing Robo1-CTF binding partners are needed to elucidate the full mechanism of transcriptional regulation by Robo1-CTFs.
In addition to intracellular cleavage, a number of axon guidance receptors, including Robo, undergo extracellular cleavages, generating protein products that regulate a number of cellular functions, such as migration. Recently, studies in Drosophila aimed at elucidating the exact mechanism of Robo activation following Slit-binding reveal a potential role for the metalloprotease-disintegrin Kuzbanian (ADAM 10 in mammals) in generating a free ectodomain by extracellular cleavage. Although Kuz/ADAM10 is expressed in both neurons and midline glia, only neuronal expression is required for Slit/Robo repulsion, suggesting that the protease acts on Robo-expressing neurons and not the glia (Coleman et al., 2010). Furthermore, it appears that extracellular cleavage of Robo is required for receptor activation following Slit stimulation, as expression of an uncleavable form of Robo is unable to rescue a Robo mutant phenotype. It was also found that cleavage of Robo by Kuz/ADAM10 is necessary for the recruitment of Son of sevenless (Sos) and other factors required for Slit/Robo-mediated repulsion at the midline (Coleman et al., 2010). Taken together, these data suggest that cleavage is an important mechanism that regulates the activation of Robo and its signal transduction, and it is likely that subsequent studies will reveal how cleavage regulates the many different functions of Slit/Robo signaling.
Structural Studies Have Defined the Interaction Between Slit and Robo
The interactions between Slit and Robo molecules are evolutionarily conserved, as evidenced by studies that show that human Slit2 is able to bind Drosophila robo1 with similar affinity as its mammalian receptor, and vice versa, that Drosophila slit successfully binds rat Robo1 and Robo2 (Brose et al., 1999). Biochemical studies also show that the interaction between this receptor/ligand pair involves the highly conserved second LRR domain (D2) of Slit and the Ig1 domain of Robo, while Ig2–Ig5 and all FN3 domains of Robo1 appear to be dispensable for binding (Figure 1C) (Chen et al., 2001; Fukuhara et al., 2008; Howitt et al., 2004; Liu et al., 2004; Morlot et al., 2007b). Thus, the binding between Slits and Robos is highly conserved and structurally well defined.
Recent insight into the structural requirements for binding between Slits and Robo1 has revealed that the complex can be stabilized by heparan sulfate glycosaminoglycans (GAGs), which are required for functional Slit/Robo signaling in both Drosophila and vertebrate neurological development (Figure 1C) (Fukuhara et al., 2008; Hu, 2001; Hussain et al., 2006; Inatani et al., 2003; Ogata-Iwao et al., 2011; Plump et al., 2002; Schulz et al., 2011; Smart et al., 2011). There appears to be dual functions for these GAGs: first, they bind to Slit in the extracellular matrix and stabilize their interaction with Robo, and second, they act on target cells to mediate Slit/Robo signaling by serving as co-receptors.
There are numerous studies demonstrating the importance of GAGs in facilitating the functional interaction between Slits and Robos. Structural studies by Hussain and colleagues show that heparin, a highly sulfated variant of heparan sulfate, binds to Slit and forms a ternary complex with Robo, resulting in a 10-fold increase in the affinity between Slit and Robo (Hussain et al., 2006). Mutational studies demonstrate a key role for the second LRR of Slit (termed D2) in binding heparin via a conserved basic patch, and binding Robo via the adjacent concave face. Further crystallographic studies reveal a contiguous HS/heparin binding surface, extending across the Slit-Robo interface and consistent with at least five HS disaccharide units, as required to support Slit/Robo signaling (Fukuhara et al., 2008). These biochemical analyses are further supported by functional studies in this and others papers, which demonstrate that enzymatic removal of heparan sulfates from neurons using heparanases results in a loss of responsiveness to Slit (Hu, 2001; Hussain et al., 2006; Piper et al., 2006). In a different strategy, mutation of exostosin, an enzyme required for heparan sulfate synthesis, leads to patterning defects at the mouse optic chiasm that phenocopy those observed in the Slit1−/−;Slit2−/− knock-out (Inatani et al., 2003; Plump et al., 2002). A similar study performed in zebrafish found that this loss of heparan sulfate synthesis phenocopies or even enhances the guidance defects observed in Robo2/astray mutants (Kastenhuber et al., 2009; Lee et al., 2004). Taken together, these studies show that heparan sulfates mediate the formation of stable Slit/Robo signaling complexes and are critical for their signaling function. However, it is unclear whether or not these GAGs constitute membrane bound proteoglycans. Genetic studies in Drosophila suggest that this may be the case, as the heparan sulfate proteoglycan, glypican, which is associated with the cell surface via a glycosylphosphatidylinositol (GPI) linkage, interacts with slit and regulates its distribution (Liang et al., 1999; Ronca et al., 2001; Smart et al., 2011; Zhang et al., 2004). Thus, heparan sulfate GAGs, either cell associated or present as free sugars in the extracellular matrix, concentrate and localize Slits, shaping the signaling environment by regulating their concentration and accessibility.
On the target cells, the heparan sulfate proteoglycan syndecan (Sdc) plays a key role as a co-receptor for Robo (Johnson et al., 2004; Rhiner et al., 2005; Steigemann et al., 2004). In the Drosophila embryo, sdc is co-expressed with robo on axons and is absent in slit-secreting midline cells. Mutations in mammalian Sdc enhance the muscle and axonal patterning phenotypes observed in loss-of-function Slit and Robo animals (Johnson et al., 2004; Steigemann et al., 2004). Moreover, cell type-specific rescue experiments in Sdc mutants reveal that axon guidance defects of the Sdc mutant are entirely rescued by Sdc expression in neurons, while there is no rescuing activity in response to Sdc expression in midline cells (Johnson et al., 2004; Steigemann et al., 2004). These findings indicate that Sdc activity does not participate in the production and/or secretion of Slit, but rather is required for the reception and/or the transmission of Slit signals in Robo-expressing target cells. Further studies in neural development in C. elegans corroborate this important role for syndecan in regulating Slit/Robo mediated axon guidance. Mutational analyses demonstrate that only the extracellular domain of Sdc is required for Slit/Robo signaling (Chanana et al., 2009; Schulz et al., 2011), and that the chondroitin sulfate modification of Sdc is necessary for its co-receptor function on target cells (Chanana et al., 2009).
Together, functional and structural evidence supports a model in which heparan sulfate proteoglycans enhance the relatively low-affinity interaction between Slits and Robos by acting as secondary receptors. Furthermore, studies in Drosophila suggest that there are different cellular requirements for proteoglycans, with syndecans acting on target cells, and glypicans acting on the slit-expressing cells. Glypicans are thought to sequester Slit and present it to the syndecan/Robo1 pair, thereby regulating the formation of the ternary signaling complex. Together, these collective interactions help to localize and fine-tune Slit/Robo signaling. While current data suggest that the interaction between Slit, Robo and GAGs represents a significant regulatory relationship, whether these heparan sulfate co-receptors are required for Slit/Robo signaling in all cell and tissues types in higher organisms, and whether they play a role in disease processes will require more in-depth studies in mammalian models.
Slit/Robo: Roles in Epithelial Tumorigenesis
The role of axon guidance molecules in cancer progression has been studied for over a decade, yet their exact function remains elusive. By their nature, axon guidance molecules are bifunctional, acting as both attractant and repellents for migrating axons and cells. As such, Slits also have this dual role; in one example, this bifunctionality is displayed in a single trajectory of mesodermal cells in the Drosophila embryo. These cells move away from the ventral midline, repelled by slit, and then migrate toward target muscles, attracted by slit (Kramer et al., 2001). Consistent with this dual role as both positive and negative cues, Slits are also capable of acting as both “friend and foe” in the progression of tumor cells; Slits have been shown to both promote and prevent tumor metastasis by suppressing or enhancing cellular attachments and migration depending on the cellular context (Tseng et al., 2010; Zhou et al., 2011). This duality of Slit function is also observed in the regulation of tumor cell proliferation and survival where they promote proliferation and angiogenesis in some contexts and prevent these same processes in others (Table 1). Thus, current evidence renders it impossible to label Slits and Robos as either tumor suppressors or oncogenes. Nevertheless, it is clear that acting in either role they play important functions during tumor progression. This makes them attractive targets for cancer therapeutics and potential candidates for diagnostic purposes.
Table 1.
The function of Slit/Robo signaling in tumor and non-tumor cell proliferation migration and angiogenesis.
| Slit/Robo members | Action | Tissue/cell type | References |
|---|---|---|---|
| Promigration/metastasis/chemotaxis | |||
| Slit2/Robol | Induces malignant transformation promotes metastasis by regulating degradation of cadherins | HEK293 cells, colorectal epithelial carcinoma cells; colorectal carcinoma xenograft model | Zhou et al. (2011) |
| Slit/Robo | Mediates migration by downregulating N-cad-herin, decreasing adhesion and increasing translocation of β-catenin to the nucleus | Axon growth cone migration, mouse fibroblast L-cells | Rhee et al. (2002, 2007) |
| Robo4 | Mediates attractant signals in endodielial cells via Rho GTPases | Embryonic zebrafish vasculature | Kaur et al. (2006) |
| Slit2/Robo4:Robol heterodimer | Mediates the chemotactic response of endothelial cells | Zebrafish vasculature | Kaur et al. (2008) |
| Slit2/Robol | Mediates the migration and vascular tube formation. Slit2 also acts as a lymphangiogenic factor | Mouse lymphatic endothelial cells | Yang et al. (2010) |
| Slit2/Robol:Robo4 heterodimer | Promotes cell migration and angiogenesis by inducing formation of filopodia | Human umbilical vein endothelial cells (HUVECs) | Sheldon et al. (2009) |
| Slit/Robo | Enhances cell migration by decreasing c-cad-herin function at the membrane | Drosophila heart tube formation | Santiago-Martinez et al. (2008) |
| Slit/Robo1 | Promotes migration by positively regulating Rho GTPases | Src-transformed epithelial cells | Khusial et al. (2010) |
| Slit2/Robol | Promotes eosinophil Chemotaxis, possibly through activation of Cdc42 | Ovalbumin airway inflammation | Yang et al. (2010), and Ye et al. (2010) |
| Slit2/Robol | Promotes directed migration and metastasis of cancer cells | Breast cancer cell lines | Schmid et al. (2007) |
| Antimigration/metasfasis/chemotaxis | |||
| Slit2/Robo3 | Prevents cell migration by stabilizing P-cadherin at the membrane | Oral mucosa, oral squamous cell carcinoma (OSCC) | Bauer et al. (2011) |
| Slit/Robo | Prevents metastasis by enhancing cell-cell adhesion via N-cadherin | Chick cranial trigeminal gangliogenesis | Shiau and Bronner-Fraser (2009) |
| Sit2/Robol | Prevents epithelial cell migration by enhancing cell-cell adhesion via PI3K and β-catenin | Breast cancer cells | Prasad et al. (2008) |
| Slit2/Robol | Prevents cell migration and promotes cell-cell adhesion by regulating E-cadhcrin expression | Human lung cancer cell lines and lung tumor samples | Tseng et al. (2010) |
| Slit2/Robo | Prevents VSMC migration by modulating cytoskeletal molecules | Vascular smooth muscle cells (VSMCs) | Liu et al. (2006) |
| Slit2/Robo | Inhibits migration of RASM cells by controlling WASP and Arp2/3 expressions | Rat airway smooth muscle (RASM) cells | Ning et al. (2011) |
| Slit2/Robo | Blocks angiocrine-induced tumor growth and migration | Endothelial cell lines, Invasive human ductal carcinoma samples | Brantley-Sieders et al. (2011) |
| Slit2/Robol | Prevents cell migration/metastasis by attenuating Cdc42 activity | Epithelial cell lines, medulloblastoma, glioma cell lines and tumor samples, mouse glioma xenograft models | Stella et al. (2009), Werbowetski-Ogilvie et al. (2006), and Yiin et al. (2009) |
| Slit2/Robo | Inhibits migration of cells toward chemoattractant stimulus by attenuating Cdc42 and Rac2 | Leukocytes, vascular smooth muscle cells (VSMCs), mouse model of chemical irritant peritonitis | Liu et al. (2006), Tole et al. (2009), and Wu et al. (2001) |
| Slit2/Robol | Regulates eosinophil/neutrophil Chemotaxis by modulating srGAP expression | Eosinophils, neutrophils in endotoxin-induced lung inflammation model | Ye et al. (2010) |
| Slit2/Robol | Prevents CXCR4/CXCL12-mediated inhibiting downstream signaling | Breast cancer cell lines | Prasad et al. (2004) |
| Slit2/Robol | Prevents metastasis of cancer cells by recruiting USP33 and redistributing Robo1 to the membrane | Breast cancer cells | Yuasa-Kawada et al. (2009) |
| Slit/Robo4 | Inhibits endothelial cell migration and proliferation | Rat endothelial cells | Suchting et al. (2005) |
| Slit/Robo4 | Prevents angiogenesis by inhibiting signaling downstream of VEGF/VEGFR | Endothelial cells of the mammary gland stroma, blood vessel endothelial cells of the corneal stroma | Marlow et al. (2010) and Mulik et al. (2011) |
| Slit2 | Decreases LPS-induced vascular permeability by increasing V/E-cadherin levels at the membrane | Blood vessel endothelium of the lung | London et al. (2010) |
| Proangiogenic | |||
| Slit2/Robol | Promotes tube formation and endothelial cell migration | Human malignant melanoma A375 cells | Wang et al. (2003) |
| Slit2/Robol | Promotes tumor angiogenesis and growth in vivo | Oral carcinogenesis | Wang et al. (2008) |
| Slit2/Robol | Promotes lymphangiogenesis and lymphatic metastasis | Lymphatic endodielial cells | Yang et al. (2010) |
| Slit2/Robo | Promotes angiogenesis in culture and in vivo | Mouse pulmonary microvascular endothelial cells | Duna way et al. (2011) |
Slit and Robo Expressions are Altered in Cancer
The progressive transformation of normal cells into malignant progeny involves the accumulation of genetic changes, such as the loss or silencing of tumor suppressor genes and the induction of oncogenes. Studies show that Slit and Robo expression is altered in a long list of cancers (Table 2 and Table 3). In examining the literature, it is interesting to note that there are examples of both up and down-regulation of these genes, suggesting that the Slit/Robo pathway can function in both promoting and suppressing tumor cell survival, proliferation and migration. Currently it is unclear whether these genes are differentially regulated based on tumor type or stage, but mounting evidence suggests that changes in the expression of these genes play important roles in regulating tumor progression.
Table 2.
A list of cancers in which expression of Slits and Robos are decreased, and the mode of regulation, if known.
| Elevated expression level
| ||
|---|---|---|
| Gene | Cancer type | References |
| Slits | Prostate cancer, nitrofen-hypoplastic lung cancer, and lobular breast carcinoma | Christgen et al. (2009), Doi et al. (2009), Latil et al. (2003), and Ma et al. (2004) |
| Slit2 | Oral cheek mucosa with oral squamous cell carcinoma | Wang et al. (2008) |
| Robo1 | Hepatocellular cancer, colorectal cancer, nonsmall cell lung cancer, Glioma cancer, monkey choroidal retinal endothelial cells, retinopathy of prematurity (ROP), and neovascularized cornea | Avci et al. (2008), Gorn et al. (2005), Grone et al. (2006), Han and Zhang (2010), Huang et al. (2009), Ito et al. (2006), Mertsch et al. (2008), and Xu et al. (2010) |
| Robo2 | Hepatocellular cancer | Avci et al. (2008) |
| Robo4 | Tumor endothelial cells, neovascularized cornea, monkey choroidal retinal endothelial cells, and colorectal cancer | Avci et al. (2008), Grone et al. (2006), Han and Zhang. (2010), Mura et al. (2011), and Seth et al. (2005) |
| Robo4 | HSV-infected endothelial cells in the corneal stroma | Mulik et al. (2011) |
Table 3.
A summary of cancers and diseases in which expression of Slits and Robos are elevated.
| Decreased expression level
| |||
|---|---|---|---|
| Gene | Mode of silencing | Cancer type | References |
| Slit2 | LOH, allelic deletion | 63% of breast carcinoma, 35% of cervical carcinoma, and >60% of small lung carcinoma and mesothelioma | Shivapurkar et al. (1999a, 1999b) and Singh et al. (2007) |
| Slit2 | Promoter hypermethylation | Breast carcinoma, nonsmall cell lung cancer, ovarian carcinoma, gliomas, hepatocellular carcinoma, colorectal carcinoma, and lymphocytic leukemia primary tumors | Dallol et al. (2005, 2003a, 2003b), Dunwell et al. (2009), Jin et al. (2009), Qiu et al. (2011), Sharma et al. (2007) |
| Slit2 | Unknown | Corneal neovascularization | Han and Zhang (2010) and Wang et al. (2008) |
| Slit, 3 | Promoter hypermethylation | 41% of breast, 33% of colorectal, and 29% of glioma tumor cell lines and primary tumors | Dallol et al. (2005) and Dickinson et al. (2004) |
| Slit3 | Unknown | Hepatocellular carcinoma | Avci et al. (2008) |
| Slit2 | Promoter hypermethylation catalyzed by polycomb group member EZH2 | Human prostate cancer samples | Yu et al. (2010) |
| Robo1 | Gene deletion | Small-cell lung cancer cell line (U2020) | Xian et al. (2001) |
| Robo1 | Promoter hypermethylation | 19% of primary invasive breast cancer, 18% of clear cell renal cell cancer, and 4% primary nonsmall cell lung cancer | Dallol et al. (2002b) |
| Robol, 2 | Promoter hypermethylation | Early dysplastic lesions of head and neck cancer | Ghosh et al. (2009) |
| Robo3 | Promoter hypermethylation | Cervical cancer | Narayan et al. (2006) |
| Robo4 | Unknown | Human breast tumor samples | Richardson et al. (2006) |
| Robo4 | Unknown | Hepatocellular carcinoma | Avci et al. (2008) |
Slit Expression is Altered in Epithelial Tumor Progression
The most frequently observed alteration of Slit expression is downregulation. This is evidenced by allelotyping studies of 44 breast carcinoma samples that show loss of heterozygosity (LOH) at several regions on chromosome 4, one of which has been identified as the Slit2 gene locus (4q25–26) (Shivapurkar et al., 1999a; Shivapurkar et al., 1999b; Singh et al., 2007). These studies report allelic deletion in 63% of breast carcinomas, 35% of cervical carcinoma and in >60% of small cell lung carcinoma and mesothelioma (Shivapurkar et al., 1999a; Shivapurkar et al., 1999b; Singh et al., 2007). Thus, it seems that a common method for alteration of Slit/Robo signaling in cancer is via Slit gene silencing. In addition to gene loss by deletion, several other mechanisms of gene silencing occur at the Slit gene locus. Of these, the most commonly encountered mechanism is hypermethylation of the promoter region. Numerous studies have shown that regions frequently hypermethylated in cancers contain the genes for Slit1, Slit2, Slit3, Robo1 and Robo3 (Dallol et al., 2005). For example, Slit2 is silenced through hypermethylation in the majority of samples from numerous tumor types including: breast, non-small cell lung, ovarian, gliomas, hepatocellular, colorectal cancers as well as lymphocytic leukemias (Dallol et al., 2003a; Dallol et al., 2003b; Dunwell et al., 2009; Jin et al., 2009; Qiu et al., 2011; Sharma et al., 2007). In further support of a role for Slits in suppressing tumor growth and, consequently, being silenced during tumor progression, re-expression of Slit2 greatly inhibits the proliferation of transformed cell lines derived from many of these tumor types (Dallol et al., 2002a; Dallol et al., 2003b; Jin et al., 2009; Qiu et al., 2011). Although Slit2 is the most frequently studied of the Slit proteins expressed in mammals, similar expression studies reveal silencing of Slit3 via hypermethylation in breast (41%), colorectal (33%) and glioma (29%) tumor cell lines, with similar frequencies of Slit3 and Slit1 promoter hypermethylation reported in these types of primary tumors (Dickinson et al., 2004).
One additional consequence of epigenetic silencing of Slit2 and Slit3 is downregulation of microRNA (miR)-218-1 and miR-218-2, which are located within intron 15 of human Slit2 and intron 14 of human Slit3, respectively (Angeloni et al., 2006; Tie et al., 2010). This miR negatively regulates Robo1 expression in gastric, head and nasopharyngeal cancers (Alajez et al., 2011; Tie et al., 2010), but its downregulation due to Slit methylation in many tumor types provides one explanation for the observation that Robo1 appears to be infrequently silenced in the majority of tumor samples (Grone et al., 2006; Ito et al., 2006; Xu et al., 2010). Interestingly, it is possible that loss of this negative feedback loop contributes to tumor progression because the expression of Robo1 in tumor cells could allow them to migrate in response to Slits that are provided by nontumor cells in the surrounding environment (Alajez et al., 2011), or by tumor cells that still secrete Slit because they have only partially silenced its expression (Tie et al., 2010). Thus, hypermethylation of the Slit gene loci in solid tumors may contribute to tumor progression by switching Slit/Robo1 signaling from autocrine to paracrine, facilitating the metastasis of tumor cells that are responding to this deregulated pathway. Given the frequency of Slit hypermethylation in human tumors and its effect on miR-218 expression, this family of genes represents attractive candidates for therapeutic strategies that reverse epigenetic silencing or re-establish miR-218 expression.
Recently, a second mechanism of Slit gene silencing was observed in human cancer samples. A genome-wide location analysis of human prostate cancer samples identified Slit2 as a target of epigenetic repression via the polycomb group (PcG) member EZH2 (Yu et al., 2010). Polycomb group (PcG) proteins are transcriptional repressors that function through multimeric chromatin-associated polycomb repressive complexes to epigenetically silence gene expression by catalyzing the methylation of specific histone residues. In prostate cancer samples, low Slit2 expression correlates with not only high EZH2 expression level, but also with the aggressiveness of the cancer and the degree of metastasis. Furthermore, treatment with either methylation inhibitors or EZH2-suppressing compounds decreases metastasis and increases Slit2 expression (Yu et al., 2010). Taken together, these studies introduce a novel mode of Slit silencing that had previously not been recognized in cancer samples.
In contrast to downregulation of Slits, which is well documented in the literature, there are relatively few papers that identify the upregulation of Slits in cancer as occurs, for example, in human ductal carcinoma samples, prostate and nitrofen-hypoplastic lung cancers (Brantley-Sieders et al., 2011; Doi et al., 2009; Latil et al., 2003). Data mining studies have also revealed their upregulation in lobular breast cancers, a type of breast carcinoma that has been ascribed only a few unique molecular characteristics (Christgen et al., 2009; Ma et al., 2004). These data suggest that at least some types of tumors are associated with Slit overexpression, but how it contributes to tumor development in these circumstances is currently unknown.
Robo Expression is Altered in Epithelial Tumor Progression
Robo1 was discovered in Drosophila as a gene required for proper midline crossing of commissural axons during development (Kidd et al., 1999). It was also found to be deleted in the small-cell lung cancer cell line U2020, hence the name Deleted in U twenty twenty, or Dutt1 (Xian et al., 2001). Robo1 and Dutt1 genes are derived from alternative promoters of the same gene and appear to have differential spatial and temporal patterns of transcriptional activity, with the Dutt1 form expressed ubiquitously, and the Robo1 form restricted primarily to embryogenesis (Clark et al., 2002). In addition to being silenced by deletion, Dutt1 is also hypermethylated in subsets of primary tumor samples, such as primary invasive breast cancer (19%), primary clear cell renal cell cancer (18%) and in primary non-small cell lung cancer (4%). Of those tumors, 80% of breast and 75% of primary clear cell renal cell carcinomas also contain allelic losses in the genomic region containing Dutt1, an observation supporting a role for Dutt1 as a tumor suppressor that obeys Knudson’s two hit hypothesis (Dallol et al., 2002b). More recently, hypermethylation at the Robo1 and Robo2 gene loci was reported in early dysplastic lesions of head and neck (Ghosh et al., 2009), as well as in cervical cancer (Narayan et al., 2006). While these data point to a tumor suppressor function for Robos, in fact the percentage of tumors displaying reduced or silenced Robo expression is much less than that seen for Slit genes. Indeed, the opposite is true, with Robo1 expression elevated in numerous cancers, including human hepatocellular carcinoma, colorectal cancer, non-small cell lung cancer, and glioma samples (Gorn et al., 2005; Grone et al., 2006; Ito et al., 2006; Mertsch et al., 2008; Xu et al., 2010). In one of these tumor samples, coordinate regulation of Slit and Robo was observed, with decreased Slit concomitant with increased Robo1 expression, as predicted by miR-218 regulation of the Slit/Robo signaling axis (Xu et al., 2010). Thus, this finding of a negative regulatory loop that upregulates Robo when Slit is silenced generates a layer of complexity to the study of Slit/Robo1 in cancer cells. While it was previously simple to label the pathway as tumor suppressive when Slits were found to be silenced, this new finding introduces the possibility that, under these circumstances, Slit/Robo signaling can function oncogenically due to upregulation of Robo in the tumor setting.
In sum, since the first hint that Slit and Robo could play a role in tumor biology, there have been numerous studies documenting changes in their level of expression in tumor samples. Recent insights demonstrate the complexity of this regulation, as evidenced by the negative feedback loop for Robo expression under the control of non-coding RNAs encoded intronically in Slit2 and Slit3. This regulatory relationship suggests that the level of both ligand and receptor must be assessed when drawing conclusions about the overall effect of Slit/Robo signaling on tumor progression. Moreover, because the Slit/Robo pathway regulates many common signaling pathways that are often deregulated in cancers, such as those mediated by the Rho family of small GTPases and β-catenin, it is becoming clear that changes in Slit and Robo expression have effects that extend beyond the roles originally identified for these proteins as instructive cues for cell migration. In fact, a growing body of literature shows that Slit/Robo signaling affects other aspects of tumor cell behavior, including their survival and growth.
Slits and Robos Mediate Tumor Cell Survival and Proliferation
Once the process of transformation has been initiated by driver mutations, the expansion and progression of premalignant cells to metastatic carcinomas depends on a multi-step process involving the evasion of pro-apoptotic signals and the reception of pro-survival and pro-proliferative signals. Thus, the early stages of cancer development involve the response of nascent tumor cells to cues such as Slits in their surrounding environment. While there are only some suggestions in the literature that Slits may regulate cell survival, our understanding of its role in regulating cell proliferation is growing. Slits appear to control cell proliferation through β-catenin, a signaling target that has distinct functions at the plasma membrane, where it mediates cell-cell contact in association with the homotypic cell adhesion protein, E-cadherin, and in the nucleus, where it regulates cell proliferation in association with Lef/Tcf transcription factors. Consequently, through this one downstream target, Slits influence two aspects of tumor cell transformation: proliferation and adhesion.
Slits and Robos: Inhibitors of Tumor Cell Death
A critical step of tumor cell transformation is achieving immortality. Cells employ several mechanisms to execute apoptosis in order to prevent the survival of rogue cells. Apoptosis can be triggered intrinsically, for example via p53, leading to the release of cytochrome c from the mitochondria and the activation of caspases via the apoptosome (Fulda and Debatin, 2006). Apoptosis can also be initiated extrinsically by signaling through Death Receptors (DRs), also culminating in the activation of caspases (Fulda and Debatin, 2006). An alternative extrinsic cell death pathway is mediated by so-called “dependence receptors” that require or “depend on” their ligand to prevent their own constitutive pro-apoptotic signaling, which occurs when their ligand falls below a critical concentration. While Robos have not been identified as dependence receptors, the receptors for the Netrin family of axon guidance cues, DCCs and UNC5s, do fit into this category of receptors. Consequently, in addition to mediating axon guidance through these receptors, Netrin also acts as a survival factor for both axons during normal development and for cancer cells during tumor progression (Delloye-Bourgeois et al., 2009; Fitamant et al., 2008; Furne et al., 2008). Intriguingly, Slits have been found to regulate Netrins by binding and sequestering them (Stein and Tessier-Lavigne, 2001). Consequently, Slits may act as pro-apoptotic factors by allowing the concentration of Netrin to fall below threshold levels for survival. Although there is no direct evidence to support a link between the regulation of Netrin by Slit, and the loss of apoptosis in cancer, this regulatory loop suggests a possible mechanism through which Slit exerts a tumor suppressive function by promoting the pro-apoptotic signaling of dependence receptors. The silencing of Slits, which occurs in many types of cancer, could, therefore, contribute to the immortality of tumor cells by disabling one of the pathways that culls rogue cells that have tumor forming potential.
Slits and Robos: Regulators of Tumor Cell Proliferation
Following survival of a few transformed cells, cancer progression requires proliferation of these cells to generate tumor mass. Investigation into the role of Slit/Robo signaling in regulating cell proliferation has revealed both positive and negative effects, as predicted by its dual function as both attractant and repellent guidance cue. In support of a role for Slit in suppressing cell proliferation, numerous studies suggest that signaling through Robo1 regulates the subcellular localization of β-catenin, inhibiting its transcriptional function in the nucleus by promoting its localization at the membrane. In a recent study, we have shown that during murine breast (mammary gland) development, Slit/Robo signaling restricts the proliferation of the outer layer of basal cells by increasing the cytoplasmic and membrane pools of β-catenin at the expense of its nuclear pool (Macias et al., 2011). This loss of growth control during early postnatal mammary gland development generates an overabundance of myoepithelial cells that produce an excess of growth factors, leading to an overall increase in cell proliferation and excessive branching morphogenesis. Eventually, these surplus myoepithelial cells invade the luminal population and disrupt cell adhesion (Strickland et al., 2006), and, along with other changes that occur, such as upregulation of CXCR4 and SDF1, spur the development of hyperplastic lesions with basal characteristics (Marlow et al., 2008).
The imbalance in growth control during early mammary gland development observed in Robo1−/− tissue provides a gratifying developmental correlate for the role of Slits in suppressing growth in models of breast and non-small cell lung cancer (Prasad et al., 2008; Tseng et al., 2010). In breast cancer cell lines, overexpression of Slits inhibits the transcriptional activity of β-catenin by activating glycogen synthase kinase (GSK)-3β through the phosphoinositol-3-kinase (PI3K)/Akt signaling pathway. These overexpressing cells display enhanced intercellular adhesion and greater co-localization of β-catenin with E-cadherin (Figure 2A). Further, studies performed in xenograft models of breast cancer show that tumors generated from Slit-overexpressing cells are significantly smaller compared to control tumors (Marlow et al., 2008; Prasad et al., 2008). A comparable but converse experiment was performed in a cell line derived from non-small cell lung cancer in which knock-down of Slit increases the metastatic potential of the cells by inhibiting GSK-3β activity, again via the PI3K/Akt pathway (Tseng et al., 2010). This, in turn, increases the levels of nuclear β-catenin and increases the expression of Snail, a crucial regulator of epithelial-mesenchymal transitions, resulting in decreased cadherin expression, reduced cell adhesion and increased cell motility (Figure 2A) (Tseng et al., 2010). Taken together, these studies show that, at least in breast and lung, Slits act as tumor suppressors promoting the adhesive role of β-catenin at the membrane at the expense of its proliferative role in the nucleus.
Figure 2. Slit/Robo Signaling Regulates Cell Proliferation and Cell-Cell Contacts by Controlling the Localization of β-catenin In the Cell.
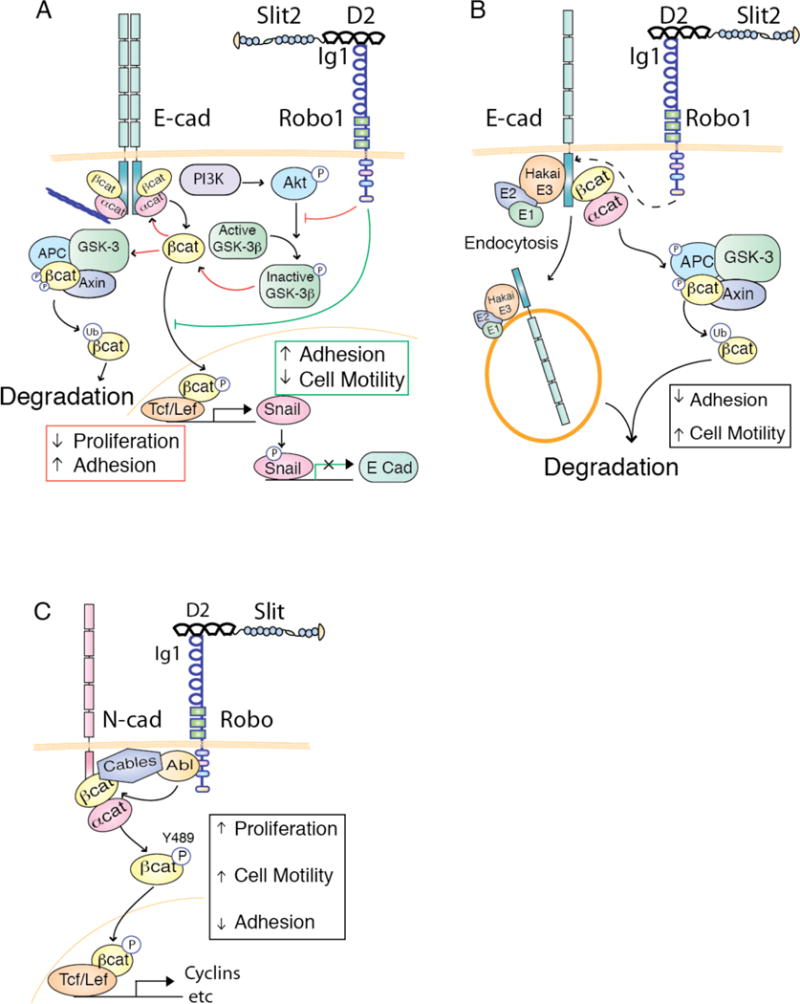
(A) As illustrated by red arrows, binding of Slit to Robo inhibits phosphatidylinositol kinase (PI3K)-induced Akt activity. Glycogen synthase kinase-3beta (GSK3β) is consequently left in its non-phosphorylated, active form, and targets β-catenin for phosphorylation, excluding it from the nucleus and thus preventing its transcriptional activity. Cytoplasmic β-catenin either becomes ubiquitinated through the GSK3β- adenomatous polyposis coli (APC)-Axin complex or transferred to the membrane where it interacts with E-cadherin, stabilizing cell-cell contacts and preventing cell migration. As illustrated by green lines, Slit2/Robo1 signaling blocks Snail expression by preventing the translocation of β-catenin to the nucleus, thus relieving the repression of E-cadherin expression and enhancing cell-cell contacts.
Slit/Robo signaling can also function to decrease cell-cell contacts and increase proliferation (B, C). (B) Slit/Robo signaling recruits the Abelson tyrosine kinase (Abl), which binds to the adaptor protein Cables. Cables links the Robo/Abl complex to the N-Cadherin/β-catenin complex, thus enabling Abl to phosphorylate β-catenin on Y489, causing its translocation to the nucleus where it activates transcription of cell proliferation genes. (C) In another context, Slit/Robo signaling drives cell migration by recruiting the ubiquitin ligase Hakai to E-cadherin and of β-catenin. This results proteasomal degradation of β-catenin and Hakai-mediated lysosomal degradation of E-cadherin, causing decreased cell-cell contacts and enhanced cell migration.
Reminiscent of the context-dependent roles that Slit plays as both attractant and repellent in cell migration (Kramer et al., 2001), the opposite role for Slit as inducer of proliferation has also been documented. In cell lines derived from colorectal carcinoma, Slit/Robo1 signaling enhances tumor growth and metastasis by regulating cadherin degradation and, thereby, increasing cell proliferation and migration (Zhou et al., 2011). Overexpression of either Slit2 or Robo1 or recombinant Slit2 treatment of Robo1-expressing colorectal epithelial carcinoma cells results in recruitment of the ubiquitin ligase, Hakai, to E-cadherin and its subsequent ubiquitination and lysosomal degradation (Figure 2B). Downregulation of E-cadherin in these cells is accompanied by an epithelial-mesenchymal transition (EMT), increased proliferation and increased migration, and in a xenograft model, this corresponds to increased tumor growth and metastasis. Clinical data corroborated these observations, showing an increase in Slit and Robo1 expression in metastatic, compared to non-metastatic, human colorectal carcinoma samples. This increase inversely correlates with the overall survival of patients, supporting the idea that in some tumor contexts, Slit/Robo signaling can function oncogenically to promote cell growth and migration (Zhou et al., 2011).
Studies on embryonic chick neural retinal cells have identified a second mechanism for down-regulating cadherin through Slit/Robo. In this setting, Slit/Robo signaling induces the recruitment of Cables to the Abelson tyrosine kinase (Abl), which is bound by Robo (Rhee et al., 2007). This causes Cables, in turn, to bind to β-catenin and form a complex with N-cadherin at the plasma membrane, which brings Abl into position to phosphorylate β-catenin on Y489. This triggers the dissociation of β-catenin from N-cadherin, compromising cell-cell adhesion and allowing translocation of Y489-phosphorylated β-catenin to the nucleus where it activates Tcf/Lef-mediated transcription (Figure 2C) (Rhee et al., 2007). Although a correlation between this Slit/Robo-mediated increase in nuclear β-catenin activity and enhanced cell proliferation is not reported in this study, an increase in proliferation in a different cell type from the retina, retinal pigment epithelial cells, has been observed in response to recombinant Slit2 treatment (Zhou et al., 2011). Thus, several signaling pathways have been identified that support an oncogenic role for Slit in reducing cell adhesion and enhancing cell proliferation.
Taken together, these studies suggest that Slit/Robo1 signaling regulates cellular proliferation by targeting both cadherins and β-catenin in order to regulate the transcriptional activity of β-catenin. In events that suppress tumor growth, Slit/Robo1 directs the subcellular localization of β-catenin through the PI3K/Akt pathway, an effect that has been documented in both non-small cell lung and breast cancer models, as well as during normal breast development (Macias et al., 2011; Prasad et al., 2008; Tseng et al., 2010). In contrast, two different mechanisms have been identified that achieve oncogenic outcomes downstream of Slit/Robo signaling. In both examples, the cadherin/β-catenin complex is disrupted, releasing β-catenin. However, in one mechanism this occurs through Slit/Robo-induced lysosomal degradation of cadherin and in the other, through targeted phosphorylation of β-catenin by an Abl/Robo complex. In conclusion, additional studies are required to determine the extent to which these pro-proliferative, pro-migratory mechanisms regulate Slit signaling in normal and disease settings.
Slits and Robos Mediate Tumor Cell Motility and Metastasis
Tumor cell metastasis requires multiple steps including: weakening associations between tumor and neighboring cells or between tumor cells and the environment, rearrangement of the actin cytoskeleton to drive actin protrusions and other structures necessary for cell motility, and sensitization of the cell to attractant signaling gradients. These changes occur while the cell is simultaneously desensitized to repellent signaling molecules in the environment, thus allowing cell migration. Slits and Robos have been implicated in each of these steps, and not surprisingly, they have been found to act as both oncogenes and tumor suppressors, enhancing and inhibiting tumor cell invasion, depending on the cellular context.
Slits and Robos: Regulators of Cell-Cell Adhesions
Cadherins are expressed in all epithelial cells and play a key role in establishing contact between a cell and its environment. Cadherin expression is often misregulated in cancer cells, which leads to decreased cell attachment and a more metastatic phenotype (Blanco et al., 2004). This allows tumor cells to migrate and invade the vasculature, leading to cancer metastasis. Slit/Robo signaling has been shown to regulate this first step towards metastasis by influencing cell adhesion through its action on cadherins and β-catenin. As discussed above, one consequence of this regulation is altered subcellular distribution of β-catenin, which increases proliferation with its translocation to the nucleus (Prasad et al., 2008; Rhee et al., 2007; Rhee et al., 2002; Tseng et al., 2010; Zhou et al., 2011). There is additional evidence, mostly genetic and collected in developmental settings, that further demonstrates a role for Slit and Robo in regulating cadherin-mediated cell-cell adhesion. Again, unsurprisingly, given Slits function as attractant and repellent in axon guidance, both an increase and decrease in cell adhesion have been attributed to Slit/Robo signaling, dependent on the biological context. For example, a positive role for Slit/Robo in enhancing cell-cell adhesion is observed during chick cranial trigeminal gangliogenesis when cells derived from neural crest and ectodermal placodes interact to generate ganglionic structures (Shiau and Bronner-Fraser, 2009). Trigeminal placode cells express N-cadherin and Robo1, while the intermingling neural crest cells express Slit1. Loss of either N-cadherin or Robo1 results in dispersed and disorganized placodal neurons within the trigeminal ganglion, suggesting that N-cadherin and Robo1 function in collaboration to mediate the proper coalescence of placode-derived neurons (Shiau and Bronner-Fraser, 2009). In concordant studies, overexpression of either Slit or Robo results in both the post-translational upregulation of N-cadherin and its redistribution to the placodal cell membrane, again leading to a model in which Slit/Robo signaling stabilizes sites of cell-cell contact by influencing the subcellular localization of cadherin (Shiau and Bronner-Fraser, 2009). It is important to note, however, that in this study no changes in the level or distribution of β-catenin were reported. In a second example of Slit mediating increased cell adhesion in collaboration with a cadherin, P-cadherin is shown to co-localize with Slit in the basal cell layers of normal oral mucosa, with this expression down-regulated in oral squamous cell carcinoma (OSCC) (Bauer et al., 2011). In an OSCC cell line that overexpresses P-cadherin, a complex of P-cadherin and Robo3 is detected, and treatment of these cells with Slit results in a dose-dependent down-regulation of cell migration that could be relieved using a small interfering RNA that reduces Robo3 expression (Bauer et al., 2011). Taken together, these studies support a tumor suppressive role for the Slit/Robo signaling axis in maintaining cell-cell adhesion and, consequently, a non-invasive cellular state by enhancing cadherin function.
Conversely, slit/robo signaling has also been shown to inhibit cadherin function at the membrane during Drosophila heart tube formation, resulting in decreased cell-cell adhesion (Santiago-Martinez et al., 2008). Again, genetic evidence suggests that robo and e-cadherin/shotgun (shg) function together in modulating cardioblast adhesion, but in this biological context, their actions oppose one another. This is evidenced by the observation that robo loss-of-function phenocopies e-cadherin/shg gain-of-function, generating embryos with no lumen due to enhanced cardioblast adhesion (Santiago-Martinez et al., 2008). Similarly robo gain-of-function phenocopies e-cadherin/shg loss-of-function, but in this circumstance lumen formation was blocked due to insufficient cardioblast adhesion. These studies support a role for Robo antagonizing E-cadherin/Shg function, with Robo mediating a repulsive or anti-adhesive signal that functions in opposition of the pro-adhesive actions of E-cadherin/Shg.
By targeting cadherins and the cadherin/β-catenin complex, Slit/Robo signaling regulates two of the crucial steps in tumor progression: cell proliferation and cell adhesion. A challenge for researchers is to understand the circumstances that determine whether this signaling pathway acts positively to enable cell contacts, or negatively to deter them. For other guidance families, different complexes of receptors specify attraction versus repulsion. For example, attraction via Netrin is mediated by DCC in a complex with DSCAM, whereas repulsion requires an UNC5 receptor that acts either together with DCC or alone (Moore et al., 2007). In contrast, no co-receptors have been identified that specifically regulate the attractant or repellent functions of Robo, although perhaps its interaction with cadherin, albeit indirect, serves this role. Regardless of these events at the plasma membrane, a central requirement for either the positive or negative response of a cell to Slit is the interaction of Robo with the actin cytoskeleton, a topic that is discussed in the next section.
Slits and Robos: Regulators of the Actin Cytoskeleton
Following detachment of cells from the surrounding tissue, tumor progression requires enhanced cell motility, which is accompanied by increased actin polymerization and the enhanced activity of proteins that optimize its turnover. Developmental studies show that Slit/Robo signaling affects cell motility by controlling the activity of several proteins involved in reorganizing the actin cytoskeleton, including the small GTPases comprising the Rho-family (Rac, Cdc42, and RhoA), and other key regulators of the actin cytoskeleton, such as the non-receptor tyrosine kinase, Abl, and Ena/Vasp proteins.
Rho GTPases
Many studies have shown that Rho GTPases play an important role in modulating the downstream action of Slit/Robo1 signaling. These proteins switch between active and inactive states, and are regulated by GEFs (guanine nucleotide exchange factors) and GAPs (GTPase activating proteins): the former stimulate, and the latter inhibit GTPase function. During Drosophila neural development, slit/robo-mediated repulsion of commissural neurons at the midline requires the activation of rac (Fan et al., 2003), and recruitment of both a rac GEF, called son of sevenless (sos), and a rac GAP, called vilse/crGAP (Hu et al., 2005; Lundstrom et al., 2004; Yang and Bashaw, 2006). Sos binds to robo through the adaptor protein dock (Nck in mammals) (Fan et al., 2003; Yang and Bashaw, 2006), whereas vilse/crGAP interacts directly with robo (Hu et al., 2005; Lundstrom et al., 2004) (Figure 3A). Studies show that the activities of both this GAP and GEF support slit/robo-mediated repulsion, as both sos and vilse/crGAP mutants display mild defects in midline repulsion that can be significantly enhanced through loss of one copy of either slit or robo (Figure 3A) (Hu et al., 2005; Yang and Bashaw, 2006). This raises the question; how does both the activation and inhibition of Rac lead to axonal repulsion? One possibility is that these GAPs and GEFs function in distinct steps, with each required for different molecular actions that support repulsion. Alternatively, it could be that Rac cycling alone is sufficient for repulsion, which may not depend on the maintenance of a specific level of Rac-GTP. In any case, these studies demonstrate the importance of GAPs and GEFs in regulating Slit/Robo signaling by controlling the activity state of small GTPases.
Figure 3. Slit/Robo Signaling Regulates Cell Migration by Controlling the Activation State of Actin Cytoskeleton Modulators.
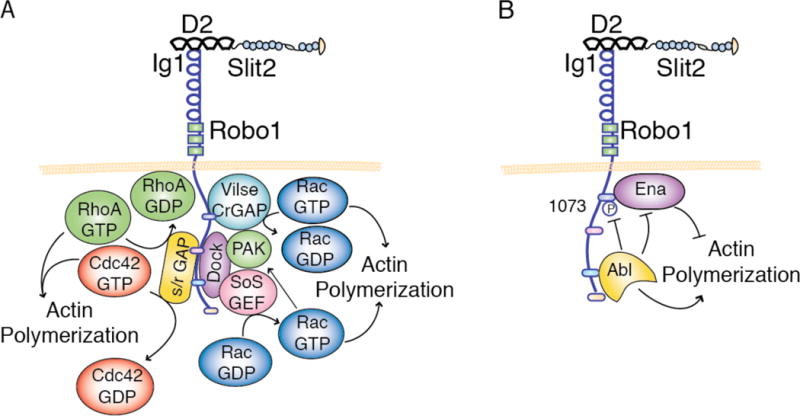
(A) Slit/Robo signaling regulates actin polymerization, and thus cell migration, by controlling the activity level of Rho GTPases. Slit/Robo signaling prevents cell migration by recruiting Slit/Robo (s/r)GAPs to the CC1 and CC2 domains, which inactivate the small Rho GTPases RhoA and Cdc42, and Vilse/CrGAP to the CC0 domain, which exchanges RacGTP for RacGDP. In other contexts, Slit/Robo signaling drives actin polymerization by recruiting Dock, which in turn recruits son of sevenless (SoS) GEF and PAK p21-activated kinase. SoS GEF activates the small Rho GTPase Rac by exchanging GDP for GTP, leading to actin polymerization. RacGTP in turn activates PAK, which also drives actin polymerization. (B) The anti-migratory function of Slit/Robo signaling is regulated by the Abl, which attenuates Robo signaling via phosphorylation of Robo at Y1073 near CC0, possibly preventing substrate binding, and by directly targeting the Robo effector protein Enabled (Ena). In the absence of Abl, Ena binds to CC0 and functions to inhibit cell migration by preventing actin polymerization.
With their central role in regulating the actin cytoskeleton, Rho GTPases are in the unique position to regulate cell motility in response to Slit. Accordingly, a number of studies demonstrate the importance of Cdc42 in mediating the aggressive spread of tumor cells and the role of Slit/Robo1 signaling in inhibiting this invasion by attenuating Cdc42 activation. For example, a study on medulloblastoma reveals expression of Slit and Robo in a variety of tumor samples and cell lines, with no evidence that Slit is silenced by methylation (Werbowetski-Ogilvie et al., 2006). Treatment of cells derived from these tumors with Slit2 inhibits their invasiveness, without affecting the direction of their migration or their proliferation. The authors suggest that these effects are due to a reduction in the activity of Cdc42 (Werbowetski-Ogilvie et al., 2006). This result is also observed in two studies on glioma cell lines and tumor samples which report reduced expression of Slit in primary human glioma specimens and invasive glioma cells, compared to normal brain cells and astrocytes (Parsons et al., 2008; Yiin et al., 2009). Furthermore, treatment of these glioma cells with recombinant Slit2 or its overexpression in these cell lines inhibits cell migration and reduces invasion by decreasing Cdc42 activity, an effect that is prevented by siRNA-mediated reduction of either Slit or Robo1 expression. This inhibition of cell invasion was further confirmed in xenograft studies that demonstrate decreased infiltration of Slit2-expressing glioma cells implanted into the brains of mice. Interestingly, there is no evidence that these effects are mediated through the regulation of β-catenin by Slit as neither the level of β-catenin, its phosphorylation status nor its association with N-cadherin is altered in Slit-expressing glioma cells (Yiin et al., 2009). Taken together these studies on two different types of neural tumors demonstrate that the small GTPase, Cdc42, is subject to negative regulation by Slit/Robo signaling.
Slit/Robo (s/r) GAP is a strong candidate for the GAP that inhibits Cdc42 activity downstream of Slit and Robo. s/rGAP has been shown to reduce the activation of both Cdc42 and Rho, but not Rac (Wong et al., 2001). Studies demonstrate that a dominant negative form of this GAP blocks the inactivation of Cdc42 by Slit and also the migration of cells in response to Slit (Wong et al., 2001). Furthermore, a study in tumor and nontransformed cultured epithelial cells has shown that Slit2/Robo1 counteracts Hepatocyte Growth Factor (HGF)-induced migration by directly targeting and inhibiting Cdc42 and, as a consequence, actin-based protrusive forces (Figure 3A) (Stella et al., 2009). Taken together, these data show that Slits inhibit the motility of tumor cells by negatively regulating Cdc42 Rho GTPase, and that one of the consequences of losing Slit expression during tumor progression is inappropriate cellular migration due to this deregulation.
Abl and Ena
In addition to the Rho family of small GTPases, Slit binding to Robo also leads to the recruitment of at least one kinase that regulates both actin cytoskeletal rearrangements and the activity of Robo itself. Structural and genetic studies show that the Abelson tyrosine kinase (Abl) and its substrate Enabled (Mena in mammals) interact directly with the cytoplasmic domains of Robo (Figure 3B) (Bashaw et al., 2000). Genetic studies in Drosophila demonstrate opposing roles for ena and abl in robo-mediated axonal repulsion, whereby abl antagonizes repulsive robo signaling and ena enhances it (Bashaw et al., 2000). In a series of studies, it has been shown that reducing the level of abl suppresses robo loss-of-function phenotypes, while its overexpression inhibits robo function. The opposite is true for ena, as reducing its levels enhances robo loss-of-function phenotypes and suppresses robo gain-of-function. Furthermore, Abl phosphorylates robo to inhibit its function because a Y-F mutation in a conserved tyrosine that is targeted by abl generates a hyperactive robo receptor. In contrast, deleting the cytoplasmic domain of robo that binds ena reduces the ability of this robo mutant to rescue robo loss-of-function phenotypes (Bashaw et al., 2000).
While these studies demonstrate the consequences of Abl and Ena interactions with Robo, the molecular mechanism by which these proteins mediate their effect on the directional outgrowth of an axon through Robo is still poorly understood. For Abl, one possibility is that it binds to and phosphorylates unliganded Robo, inhibiting the ability of signaling proteins to interact with their docking sites on the Robo cytoplasmic domain, until Slit binds Robo and relieves the inhibition. Abl is known to phosphorylate Ena, but the significance of this phosphorylation is poorly understood. The Ena proteins (Mena, Vasp and EVL in vertebrates) contain N and C terminal Ena/Vasp homology domains that flank a proline-rich central region. They are generally thought of as positive regulators of actin assembly that function in promoting the growth of long, sparsely branched actin filament networks. Consequently, it is still unclear how Ena, which enhances actin polymerization and filopodial/lamellipodial protrusion, plays a role in repulsive axon guidance downstream of Robo (Bear and Gertler, 2009), except that it may direct the growth of the cell away from Slit by promoting assembly at sites distal to high ligand concentration. Moreover, few studies have been published on how Ena and Abl contribute to the migration of tumor cells in response to Slit. It has been shown that transformation of cells with the Src oncogene results in Abl activation, which stabilizes Robo1 at the plasma membrane, leading to the activation of both Cdc42 and Rac, as well as Slit-independent cell migration (Khusial et al., 2010). Thus, under these circumstances, the activation of Robo1 positively regulates downstream Rho GTPases and induces migration, whereas during central nervous system development and in other tumor types, the opposite effect is observed. Taken together, these studies demonstrate that Slit/Robo signaling communicates with the actin cytoskeleton to regulate cell motility, although the nature of the response depends upon the developmental and disease context. It is clear from many studies that Rho GTPases, Abl and Ena all play important roles in promoting tumor cell metastasis (Allington and Schiemann, 2011; Gertler and Condeelis, 2011; Hall, 2009). However, additional research is required to determine how Slits regulate these cytoskeletal effectors in tumor cells and whether Slits could be potential therapeutic targets for hindering tumor cell motility by interfering with these signaling routines.
Slits and Robos: Regulators of Cell Chemotaxis
Cells must decipher and integrate a complex set of signals in order to migrate toward targets. Indeed, even the metastatic migration of tumor cells is not a random walk and many types of cancers preferentially target specific organs. While Slit is one of the cues cells respond to in the extracellular environment, there are many others, such as chemokines. Compared to large, extracellular matrix-associated Slits, chemokines are small (8–10 kD) soluble factors, first identified in the immune system, but now with documented roles in regulating the migration of many cell types, including tumor cells. A number of studies dxamine how Slits affect the motility of immune and tumor cells in response to other extracellular factors, notably chemokines, but also growth factors such as HGF and Platelet-Derived Growth Factor (PDGF). Although the preponderance of data supports a role for Slits in inhibiting the migration of cells responding to stimulant, there are two examples of Slit increasing cell migration in response to chemokines (Schmid et al., 2007; Ye et al., 2010).
The first of these studies was published over a decade ago. Standard Transwell assays were used to evaluate the effects of Slit on leukocyte migration from the upper to lower chamber in response the chemokine CXCL12 (SDF1) or bacterial chemotactic factor, N-formyl peptide f-Met-Leu-Phe (fMLP). It was found that Slit reduces the chemotactic migration of leukocytes when added to either the upper, lower or both chambers, indicating that Slit reduces the overall motility of cells, rather than acting as a repulsive cue to guide their migration (Wu et al., 2001). Further studies have refined our understanding of the underlying molecular mechanisms. Videomicroscopic live cell tracking demonstrates that Slit2 selectively impairs chemotaxis, defined as the directional migration of cells, but not chemokinesis, which is the random movement of neutrophils in response to stimulant (Tole et al., 2009). Slit2 achieves this effect by suppressing the activation of Cdc42 and Rac2 that would normally occur in response to stimulation, with consequent disruption of actin free barbed end formation. A similar inhibition of migration and downregulation of Rac activity is observed in vascular smooth muscle cells in response to platelet derived growth factor (Liu et al., 2006).
These observations were translated in vivo using a mouse model of chemical irritant peritonitis. Preadministration of Slit2 either intraperitoneally or by tail vein injection significantly reduces the recruitment of neutrophils to the site of inflammation (Tole et al., 2009). These data suggest that localized or systemic delivery of Slit2 reduces leukocyte recruitment and, consequently, the tissue damage associated with inflammation. This finding is in accordance with observations from other inflammation models, including glomerulonephritis-associated kidney injury, global cerebral ischemia, and skin sensitization to allergin (Altay et al., 2007; Guan et al., 2003; Kanellis et al., 2004), in which Slit functions similarly in an anti-inflammatory manner. However, a recently published study using two different models of allergic airway inflammation suggests that Slits have a more complex role in the immune system. In the first model of ovalbumin (OVA) airway inflammation, Slits enhance eosinophil chemotaxis, while in the second model of endotoxin-induced lung inflammation, Slits suppress neutrophil chemotaxis (Ye et al., 2010). Eosinophils and neutrophils both express Robo1, while Clara cells in the bronchial epithelium secrete Slit2. Aerosol challenge of wildtype mice with OVA triggers leukocytes, primarily eosinophils, to infiltrate into lung. This infiltration is significantly enhanced in Slit2 transgenic (Slit2-Tg) mice, which overexpress Slit2 under the control of the cPMV promoter (Yang et al., 2010). These data suggest that Slit augments eosinophil recruitment. Similarly aerosol challenge of wildtype mice with endotoxin again triggers leukocyte infiltration into lung, but in this case primarily neutrophils are mobilized. With endotoxin challenge, however, significantly fewer neutrophils are observed in the lungs of Slit2-Tg mice, an effect that is reversed by the application of a function-blocking antibody directed against the extracellular domain of Robo1. These data suggest that Slit inhibits neutrophil recruitment. Together with in vitro studies that demonstrate enhanced chemokine-induced eosinophil migration in response to Slit, but reduced neutrophil migration (Yang et al., 2010), these data suggest that, depending on the cellular circumstance, Slits can have differential effects on leukocytes. The molecular basis for these distinct responses can be traced to levels of s/rGAP expression, with eosinophils containing significantly lower levels of this Slit/Robo effector compared to neutrophils. This results in the activation of Cdc42 in eosinophils, rather than inhibition, which occurs in neutrophils when s/rGAP is present (Yang et al., 2010). Thus, even though almost a decade of work has pointed to a single role for Slit as an inhibitory factor in the immune system, with this recent finding, it appears that this is not the case and that, once again, depending on the cellular context, Slit has a dual role as activator and inhibitor of cellular response.
The role of Slit as an inhibitor of inflammation has potentially far-reaching implications in terms of its role in cancer biology as a tumor suppressor. In normal tissue during wound healing, removing the irritant or completing the repair limits the inflammatory response. In contrast, tumors become essentially unhealed wounds, characterized by chronic inflammation, which promotes rather than suppresses tumor growth by releasing growth and survival factors, creating genomic instability, promoting angiogenesis and remodeling the extracellular matrix to facilitate invasion. That Slits inhibit the infiltration of not only leukocytes, but also dendritic cells (Guan et al., 2003), T lymphocytes and monocytes (Prasad et al., 2007), could be harnessed therapeutically to normalize the inflammatory network and restrict infiltrating cells with tumor-promoting properties, while attracting those cells with tumor-suppressing properties.
Another way that Slit could function as a therapeutic agent in the war on cancer is by inhibiting metastasizing cells. This has been evidenced by a number of studies using breast cancer models that demonstrate the ability of Slit/Robo signaling to counter the pro-migratory, pro-metastatic consequences of the CXCL12 /CXCR4 chemokine axis. A study by Muller and colleagues a decade ago demonstrates that the pattern of breast cancer metastases is governed, at least in part, by this chemokine axis (Muller et al., 2001). CXCR4 is upregulated in breast cancer cells (Salvucci et al., 2006), and, upon metastasis, guides these cells to organ sites with high CXCL12 levels such as the lung, liver and bone. The involvement of CXCR4 in metastasis is not confined to breast cancer, as it is also expressed in other tumor cell lines that respond to CXCL12, such as astrogliomas, prostate carcinomas, B-cell lymphomas and chronic lymphocytic leukemias (Moore, 2001). Using Transwell filters, two studies have demonstrated that Slit2 has the capacity to counteract CXCL12-induced chemotaxis of breast cancer cell lines that express both Robo1 and CXCR4 (Prasad et al., 2004; Schmid et al., 2007). By signaling through Robo1, Slit2 inhibits a number of downstream effectors that are activated by CXCR4, such as the focal adhesion components RAFTK/Pyk2, focal adhesion kinase, paxillin, PI3K, p44/42 MAP kinase, and metalloproteases 2 and 9 (Prasad et al., 2004). In the absence of Slit2, which is downregulated in over 50% of sampled breast tumors, the expression of both CXCL12 and CXCR4 is upregulated (Marlow et al., 2010), contributing to the development of hyperplastic lesions in Slit2 and Robo1 knockout mammary glands. Another study on the role of Slit/Robo signaling in inhibiting breast cancer cell migration in response to CXCL12 implicates ubiquitin-specific protease 33 (USP33), a deubiquitinating enzyme. The authors provide evidence that Slit stalls the chemotaxis of breast cancer cells by inducing the redistribution of Robo to the plasma membrane, a process that is dependent on USP33 (Yuasa-Kawada et al., 2009). Taken together, these data once again raise the possibility that Slits could function therapeutically, in this case to combat tumor metastasis by inhibiting tumor cell migration in response to CXCL12.
Robo4 is an Unconventional Robo Receptor
Robo4 was a late addition to the family of Robo receptors due to the lack of structural homology between it and the other Robos. Nevertheless, studies over the last decade have shown that Robo4 is a key member of the Slit/Robo signaling axis, especially in the vasculature, where it is expressed on the surface of endothelial cells and functions in regulating angiogenesis (Huminiecki et al., 2002; Park et al., 2003). Robo4 was considered an endothelial-specific member of the Robo family until very recently with the publication of a study showing its expression in the developing brain, where it appears to regulate the radial migration of newborn neurons (Zheng et al., 2011). These new data raise the possibility that Robo4 has, as yet undiscovered, roles in different organs.
Robo4 is structurally unique compared to the other receptors in the Robo family. In contrast to the five Ig and three FN3 domains in the extracellular region of Robo1–3, Robo4 contains only two extracellular Ig and FN3 domains, and only two intracellular CC domains, CC0 and CC2 (Huminiecki and Bicknell, 2000; Huminiecki et al., 2002). Moreover, while strong evidence for an interaction between the extracellular domain of Robo1–3 and the D2 domain of Slits exists, current data do not support a direct interaction between Slits and Robo4. Biochemical and structural studies carried out by Morlot and colleagues have identified the critical amino acids in the Robo ectodomain required for Slit binding, and Robo4 lacks these amino acids (Morlot et al., 2007b). BiaCore analysis on recombinant proteins also fails to provide evidence for a Slit/Robo4 interaction (Suchting et al., 2005), even though Slit2 and Robo4 can be co-immunoprecipitated from cell lysates and positive immunostaining is observed on cells incubated with recombinant Slits (Park et al., 2003; Zhang et al., 2009). One explanation for the apparent association between Slits and Robo4 when they are in a cellular context, but not in purified forms, is that they exist in a protein complex on the cell and that a co-receptor is present to transmit Slit binding into Robo4 activation. Candidates for this co-receptor are transmembrane heparin sulfate proteoglycans such as Syndecan (Hu, 2001; Steigemann et al., 2004). Indeed, in vitro studies by Hussain and colleagues found that heparan sulfate is required for functional Slit-Robo4 signaling (Hohenester et al., 2006). Additionally, a nonexclusive alternative is that Robo1 fulfills this co-receptor function as Robo1/Robo4 complexes have been documented, as well as a requirement for this interaction in inhibiting endothelial chemotaxis in response to Slit2 (Kaur et al., 2008; Sheldon et al., 2009). More recently, UNC5B, a chemorepellent receptor for Netrin1 (Leonardo et al., 1997), was identified in a protein-protein interaction screen as an alternative binding partner for Robo4 in the vasculature (Koch et al., 2011). While additional studies are required to investigate this novel interaction, it reveals a new layer of complexity in Robo4 function. The role of Robo4 as a regulator of angiogenesis will be described in more detail below, but suffice it to say that, while it is unlikely that Robo4 interacts directly with Slits, there is ample evidence that it transduces Slit signaling, possibly via co-receptors such as heparan sulfate proteoglycans or Robo1, or even via reverse signaling through UNC5B.
Slit/Robo: Roles in Tumor Angiogenesis
Angiogenesis describes the process of new vessel growth from mature, pre-existing vessels. This process occurs naturally throughout development and later in the adult during both wound healing and pregnancy, both times of increased tissue remodeling. Angiogenesis involves several steps: first, endothelial cells migrate towards a pro-angiogenic stimulus, such as vascular endothelial growth factor (VEGF), released extracellularly into the environment; and second, endothelial cells become migratory and congregate at the source of the angiogenic stimulus to form loops, and later vessels, as more and more cells arrive (Potente et al., 2011). Tumor cells often hijack several aspects of this process to enable additional tumor growth and metastasis, both of which require an increased supply of oxygen and other nutrients. Without increased angiogenesis, tumors are limited in their growth potential and become necrotic. Thus, tumor cells must induce angiogenesis to transition from a small group of cells to a large, malignant tumor, and to ultimately metastasize to other tissues. These processes require intimate communication between tumor cells and endothelial cells, and studies from the last decade provide strong support for the Slit/Robo pathway in mediating this crosstalk. However, how Slit/Robo signaling affects tumor angiogenesis remains unclear, as reports demonstrate both pro-and anti-angiogenic functions in pathological systems (Table 1). What is clear, however, is that Slits and Robos play key roles in regulating the process of tumor angiogenesis.
Vascular Expression of Slits and Robos
Of the three Slit proteins expressed in vertebrates, multiple studies have reported Slit2 and Slit3 in the vasculature of normal tissues, expressed by vasculature smooth muscle cells/pericytes that encircle blood vessels (Jones et al., 2009; Liu et al., 2006; Marlow et al., 2010), and also by endothelial cells (Brantley-Sieders et al., 2011; Zhang et al., 2009). In contrast, only one study has reported detection of Slit1 in the vasculature (Abdollahi et al., 2007), suggesting that it may not play a key role in developmental and tumor angiogenesis, although future studies may change this view. As for Slit receptors, Robo4 appears to be expressed by all endothelial cells, whereas Robo1 may only be expressed on some types of blood vessels (Huminiecki et al., 2002; Legg et al., 2008; Mura et al., 2011; Park et al., 2003; Sheldon et al., 2009; Verissimo et al., 2009).
Function of Slits and Robos in the Vasculature
Robo4 expression was initially identified by bioinformatic data mining, with the accompanying analysis of its expression revealing its presence exclusively at sites of active angiogenesis, notably tumor vessels (Huminiecki et al., 2002). Robo4 was also independently identified in a study aimed at identifying genes whose expression is perturbed in an Activin receptor-like (Alk) mutant mouse model (Park et al., 2003). Alk is a member of the TGF-β superfamily of receptors and is involved in the normal development of the vasculature (Johnson et al., 1996). Mice lacking Alk expression develop abnormal connections between arterial and venous vascular beds and die at midgestation (Urness et al., 2000). Such a developmental role for robo4 was supported by studies in zebrafish in which either morpholino knockdown of robo4 or its overexpression results in stunted or absent intersomitic vessels, although normal patterning of axial vessels are seen, suggesting both primary and redundant roles for robo4 in this system (Bedell et al., 2005). Furthermore, numerous studies show that Slits affect the migration of endothelial cells, acting as both a chemoattractant (Howitt et al., 2004; Kaur et al., 2006; Wang et al., 2003; Wang et al., 2008) and chemorepellent (Marlow et al., 2010; Park et al., 2003; Seth et al., 2005; Zhang et al., 2009). More recently, a study aimed at identifying novel tumor endothelial markers that can be used as targets by anti-angiogenic therapeutics found that Robo4 is highly expressed on tumor vessels compared to normal tissue vessels, which display little or no immunostaining (Mura et al., 2011). Taken together, these studies support a role for Robo4 in both normal development of the vasculature and in tumor angiogenesis. It was therefore surprising when it was discovered that Robo4−/− mice display normal vessel patterning in a variety of contexts: intersomitic and cephalic vessels during early embryogenesis, stereotypical nerve-artery alignment in embryonic limb skin, and normal vascularization of the mammary gland during postnatal organogenesis (Jones et al., 2008; Marlow et al., 2010). These data raise the question as to the role of Robo4 in mammalian endothelium.
Although a comprehensive understanding of the function of Robo4 in the vasculature has been hindered by contradictory findings, accumulating evidence suggests that Slit/Robo4 signaling functions to downregulate VEGF signaling in the mature vasculature, thus restraining angiogenesis during pathological neovascular processes (Han and Zhang, 2010; Huang et al., 2009a; Jones et al., 2008; Jones et al., 2009; Koch et al., 2011; London and Li, 2011; Mulik et al., 2011), as well as during normal periods of robust sprouting angiogenesis such as occurs during pregnancy (Marlow et al., 2010). The analysis of Robo4−/− mice provided the first insight into this function by showing that in wildtype, but not knockout animals, Slit2 suppresses VEGF-induced hyperpermeability of retinal endothelium (Jones et al., 2009). Further experiments employed two models of vascular disease, oxygen-induced retinopathy and laser-induced choroidal neovascularization, both of which result in pathological angiogenesis (Jones et al., 2009). In both cases, intravitreal administration of Slit2 reduces angiogenesis in wildtype, but not Robo4−/−, mice. Moreover, the opposite effect of elevated angiogenesis was observed in concordant experiments looking at breast development and cancer. In these contexts, loss of Slit/Robo4 signaling results in excessive angiogenesis only when there is increased VEGF expression in the gland, which occurs during pregnancy and preneoplasia (Marlow et al., 2010). There is no Robo4−/− mammary gland phenotype in the absence of pro-angiogenic stimulation (Marlow et al., 2010). In all these examples, Slit/Robo4 regulates angiogenesis by inhibiting signaling downstream of VEGF/VEGFR, including the activation of Src, Rac and FAK (Jones et al., 2008; Marlow et al., 2010). Further analysis of the pathway has shown that Robo4 interacts directly with paxillin and the ArfGAP (ADP-ribosylation factor-directed GTPase activating proteins), GIT1, to block the activation of Arf6 in response to VEGF and fibronectin (Figure 4A) (Jones et al., 2009).
Figure 4. Slit/Robo Signaling Regulates the Process of Tumor Angiogenesis.
(A) Slit2 binds to Robo4/proteoglycan complex and signals to block pro-angiogenic signaling downstream of VEGF/VEGFR. (B) Slit promotes angiogenesis by binding to a Robo1/Robo4 heterodimer and driving endothelial migration. (C) Robo4 binds UNC5B, and signals to block pro-angiogenic signaling downstream of VEGF/VEGFR.
Additional studies on disease models support the role of Slit/Robo4 signaling in inhibiting angiogenesis and enhancing vascular stability. One study used infection with herpes simplex virus (HSV) to generate chronic inflammatory lesions, called stromal keratitis, in the cornea. This pathological condition is associated with enhanced angiogenesis that is driven by VEGF (Mulik et al., 2011; Suryawanshi et al., 2011). Following HSV infection, Robo4 is upregulated in endothelial cells of corneal stroma. However, a corresponding increase in Slit2 is not observed, suggesting that the Slit/Robo4 signaling axis is unable to control angiogenesis in response to this infection because Slit production is limited. To investigate, Slit2 was subconjunctivally administered. This treatment resulted in reduced neovascularization, a result that is not observed when Slit expression is knocked down using shRNA. The researchers also examined the activity status of Arf6 and Rac, and found that they were reduced after Slit2 treatment, supporting previous studies that Slit/Robo4 signaling opposes VEGF/VEGFR signaling by modulating downstream signaling pathways. In other studies, an inflammatory reaction was triggered by the administration of lipopolysaccharide (LPS), which induces endothelial hyperpermeability (London et al., 2010). In this context, Slit2 treatment reduces LPS-induced vascular permeability by increasing the localization of V/E cadherin to cell/cell contacts and stabilizing its interaction with p120-catenin. Taken together, these studies have paved the way to a new view on the function of Robo4, in which it functions as a guardian of blood vessel stability by countering pro-angiogenic and pro-inflammatory signals.
There are, nevertheless, several aspects to this model of Robo4 function that remain unresolved. First, is the question concerning the role of Robo1, which is (1) expressed on at least some endothelial cells; (2) heterodimerizes with Robo4 (Kaur et al., 2006; Kaur et al., 2008; Sheldon et al., 2009); and (3) is required for some (Sheldon et al., 2009), but not all (Marlow et al., 2010), of Robo4 functions. The papers summarized above describing the anti-angiogenic and anti-inflammatory roles of Robo4 do not address the function of Robo1 in these processes. However, there is some evidence that Robo1 binds Robo4 and may be required to transduce the Slit signal. Because Robo4 lacks many of the amino acids critical for Slit2 binding (Morlot et al., 2007b), it is considered unlikely that it binds Slit directly. Instead, Robo1 may serve as a co-receptor for Robo4, and in this circumstance, the heterodimer may function to promote endothelial cell migration and angiogenesis (Figure 4B) (Kaur et al., 2006; Kaur et al., 2008; Sheldon et al., 2009). The notion that Robo1 plays a functional role in angiogenesis, either alone or in complex with Robo4, is supported by a number of studies, including one on tumor angiogenesis (Wang et al., 2003). In addition, the analysis of gene expression levels during retinal development reveals fluctuations in Robo1 levels that coincide with times of active retinal vascular development (Huang et al., 2009b). Further, these researchers show that loss of Robo1 in monkey choroidal retinal endothelial cells perturbs tube formation, in addition to lowering cell proliferation and migration (Huang et al., 2009b). Elevated levels of Robo1 are seen in several pathological animal models, such as retinopathy of prematurity and neovascularized corneas (Han and Zhang, 2010; Huang et al., 2009a). Robo1 was also identified as a putative pro-angiogenic gene in assays to elucidate human genes whose expression correlates with either increased or decreased angiogenesis (Abdollahi et al., 2007). Taken together these data suggest that Robo1 functions to restrict angiogenesis, either on its own or in a complex with Robo4, but additional studies are required to fully elucidate its role during developmental and pathological angiogenesis.
A second issue concerning Robo4 and its role in angiogenesis is the uncertain status of Slit as ligand. The discovery by Koch and colleagues that Robo4 binds UNC5B suggests that there may be an alternative mechanism for Robo4 signaling in the vasculature (Koch et al., 2011). Further characterization of this interaction revealed the surprising finding that Robo4 acts as the ligand, not the receptor, in this relationship. Soluble Robo4 protein rescues vessel hyperpermeability in the Robo4−/− mice and reduces VEGF-induced hyperpermeability in wildtype mice, but not in mice treated with anti-UNC5B, supporting a model whereby Robo4 maintains vessel integrity by binding and signaling through UNC5B. In these studies, Robo4/UNC5B signaling was shown to counter the activation of Src kinase by VEGF/VEGFR (Koch et al., 2011), the same mechanism that was previously identified for Slit/Robo4 signaling (Figure 4C) (Jones et al., 2009). There is, however, no evidence that the vascular phenotypes of Robo4−/− and Unc5b−/− mice are similar, a result that may be expected for proteins in a ligand/receptor relationship. Altogether, these data support a role for Robo4 in blocking signaling pathways downstream of VEGF/VEGFR and, consequently, in inhibiting VEGF-induced changes in blood vessels, but it is currently unclear whether this requires Slit, UNC5B or both (Figure 4A).
Slit and Robo Expression is Altered in Tumor Angiogenesis
Like other members of the Robo family of receptors, there are documented changes in Robo4 expression in samples from tumors and other diseases. Increased Robo4 expression on endothelial cells has been reported in a number of pathologies, including tumor samples that display increased angiogenesis (Grone et al., 2006; Han and Zhang, 2010; Huang et al., 2009c; Mura et al., 2011; Seth et al., 2005)(Table 2). Conversely, decreased Robo4 expression has also been observed in hepatocellular carcinoma samples analyzed by quantitative RT-PCR (Avci et al., 2008), and in datasets from microarray analyses on human breast tumor samples (Richardson et al., 2006)(Table 3). Thus, while it is possible that Robo4 is both up- and down-regulated in disease models, at this point the data suggest that it is most often increased, suggesting a link between Slit/Robo4 signaling and new vessel formation, at least in a tumorigenic setting. There is some documentation of Robo1 expression on endothelial cells in tumors and other tissues undergoing neovascularization (Han and Zhang, 2010; Wang et al., 2003), although whether Robo1 is expressed and how it functions in blood vessels remains controversial. The regulation of Slits in tissues surrounding blood vessels has already been discussed, with examples of both up and down regulation in tumors and other tissues undergoing neovascularization (Han and Zhang, 2010; Wang et al., 2008; Yang et al., 2010). There is also some data suggesting that Slit expression is regulated by EphA2 tyrosine kinase, and that loss of EphA2 expression in a tumor setting elevates Slit expression, which acts in a pro-angiogenic manner (Brantley-Sieders et al., 2011).
Slits and Robos: Regulators of Tumor Angiogenesis
Tumor growth and metastasis requires increased supplies of oxygen and nutrients, and this requirement can be met by enhanced angiogenesis in the surrounding vasculature (Potente et al., 2011). In order to achieve increased angiogenesis, tumor cells must downregulate anti-angiogenic signals and then secrete pro-angiogenic cues to increase blood vessel growth (Kerbel, 2008). Currently there are no studies that directly examine the functional role of Robo4 in tumor angiogenesis, although in a preneoplastic setting in breast, loss of Robo4 results in enhanced angiogenesis in response to epithelial-derived VEGF and CXCL12 (Marlow et al., 2010). These data are in line with the documented anti-angiogenic role of Robo4 in stabilizing the vasculature during pathological angiogenesis as described above.
In contrast, Wang and colleagues have shown that Robo1 is expressed on tumor endothelial cells and plays a role in new vessel formation in tumors that upregulate Slit expression (Wang et al., 2003; Wang et al., 2008). In these studies, the extracellular domain of Robo1, or a function-blocking antibody called R5, are shown to inhibit both the chemoattractive migration of endothelial cells and their tube formation in response to Slit2, as well as tumor angiogenesis in xenograft models, suggesting that Slit2 functions pro-angiogenically in tumors that overexpress it, and further, that its signal is transduced via Robo1, and not Robo4 (Wang et al., 2003; Wang et al., 2008). Furthermore, overexpression of Slit2 in a non-metastatic pancreatic islet cell model of carcinogenesis (RIP1-Tag2) is reported by the same group to promote tumor lymphangiogenesis and lymphatic metastasis by signaling through Robo1, expressed on the lymphatic endothelial cells. Again, administration of the R5 function-blocking antibody reverses the enhanced angiogenesis and decreases tumor formation (Yang et al., 2010).
A possible explanation for the apparent pro- versus anti-angiogenic functions of Slit2/Robo signaling comes from a study by Dunaway and colleagues in which Slit2 promotes angiogenesis on its own, but inhibits it in the presence of ephrin-A1 (Dunaway et al., 2011). Eprhin-A1 is the primary GPI-linked ligand for the EphA2 receptor, whose expression was shown by these researchers in a previous paper to inversely correlate with Slit expression (Brantley-Sieders et al., 2011). The present study, however, shows that Slit2 stimulates angiogenesis through the activation of Akt and Rac GTPase, an effect that is inhibited in the presence of ephrin-A1. Thus, this study echoes the common theme that, because Slit/Robo signaling targets common signaling pathways, the context in which it signals determines the outcome. Clearly, additional studies are required to understand the complex and intertwined signaling that regulates neovascularization during disease processes. The current data are incomplete and conflicting, but there is one take home message from all these studies: Slit plays a central role in endothelial cell biology. As such, it holds promise as a therapeutic agent that could be used to treat pathologies and cancers that are made worse by enhanced angiogenesis.
Conclusion
Since their early discovery as key regulators of axon migration in the developing nervous system, the Slit/Robo signaling pair has been implicated in a wide variety of developmental and pathological processes. More specifically, Slit/Robo signaling has been found to have a significant impact on a cell’s behavior, from regulating cell migration to controlling cell growth, both processes critical in tumor cell progression. When activated aberrantly, these signaling events can promote tumor cell growth and migration, contributing to tumor metastasis and poor patient prognosis. As such, Slits and Robos are promising candidates for anti-cancer therapeutics, but care must be exercised because the bifunctionality of this signaling axis could result in both tumor suppressive and oncogenic outcomes. For example, in its pro-migratory role, Slit-induced Robo signaling causes decreased cell-cell attachments and initiates pro-migratory pathways. In this context, treatment with RoboN, the ectodomain of Robo1, causes sequestration of Slit in the environment, thus stabilizing cell-cell attachments and preventing metastasis. However, in other contexts, Slit/Robo signaling has the opposite effect, enhancing cell-cell contacts by increasing E-cadherin stability and downregulating transcriptional programs that promote proliferation. In this case, sequestering Slit by RoboN treatment would potentially weaken cell-cell contacts, enabling, rather than inhibiting, pro-migratory signals and tumor cell metastasis. Thus, since the downstream effects of Slit/Robo signaling vary greatly depending on both the extracellular and intracellular milieu, development of effective therapeutics will require a better understanding of their disparate signaling consequences. Furthermore, it will be necessary to design therapeutics with elaborate delivery mechanisms that ensure delivery of the drug to specific tissues in order to prevent unintended effects caused by changing normal Slit/Robo signaling in nearby tissues, as loss of Slit/Robo signaling in normal tissues and cells could cause deleterious effects. Despite these cautions, it is increasingly clear that Slits and Robos are key regulators of a wide variety of developmental and adult processes, from the epithelium to the vasculature, and that they hold promise as therapeutic targets in the fight against cancer and other diseases.
Acknowledgments
This work was supported by funds from the NIH RO1 (GM-098897, L.H.), Santa Cruz Cancer Benefit Group (L.H.) and the California Institute of Regenerative Medicine pre-doctoral fellowship program (TG2-01157, M.B.)
References
- Abdollahi A, Schwager C, Kleeff J, Esposito I, Domhan S, Peschke P, Hauser K, Hahnfeldt P, Hlatky L, Debus J, Peters JM, Friess H, Folkman J, Huber PE. Transcriptional network governing the angiogenic switch in human pancreatic cancer. Proc Natl Acad Sci U S A. 2007;104:12890–12895. doi: 10.1073/pnas.0705505104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alajez NM, Lenarduzzi M, Ito E, Hui AB, Shi W, Bruce J, Yue S, Huang SH, Xu W, Waldron J, O’Sullivan B, Liu FF. MiR-218 suppresses nasopharyngeal cancer progression through downregulation of survivin and the SLIT2-ROBO1 pathway. Cancer Res. 2011;71:2381–2391. doi: 10.1158/0008-5472.CAN-10-2754. [DOI] [PubMed] [Google Scholar]
- Allington TM, Schiemann WP. The Cain and Abl of epithelial-mesenchymal transition and transforming growth factor-beta in mammary epithelial cells. Cells Tissues Organs. 2011;193:98–113. doi: 10.1159/000320163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altay T, McLaughlin B, Wu JY, Park TS, Gidday JM. Slit modulates cerebrovascular inflammation and mediates neuroprotection against global cerebral ischemia. Exp Neurol. 2007;207:186–194. doi: 10.1016/j.expneurol.2007.06.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Angeloni D, ter Elst A, Wei MH, van der Veen AY, Braga EA, Klimov EA, Timmer T, Korobeinikova L, Lerman MI, Buys CH. Analysis of a new homozygous deletion in the tumor suppressor region at 3p12.3 reveals two novel intronic noncoding RNA genes. Genes Chromosomes Cancer. 2006;45:676–691. doi: 10.1002/gcc.20332. [DOI] [PubMed] [Google Scholar]
- Avci ME, Konu O, Yagci T. Quantification of SLIT-ROBO transcripts in hepatocellular carcinoma reveals two groups of genes with coordinate expression. BMC Cancer. 2008;8:392. doi: 10.1186/1471-2407-8-392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bashaw GJ, Kidd T, Murray D, Pawson T, Goodman CS. Repulsive axon guidance: Abelson and Enabled play opposing roles downstream of the roundabout receptor. Cell. 2000;101:703–715. doi: 10.1016/s0092-8674(00)80883-1. [DOI] [PubMed] [Google Scholar]
- Bauer K, Dowejko A, Bosserhoff AK, Reichert TE, Bauer R. Slit-2 facilitates interaction of P-cadherin with Robo-3 and inhibits cell migration in an oral squamous cell carcinoma cell line. Carcinogenesis. 2011;32:935–943. doi: 10.1093/carcin/bgr059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bear JE, Gertler FB. Ena/VASP: towards resolving a pointed controversy at the barbed end. J Cell Sci. 2009;122:1947–1953. doi: 10.1242/jcs.038125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bedell VM, Yeo SY, Park KW, Chung J, Seth P, Shivalingappa V, Zhao J, Obara T, Sukhatme VP, Drummond IA, Li DY, Ramchandran R. roundabout4 is essential for angiogenesis in vivo. Proc Natl Acad Sci U S A. 2005;102:6373–6378. doi: 10.1073/pnas.0408318102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blanco D, Vicent S, Elizegi E, Pino I, Fraga MF, Esteller M, Saffiotti U, Lecanda F, Montuenga LM. Altered expression of adhesion molecules and epithelial-mesenchymal transition in silica-induced rat lung carcinogenesis. Lab Invest. 2004;84:999–1012. doi: 10.1038/labinvest.3700129. [DOI] [PubMed] [Google Scholar]
- Brantley-Sieders DM, Dunaway CM, Rao M, Short S, Hwang Y, Gao Y, Li D, Jiang A, Shyr Y, Wu JY, Chen J. Angiocrine factors modulate tumor proliferation and motility through EphA2 repression of Slit2 tumor suppressor function in endothelium. Cancer Res. 2011;71:976–987. doi: 10.1158/0008-5472.CAN-10-3396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brose K, Bland KS, Wang KH, Arnott D, Henzel W, Goodman CS, Tessier-Lavigne M, Kidd T. Slit proteins bind Robo receptors and have an evolutionarily conserved role in repulsive axon guidance. Cell. 1999;96:795–806. doi: 10.1016/s0092-8674(00)80590-5. [DOI] [PubMed] [Google Scholar]
- Challa AK, Beattie CE, Seeger MA. Identification and characterization of roundabout orthologs in zebrafish. Mech Dev. 2001;101:249–253. doi: 10.1016/s0925-4773(00)00570-0. [DOI] [PubMed] [Google Scholar]
- Chanana B, Steigemann P, Jackle H, Vorbruggen G. Reception of Slit requires only the chondroitin-sulphate-modified extracellular domain of Syndecan at the target cell surface. Proc Natl Acad Sci U S A. 2009;106:11984–11988. doi: 10.1073/pnas.0901148106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen JH, Wen L, Dupuis S, Wu JY, Rao Y. The N-terminal leucine-rich regions in Slit are sufficient to repel olfactory bulb axons and subventricular zone neurons. J Neurosci. 2001;21:1548–1556. doi: 10.1523/JNEUROSCI.21-05-01548.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christgen M, Bruchhardt H, Hadamitzky C, Rudolph C, Steinemann D, Gadzicki D, Hasemeier B, Romermann D, Focken T, Krech T, Ballmaier M, Schlegelberger B, Kreipe H, Lehmann U. Comprehensive genetic and functional characterization of IPH-926: a novel CDH1-null tumour cell line from human lobular breast cancer. J Pathol. 2009;217:620–632. doi: 10.1002/path.2495. [DOI] [PubMed] [Google Scholar]
- Clark K, Hammond E, Rabbitts P. Temporal and spatial expression of two isoforms of the Dutt1/Robo1 gene in mouse development. FEBS Lett. 2002;523:12–16. doi: 10.1016/s0014-5793(02)02904-6. [DOI] [PubMed] [Google Scholar]
- Coleman HA, Labrador JP, Chance RK, Bashaw GJ. The Adam family metalloprotease Kuzbanian regulates the cleavage of the roundabout receptor to control axon repulsion at the midline. Development. 2010;137:2417–2426. doi: 10.1242/dev.047993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dallol A, Da Silva NF, Viacava P, Minna JD, Bieche I, Maher ER, Latif F. SLIT2, a human homologue of the Drosophila Slit2 gene, has tumor suppressor activity and is frequently inactivated in lung and breast cancers. Cancer Res. 2002a;62:5874–5880. [PubMed] [Google Scholar]
- Dallol A, Dickinson RE, Latif F. Epigenetic Disruption of the SLIT-ROBO Interactions in Human Cancer. Springer; New York, NY: 2005. [Google Scholar]
- Dallol A, Forgacs E, Martinez A, Sekido Y, Walker R, Kishida T, Rabbitts P, Maher ER, Minna JD, Latif F. Tumour specific promoter region methylation of the human homologue of the Drosophila Roundabout gene DUTT1 (ROBO1) in human cancers. Oncogene. 2002b;21:3020–3028. doi: 10.1038/sj.onc.1205421. [DOI] [PubMed] [Google Scholar]
- Dallol A, Krex D, Hesson L, Eng C, Maher ER, Latif F. Frequent epigenetic inactivation of the SLIT2 gene in gliomas. Oncogene. 2003a;22:4611–4616. doi: 10.1038/sj.onc.1206687. [DOI] [PubMed] [Google Scholar]
- Dallol A, Morton D, Maher ER, Latif F. SLIT2 axon guidance molecule is frequently inactivated in colorectal cancer and suppresses growth of colorectal carcinoma cells. Cancer Res. 2003b;63:1054–1058. [PubMed] [Google Scholar]
- Delloye-Bourgeois C, Fitamant J, Paradisi A, Cappellen D, Douc-Rasy S, Raquin MA, Stupack D, Nakagawara A, Rousseau R, Combaret V, Puisieux A, Valteau-Couanet D, Benard J, Bernet A, Mehlen P. Netrin-1 acts as a survival factor for aggressive neuroblastoma. J Exp Med. 2009;206:833–847. doi: 10.1084/jem.20082299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dickinson RE, Dallol A, Bieche I, Krex D, Morton D, Maher ER, Latif F. Epigenetic inactivation of SLIT3 and SLIT1 genes in human cancers. Br J Cancer. 2004;91:2071–2078. doi: 10.1038/sj.bjc.6602222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dickson BJ, Gilestro GF. Regulation of commissural axon pathfinding by slit and its Robo receptors. Annu Rev Cell Dev Biol. 2006;22:651–675. doi: 10.1146/annurev.cellbio.21.090704.151234. [DOI] [PubMed] [Google Scholar]
- Doi T, Hajduk P, Puri P. Upregulation of Slit-2 and Slit-3 gene expressions in the nitrofen-induced hypoplastic lung. J Pediatr Surg. 2009;44:2092–2095. doi: 10.1016/j.jpedsurg.2009.02.068. [DOI] [PubMed] [Google Scholar]
- Dunaway CM, Hwang Y, Lindsley CW, Cook RS, Wu JY, Boothby M, Chen J, Brantley-Sieders DM. Cooperative signaling between Slit2 and Ephrin-A1 regulates a balance between angiogenesis and angiostasis. Mol Cell Biol. 2011;31:404–416. doi: 10.1128/MCB.00667-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunwell TL, Dickinson RE, Stankovic T, Dallol A, Weston V, Austen B, Catchpoole D, Maher ER, Latif F. Frequent epigenetic inactivation of the SLIT2 gene in chronic and acute lymphocytic leukemia. Epigenetics. 2009;4:265–269. doi: 10.4161/epi.9137. [DOI] [PubMed] [Google Scholar]
- Evans TA, Bashaw GJ. Functional diversity of Robo receptor immunoglobulin domains promotes distinct axon guidance decisions. Curr Biol. 2010;20:567–572. doi: 10.1016/j.cub.2010.02.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan X, Labrador JP, Hing H, Bashaw GJ. Slit stimulation recruits Dock and Pak to the roundabout receptor and increases Rac activity to regulate axon repulsion at the CNS midline. Neuron. 2003;40:113–127. doi: 10.1016/s0896-6273(03)00591-9. [DOI] [PubMed] [Google Scholar]
- Fitamant J, Guenebeaud C, Coissieux MM, Guix C, Treilleux I, Scoazec JY, Bachelot T, Bernet A, Mehlen P. Netrin-1 expression confers a selective advantage for tumor cell survival in metastatic breast cancer. Proc Natl Acad Sci U S A. 2008;105:4850–4855. doi: 10.1073/pnas.0709810105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukuhara N, Howitt JA, Hussain SA, Hohenester E. Structural and functional analysis of slit and heparin binding to immunoglobulin-like domains 1 and 2 of Drosophila Robo. J Biol Chem. 2008;283:16226–16234. doi: 10.1074/jbc.M800688200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fulda S, Debatin KM. Extrinsic versus intrinsic apoptosis pathways in anticancer chemotherapy. Oncogene. 2006;25:4798–4811. doi: 10.1038/sj.onc.1209608. [DOI] [PubMed] [Google Scholar]
- Furne C, Rama N, Corset V, Chedotal A, Mehlen P. Netrin-1 is a survival factor during commissural neuron navigation. Proc Natl Acad Sci U S A. 2008;105:14465–14470. doi: 10.1073/pnas.0803645105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gertler F, Condeelis J. Metastasis: tumor cells becoming MENAcing. Trends Cell Biol. 2011;21:81–90. doi: 10.1016/j.tcb.2010.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosh S, Ghosh A, Maiti GP, Alam N, Roy A, Roychoudhury S, Panda CK. Alterations of ROBO1/DUTT1 and ROBO2 loci in early dysplastic lesions of head and neck: clinical and prognostic implications. Hum Genet. 2009;125:189–198. doi: 10.1007/s00439-008-0610-9. [DOI] [PubMed] [Google Scholar]
- Gorn M, Anige M, Burkholder I, Muller B, Scheffler A, Edler L, Boeters I, Panse J, Schuch G, Hossfeld DK, Laack E. Serum levels of Magic Roundabout protein in patients with advanced non-small cell lung cancer (NSCLC) Lung Cancer. 2005;49:71–76. doi: 10.1016/j.lungcan.2004.12.005. [DOI] [PubMed] [Google Scholar]
- Greenberg JM, Thompson FY, Brooks SK, Shannon JM, Akeson AL. Slit and robo expression in the developing mouse lung. Dev Dyn. 2004;230:350–360. doi: 10.1002/dvdy.20045. [DOI] [PubMed] [Google Scholar]
- Grone J, Doebler O, Loddenkemper C, Hotz B, Buhr HJ, Bhargava S. Robo1/Robo4: differential expression of angiogenic markers in colorectal cancer. Oncol Rep. 2006;15:1437–1443. [PubMed] [Google Scholar]
- Guan H, Zu G, Xie Y, Tang H, Johnson M, Xu X, Kevil C, Xiong WC, Elmets C, Rao Y, Wu JY, Xu H. Neuronal repellent Slit2 inhibits dendritic cell migration and the development of immune responses. J Immunol. 2003;171:6519–6526. doi: 10.4049/jimmunol.171.12.6519. [DOI] [PubMed] [Google Scholar]
- Hall A. The cytoskeleton and cancer. Cancer Metastasis Rev. 2009;28:5–14. doi: 10.1007/s10555-008-9166-3. [DOI] [PubMed] [Google Scholar]
- Han X, Zhang MC. Potential anti-angiogenic role of Slit2 in corneal neovascularization. Exp Eye Res. 2010;90:742–749. doi: 10.1016/j.exer.2010.03.007. [DOI] [PubMed] [Google Scholar]
- Hinck L. The versatile roles of “axon guidance” cues in tissue morphogenesis. Dev Cell. 2004;7:783–793. doi: 10.1016/j.devcel.2004.11.002. [DOI] [PubMed] [Google Scholar]
- Hohenester E. Structural insight into Slit-Robo signalling. Biochem Soc Trans. 2008;36:251–256. doi: 10.1042/BST0360251. [DOI] [PubMed] [Google Scholar]
- Hohenester E, Hussain S, Howitt JA. Interaction of the guidance molecule Slit with cellular receptors. Biochem Soc Trans. 2006;34:418–421. doi: 10.1042/BST0340418. [DOI] [PubMed] [Google Scholar]
- Howitt JA, Clout NJ, Hohenester E. Binding site for Robo receptors revealed by dissection of the leucine-rich repeat region of Slit. EMBO J. 2004;23:4406–4412. doi: 10.1038/sj.emboj.7600446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu H. Cell-surface heparan sulfate is involved in the repulsive guidance activities of Slit2 protein. Nat Neurosci. 2001;4:695–701. doi: 10.1038/89482. [DOI] [PubMed] [Google Scholar]
- Hu H, Li M, Labrador JP, McEwen J, Lai EC, Goodman CS, Bashaw GJ. Cross GTPase-activating protein (CrossGAP)/Vilse links the Roundabout receptor to Rac to regulate midline repulsion. Proc Natl Acad Sci U S A. 2005;102:4613–4618. doi: 10.1073/pnas.0409325102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang L, Xu Y, Yu W, Li X, Liqun C, He X, Peiying H. Robo1: a potential role in ocular angiogenesis. Curr Eye Res. 2009a;34:1019–1029. doi: 10.3109/02713680903308495. [DOI] [PubMed] [Google Scholar]
- Huang L, Yu W, Li X, Niu L, Li K, Li J. Robo1/robo4: different expression patterns in retinal development. Exp Eye Res. 2009b;88:583–588. doi: 10.1016/j.exer.2008.11.016. [DOI] [PubMed] [Google Scholar]
- Huang L, Yu W, Li X, Xu Y, Niu L, He X, Dong J, Yan Z. Expression of Robo4 in the fibrovascular membranes from patients with proliferative diabetic retinopathy and its role in RF/6A and RPE cells. Mol Vis. 2009c;15:1057–1069. [PMC free article] [PubMed] [Google Scholar]
- Huminiecki L, Bicknell R. In silico cloning of novel endothelial-specific genes. Genome Res. 2000;10:1796–1806. doi: 10.1101/gr.150700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huminiecki L, Gorn M, Suchting S, Poulsom R, Bicknell R. Magic roundabout is a new member of the roundabout receptor family that is endothelial specific and expressed at sites of active angiogenesis. Genomics. 2002;79:547–552. doi: 10.1006/geno.2002.6745. [DOI] [PubMed] [Google Scholar]
- Hussain SA, Piper M, Fukuhara N, Strochlic L, Cho G, Howitt JA, Ahmed Y, Powell AK, Turnbull JE, Holt CE, Hohenester E. A molecular mechanism for the heparan sulfate dependence of slit-robo signaling. J Biol Chem. 2006;281:39693–39698. doi: 10.1074/jbc.M609384200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inatani M, Irie F, Plump AS, Tessier-Lavigne M, Yamaguchi Y. Mammalian brain morphogenesis and midline axon guidance require heparan sulfate. Science. 2003;302:1044–1046. doi: 10.1126/science.1090497. [DOI] [PubMed] [Google Scholar]
- Ito H, Funahashi S, Yamauchi N, Shibahara J, Midorikawa Y, Kawai S, Kinoshita Y, Watanabe A, Hippo Y, Ohtomo T, Iwanari H, Nakajima A, Makuuchi M, Fukayama M, Hirata Y, Hamakubo T, Kodama T, Tsuchiya M, Aburatani H. Identification of ROBO1 as a novel hepatocellular carcinoma antigen and a potential therapeutic and diagnostic target. Clin Cancer Res. 2006;12:3257–3264. doi: 10.1158/1078-0432.CCR-05-2787. [DOI] [PubMed] [Google Scholar]
- Jin J, You H, Yu B, Deng Y, Tang N, Yao G, Shu H, Yang S, Qin W. Epigenetic inactivation of SLIT2 in human hepatocellular carcinomas. Biochem Biophys Res Commun. 2009;379:86–91. doi: 10.1016/j.bbrc.2008.12.022. [DOI] [PubMed] [Google Scholar]
- Johnson DW, Berg JN, Baldwin MA, Gallione CJ, Marondel I, Yoon SJ, Stenzel TT, Speer M, Pericak-Vance MA, Diamond A, Guttmacher AE, Jackson CE, Attisano L, Kucherlapati R, Porteous ME, Marchuk DA. Mutations in the activin receptor-like kinase 1 gene in hereditary haemorrhagic telangiectasia type 2. Nat Genet. 1996;13:189–195. doi: 10.1038/ng0696-189. [DOI] [PubMed] [Google Scholar]
- Johnson KG, Ghose A, Epstein E, Lincecum J, O’Connor MB, Van Vactor D. Axonal heparan sulfate proteoglycans regulate the distribution and efficiency of the repellent slit during midline axon guidance. Curr Biol. 2004;14:499–504. doi: 10.1016/j.cub.2004.02.005. [DOI] [PubMed] [Google Scholar]
- Jones CA, London NR, Chen H, Park KW, Sauvaget D, Stockton RA, Wythe JD, Suh W, Larrieu-Lahargue F, Mukouyama YS, Lindblom P, Seth P, Frias A, Nishiya N, Ginsberg MH, Gerhardt H, Zhang K, Li DY. Robo4 stabilizes the vascular network by inhibiting pathologic angiogenesis and endothelial hyperpermeability. Nat Med. 2008;14:448–453. doi: 10.1038/nm1742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones CA, Nishiya N, London NR, Zhu W, Sorensen LK, Chan AC, Lim CJ, Chen H, Zhang Q, Schultz PG, Hayallah AM, Thomas KR, Famulok M, Zhang K, Ginsberg MH, Li DY. Slit2-Robo4 signalling promotes vascular stability by blocking Arf6 activity. Nat Cell Biol. 2009;11:1325–1331. doi: 10.1038/ncb1976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanellis J, Garcia GE, Li P, Parra G, Wilson CB, Rao Y, Han S, Smith CW, Johnson RJ, Wu JY, Feng L. Modulation of inflammation by slit protein in vivo in experimental crescentic glomerulonephritis. Am J Pathol. 2004;165:341–352. doi: 10.1016/S0002-9440(10)63301-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kastenhuber E, Kern U, Bonkowsky JL, Chien CB, Driever W, Schweitzer J. Netrin-DCC, Robo-Slit, and heparan sulfate proteoglycans coordinate lateral positioning of longitudinal dopaminergic diencephalospinal axons. J Neurosci. 2009;29:8914–8926. doi: 10.1523/JNEUROSCI.0568-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaur S, Castellone MD, Bedell VM, Konar M, Gutkind JS, Ramchandran R. Robo4 signaling in endothelial cells implies attraction guidance mechanisms. J Biol Chem. 2006;281:11347–11356. doi: 10.1074/jbc.M508853200. [DOI] [PubMed] [Google Scholar]
- Kaur S, Samant GV, Pramanik K, Loscombe PW, Pendrak ML, Roberts DD, Ramchandran R. Silencing of directional migration in roundabout4 knockdown endothelial cells. BMC Cell Biol. 2008;9:61. doi: 10.1186/1471-2121-9-61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerbel RS. Tumor angiogenesis. N Engl J Med. 2008;358:2039–2049. doi: 10.1056/NEJMra0706596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khusial PR, Vadla B, Krishnan H, Ramlall TF, Shen Y, Ichikawa H, Geng JG, Goldberg GS. Src activates Abl to augment Robo1 expression in order to promote tumor cell migration. Oncotarget. 2010;1:198–209. doi: 10.18632/oncotarget.126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kidd T, Bland KS, Goodman CS. Slit is the midline repellent for the robo receptor in Drosophila. Cell. 1999;96:785–794. doi: 10.1016/s0092-8674(00)80589-9. [DOI] [PubMed] [Google Scholar]
- Koch AW, Mathivet T, Larrivee B, Tong RK, Kowalski J, Pibouin-Fragner L, Bouvree K, Stawicki S, Nicholes K, Rathore N, Scales SJ, Luis E, del Toro R, Freitas C, Breant C, Michaud A, Corvol P, Thomas JL, Wu Y, Peale F, Watts RJ, Tessier-Lavigne M, Bagri A, Eichmann A. Robo4 maintains vessel integrity and inhibits angiogenesis by interacting with UNC5B. Dev Cell. 2011;20:33–46. doi: 10.1016/j.devcel.2010.12.001. [DOI] [PubMed] [Google Scholar]
- Kramer SG, Kidd T, Simpson JH, Goodman CS. Switching repulsion to attraction: changing responses to slit during transition in mesoderm migration. Science. 2001;292:737–740. doi: 10.1126/science.1058766. [DOI] [PubMed] [Google Scholar]
- Latil A, Bieche I, Chene L, Laurendeau I, Berthon P, Cussenot O, Vidaud M. Gene expression profiling in clinically localized prostate cancer: a four-gene expression model predicts clinical behavior. Clin Cancer Res. 2003;9:5477–5485. [PubMed] [Google Scholar]
- Lee JS, Ray R, Chien CB. Cloning and expression of three zebrafish roundabout homologs suggest roles in axon guidance and cell migration. Dev Dyn. 2001;221:216–230. doi: 10.1002/dvdy.1136. [DOI] [PubMed] [Google Scholar]
- Lee JS, von der Hardt S, Rusch MA, Stringer SE, Stickney HL, Talbot WS, Geisler R, Nusslein-Volhard C, Selleck SB, Chien CB, Roehl H. Axon sorting in the optic tract requires HSPG synthesis by ext2 (dackel) and extl3 (boxer) Neuron. 2004;44:947–960. doi: 10.1016/j.neuron.2004.11.029. [DOI] [PubMed] [Google Scholar]
- Legg JA, Herbert JM, Clissold P, Bicknell R. Slits and Roundabouts in cancer, tumour angiogenesis and endothelial cell migration. Angiogenesis. 2008;11:13–21. doi: 10.1007/s10456-008-9100-x. [DOI] [PubMed] [Google Scholar]
- Leonardo ED, Hinck L, Masu M, Keino-Masu K, Ackerman SL, Tessier-Lavigne M. Vertebrate homologues of C. elegans UNC-5 are candidate netrin receptors. Nature. 1997;386:833–838. doi: 10.1038/386833a0. [DOI] [PubMed] [Google Scholar]
- Liang Y, Annan RS, Carr SA, Popp S, Mevissen M, Margolis RK, Margolis RU. Mammalian homologues of the Drosophila slit protein are ligands of the heparan sulfate proteoglycan glypican-1 in brain. J Biol Chem. 1999;274:17885–17892. doi: 10.1074/jbc.274.25.17885. [DOI] [PubMed] [Google Scholar]
- Liu D, Hou J, Hu X, Wang X, Xiao Y, Mou Y, De Leon H. Neuronal chemorepellent Slit2 inhibits vascular smooth muscle cell migration by suppressing small GTPase Rac1 activation. Circ Res. 2006;98:480–489. doi: 10.1161/01.RES.0000205764.85931.4b. [DOI] [PubMed] [Google Scholar]
- Liu Z, Patel K, Schmidt H, Andrews W, Pini A, Sundaresan V. Extracellular Ig domains 1 and 2 of Robo are important for ligand (Slit) binding. Mol Cell Neurosci. 2004;26:232–240. doi: 10.1016/j.mcn.2004.01.002. [DOI] [PubMed] [Google Scholar]
- London NR, Li DY. Robo4-dependent Slit signaling stabilizes the vasculature during pathologic angiogenesis and cytokine storm. Curr Opin Hematol. 2011;18:186–190. doi: 10.1097/MOH.0b013e328345a4b9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- London NR, Zhu W, Bozza FA, Smith MC, Greif DM, Sorensen LK, Chen L, Kaminoh Y, Chan AC, Passi SF, Day CW, Barnard DL, Zimmerman GA, Krasnow MA, Li DY. Targeting Robo4-dependent Slit signaling to survive the cytokine storm in sepsis and influenza. Sci Transl Med. 2010;2:23ra19. doi: 10.1126/scitranslmed.3000678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lundstrom A, Gallio M, Englund C, Steneberg P, Hemphala J, Aspenstrom P, Keleman K, Falileeva L, Dickson BJ, Samakovlis C. Vilse, a conserved Rac/Cdc42 GAP mediating Robo repulsion in tracheal cells and axons. Genes Dev. 2004;18:2161–2171. doi: 10.1101/gad.310204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma XJ, Wang Z, Ryan PD, Isakoff SJ, Barmettler A, Fuller A, Muir B, Mohapatra G, Salunga R, Tuggle JT, Tran Y, Tran D, Tassin A, Amon P, Wang W, Enright E, Stecker K, Estepa-Sabal E, Smith B, Younger J, Balis U, Michaelson J, Bhan A, Habin K, Baer TM, Brugge J, Haber DA, Erlander MG, Sgroi DC. A two-gene expression ratio predicts clinical outcome in breast cancer patients treated with tamoxifen. Cancer Cell. 2004;5:607–616. doi: 10.1016/j.ccr.2004.05.015. [DOI] [PubMed] [Google Scholar]
- Macias H, Moran A, Samara Y, Moreno M, Compton JE, Harburg G, Strickland P, Hinck L. SLIT/ROBO1 signaling suppresses mammary branching morphogenesis by limiting basal cell number. Dev Cell. 2011;20:827–840. doi: 10.1016/j.devcel.2011.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marlow R, Binnewies M, Sorensen LK, Monica SD, Strickland P, Forsberg EC, Li DY, Hinck L. Vascular Robo4 restricts proangiogenic VEGF signaling in breast. Proc Natl Acad Sci U S A. 2010;107:10520–10525. doi: 10.1073/pnas.1001896107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marlow R, Strickland P, Lee JS, Wu X, Pebenito M, Binnewies M, Le EK, Moran A, Macias H, Cardiff RD, Sukumar S, Hinck L. SLITs suppress tumor growth in vivo by silencing Sdf1/Cxcr4 within breast epithelium. Cancer Res. 2008;68:7819–7827. doi: 10.1158/0008-5472.CAN-08-1357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medioni C, Bertrand N, Mesbah K, Hudry B, Dupays L, Wolstein O, Washkowitz AJ, Papaioannou VE, Mohun TJ, Harvey RP, Zaffran S. Expression of Slit and Robo genes in the developing mouse heart. Dev Dyn. 2010;239:3303–3311. doi: 10.1002/dvdy.22449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mertsch S, Schmitz N, Jeibmann A, Geng JG, Paulus W, Senner V. Slit2 involvement in glioma cell migration is mediated by Robo1 receptor. J Neurooncol. 2008;87:1–7. doi: 10.1007/s11060-007-9484-2. [DOI] [PubMed] [Google Scholar]
- Moore MA. The role of chemoattraction in cancer metastases. Bioessays. 2001;23:674–676. doi: 10.1002/bies.1095. [DOI] [PubMed] [Google Scholar]
- Moore SW, Tessier-Lavigne M, Kennedy TE. Netrins and their receptors. Adv Exp Med Biol. 2007;621:17–31. doi: 10.1007/978-0-387-76715-4_2. [DOI] [PubMed] [Google Scholar]
- Morlot C, Hemrika W, Romijn RA, Gros P, Cusack S, McCarthy AA. Production of Slit2 LRR domains in mammalian cells for structural studies and the structure of human Slit2 domain 3. Acta Crystallogr D Biol Crystallogr. 2007a;63:961–968. doi: 10.1107/S0907444907035470. [DOI] [PubMed] [Google Scholar]
- Morlot C, Thielens NM, Ravelli RB, Hemrika W, Romijn RA, Gros P, Cusack S, McCarthy AA. Structural insights into the Slit-Robo complex. Proc Natl Acad Sci U S A. 2007b;104:14923–14928. doi: 10.1073/pnas.0705310104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mulik S, Sharma S, Suryawanshi A, Veiga-Parga T, Reddy PB, Rajasagi NK, Rouse BT. Activation of endothelial roundabout receptor 4 reduces the severity of virus-induced keratitis. J Immunol. 2011;186:7195–7204. doi: 10.4049/jimmunol.1100014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller A, Homey B, Soto H, Ge N, Catron D, Buchanan ME, McClanahan T, Murphy E, Yuan W, Wagner SN, Barrera JL, Mohar A, Verastegui E, Zlotnik A. Involvement of chemokine receptors in breast cancer metastasis. Nature. 2001;410:50–56. doi: 10.1038/35065016. [DOI] [PubMed] [Google Scholar]
- Mura M, Swain RK, Zhuang X, Vorschmitt H, Reynolds G, Durant S, Beesley JF, Herbert JM, Sheldon H, Andre M, Sanderson S, Glen K, Luu NT, McGettrick HM, Antczak P, Falciani F, Nash GB, Nagy ZS, Bicknell R. Identification and angiogenic role of the novel tumor endothelial marker CLEC14A. Oncogene. 2011 doi: 10.1038/onc.2011.233. [DOI] [PubMed] [Google Scholar]
- Narayan G, Goparaju C, Arias-Pulido H, Kaufmann AM, Schneider A, Durst M, Mansukhani M, Pothuri B, Murty VV. Promoter hypermethylation-mediated inactivation of multiple Slit-Robo pathway genes in cervical cancer progression. Mol Cancer. 2006;5:16. doi: 10.1186/1476-4598-5-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen Ba-Charvet KT, Brose K, Ma L, Wang KH, Marillat V, Sotelo C, Tessier-Lavigne M, Chedotal A. Diversity and specificity of actions of Slit2 proteolytic fragments in axon guidance. J Neurosci. 2001;21:4281–4289. doi: 10.1523/JNEUROSCI.21-12-04281.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogata-Iwao M, Inatani M, Iwao K, Takihara Y, Nakaishi-Fukuchi Y, Irie F, Sato S, Furukawa T, Yamaguchi Y, Tanihara H. Heparan sulfate regulates intraretinal axon pathfinding by retinal ganglion cells. Invest Ophthalmol Vis Sci. 2011;52:6671–6679. doi: 10.1167/iovs.11-7559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park KW, Morrison CM, Sorensen LK, Jones CA, Rao Y, Chien CB, Wu JY, Urness LD, Li DY. Robo4 is a vascular-specific receptor that inhibits endothelial migration. Dev Biol. 2003;261:251–267. doi: 10.1016/s0012-1606(03)00258-6. [DOI] [PubMed] [Google Scholar]
- Parsons DW, Jones S, Zhang X, Lin JC, Leary RJ, Angenendt P, Mankoo P, Carter H, Siu IM, Gallia GL, Olivi A, McLendon R, Rasheed BA, Keir S, Nikolskaya T, Nikolsky Y, Busam DA, Tekleab H, Diaz LA, Jr, Hartigan J, Smith DR, Strausberg RL, Marie SK, Shinjo SM, Yan H, Riggins GJ, Bigner DD, Karchin R, Papadopoulos N, Parmigiani G, Vogelstein B, Velculescu VE, Kinzler KW. An integrated genomic analysis of human glioblastoma multiforme. Science. 2008;321:1807–1812. doi: 10.1126/science.1164382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piper M, Anderson R, Dwivedy A, Weinl C, van Horck F, Leung KM, Cogill E, Holt C. Signaling mechanisms underlying Slit2-induced collapse of Xenopus retinal growth cones. Neuron. 2006;49:215–28. doi: 10.1016/j.neuron.2005.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piper M, Georgas K, Yamada T, Little M. Expression of the vertebrate Slit gene family and their putative receptors, the Robo genes, in the developing murine kidney. Mech Dev. 2000;94:213–217. doi: 10.1016/s0925-4773(00)00313-0. [DOI] [PubMed] [Google Scholar]
- Plump AS, Erskine L, Sabatier C, Brose K, Epstein CJ, Goodman CS, Mason CA, Tessier-Lavigne M. Slit1 and Slit2 cooperate to prevent premature midline crossing of retinal axons in the mouse visual system. Neuron. 2002;33:219–232. doi: 10.1016/s0896-6273(01)00586-4. [DOI] [PubMed] [Google Scholar]
- Potente M, Gerhardt H, Carmeliet P. Basic and therapeutic aspects of angiogenesis. Cell. 2011;146:873–887. doi: 10.1016/j.cell.2011.08.039. [DOI] [PubMed] [Google Scholar]
- Prasad A, Fernandis AZ, Rao Y, Ganju RK. Slit protein-mediated inhibition of CXCR4-induced chemotactic and chemoinvasive signaling pathways in breast cancer cells. J Biol Chem. 2004;279:9115–9124. doi: 10.1074/jbc.M308083200. [DOI] [PubMed] [Google Scholar]
- Prasad A, Paruchuri V, Preet A, Latif F, Ganju RK. Slit-2 induces a tumor-suppressive effect by regulating beta-catenin in breast cancer cells. J Biol Chem. 2008;283:26624–26633. doi: 10.1074/jbc.M800679200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prasad A, Qamri Z, Wu J, Ganju RK. Slit-2/Robo-1 modulates the CXCL12/CXCR4-induced chemotaxis of T cells. J Leukoc Biol. 2007;82:465–476. doi: 10.1189/jlb.1106678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiu H, Zhu J, Yu J, Pu H, Dong R. SLIT2 is epigenetically silenced in ovarian cancers and suppresses growth when activated. Asian Pac J Cancer Prev. 2011;12:791–795. [PubMed] [Google Scholar]
- Rhee J, Buchan T, Zukerberg L, Lilien J, Balsamo J. Cables links Robo-bound Abl kinase to N-cadherin-bound beta-catenin to mediate Slit-induced modulation of adhesion and transcription. Nat Cell Biol. 2007;9:883–892. doi: 10.1038/ncb1614. [DOI] [PubMed] [Google Scholar]
- Rhee J, Mahfooz NS, Arregui C, Lilien J, Balsamo J, VanBerkum MF. Activation of the repulsive receptor Roundabout inhibits N-cadherin-mediated cell adhesion. Nat Cell Biol. 2002;4:798–805. doi: 10.1038/ncb858. [DOI] [PubMed] [Google Scholar]
- Rhiner C, Gysi S, Frohli E, Hengartner MO, Hajnal A. Syndecan regulates cell migration and axon guidance in C. elegans. Development. 2005;132:4621–4633. doi: 10.1242/dev.02042. [DOI] [PubMed] [Google Scholar]
- Richardson AL, Wang ZC, De Nicolo A, Lu X, Brown M, Miron A, Liao X, Iglehart JD, Livingston DM, Ganesan S. X chromosomal abnormalities in basal-like human breast cancer. Cancer Cell. 2006;9:121–132. doi: 10.1016/j.ccr.2006.01.013. [DOI] [PubMed] [Google Scholar]
- Ronca F, Andersen JS, Paech V, Margolis RU. Characterization of Slit protein interactions with glypican-1. J Biol Chem. 2001;276:29141–29147. doi: 10.1074/jbc.M100240200. [DOI] [PubMed] [Google Scholar]
- Salvucci O, Bouchard A, Baccarelli A, Deschenes J, Sauter G, Simon R, Bianchi R, Basik M. The role of CXCR4 receptor expression in breast cancer: a large tissue microarray study. Breast Cancer Res Treat. 2006;97:275–283. doi: 10.1007/s10549-005-9121-8. [DOI] [PubMed] [Google Scholar]
- Santiago-Martinez E, Soplop NH, Patel R, Kramer SG. Repulsion by Slit and Roundabout prevents Shotgun/E-cadherin-mediated cell adhesion during Drosophila heart tube lumen formation. J Cell Biol. 2008;182:241–248. doi: 10.1083/jcb.200804120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmid BC, Rezniczek GA, Fabjani G, Yoneda T, Leodolter S, Zeillinger R. The neuronal guidance cue Slit2 induces targeted migration and may play a role in brain metastasis of breast cancer cells. Breast Cancer Res Treat. 2007;106:333–342. doi: 10.1007/s10549-007-9504-0. [DOI] [PubMed] [Google Scholar]
- Schulz JG, Ceulemans H, Caussinus E, Baietti MF, Affolter M, Hassan BA, David G. Drosophila syndecan regulates tracheal cell migration by stabilizing Robo levels. EMBO Rep. 2011;12:1039–1046. doi: 10.1038/embor.2011.153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seki M, Watanabe A, Enomoto S, Kawamura T, Ito H, Kodama T, Hamakubo T, Aburatani H. Human ROBO1 is cleaved by metalloproteinases and gamma-secretase and migrates to the nucleus in cancer cells. FEBS Lett. 2010;584:2909–2915. doi: 10.1016/j.febslet.2010.05.009. [DOI] [PubMed] [Google Scholar]
- Seth P, Lin Y, Hanai J, Shivalingappa V, Duyao MP, Sukhatme VP. Magic roundabout, a tumor endothelial marker: expression and signaling. Biochem Biophys Res Commun. 2005;332:533–541. doi: 10.1016/j.bbrc.2005.03.250. [DOI] [PubMed] [Google Scholar]
- Sharma G, Mirza S, Prasad CP, Srivastava A, Gupta SD, Ralhan R. Promoter hypermethylation of p16INK4A, p14ARF, CyclinD2 and Slit2 in serum and tumor DNA from breast cancer patients. Life Sci. 2007;80:1873–1881. doi: 10.1016/j.lfs.2007.02.026. [DOI] [PubMed] [Google Scholar]
- Sheldon H, Andre M, Legg JA, Heal P, Herbert JM, Sainson R, Sharma AS, Kitajewski JK, Heath VL, Bicknell R. Active involvement of Robo1 and Robo4 in filopodia formation and endothelial cell motility mediated via WASP and other actin nucleation-promoting factors. FASEB J. 2009;23:513–522. doi: 10.1096/fj.07-098269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shiau CE, Bronner-Fraser M. N-cadherin acts in concert with Slit1-Robo2 signaling in regulating aggregation of placode-derived cranial sensory neurons. Development. 2009;136:4155–4164. doi: 10.1242/dev.034355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shivapurkar N, Sood S, Wistuba II, Virmani AK, Maitra A, Milchgrub S, Minna JD, Gazdar AF. Multiple regions of chromosome 4 demonstrating allelic losses in breast carcinomas. Cancer Res. 1999a;59:3576–3580. [PubMed] [Google Scholar]
- Shivapurkar N, Virmani AK, Wistuba II, Milchgrub S, Mackay B, Minna JD, Gazdar AF. Deletions of chromosome 4 at multiple sites are frequent in malignant mesothelioma and small cell lung carcinoma. Clin Cancer Res. 1999b;5:17–23. [PubMed] [Google Scholar]
- Singh RK, Indra D, Mitra S, Mondal RK, Basu PS, Roy A, Roychowdhury S, Panda CK. Deletions in chromosome 4 differentially associated with the development of cervical cancer: evidence of slit2 as a candidate tumor suppressor gene. Hum Genet. 2007;122:71–81. doi: 10.1007/s00439-007-0375-6. [DOI] [PubMed] [Google Scholar]
- Smart AD, Course MM, Rawson J, Selleck S, Van Vactor D, Johnson KG. Heparan sulfate proteoglycan specificity during axon pathway formation in the Drosophila embryo. Dev Neurobiol. 2011;71:608–618. doi: 10.1002/dneu.20854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spitzweck B, Brankatschk M, Dickson BJ. Distinct protein domains and expression patterns confer divergent axon guidance functions for Drosophila Robo receptors. Cell. 2010;140:409–420. doi: 10.1016/j.cell.2010.01.002. [DOI] [PubMed] [Google Scholar]
- Steigemann P, Molitor A, Fellert S, Jackle H, Vorbruggen G. Heparan sulfate proteoglycan syndecan promotes axonal and myotube guidance by slit/robo signaling. Curr Biol. 2004;14:225–230. doi: 10.1016/j.cub.2004.01.006. [DOI] [PubMed] [Google Scholar]
- Stein E, Tessier-Lavigne M. Hierarchical organization of guidance receptors: silencing of netrin attraction by slit through a Robo/DCC receptor complex. Science. 2001;291:1928–1938. doi: 10.1126/science.1058445. [DOI] [PubMed] [Google Scholar]
- Stella MC, Trusolino L, Comoglio PM. The Slit/Robo system suppresses hepatocyte growth factor-dependent invasion and morphogenesis. Mol Biol Cell. 2009;20:642–657. doi: 10.1091/mbc.E08-03-0321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strickland P, Shin GC, Plump A, Tessier-Lavigne M, Hinck L. Slit2 and netrin 1 act synergistically as adhesive cues to generate tubular bi-layers during ductal morphogenesis. Development. 2006;133:823–832. doi: 10.1242/dev.02261. [DOI] [PubMed] [Google Scholar]
- Suchting S, Heal P, Tahtis K, Stewart LM, Bicknell R. Soluble Robo4 receptor inhibits in vivo angiogenesis and endothelial cell migration. FASEB J. 2005;19:121–123. doi: 10.1096/fj.04-1991fje. [DOI] [PubMed] [Google Scholar]
- Suryawanshi A, Mulik S, Sharma S, Reddy PB, Sehrawat S, Rouse BT. Ocular neovascularization caused by herpes simplex virus type 1 infection results from breakdown of binding between vascular endothelial growth factor A and its soluble receptor. J Immunol. 2011;186:3653–3665. doi: 10.4049/jimmunol.1003239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tie J, Pan Y, Zhao L, Wu K, Liu J, Sun S, Guo X, Wang B, Gang Y, Zhang Y, Li Q, Qiao T, Zhao Q, Nie Y, Fan D. MiR-218 inhibits invasion and metastasis of gastric cancer by targeting the Robo1 receptor. PLoS Genet. 2010;6:e1000879. doi: 10.1371/journal.pgen.1000879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tole S, Mukovozov IM, Huang YW, Magalhaes MA, Yan M, Crow MR, Liu GY, Sun CX, Durocher Y, Glogauer M, Robinson LA. The axonal repellent, Slit2, inhibits directional migration of circulating neutrophils. J Leukoc Biol. 2009;86:1403–1415. doi: 10.1189/jlb.0609391. [DOI] [PubMed] [Google Scholar]
- Tseng RC, Lee SH, Hsu HS, Chen BH, Tsai WC, Tzao C, Wang YC. SLIT2 attenuation during lung cancer progression deregulates beta-catenin and E-cadherin and associates with poor prognosis. Cancer Res. 2010;70:543–551. doi: 10.1158/0008-5472.CAN-09-2084. [DOI] [PubMed] [Google Scholar]
- Urness LD, Sorensen LK, Li DY. Arteriovenous malformations in mice lacking activin receptor-like kinase-1. Nat Genet. 2000;26:328–331. doi: 10.1038/81634. [DOI] [PubMed] [Google Scholar]
- Verissimo AR, Herbert JM, Heath VL, Legg JA, Sheldon H, Andre M, Swain RK, Bicknell R. Functionally defining the endothelial transcriptome, from Robo4 to ECSCR. Biochem Soc Trans. 2009;37:1214–1217. doi: 10.1042/BST0371214. [DOI] [PubMed] [Google Scholar]
- Wang B, Xiao Y, Ding BB, Zhang N, Yuan X, Gui L, Qian KX, Duan S, Chen Z, Rao Y, Geng JG. Induction of tumor angiogenesis by Slit-Robo signaling and inhibition of cancer growth by blocking Robo activity. Cancer Cell. 2003;4:19–29. doi: 10.1016/s1535-6108(03)00164-8. [DOI] [PubMed] [Google Scholar]
- Wang KH, Brose K, Arnott D, Kidd T, Goodman CS, Henzel W, Tessier-Lavigne M. Biochemical purification of a mammalian slit protein as a positive regulator of sensory axon elongation and branching. Cell. 1999;96:771–784. doi: 10.1016/s0092-8674(00)80588-7. [DOI] [PubMed] [Google Scholar]
- Wang LJ, Zhao Y, Han B, Ma YG, Zhang J, Yang DM, Mao JW, Tang FT, Li WD, Yang Y, Wang R, Geng JG. Targeting Slit-Roundabout signaling inhibits tumor angiogenesis in chemical-induced squamous cell carcinogenesis. Cancer Sci. 2008;99:510–517. doi: 10.1111/j.1349-7006.2007.00721.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Werbowetski-Ogilvie TE, Seyed Sadr M, Jabado N, Angers-Loustau A, Agar NY, Wu J, Bjerkvig R, Antel JP, Faury D, Rao Y, Del Maestro RF. Inhibition of medulloblastoma cell invasion by Slit. Oncogene. 2006;25:5103–5112. doi: 10.1038/sj.onc.1209524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong K, Ren XR, Huang YZ, Xie Y, Liu G, Saito H, Tang H, Wen L, Brady-Kalnay SM, Mei L, Wu JY, Xiong WC, Rao Y. Signal transduction in neuronal migration: roles of GTPase activating proteins and the small GTPase Cdc42 in the Slit-Robo pathway. Cell. 2001;107:209–221. doi: 10.1016/s0092-8674(01)00530-x. [DOI] [PubMed] [Google Scholar]
- Wu JY, Feng L, Park HT, Havlioglu N, Wen L, Tang H, Bacon KB, Jiang Z, Zhang X, Rao Y. The neuronal repellent Slit inhibits leukocyte chemotaxis induced by chemotactic factors. Nature. 2001;410:948–952. doi: 10.1038/35073616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xian J, Clark KJ, Fordham R, Pannell R, Rabbitts TH, Rabbitts PH. Inadequate lung development and bronchial hyperplasia in mice with a targeted deletion in the Dutt1/Robo1 gene. Proc Natl Acad Sci U S A. 2001;98:15062–15066. doi: 10.1073/pnas.251407098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Y, Li WL, Fu L, Gu F, Ma YJ. Slit2/Robo1 signaling in glioma migration and invasion. Neurosci Bull. 2010;26:474–478. doi: 10.1007/s12264-010-0730-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang L, Bashaw GJ. Son of sevenless directly links the Robo receptor to rac activation to control axon repulsion at the midline. Neuron. 2006;52:595–607. doi: 10.1016/j.neuron.2006.09.039. [DOI] [PubMed] [Google Scholar]
- Yang XM, Han HX, Sui F, Dai YM, Chen M, Geng JG. Slit-Robo signaling mediates lymphangiogenesis and promotes tumor lymphatic metastasis. Biochem Biophys Res Commun. 2010;396:571–577. doi: 10.1016/j.bbrc.2010.04.152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye BQ, Geng ZH, Ma L, Geng JG. Slit2 regulates attractive eosinophil and repulsive neutrophil chemotaxis through differential srGAP1 expression during lung inflammation. J Immunol. 2010;185:6294–6305. doi: 10.4049/jimmunol.1001648. [DOI] [PubMed] [Google Scholar]
- Yiin JJ, Hu B, Jarzynka MJ, Feng H, Liu KW, Wu JY, Ma HI, Cheng SY. Slit2 inhibits glioma cell invasion in the brain by suppression of Cdc42 activity. Neuro Oncol. 2009;11:779–789. doi: 10.1215/15228517-2008-017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu J, Cao Q, Wu L, Dallol A, Li J, Chen G, Grasso C, Cao X, Lonigro RJ, Varambally S, Mehra R, Palanisamy N, Wu JY, Latif F, Chinnaiyan AM. The neuronal repellent SLIT2 is a target for repression by EZH2 in prostate cancer. Oncogene. 2010;29:5370–5380. doi: 10.1038/onc.2010.269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuasa-Kawada J, Kinoshita-Kawada M, Rao Y, Wu JY. Deubiquitinating enzyme USP33/VDU1 is required for Slit signaling in inhibiting breast cancer cell migration. Proc Natl Acad Sci U S A. 2009;106:14530–14535. doi: 10.1073/pnas.0801262106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang B, Dietrich UM, Geng JG, Bicknell R, Esko JD, Wang L. Repulsive axon guidance molecule Slit3 is a novel angiogenic factor. Blood. 2009;114:4300–4309. doi: 10.1182/blood-2008-12-193326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang F, Ronca F, Linhardt RJ, Margolis RU. Structural determinants of heparan sulfate interactions with Slit proteins. Biochem Biophys Res Commun. 2004;317:352–357. doi: 10.1016/j.bbrc.2004.03.059. [DOI] [PubMed] [Google Scholar]
- Zheng W, Geng AQ, Li PF, Wang Y, Yuan XB. Robo4 Regulates the Radial Migration of Newborn Neurons in Developing Neocortex. Cereb Cortex. 2011 doi: 10.1093/cercor/bhr330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou WJ, Geng ZH, Chi S, Zhang W, Niu XF, Lan SJ, Ma L, Yang X, Wang LJ, Ding YQ, Geng JG. Slit-Robo signaling induces malignant transformation through Hakai-mediated E-cadherin degradation during colorectal epithelial cell carcinogenesis. Cell Res. 2011;21:609–626. doi: 10.1038/cr.2011.17. [DOI] [PMC free article] [PubMed] [Google Scholar]