

Abstract
Abnormal expression of solute carrier family 34 (sodium phosphate), member 2 (SLC34A2) in the lung may induce abnormal alveolar type II (AT II) cells to transform into lung adenocarcinoma cells, and may also be important in biological process of lung adenocarcinoma. However, at present, the effects and molecular mechanisms of SLC34A2 in the initiation and progression of lung cancer remain to be elucidated. To the best of our knowledge, the present study revealed for the first time that the expression levels of SLC34A2 were downregulated in the A549 and H1299 lung adenocarcinoma cell lines. Further investigation demonstrated that the elevated expression of SLC34A2 in A549 cells was able to significantly inhibit cell viability and invasion in vitro. In addition, 10 upregulated genes between the A549-P-S cell line stably expressing SLC34A2 and the control cell line A549-P were identified by microarray analysis and quantitative polymerase chain reaction, including seven tumor suppressor genes and three complement genes. Furthermore, the upregulation of complement gene C3 and complement 4B preproprotein (C4b) in A549-P-S cells was confirmed by ELISA analysis and was identified to be correlated with recovering Pi absorption in A549 cells by the phosphomolybdic acid method by enhancing the expression of SLC34A2. Therefore, it was hypothesized that the mechanisms underlying the effect of SLC34A2 on A549 cells might be associated with the activation of the complement alternative pathway (C3 and C4b) and upregulation of the expression of selenium binding protein 1, thioredoxin-interacting protein, PDZK1-interacting protein 1 and dual specificity protein phosphatase 6. Downregulation of SLC34A2 may primarily cause abnormal AT II cells to escape from complement-associated immunosurveillance and abnormally express certain tumor-suppressor genes inducing AT II cells to develop into lung adenocarcinoma. The present study further elucidated the effects and mechanisms of SLC34A2 in the generation and development of lung cancer.
Keywords: complement, initiation and progression, molecular mechanism, A549 lung adenocarcinoma cells, SLC34A2, viability and invasion
Introduction
As the incidence of lung adenocarcinoma has rapidly increased, it has become a major pathological type of lung cancer (1). Revealing the molecular pathogenesis of lung adenocarcinoma may enable improvement in the diagnosis and treatment of this disease.
The gene solute carrier family 34 (sodium phosphate), member 2 (SLC34A2) is a member of the SLC34 family located on chromosome 4p15–p16. The full-length cDNA of SLC34A2 is 4,167 bp with an open reading frame that encodes a 689-amino-acid protein. The SLC34A2 gene encodes the type 2b sodium-phosphate cotransporter NaPi-IIb (2,3), which is responsible for the transcellular absorption of Pi in an apical membrane (4–6). According to previous studies, mutations in SLC34A2 led to the occurrence of pulmonary alveolar microlithiasis, testicular microlithiasis and hypophosphatemia (7–9). Previous studies have suggested that the tumorigenesis of several types of cancer might be associated with abnormal expression of SLC34A2, including papillary thyroid, ovarian and breast cancer (10).
In 1999, Xu et al (6) revealed that SLC34A2 was expressed in numerous human tissues, with adult and fetal lungs demonstrating the highest levels of expression. Shibasaki et al (11) confirmed that targeted deletion of the SLC34A2 gene resulted in early embryonic lethality, and suggested that SLC34A2 was a vital gene in early embryonic development. Simultaneously, a study by Kopantzev et al (12) confirmed that the mRNA expression level of SLC34A2 was increased during human lung embryogenesis; however, was decreased in non-small cell lung carcinoma (NSCLC). These studies proposed that SLC34A2 might be a novel candidate for a molecular marker of NSCLC. It is widely accepted that the decreased expression of a gene in lung cancer tends to exhibit a monotonically increased expression during lung development. By contrast, upregulated genes in various types of lung cancer tend to exhibit a monotonically downregulated expression during lung development (13–15). For example, a key gene of lung embryogenesis (16,17), caveolin 1 (CAV1), which has the opposite trend of expression between embryogenesis and tumorigenesis (15), has been implicated in oncogenic cell transformation, tumorigenesis and metastasis (18). Furthermore, downregulation of CAV1 has been confirmed to be a promoting factor in the development of lung cancer (19,20). These studies prompted us to hypothesize that SLC34A2 might be linked to the onset of lung cancer, and further motivated the investigation of the effects and molecular mechanisms of SLC34A2 in the initiation and progression of lung cancer.
In the lung, SLC34A2 is expressed primarily in alveolar type II (AT II) cells (21). The AT II cells are not only responsible for the production of surfactant fluids, but are also the potential pulmonary alveolar epithelium stem cells, which are able to differentiate into alveolar type I (AT I) cells and is capable of self renewal (21–23). Previous studies demonstrated that the AT II cells were a progenitor cell of lung adenocarcinoma and bronchioloalveolar carcinoma (24,25). In addition, Kitinya et al (26) and Gazdar et al (27) also found that AT II cells might be the progenitor cells of several types of lung carcinoma, including large cell carcinoma, adenocarcinoma and squamous cell carcinoma, particularly lung adenocarcinoma. In addition, previous studies verified that long-term exposure to carcinogenic factors was able to cause AT II cells to transform into lung cancer cells (28,29). In 2009, Xu et al (30) found that a diet low in Pi might affect normal lung development by disturbing the Akt-FGF-2 signals associated with tumor progression. Xu et al also indicated that pulmonary NaPi-IIb was critical in Pi metabolism. These studies highlighted that a lack of Pi might be associated with the pathogenesis of lung cancer. Thus, it was hypothesized that a lower expression of SLC34A2 in AT II cells might lead to the deficiency in Pi, which might cause the hyperproliferation and lack of differentiation of AT II cells, and then cause these abnormal AT II cells to transform into lung adenocarcinoma. SLC34A2 might therefore be important in the development of lung adenocarcinoma.
To examine this hypothesis, the expression of SLC34A2 in A549 and H1299 lung adenocarcinoma cells compared with normal human bronchial epithelial (HBE) cells was first detected by quantitative polymerase chain reaction (qPCR). The AT II cell-like A549 human lung adenocarcinoma cell line was then selected for further identification of the biological functions of SLC34A2 in lung cancer cells. The present study preliminarily revealed the effects and mechanisms of SLC34A2 against A549 lung adenocarcinoma cells in vitro, and provided insights into the effects and mechanisms of SLC34A2 in the generation and development of lung cancer.
Materials and methods
Cell culture
The HBE human bronchial epithelial cell line obtained from the American Type Culture Collection (ATCC, Arlington, VA, USA) was cultured in Dulbecco’s modified Eagle’s medium supplemented with 10% fetal bovine serum (FBS; Gibco-BRL, Carlsbad, CA, USA). The cells from the primary explants in their first passage were infected with the recombinant retrovirus LXSN16E6E7 containing the human papilloma virus E6E7 gene. The cells were selected in the presence of 0.4 mg/ml G418 (31). The A549 human lung adenocarcinoma cell line (32) and H1299 cell line obtained from the ATCC were maintained in RPMI-1640 supplemented with 10% FBS. For all experiments described, the cells were incubated in the aforementioned medium at 5% CO2 and at 37°C.
Plasmid construction
According to the SLCS4A2 full-length coding region (Gene Bank serial no. NM_006424), the sense primer, containing a BamHI site at its 5′-end (SLCS4A2 forward: 5′-GCGGATCCTAATGGCTCCCTGGCCTGAAT-3′) and an antisense primer, containing an EcoRI site at its 5′-end (reverse: 5′-GCGAATTCCTACAAGGCCGTGCATTCG-3′) were used to clone the coding DNA sequence of SLCS4A2 from the pcmv-sport6-SLCS4A2 (Open Biosystems, Inc., Huntsville, AL, USA) using a PrimeSTAR HS PCR kit (Takara, Dalian, China). The cloned cDNA sequence was connected to the pcDNA3.1 plasmid vector (InvivoGen, San Diego, CA, USA) using a DNA Ligation kit Ver. 2.0 (Takara). Pure pcDNA3.1 and pcDNA3.1-SLC34A2 plasmids were prepared using an EndofreeTM Plasmid Giga kit (Qiagen, Chatsworth, CA, USA) for the following experiments.
SLC34A2 gene expression analysis by qPCR
Total RNA from cultured cells was isolated using TRIzol reagent (Invitrogen Life Technologies) according to the manufacturer’s instructions. Synthesis of cDNA with reverse transcriptase was performed using a PrimeScript RT reagent kit (Perfect Real Time) (Takara). For SLC34A2 gene expression analysis, qPCR analysis was performed in the iCycler iQ5 real-time PCR detection system (Bio-Rad Laboratories, Hercules, CA, USA) with SYBR-Green reagents (Bio-Rad Laboratories), SLC34A2-specific primers and a probe (Invitrogen Life Technologies). The human GAPDH primer and probe reagents were used as the normalization control in subsequent quantitative analysis.
Stable transfection of A549 cells with SLC34A2
A549 cells were seeded on 6-well plates at 2×105/well. When the cell density reached 60–70%, they were transfected with the pcDNA3.1-SLC34A2 and pcDNA3.1 plasmids using Lipofectamine 2000 (InvivoGen). Following 48 h, the transfected cells were replaced with a medium containing G418 (800 μg/ml; Sigma, St. Louis, MO, USA) to eliminate nontransfected cells. Individual colonies appeared following 2 weeks. Then, G418-resistant colonies were isolated into a 96-well plate to be expanded. Following 2–3 weeks, when the quantity of positive transfected cells was enough, they were cultured in 6-well plates with 500 μg/ml G418. Following this, the positive transfected cells were identified by determining whether SLC34A2 was expressed stably by qPCR and western blot analysis. Stable transfectants were maintained in the medium containing G418 (500 μg/ml). A549 cells stably expressing pcDNA3.1-SLC34A2 and pcDNA3.1 were designated A549-pcDNA3.1-SLC34A2 (A549-P-S) and A549-pcDNA3.1 (A549-P), respectively, for further analysis.
Growth curve
The cells were plated into 96-well plates at 2.0×104 cells/well. The effect of SLC34A2 on the cell viability of A549 cells was determined using an 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay. At the indicated time points (1, 2, 3, 4, 5, 6 and 7 days), the culture medium was replaced with 180 μl of fresh medium, and 20 μl MTT solution (5 mg/ml; Sigma) was added to the culture medium and incubated for another 4 h at 37°C. Following 4 h, the unreacted dye was removed by aspiration. Blue formazan crystals were observed in the well when examined under a microscope (Olympus IX51; Olympus, Tokyo, Japan). Following this, 150 ml dimethylsulfoxide was added to each well and then incubated on a shaker for 10 min at room temperature to dissolve the blue crystal. The absorbance was measured using a microplate reader (Bio-Rad Laboratories) at a wavelength of 490 nm. The percentage cell viability was then calculated using the following formula: [optical density (OD)490 (treated cells) / OD490 (control)] × 100.
Colony formation assay
The cells were seeded into 6-well plates in triplicates at a density of 300 cells/well in 2 ml of medium containing 10% FBS. The cultures were regularly replaced with fresh medium in a 37°C humidified atmosphere containing 5% CO2 and grown for 3 weeks. The cell clones were fixed with pure methanol and stained for 15 min with a solution containing 0.05% crystal violet, then followed by three rinses with double distilled water to remove excess dye. The colony numbers were counted using a gel documentation system (ImagePro Plus software; Media Cybernetics, Rockville, MD, USA). Additionally, the colony formation rates were calculated in terms of the number of A549-P-S cells and A549-P cells relative to A549 cells.
Cell invasion analysis
The cell invasion assay was performed using a Boyden chamber (Millipore, Billerica, MA, USA) with BD Matrigel™ (BD Biosciences, Franklin Lakes, NJ, USA). The filters in the upper compartment were loaded with 400 μl serum-free RPMI-1640 containing 5×104 cells, and filters in the lower compartment were filled with 600 μl RPMI-1640 containing 10% FBS. The chamber was then cultured in 5% CO2 at 37°C for 24 h. Then, the Matrigel and cells in the upper chamber were removed, and the attached cells in the lower section were fixed with pure methanol and stained with 0.05% crystal violet. The number of migrated cells was counted in five randomly selected power fields (magnification, ×200) under a light microscope. The invasion rates were calculated in terms of the number of A549-P-S cells and A549-P cells relative to A549 cells.
Microarray analysis
Total RNA extraction from A549-P and A549-P-S using TRIzol reagent (Invitrogen Life Technologies, Gaithersburg, MD, USA) and purification of the RNA using a NucleoSpin® RNA clean-up kit (Macherey-Nagel, Düren, Germany) were completed and prepared for microarray analysis. The commercially available 35K Human Genome Array, including 25,000 human genes, was obtained from CapitalBio Corporation (Beijing, China). Double-stranded cDNAs were synthesized from 1 μg total RNA using the CbcScript reverse transcriptase with the T7 Oligo (dT). The dsDNA was transcribed into cRNA in vitro transcription reactions at 37°C for 4–14 h using a T7 enzyme mix. Next, 2 μg of cRNA was reverse transcribed to generate cDNA using CbcScript II reverse transcriptase. The cDNA was fluorescently labeled by Cy5 or Cy3 CPT with the Klenow enzyme following reverse transcription. The cDNA of A549-P-S cells was labeled with Cy3-CPT and the cDNA of A549-P cells was labeled with Cy5-CPT as a control. Array hybridization was performed in a CapitalBio BioMixerTM II Hybridization station overnight at a rotation speed of 8 rpm and a temperature of 42°C, and subsequently washed with two consecutive solutions (0.2% sodium dodecyl sulfate, 2X saline-sodium citrate at 42°C for 5 min and 0.2X saline-sodium citrate for 5 min at room temperature). These arrays were scanned with a confocal LuxScan™ scanner and the images obtained were then analyzed using LuxScan 3.0 software (CapitalBio Corporation). The obtained images were analyzed using LuxScan 3.0 (CapitalBio Corporation), which employed the LOWESS normalization method (33). The differentially expressed genes that exhibited an average ratio in triplicate tests >2.0-fold upregulated or <0.5-fold downregulated were obtained. Significance Analysis of Microarrays (LightCycler software version 3.02) was performed.
Identification of differentially expressed genes by qPCR
To further identify the results of the cDNA microarray data, qPCR was performed in the iCycler iQ5 real-time PCR detection system (Bio-Rad Laboratories) using EQ SYBR-Green dye (Bio-Rad Laboratories) according to the manufacturer’s instructions. The comparative threshold cycle (CT) method was used to calculate the amplification fold. β-actin (forward: 5′-CTTAGTTGCGTTACACCCTTTCTTG-3′ and reverse: 5′-CTTAGTTGCGTTACACCCTTTCTTG-3′) was used as a reference control gene to normalize the expression value of each gene. The primer sequences are listed in Table I.
Table I.
Primer sequences and detection results of differentially expressed genes identified by qPCR.
| Gene name | Primer direction | Primer sequence (5′-3′) | Product (bp) | Upregulation fold |
|---|---|---|---|---|
| C3 | F-1 | AAAGATAAGAACCGCTGGGAGG | 117 | 20.88 |
| R-1 | CACGACGGGAGGCACAAA | |||
| C5 | F-1 | CCCACTACAGAGGCTACG | 305 | 3.18 |
| R-1 | AGGACTGAGAAACCCAAC | |||
| C4b | F-1 | CTGTCTGCCTACTGGATTGC | 205 | 12.15 |
| R-1 | CCTTCAGGGTTCCTTTGC | |||
| FGA | F-1 | AGGCAACACTTACCACTG | 176 | 26.15 |
| R-1 | GTAATCTCATTTCCACCAG | |||
| FGG | F-1 | ACATTGCCAATAAGGGAG | 209 | 10.15 |
| R-1 | TGTTGTGCCAGTAGGAGA | |||
| FGB | F-1 | TTGCCCATAGAAACGAGG | 153 | 14.82 |
| R-1 | GTGGCGACTTGGAGTGAA | |||
| SELENBP1 | F-1 | CAAAGTATGGCTACAGGG | 84 | 10.16 |
| R-1 | AGTGGCTCTAAGACGATT | |||
| TXNIP | F-1 | CCACCGTCATTTCTAACT | 147 | 20.90 |
| R-1 | ACACCTCCACTATCACCC | |||
| PDZK1IP1 | F-1 | TTTCAGGCGGACACCAAT | 217 | 8.41 |
| R-1 | ACGAGGACCAGGAACACG | |||
| DUSP6 | F-1 | AGCGACTGGAACGAGAAT | 297 | 7.01 |
| R-1 | GTTGGACAGCGGACTACC |
F, forward; R, reverse; C3, complement C3 precursor; C5, complement C5 precursor; C4b, complement 4B preproprotein; FGA, fibrinogen α chain precursor; FGG, fibrinogen γ chain precursor; FGB, fibrinogen β chain precursor; SELENBP1, selenium binding protein 1; TXNIP, thioredoxin-interacting protein; PDZK1IP1, PDZK1-interacting protein 1; DUSP6, dual specificity protein phosphatase 6.
Measurement of C3 and C4b concentrations by ELISA
The cells were plated onto 96-well plates at 4.0×104 cells/well with three duplicate wells. Then, the supernatants of each group of cells at 24, 48, 72 and 96 h were respectively collected to measure human complement factor C3 using the C3 ELISA kit (Cusabio, Wuhan, China) and C4b concentration using the C4b ELISA kit (Cusabio) according to the manufacturer’s instructions (34). The protein concentrations of C3 and C4b were quantified in the media supernatants.
Determination of extracellular phosphate ion concentrations using the phosphomolybdic acid method
The same supernatants of each group of cells at 24, 48, 72 and 96 h were used to detect extracellular phosphate ion concentration by the phosphomolybdic acid method (Nanjing Jiancheng Bioengineering Institute, Nanjing, China) according to the manufacturer’s instructions (35). The quantities of Pi were quantified in the media supernatants.
Statistical analysis
Each experiment was performed at least three times. Statistical differences between the A549-P-S groups and A549-P groups (or A549 groups) were assessed using one-way analysis of variance and an unpaired Student’s t-test. All analyses were performed using SPSS software, version 19.0 (IBM, Armonk, NY, USA). P<0.05 was considered to indicate a statistically significant difference.
Results
Downregulation of SLC34A2 in A549 and H1299 lung adenocarcinoma cell lines
qPCR was performed to investigate whether the expression of SLC34A2 was different in A549 and H1299 lung adenocarcinoma cells compared with normal human bronchial HBE cells (Fig. 1A). The results indicated that the expression levels of SLC34A2 were downregulated 66.51-fold in the H1299 cell line and 104.98-fold in the A549 cell line compared with the HBE cell line (P<0.01). This suggested that SLC34A2 might be associated with the initiation and progression of lung adenocarcinoma.
Figure 1.
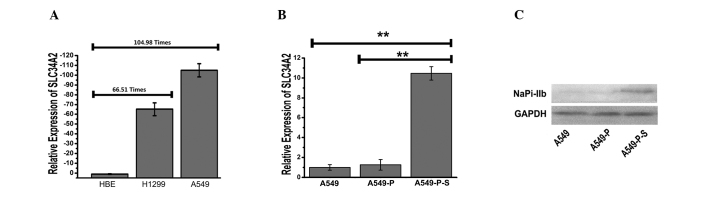
Expression of SLC34A2 between normal lung cells and lung adenocarcinoma cells and detection of a stable transfection cell line. (A) qPCR analysis was used to assess the mRNA levels of SLC34A2 expression in A549 and H1299 lung adenocarcinoma cells compared with normal HBE lung cells. (B) mRNA expression levels of SLC34A2 in the stably transfected cell lines A549, A549-P and A549-P-S were detected by qPCR analysis. (C) Protein expression levels of SLC34A2 in the stably transfected cell lines A549, A549-P and A549-P-S were determined by western blotting. The values are expressed as the mean ± standard error of the mean of three independent sets of data. **P<0.01. qPCR, quantitative polymerase chain reaction; SLC34A2, solute carrier family 34 (sodium phosphate), member 2.
Generation of A549 cells with stably expressing SLC34A2
In order to investigate the effects of SLC34A2 upregulation in human lung adenocarcinoma, A549 cells stably expressing SLC34A2 (A549-P-S) or PcDNA3.1 vector (A549-P) were initially selected by G418. qPCR and western blot analysis were performed to detect the expression of SLC34A2 in A549-P-S compared with A549-P and A549 cells. As shown in Fig. 1B, the expression level of SLC34A2 in the A549-P-S group was upregulated 10.46-fold compared with the A549 cell line (P<0.01). As shown in Fig. 1C, the SLC34A2 protein (NaPi-IIb) was identified in the A549-P-S group and was also functionally expressed compared with the A549-P group. The results confirmed that the A549 cell line with stable transfection of SLC34A2 was constructed successfully.
Effects of SLC34A2 on the viability and invasion of A549 cells
The cell viability of A549, A549-P and A549-P-S cells was examined at consecutive time points (1, 2, 3, 4, 5, 6 and 7 days) using the MTT assay and colony forming assay. As shown in Fig. 2A, the cell viability of the A549-P-S group was the lowest at each time point. As shown in Fig. 2B, the colony formation rate of the A549-P group was 83%; however, it was only 53% in the A549-P-S group (P<0.01). The data were consistent with the results of the growth curve of SLC34A2. In addition, a transwell assay was performed to detect the cell invasion ability of SLC34A2 (Fig. 2C). It was revealed that the quantity of the cells in the chamber decreased by 7% in the A549-P group, however, decreased by 42% in the A549-P-S group (P<0.01). These results indicated that enhancing the expression of SLC34A2 significantly inhibited the viability and invasion of A549 cells.
Figure 2.

Effects of SLC34A2 on the viability and invasion of A549 cells. (A) 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide analysis detected the different cell viability of the stably transfected cell line A549-P-S compared with A549 and A549-P-S cells for seven consecutive days. (B) Colony formation experiment confirmed the different cell growth rate of the stably transfected cell line A549-P-S compared with A549 and A549-P-S cells following 2 weeks. (C) A transwell assay demonstrated the different cell invasion of the stably transfected cell line A549-P-S compared with A549 and A549-P-S following 24 h (magnification, ×200). The values are expressed as the mean ± standard error of the mean of three independent sets of data. **P<0.01. SLC34A2, solute carrier family 34 (sodium phosphate), member 2.
Microarray data analysis and identification of differentially expressed genes
In order to investigate the molecular mechanisms underlying the effects of SLC34A2 on the viability and invasion of A549 cells, an oligonucleotide microarray was used to screen differentially expressed genes between A549-P-S and A549-P cells (Fig. 3A). As shown in Table II, the expression levels of 10 genes were higher in the A549-P-S group compared with the A549-P group. The upregulated genes included complement genes (C3, C4b and C5), complement-associated genes [fibrinogen α chain precursor (FGA), fibrinogen β chain precursor (FGB) and fibrinogen γ chain precursor (FGG)] and tumor suppressor genes [selenium binding protein 1 (SELENBP1), thioredoxin-interacting protein (TXNIP), PDZK1-interacting protein 1 (PDZK1IP) and dual specificity protein phosphatase 6 (DUSP6)]. To further validate the precision of microarray data, qPCR was performed using the same RNA sample as that used in the microarray. As shown in Fig. 3B, the results of qPCR were marginally different to the microarray; however, the upregulation observed was consistent with the microarray. Therefore, the results suggest that the effects of SLC34A2 on A549 cells might be associated with alterations in these genes.
Figure 3.
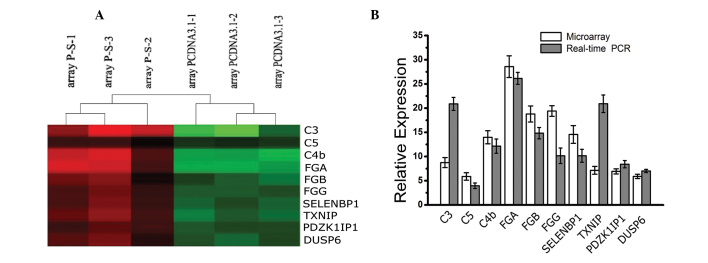
Differentially expressed genes between A549-P-S and A549-P cells by microarray assay and qPCR validation. (A) Each group in triplicate was shown on the same chip. Color intensity was assigned to ratios of gene expression; shades of red indicate genes that were upregulated; shades of green indicate genes that were downregulated. (B) 10 upregulated genes were identified by qPCR, which was consistent with cDNA microarray data. qPCR, quantitative polymerase chain reaction; C3, complement C3 precursor; C4b, complement 4B preproprotein; FGA, fibrinogen α chain precursor; FGB, fibrinogen β chain precursor; FGG, fibrinogen γ chain precursor; SELENBP1, selenium binding protein 1; TXNIP, thioredoxin-interacting protein; PDZK1IP1, PDZK-interacting protein 1; DUSP6, dual specificity protein phosphatase 6.
Table II.
Microarray analysis to determing overexpressed genes in an A549 cell line stably expressing SLC34A2.
| GenBank acc. no. | Average ratio (Cy5/Cy3) | GenBank identity | Gene ontology molecular function |
|---|---|---|---|
| NM_001002029 | 13.98 | Homo sapiens complement component 4B preproprotein (C4B) | Complement and coagulation cascades |
| NM_000064 | 8.75 | Homo sapiens complement C3 precursor (C3) | Complement and coagulation cascades |
| NM_001735 | 5.88 | Homo sapiens complement C5 precursor (C5) | Complement and coagulation cascades |
| NM_021871 | 28.58 | Homo sapiens fibrinogen α chain precursor (FGA) | Complement and coagulation cascades |
| NM_000509 | 19.41 | Homo sapiens fibrinogen γ chain precursor (FGG) | Complement and coagulation cascades |
| NM_005141 | 18.80 | Homo sapiens fibrinogen β chain precursor (FGB) | Complement and coagulation cascades |
| NM_005025 | 14.56 | Homo sapiens selenium binding protein 1 (SELENBP1) | Contributed - metabolic_process--Hs_Selenoproteins |
| NM_006472 | 7.15 | Homo sapiens Thioredoxin-interacting protein (TXNIP) | Tumor suppressor and thioredoxin pathway |
| NM_005764 | 6.93 | Homo sapiens PDZK1-interacting protein 1 (PDZK1IP1) | Tumor suppressor |
| NM_001946 | 5.90 | Homo sapiens dual specificity protein phosphatase 6 (DUSP6) | Activates extracellular signal-regulated kinase 2 (ERK2) |
SLC34A2, solute carrier family 34 (sodium phosphate), member 2.
Effect of SLC34A2 on C3 and C4b production in A549 cells
To further evaluate whether enhancing the expression of SLC34A2 was able to increase the secretion of C3 and C4b in A549 cells, cell culture supernatants of A549, A549-P and A549-P-S cells were used to detect the concentration of C3 and C4b by ELISA at consecutive time points (24, 48, 72 and 96 h). As shown in Fig. 4A, the C3 concentration in cell culture supernatants of the A549-P-S group respectively increased to 1.40 ng/ml (1.26 ng/ml), 4.58 ng/ml (3.90 ng/ml), 5.87 ng/ml (7.24 ng/ml) and 8.59 ng/ml (9.30 ng/ml) at the continuous time points compared with the A549 group (A549-P group; P<0.01). Simultaneously, the other results, shown in Fig. 4B, demonstrated that C4b concentration in the cell culture supernatants of the A549-P-S group respectively increased to 0.30 ng/ml (0.27 ng/ml), 0.71 ng/ml (0.66 ng/ml), 0.92 ng/ml (0.61 ng/ml), 1.16 ng/ml (0.99 ng/ml) at the continuous time points compared with the A549 group (A549-P group; P<0.01). These data further indicated that enhancing the expression of SLC34A2 was able to significantly increase the secretion of the complement factor C3 and C4b in A549 cells.
Figure 4.
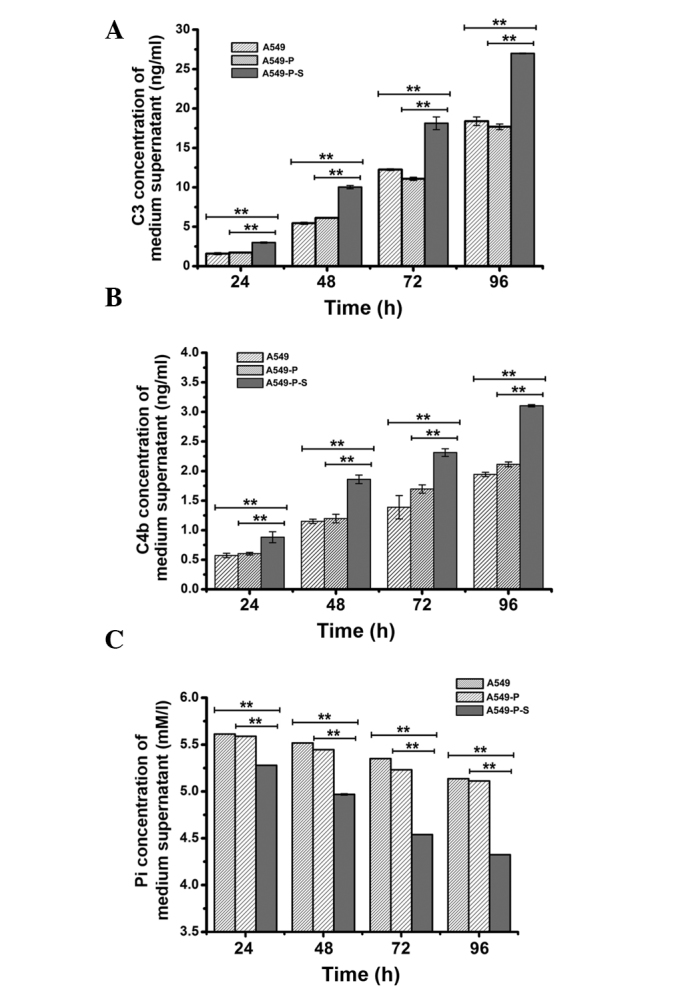
Effects of SLC34A2 on C3, C4b production and Pi absorption of A549 cells. (A) Effect of SLC34A2 on C3 production of A549 cells was detected by ELISA assay at consecutive time points. (B) Effect of SLC34A2 on the C4b production of A549 cells was detected by an ELISA assay at consecutive time points. (C) Effect of SLC34A2 on the absorption of Pi in A549 cells was assayed by the phosphomolybdic acid method at consecutive time points. The values are expressed as the mean ± standard error of the mean of three independent sets of data. **P<0.01. C3, complement C3 precursor; C4b, complement 4B preproprotein; SLC34A2, solute carrier family 34 (sodium phosphate), member 2.
Effect of SLC34A2 on Pi absorption in A549 cells
To determine whether enhancing the expression of SLC34A2 in A549 cells was able to increase the absorption of Pi in A549 cells and whether increasing Pi absorption was associated with the effect of SLC34A2 on the secretion of C3 and C4b, cell culture supernatants used in the detection of C3 and C4b concentration were also used to detect Pi concentration using the phosphomolybdic acid method. As shown in Fig. 4C, the Pi concentration in the cell culture supernatants of the A549-P-S group decreased to 0.33 mmol/l (0.31 mmol/l), 0.54 mmol/l (0.47 mmol/l), 0.811 mmol/l (0.69 mmol/l) and 0.812 mmol/l (0.78 mmol/l), respectively, at the continuous time points compared with the A549 group (A549-P group; P<0.01). The Pi concentration and the secretion of C3 and C4b in the A549-P-S group was greater than in the other two groups. The results suggested that not only was enhancing the expression of SLC34A2 in A549 cells able to increase Pi absorption of A549 cells, but also enhancing Pi absorption capacity may have a certain connection with the effect of SLC34A2 on the secretion of C3 and C4b in A549 cells.
Discussion
The role of SLC34A2 in carcinogenesis has not been fully elucidated, however, previous studies have indicated that this gene might be a potential molecular marker in carcinogenesis. Gaiłza et al (36) found that the expression of SLC34A2 was increased in papillary thyroid carcinoma, and suggested that this gene might be used as a potential biomarker in the diagnosis of papillary thyroid carcinoma. Blanchard et al (37) revealed that the expression of SLC34A2 was decreased in breast cancer, suggesting that this gene had a certain association with breast cancer. Yin et al (38) found that SLC34A2 was able to be used as a target for MX35 to treat ovarian cancer. Previous studies have demonstrated that the expression of SLC34A2 between normal lung tissue and lung cancer tissue was significantly different (12). To the best of our knowledge, the present study provided the first evidence, that the expression levels of the SLC34A2 gene in A549 and H1299 cells are clearly different compared with HBE cells by qPCR. Furthermore, the results demonstrate that enhancing the expression of SLC34A2 in A549 cells was able to significantly suppress the viability and invasion of A549 cells in vitro. These results imply that downregulation of SLC34A2 may be associated with the initiation and progression of lung adenocarcinoma.
Microarray analysis is able to simultaneously analyze almost 10,000 genes in a chip with added benefits, including high-flux and high-sensitivity (39). For further examining the potential mechanisms of SLC34A2 in lung adenocarcinoma, differentially expressed genes between A549-P and A549-P-S cells were screened using microarray analysis. A total of 10 upregulated genes, including complement genes (C3, C4b and C5), complement associated genes (FGA, FGB and FGG) and tumor suppressor genes (SELENBP1, TXNIP, PDZK1IP1 and DUSP6) were identified by microarray and qPCR. The results suggested that the effects of SLC34A2 on A549 cells might be associated with the expression changes of these genes.
AT II cells possess immune functions. They can directly synthesize factors of immune regulation, including the complement C2, C3, C4 and C5 factors and interleukin (IL)-3 (40). It is well known that the complement system (C1–C9) is important in immunosurveillance in the initial stage of tumorigenesis (41). Usually, the antigen-antibody complex formed through an antibody combining with a tumor cell antigen is able to activate C1 and C4, thereby further activating C3, and then activating a complement cascade to kill tumor cells in vivo (42). Another study indicated that the tumor cells themselves could also directly activate complement C3, resulting in activation of the complement alternative pathway to kill tumor cells (43). In addition, the complement alternative pathway was also activated when complement C3 bound to the tumor cell receptor in vitro, resulting in the formation of the membrane attack complex (C5b-9) to destroy the tumor cell membrane lipid bilayer and cause the tumor cell to be lysed (44). As the key to activating the complement alternative pathway, C3 is the most important complement factor in the complement alternative pathway. In addition, C4b is an active fragment of C4, which is involved in activating C3 (43,44). C5 is a key complement factor involved in the formation of the membrane attack complex. The present study revealed that enhancing the expression of SLC34A2 was able to increase mRNA expression levels of C3, C4b and C5 in A549 cells by microarray analysis. Furthermore, the present study confirmed that enhancing the expression of SLC34A2 was also able to stimulate the secretion of C3 and C4b in A549 cells by an ELISA assay. Rothman et al (45,46) found that cytokines IL-1, IL-2 as well as lipopolysaccharide increased C3 production in A549 cells. In addition, dexamethasone and interferon-γ had the same effect on A549 cells (34). Duerst et al (47) found that monoclonal antibodies (mAbs) were able to activate C3 to induce complement-dependent cytotoxicity, which led to tumor cells lysis. With the exception of mAbs, corresponding inhibitors of membrane complement regulatory proteins were also able to activate the complement alternative pathway through activating C3 (48). However, at present, there is no report of a relevant gene that is capable of activating C3 production in vitro. To the best of our knowledge, the present study was the first to reveal that SLC34A2 was able to increase the secretion of C3 and C4b in A549 cells.
In AT II cells, SLC34A2 is responsible for the synthesis of phosphate, which is the main component of surfactant in AT II cells. The surfactant is able to increase the activity of membrane proteins and the mobility of phospholipids (49). Previous studies found that improving the activity of cell surface proteins was useful for activating C3 to trigger the complement alternative pathway (50,51). The present study confirmed that enhancing the expression of SLC34A2 was able to significantly strengthen the ability to absorb Pi in the A549 cell line. The results of the present study also demonstrated that the effect of SLC34A2 on promoting the secretion of C3 and C4b may be associated with a time-dependent increase of Pi absorption. Based on the these results, it was hypothesized that the effect of the inhibition of SLC34A2 on the viability and invasion of A549 cells in vitro might be attributed to the activation of the complement alternative pathway by C3 activation and increased Pi absorption. It was also suggested that the lack of Pi might aid the escape of abnormal AT II cells from complement-associated immunosurveillance in the initial stages of lung adenocarcinoma development, when downregulation of SLC34A2 induces aberrant Pi transport. Therefore, downregulation of SLC34A2 might cause abnormal AT II cells to develop into lung adenocarcinoma cells. However, in the future, investigation of the effects of inhibiting C3 and C4b on the proliferation of lung cancer cells is required.
Adequate phosphate absorption is important for the maintenance of cellular metabolism (52). The study by Xu et al (30) demonstrated that the low Pi environment was able to activate certain cell signaling pathways associated with tumorigenesis in the normal lung cells, including AKT signaling pathways. AKT signaling pathways were generally active in lung cancer cells (53). A total of four tumor suppressor genes were identified including, SELENBP1, TXNIP, PDZK1IP1 and DUSP, which were upregulated in A549-P-S cells compared with A549-P cells. DUSP6 is an upstream inhibitor of ERK2, which is an important signaling protein in the AKT signaling pathway. Overexpression of DUSP6 in NSCLC may inactivate ERK2 and further act as a natural terminator of AKT/MAPK signal transduction (54). The present study found that increasing the expression of SLC34A2 in A549 cells was able to increase DUSP6 expression. Furthermore, Pi absorption in A549 cells increased. Therefore, it was hypothesized that the effects of SLC34A2 on the viability and invasion of A549 cells might be associated with the upregulation of DUSP6 in the AKT/ERK2 signaling pathway. However, further investigation is in progress. As a member of the selenium-containing protein family, the expression of SELENBP1 is commonly decreased in numerous types of human epithelial cancer, and this decrease is correlated with a poor prognosis. Previous studies have suggested that SELENBP1 exerts a tumor suppressor function by inhibition of proliferation and was identified as a lung cancer suppressor HIF-1 target gene (55–58). TXNIP is a member of the thioredoxin pathway and a tumor metastasis suppressor gene (59). PDZK1IP1 exhibited a tumor-suppressor phenotype in cultured colon cancer cells by negatively affecting proliferation and tumor growth (60). Therefore, our data suggested that a low Pi environment induced by downregulation of SLC34A2 in A549 cells may cause expression changes of these genes associated with tumorigenesis signaling pathways, resulting in AT II cells that develop into lung adenocarcinoma cells.
The present study first determined that the expression levels of SLC34A2 were downregulated in A549 and H1299 lung adenocarcinoma cells, then further revealed that the elevated expression of SLC34A2 was able to significantly inhibit the viability and invasion of A549 cells in vitro. These results indicated that SLC34A2 may be important in the initiation and progression of lung adenocarcinoma. The present study also indicated that the relative mechanisms of SLC34A2 in A549 lung cancer may be associated with the activation of the complement alternative pathway (C3 and C4b) and upregulation of the expression of SELENBP1, TXNIP, PDZK1IP1 and DUSP6. These incidents might be attributed to enhancing Pi transport in A549 cells by elevating the expression of SLC34A2. From this it was hypothesized that the downregulation of SLC34A2, expressed primarily in AT II cells, might cause abnormal AT II cells to escape from complement-associated immunosurveillance and abnormally express certain tumor-suppressor genes, inducing development of these cells into lung adenocarcinoma. The present study has provided further insights into the effects and mechanisms of SLC34A2 in lung cancer.
Acknowledgements
This study was partly supported by the National Science and Technology Major Projects of New Drugs (grant no. 2012ZX09103301-009).
Abbreviations
- SLC34A2
solute carrier family 34 (sodium phosphate), member 2
- NSCLC
non-small cell lung cancer
- CAV1
caveolin 1
- C3
complement C3 precursor
- C4b
complement 4B preproprotein
- C5
complement C5 precursor
- FGA
fibrinogen α chain precursor
- FGB
fibrinogen β chain precursor
- FGG
fibrinogen γ chain precursor
- SELENBP1
selenium binding protein 1
- TXNIP
thioredoxin-interacting protein
- PDZK1IP1
PDZK1-interacting protein 1
- DUSP6
dual specificity protein phosphatase 6
- MTT
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide
- qPCR
quantitative polymerase chain reaction
- AT II
alveolar type II
- mAbs
monoclonal antibodies
- OD
optical density
References
- 1.Jemal A, Siegel R, Ward E, Hao Y, Xu J, Thun MJ. Cancer statistics, 2009. CA Cancer J Clin. 2009;59:225–249. doi: 10.3322/caac.20006. [DOI] [PubMed] [Google Scholar]
- 2.Murer H, Forster I, Biber J. The sodium phosphate cotransporter family SLC34. Pfugers Arch. 2004;447:763–767. doi: 10.1007/s00424-003-1072-5. [DOI] [PubMed] [Google Scholar]
- 3.Zheng X, Kammerer CM, Cox LA, Morrison A, Turner ST, Ferrell RE. Association of SLC34A2 variation and sodium-lithium countertransport activity in humans and baboons. Am J Hypertens. 2009;22:288–293. doi: 10.1038/ajh.2008.355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lituiev DS, Kiyamova RG. Mutations in the gene of human type IIb sodium-phosphate cotransporter SLC34A2. Biopolym Cell. 2010;26:13–22. [Google Scholar]
- 5.Tenenhouse HS. Phosphate transport: Molecular basis, regulation and pathophysiology. J Steroid Biochem Mol Biol. 2007;103:572–577. doi: 10.1016/j.jsbmb.2006.12.090. [DOI] [PubMed] [Google Scholar]
- 6.Xu H, Bai L, Collins JF, Ghishan FK. Molecular cloning, functional characterization, tissue distribution, and chromosomal localization of a human, small intestinal sodium-phosphate (Na+-Pi) transporter (SLC34A2) Genomics. 1999;62:281–284. doi: 10.1006/geno.1999.6009. [DOI] [PubMed] [Google Scholar]
- 7.Corut A, Senyigit A, Ugur S, Altin S, Ozcelik U, Calisir H, Yildirim Z, Gocmen A, Tolun A. Mutations in SLC34A2 cause pulmonary alveolar microlithiasis and are possibly associated with testicular microlithiasis. Am J Hum Genet. 2006;79:650–656. doi: 10.1086/508263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Vachon L, Fareau GE, Wilson MG, Chan LS. Testicular microlithiasis in patients with Down syndrome. J Pediatr. 2006;149:233–236. doi: 10.1016/j.jpeds.2006.03.051. [DOI] [PubMed] [Google Scholar]
- 9.Hernando N, Sheikh S, Karim-Jimenez Z, Galliker H, Forgo J, Biber J. Asymmetrical targeting of type II Na-P(i) cotransporters in renal and intestinal epithelial cell lines. Am J Physiol Renal Physiol. 2000;278:F361–F368. doi: 10.1152/ajprenal.2000.278.3.F361. [DOI] [PubMed] [Google Scholar]
- 10.Cerri MF, Rezende LC, Paes FM, Silva IV, Rangel LBA. The cotransporter NaPi-IIb: characteristics, regulation and its role in carcinogenesis. Applied Cancer Res. 2010;30:197–203. [Google Scholar]
- 11.Shibasaki Y, Etoh N, Hayasaka M. Targeted deletion of the tybe IIb Na+dependent Pi-co-transporter, NaPi-IIb, results in early embryonic lethality. Biochem Biophys Res Commun. 2009;381:482–486. doi: 10.1016/j.bbrc.2009.02.067. [DOI] [PubMed] [Google Scholar]
- 12.Kopantzev EP, Monastyrskaya GS, Vinogradova TV, et al. Differences in gene expression levels between early and later stages of human lung development are opposite to those between normal lung tissue and non-small lung cell carcinoma. Lung Cancer. 2008;62:23–34. doi: 10.1016/j.lungcan.2008.02.011. [DOI] [PubMed] [Google Scholar]
- 13.el-Deiry WS, Nelkin BD, Celano P, Yen RW, Falco JP, Hamilton SR, Baylin SB. High expression of the DNA methyltransferase gene characterizes human neoplastic cells and progression stages of colon cancer. Proc Natl Acad Sci USA. 1991;88:3470–3474. doi: 10.1073/pnas.88.8.3470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lengauer C, Kinzler KW, Vogelstein B. DNA methylation and genetic instability in colorectal cancer cells. Proc Natl Acad Sci USA. 1997;94:2545–2550. doi: 10.1073/pnas.94.6.2545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Liu H, Kho AT, Kohane IS, Sun Y. Predicting survival within the lung cancer histopathological hierarchy using a multi-scale genomic model of development. PLoS Med. 2006;3:e232. doi: 10.1371/journal.pmed.0030232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ramirez MI, Pollack L, Millien G, Cao YX, Hinds A, Williams MC. The alpha-isoform of caveolin-1 is a marker of vasculogenesis in early lung development. J Histochem Cytochem. 2002;50:33–42. doi: 10.1177/002215540205000104. [DOI] [PubMed] [Google Scholar]
- 17.Hnasko R, Ben-Jonathan N. Developmental regulation of PV-1 in rat lung: association with the nuclear envelope and limited colocalization with Cav-1. Am J Physiol Lung Cell Mol Physiol. 2005;288:L275–L284. doi: 10.1152/ajplung.00236.2004. [DOI] [PubMed] [Google Scholar]
- 18.Wiliams TM, Lisanti MP. Caveolin-1 in oncogenic transformation, cancer, and metastasis. Am J Physiol Cell Physiol. 2005;288:C494–C506. doi: 10.1152/ajpcell.00458.2004. [DOI] [PubMed] [Google Scholar]
- 19.Sunaga N, Miyajima K, Suzuki M, Sato M, White MA, Ramirez RD, Shay JW, Gazdar AF, Minna JD. Different roles for caveolin-1 in the development of non-small cell lung cancer versus small cell lung cancer. Cancer Res. 2004;64:4277–4285. doi: 10.1158/0008-5472.CAN-03-3941. [DOI] [PubMed] [Google Scholar]
- 20.Bélanger MM, Gaudreau M, Roussel E, Couet J. Role of caveolin-1 in etoposide resistance development in A549 lung cancer cells. Cancer Biol Ther. 2004;3:954–959. doi: 10.4161/cbt.3.10.1112. [DOI] [PubMed] [Google Scholar]
- 21.Traebert M. Expression of type II Na-P(i) cotransporterin alveolar type II cells. Am J Physiol. 1999;277:L868–L873. doi: 10.1152/ajplung.1999.277.5.L868. [DOI] [PubMed] [Google Scholar]
- 22.Griffiths MJ, Bonnet D, Janes SM. Stem cells of the alveolar epithelium. Lancet. 2005;366:249–260. doi: 10.1016/S0140-6736(05)66916-4. [DOI] [PubMed] [Google Scholar]
- 23.Kinnard WV, Tuder R, Papst P, Fisher JH. Regulation of alveolar type II cell differentiation and proliferation in adult rat lung explants. Am J Respir Cell Mol Biol. 1994;11:416–425. doi: 10.1165/ajrcmb.11.4.7917310. [DOI] [PubMed] [Google Scholar]
- 24.Ten Have-Opbroek AA, Benfield JR, Hammond WG, Dijkman JH. Alveolar stem cells in canine bronchial carcinogenesis. Cancer Lett. 1996;101:211–217. doi: 10.1016/0304-3835(96)04137-7. [DOI] [PubMed] [Google Scholar]
- 25.Ten Have-Opbroek AA, Benfield JR, van Krieken JH, Dijkman JH. The alveolar type II cell is a pluripotential stem cell in the genesis of human adenocarcinomas and squamous cell carcinomas. Histol Histopathol. 1997;12:319–336. [PubMed] [Google Scholar]
- 26.Kitinya JN, Sueishi K, Tanaka K, Katsuda Y. Immunoreactivity of surfactant-apoprotein adenocarcinomas, large cell and small cell carcinomas of the lung. Acta Pathol Jpn. 1986;36:1271–1278. doi: 10.1111/j.1440-1827.1986.tb02848.x. [DOI] [PubMed] [Google Scholar]
- 27.Gazdar FA, Linnoila R, Kurita Y, et al. Peripheral airway cell differentiation in human lung cancer. Cancer Res. 1990;50:5481–5487. [PubMed] [Google Scholar]
- 28.Holm BA, Matalon S, Finkelstein JN, Notter RH. Type II pneumocyte changes during hyperoxic lung injury and recovery. J Appl Physiol. 1985;1988;65:2672–2678. doi: 10.1152/jappl.1988.65.6.2672. [DOI] [PubMed] [Google Scholar]
- 29.Kim CF, Jackson EL, Woolfenden AE, et al. Identification of bronchioalveolar stem cells in normal lung and lung cancer. Cell. 2005;121:823–835. doi: 10.1016/j.cell.2005.03.032. [DOI] [PubMed] [Google Scholar]
- 30.Xu CX, Jin H, Chung YS, et al. Low dietary inorganic phosphate affects the lung growth of developing mice. J Vet Sci. 2009;10:105–113. doi: 10.4142/jvs.2009.10.2.105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tsao MS, Zhu H, Viallet J. Autocrine growth loop of the epidermal growth factor receptor in normal and immortalized human bronchial epithelial cells. Exp Cell Res. 1996;223:268–273. doi: 10.1006/excr.1996.0081. [DOI] [PubMed] [Google Scholar]
- 32.Lieber M, Smith B, Szakal A, Nelson-Rees W, Todaro G. A continuous tumor-cell line from a human lung carcinoma with properties of type II alveolar epithelial cells. Int J Cancer. 1976;17:62–70. doi: 10.1002/ijc.2910170110. [DOI] [PubMed] [Google Scholar]
- 33.Xu Q, Lu A, Xiao G, et al. Transcriptional profiling of midgut immunity response and degeneration in the wandering silkworm, Bombyx mori. Plos one. 2012;7:e43769. doi: 10.1371/journal.pone.0043769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hill LD, Sun L, Leuschen MP, Zach TL. C3 synthesis by A549 alveolar epithelial cells is increased by interferon-γ and dexamethasone. Immunology. 1993;79:236–240. [PMC free article] [PubMed] [Google Scholar]
- 35.Neal C, Neal M, Wickham H. Phosphate measurement in natural waters: two examples of analytical problems associated with silica interference using phosphomolybdic acid methodologies. Sci Total Environ. 2000;252:511–512. doi: 10.1016/s0048-9697(00)00402-2. [DOI] [PubMed] [Google Scholar]
- 36.Gałeza M, Zebracka J, Szpak-Ulczok S, et al. Expression of selected genes involved in transport of ions in papillary thyroid carcinoma. Endokrynol Pol. 2006;57:26–31. (In Polish) [PubMed] [Google Scholar]
- 37.Blanchard A, Shiu R, Booth S, et al. Gene expression profiling of early involuting mammary gland reveals novel genes potentially relevant to human breast cancer. Front Biosci. 2007;12:2221–2232. doi: 10.2741/2225. [DOI] [PubMed] [Google Scholar]
- 38.Yin BW, Kiyamova R, Chua R, Caballero OL, et al. Monoclonal antibody MX35 detects the membrane transporter NaPi2b (SLC34A2) in human carcinomas. Cancer Immun. 2008;8:3. [PMC free article] [PubMed] [Google Scholar]
- 39.Cuzin M. DNA chips: a new tool for genetic analysis and diagnostics. Transfus Clin Biol. 2001;8:291–296. doi: 10.1016/s1246-7820(01)00141-0. [DOI] [PubMed] [Google Scholar]
- 40.Strunk RC, Eidlen DM, Mason RJ. Pulmonary alveolar type II epithelial cells synthesize and secrete proteins of the classical and alternative complement pathways. J Clin Invest. 1988;81:1419–1426. doi: 10.1172/JCI113472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Niculescu F, Rus HG, Retegan M, Vlaicu R. Persistent complement activation on tumor cells in breast cancer. Am J Pathol. 1992;140:1039–1043. [PMC free article] [PubMed] [Google Scholar]
- 42.Magyarlaki T, Mosolits S, Baranyay F, Buzogany I. Immunohistochemistry of complement response on human renal cell carcinoma biopsies. Tumori. 1996;82:473–479. doi: 10.1177/030089169608200513. [DOI] [PubMed] [Google Scholar]
- 43.Clynes RA, Towers TL, Presta LG, Ravetch JV. Inhibitory Fc receptors modulate in vivo cytoxicity against tumor targets. Nat Med. 2000;6:443–446. doi: 10.1038/74704. [DOI] [PubMed] [Google Scholar]
- 44.Kierszenbaum F, Budzko DB. Cytotoxic effects of normal sera on lymphoid cells. I. Antibody-independent killing of heterologous thymocytes by guinea pig, rabbit, and human sera: Role of the alternative pathway of complement activation. Cell Immunol. 1977;29:137–146. doi: 10.1016/0008-8749(77)90282-9. [DOI] [PubMed] [Google Scholar]
- 45.Rothman BL, Despins AW, Kreutzer DL. Cytokine regulation of C3 and C5 production by the human type II pneumocyte cell line, A549. J Immunol. 1990;145:592–598. [PubMed] [Google Scholar]
- 46.Rothman BL, Merrow M, Despins A, Kennedy T, Kreutzer DL. Effect of lipopolysaccharide on C3 and C5 production by human lung cells. J Immunol. 1989;143:196–202. [PubMed] [Google Scholar]
- 47.Duerst RE, Ryan DH, Frantz CN. Variables affecting the killing of cultured human neuroblastoma cells with monoclonal antibody and complement. Cancer Res. 1986;46:3420–3425. [PubMed] [Google Scholar]
- 48.Jima DD, Shaha RN, Orcutta TM. Enhanced transcription of complement and coagulation genes in the absence of adaptive immunity. Mol Immunol. 2009;46:1505–1516. doi: 10.1016/j.molimm.2008.12.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hickman-Davis JM, Fang FC, Nathan C. Lung surfactant and reactive oxygen-nitrogen species: antimicrobial activity and host-pathogen interactions. Am J Physiol Lung Cell Mol Physiol. 2001;281:L517–L523. doi: 10.1152/ajplung.2001.281.3.L517. [DOI] [PubMed] [Google Scholar]
- 50.Malmsten M, Lassen B, James MV, Nilsson UR. Adsorption of complement proteins C3 and C1q. J Colloid Interface Sci. 1996;178:123–134. [Google Scholar]
- 51.Mold C. Effect of membrane phospholipids on activation of the alternative complement pathway. J Immunol. 1989;143:1663–1668. [PubMed] [Google Scholar]
- 52.Takeda E, Yamamoto H, Nashiki K, Sato T, Arai H, Taketani Y. Inorganic phosphate homeostasis and the role of dietary phosphorus. J Cell Mol Med. 2004;8:191–200. doi: 10.1111/j.1582-4934.2004.tb00274.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Furukawa T, Sunamura M, Motoi F, Matsuno S, Horii A. Potential tumor suppressive pathway involving DUSP6/MKP-3 in pancreatic cancer. Am J Pathol. 2003;162:1807–1815. doi: 10.1016/S0002-9440(10)64315-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Zhang Z, Kobayashi S, Borczuk AC, Leidner RS, Laframboise T, Levine AD, Halmos B. Dual specificity phosphatase 6 (DUSP6) is an ETS-regulated negative feedback mediator of oncogenic ERK signaling in lung cancer cells. Carcinogenesis. 2010;31:577–586. doi: 10.1093/carcin/bgq020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Li T, Yang W, Li M. Expression of selenium binding protein 1 characterizes intestinal cell maturation and predicts survival for patients with colorectal cancer. Mol Nutr Food Res. 2008;52:1289–1299. doi: 10.1002/mnfr.200700331. [DOI] [PubMed] [Google Scholar]
- 56.Huang KC, Park DC, Ng SK, et al. Selenium binding protein 1 in ovarian cancer. Int J Cancer. 2006;118:2433–2440. doi: 10.1002/ijc.21671. [DOI] [PubMed] [Google Scholar]
- 57.Chen G, Wang H, Miller CT. Reduced selenium-binding protein 1 expression is associated with poor outcome in lung adenocarcinomas. J Pathol. 2004;202:321–329. doi: 10.1002/path.1524. [DOI] [PubMed] [Google Scholar]
- 58.Yang M, Sytkowski AJ. Differential expression and androgen regulation of the human selenium-binding protein gene hSP56 in prostate cancer cells. Cancer Res. 1998;58:3150–3153. [PubMed] [Google Scholar]
- 59.Parikh H, Carlsson E, Chutkow WA, Johansson LE. TXNIP regulates peripheral glucose metabolism in humans. PLoS Med. 2007;4:e158. doi: 10.1371/journal.pmed.0040158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Capuano P, Bacic D, Stange G. Expression and regulation of the renal Na/phosphate cotransporter NaPi-IIa in a mouse model deficient for the PDZ protein PDZK1. Pflugers Arch. 2005;449:392–402. doi: 10.1007/s00424-004-1351-9. [DOI] [PubMed] [Google Scholar]