

Abstract
Nephrotic syndrome (NS) is a genetically heterogeneous group of diseases that are divided into steroid-sensitive NS (SSNS) and steroid-resistant NS (SRNS). SRNS inevitably leads to end-stage kidney disease, and no curative treatment is available. To date, mutations in more than 24 genes have been described in Mendelian forms of SRNS; however, no Mendelian form of SSNS has been described. To identify a genetic form of SSNS, we performed homozygosity mapping, whole-exome sequencing, and multiplex PCR followed by next-generation sequencing. We thereby detected biallelic mutations in EMP2 (epithelial membrane protein 2) in four individuals from three unrelated families affected by SRNS or SSNS. We showed that EMP2 exclusively localized to glomeruli in the kidney. Knockdown of emp2 in zebrafish resulted in pericardial effusion, supporting the pathogenic role of mutated EMP2 in human NS. At the cellular level, we showed that knockdown of EMP2 in podocytes and endothelial cells resulted in an increased amount of CAVEOLIN-1 and decreased cell proliferation. Our data therefore identify EMP2 mutations as causing a recessive Mendelian form of SSNS.
Main Text
Nephrotic syndrome (NS) is a disorder characterized by proteinuria caused by disruption of the glomerular filtration barrier. It affects 16 per 100,000 children.1 Although most affected individuals have steroid-sensitive NS (SSNS), approximately 10%–20% of children and 40% of adults do not achieve sustained remission after steroid therapy.2 This steroid-resistant NS (SRNS), which typically manifests histologically as focal segmental glomerulosclerosis, eventually results in end-stage kidney disease through the progressive loss of the filtration barrier and remains one of the most intractable kidney diseases.3 The identification of genes mutated in SRNS and subsequent research on the function of those genes have helped in the assembly of essential components of glomerular podocyte function.4 Podocytes are neuron-like cells with a complex cellular organization consisting of a cell body, major processes, and foot processes (FPs).5 The FPs assemble into an interdigitating pattern with FPs of neighboring podocytes and form the glomerular slit diaphragm, which is critical for the filtration barrier and the retention of proteins in the blood.6 The loss of the slit diaphragm is a hallmark of NS.
SRNS is genetically heterogeneous, and mutations in more than 24 distinct genes have been linked to this disorder.7 Mutations in these genes account for two-thirds of the SRNS cases that manifest in the first year of life.8 However, genetic causes of a significant proportion of childhood- and young-adult-onset SRNS are still molecularly unidentified. In addition, genetic-mapping data strongly suggest that there are a multitude of additional loci that are linked to late-onset SRNS.9 Furthermore, no Mendelian form of SSNS has been described yet, even though SSNS and SRNS might be part of a clinical spectrum in which prior steroid sensitivity might evolve into steroid resistance.
To identify additional genes mutated in NS, we applied homozygosity mapping and whole-exome sequencing (WES) to 67 families affected by NS. We obtained blood samples and pedigrees after acquiring informed consent from individuals with NS and their family members. Approval for human subjects research was obtained from the institutional review boards at the University of Michigan and Boston Children’s Hospital.
Homozygosity mapping in a Turkish family (A1679) with two children affected by SSNS yielded four candidate regions of homozygosity by descent with a cumulative genomic length of ∼55 Mb (Figure 1A). We performed WES in one affected child (A1679-21) by using NimbleGen SeqCap EZ Exome with consecutive next-generation sequencing (NGS) on an Illumina-Genome Analyzer II. We detected in this individual a homozygous truncating variant (c.184C>T [p.Gln62∗]) in EMP2 (epithelial membrane protein 2 [MIM 601223, RefSeq accession number NM_001424.4]) (Figures 1B–1E; Table 1; Table S1, available online). The mutation segregated with the affected status in this family and was absent from 86 Turkish healthy control individuals. NS of both affected individuals was initially treated with steroids and relapsed frequently. Therefore, cyclophosphamide was given for 3 months in 2006, and both of these individuals have been in remission until now.
Figure 1.
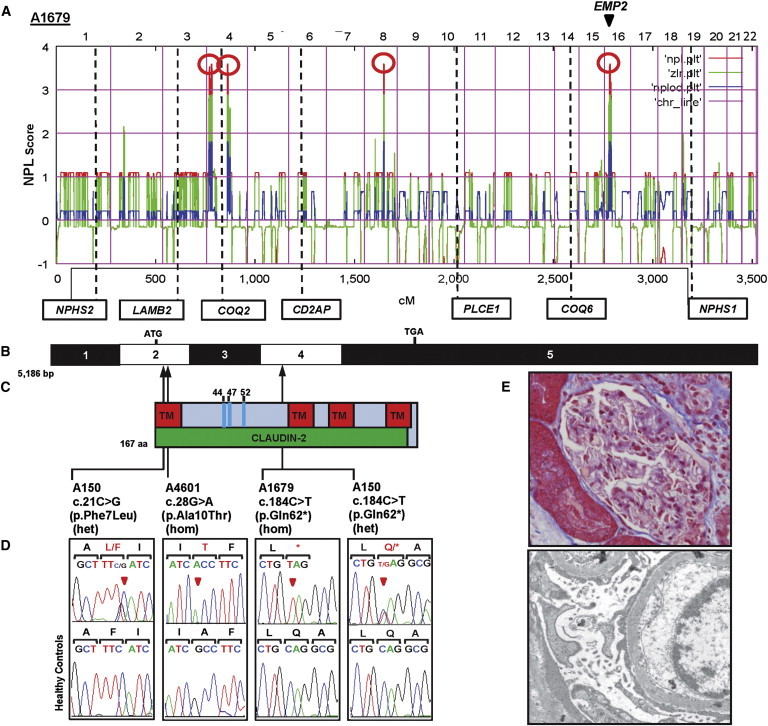
Homozygosity Mapping and WES Identify EMP2 Mutations in Families Affected by NS
(A) Homozygosity mapping identified four recessive candidate loci. A nonparametric LOD (NPL) score profile across the human genome is shown for two SSNS siblings from consanguineous family A1679. The x axis shows Affymetrix 250K StyI Array SNP positions concatenated from pter (left) to qter (right) on all human chromosomes. Four maximum NPL peaks (red circles) indicate candidate regions of homozygosity by descent. Note that none of the peaks overlapped any of seven known recessive NS-associated loci (in boxes). EMP2 (arrow head) is within one of the maximum NPL peaks on chromosome 16.
(B) Exon structure of human EMP2 cDNA. EMP2 contains five exons. The positions of the start codon (ATG) and the stop codon (TGA) are indicated.
(C) Domain structure of EMP2. The transmembrane (TM) and claudin-2 domains are depicted by colored bars in relation to the positions of the encoding exons. EMP2 has three N-linked glycosylation sites (amino acid positions 44, 47, and 52).
(D) Three homozygous or compound-heterozygous EMP2 mutations detected in three NS-affected families. Family numbers, mutations, and predicted translational changes are indicated (see Table 1). Sequence traces are shown for the mutation above the normal control individuals. Arrowheads denote altered nucleotides.
(E) Renal histology of individual A4601-21 revealed minimal change disease (upper), and electron microscopy (lower) showed diffuse FP effacement.
Table 1.
Recessive EMP2 Mutations Detected in Individuals with NS
| Individual | Ethnic Origin | Parental Consan guinity | Nucleotide Mutationa,b | Alteration in Coding Sequence | Exon (Segregation) | Amino Acid Sequence Conservationc | PolyPhen-2 Score | Age at Onset | Kidney Disease | Age at ESKD | Treatment and Renal Transplantation | Histology (Age) |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| A1679-21 | Turkish | yes | c.184C>T | p.Gln62∗ | 4 (hom, M, P) | NA | NA | 2.5 years | SSNS | none at age of > 20 years | CP | ND |
| A1679-22 | Turkish | yes | c.184C>T | p.Gln62∗ | 4 (hom, M, P) | NA | NA | 2 years | SSNS | none at age of > 20 years | CP | ND |
| A150-21 | Turkish | no | c.21C>G | p.Phe7Leu | 2 (het, M) | D. rerio | 1 | <1 year | SSNS | none at age of 5 years | ND | ND |
| c.184C>T | p.Gln62∗ | 4 (het, P) | NA | NA | ||||||||
| A4601-21 | African American | ND | c.28G>A | p.Ala10Thr | 2 (hom, M, ND) | C. intestinalis | 0.753 | 3 years | SRNS | ND | CsA | MCNS (5 years) |
Abbreviations are as follows: CP, cyclophosphamide; CsA, cyclosporine A; ESKD, end-stage kidney disease; het, heterozygous in affected individual; hom, homozygous in affected individual; M, heterozygous mutation identified in mother; MCNS, minimal change nephrotic syndrome; NA, not applicable; ND, no data or DNA available; P, heterozygous mutation identified in father; SRNS, steroid-resistant nephrotic syndrome; and SSNS, steroid-sensitive nephrotic syndrome.
All mutations were absent from >190 ethnically matched healthy control individuals and from >8,600 European control individuals in the NHLBI EVS (see Web Resources).
cDNA mutations are numbered according to human cDNA RefSeq NM_001424.4 (EMP2); +1 corresponds to the A of the ATG translation initiation codon.
The amino acid residue is continually conserved throughout evolution as indicated.
In order to investigate whether EMP2 mutations occur in additional individuals with NS, we examined a worldwide cohort of over 1,600 individuals with NS by performing exon resequencing of all four EMP2 coding exons (Figure 1B) and using an in-house-developed approach of multiplex PCR followed by NGS.10 In an individual with SSNS (A150-21), we detected compound-heterozygous mutations c.21C>G (p.Phe7Leu) and c.184C>T (p.Gln62∗) in EMP2 (Figures 1C–1E; Table 1). The c.21C>G (p.Phe7Leu) allele alters an evolutionarily conserved amino acid residue and is predicted to be deleterious to protein function by publically available software programs (Table 1). In an SRNS-affected individual (A4601-21) of African American descent, we detected a homozygous missense mutation (c.28G>A [p.Ala10Thr]) in EMP2. This mutation alters an amino acid residue conserved throughout evolution from C. intestinalis. These mutations segregated in a recessive mode with the affected status in their families and were absent from >190 ethnically matched healthy control individuals and from >4,300 European control individuals in the NHLBI Exome Sequencing Project Exome Variant Server (EVS). For exclusion of known genetic causes of SRNS, 23 known genes previously linked to SRNS were screened in this individual, but no explanatory mutations were detected. When individual A4601-21 was 5 years of age, renal histology exhibited minimal change disease, and electron microscopy showed diffusely effaced FPs of podocytes with microvillous changes (Figure 1E). In total, we identified both disease-causing alleles of EMP2 in three NS-affected families and detected three different homozygous or compound-heterozygous EMP2 mutations (Table 1; Figure 1C). We thereby identified recessive mutations in EMP2 as a cause of NS. EMP2 (also known as XMP) is a tetraspan protein that contains four transmembrane domains (Figure 1C). It was initially identified by its homology with the PMP-22 (peripheral myelin protein 22) family of proteins.11 EMP2 is highly enriched in kidney glomeruli and localizes to both podocytes and nonpodocyte glomerular cells.12 EMP2 is known to control cell-membrane localization of integrins, caveolins, and glycosylphosphatidyl-inositol-linked proteins,13–15 but little is known about its function in the kidney.
To recapitulate the human phenotype and investigate the function of EMP2 in vivo, we performed whole-mount in situ hybridization (WISH) and knockdown of the EMP2 zebrafish ortholog, emp2 (Figure 2). We did WISH on embryos at 3.5 days postfertilization (dpf), when the nephrons become mature structurally and functionally.16 Our data showed that emp2 was expressed in the arches, orbits, pectoral fins, vessels, pronephric renal tubules, and glomeruli (Figures 2A and 2B). Specifically, targeting the emp2 ortholog in zebrafish by either translation-blocking (MO1) or two splice-blocking (MO2 and MO3) morpholino oligonucleotides (MOs) caused pericardial effusion in more than 85% (78/96, 79/94, and 68/72 for MO1, MO2, and MO3, respectively) of injected embryos at 3.5 dpf (Figures 2C–2F; Table S2). We coinjected the p53 MO along with emp2 MOs to reduce toxicity of MOs.17 The efficiency of splice-blocking MOs was demonstrated by RT-PCR (Figure S1). Pericardial effusion is a disease phenotype associated with defective glomerular filtration barriers in zebrafish. It has been confirmed in multiple zebrafish models of NS, such as knockdown of nephrin, podocin, or other genes essential for podocyte function.18,19 Knockdown of emp2 in zebrafish larvae led to the disruption of the glomerular filtration barrier, consistent with NS phenotypes seen in individuals with EMP2 mutations. To demonstrate the observed phenotype specifically caused by loss of function of emp2 in zebrafish, we performed rescue experiments with human EMP2 mRNA. When wild-type (WT) EMP2 mRNA was coinjected with the emp2 MO3, the percentage of morphants that showed pericardial effusion was reduced from 94% (68/72) to 39% (24/62), indicating that emp2 is necessary for renal integrity and that there is functional consistency between human EMP2 and zebrafish emp2 (Figures 2G and 2K). Coinjection into zebrafish emp2 morphants of mRNA containing EMP2 missense variants c.21C>G (p.Phe7Leu) or c.28G>A (p.Ala10Thr) or truncating mutation c.184C>T (p.Gln62∗) (all of which we identified in individuals with NS) not only failed to rescue pericardial effusion but also caused embryonic malformation and accelerated lethality (Figures 2H–2K). After coinjection with EMP2 mRNA with c.21C>G (p.Phe7Leu), c.28G>A (p.Ala10Thr), or c.184C>T (p.Gln62∗), 87% (66/76), 95% (79/83), or 84% (70/83), respectively, of emp2 morphants still showed pericardial effusion.
Figure 2.
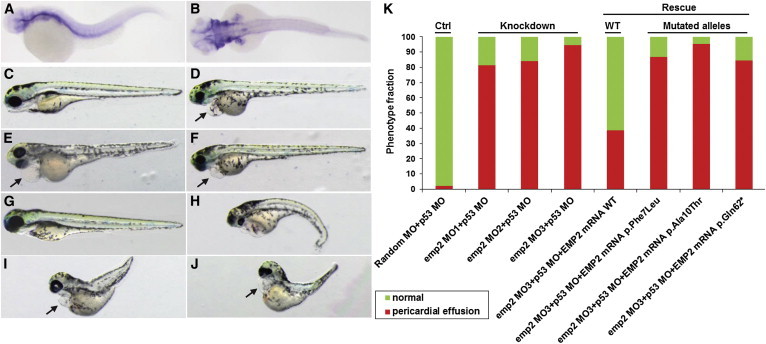
emp2 Knockdown Causes Pericardial Effusion in Zebrafish
(A and B) The expression pattern of emp2 in 3.5 dpf embryos (lateral view, A; dorsal view, B) was examined by WISH.
(C–F) emp2 knockdown in zebrafish replicated a nephrosis phenotype. Compared to control-MO-injected embryos (C), embryos injected with three different emp2 MOs (MO1, D; MO2, E; and MO3, F) showed pericardial effusion at 3.5 dpf (arrows). p53 MO coinjection was used for minimizing nonspecific apoptotic MO effects. Arrows indicate pericardial effusion.
(G–J) Coinjection of emp2 MO3 with a full-length human EMP2 mRNA (G) rescued the pericardial effusion, whereas three EMP2 mRNAs encoding p.Phe7Leu (H), p.Ala10Thr (I), or p.Gln62∗ (J) failed to rescue the nephrosis phenotype.
(K) Quantification of the percentage of pericardial effusion.
Most gene products that are altered in SRNS localize to glomerular podocytes. We therefore examined the localization of EMP2 by immunofluorescence. EMP2 is highly enriched in glomeruli of the adult rat kidney, but not in tubules (Figure 3A). In glomeruli, EMP2 localizes to the cytoplasm of podocytes, as identified by the presence of nuclear WT1 (Figure 3B). However, EMP2 is also present in other glomerular cell types, as shown previously at the transcriptional level.12 EMP2 is cytoplasmic in podocytes and does not colocalize with podocytic markers such as PODOCALYXIN, GLEPP1, or SYNAPTOPODIN (Figures 3C–3E). Immunogold electron microscopy of the adult rat kidney confirmed the presence of EMP2 in podocytes and endothelial cells (Figures 3F–3I). In podocytes, immunogold labeling was detected in both the FPs and the cell bodies (Figures 3G and 3I), whereas in endothelial cells, EMP2 labeling was detected predominantly in the nucleus (Figures 3H and 3I).
Figure 3.
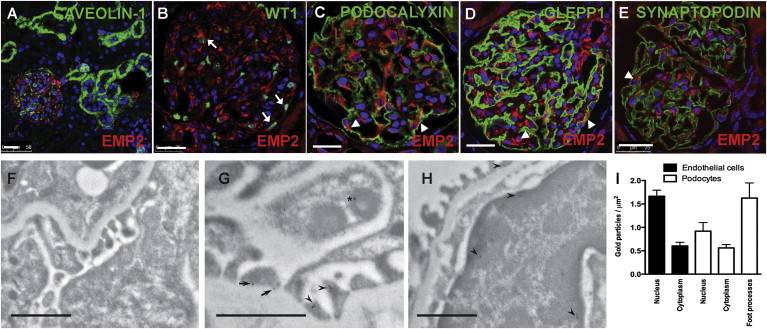
Localization of EMP2 in the Kidney and Cultured Podocytes
(A) Coimmunofluorescence of EMP2 (Sigma) with CAVEOLIN-1 (BD Transduction Laboratories). EMP2 (red) localized to glomeruli in the adult rat kidney but was not detected in tubules.
(B) Coimmunofluorescence of EMP2 with WT1 (Santa Cruz). EMP2 (red) localized to podocytes, whose nuclei are marked by WT1 (green, arrow).
(C–E) Coimmunofluorescence of EMP2 with podocytic markers PODOCALYXIN (C), GLEPP1 (D), and SYNAPTOPODIN (E) (American Research Products). EMP2 localized to the cytoplasm of podocytes (arrowheads). White scale bars in (A)–(E) represent 25 μm.
(F–H) Immunogold electron microscopy of EMP2 in the adult rat kidney. A control individual without a primary antibody is shown in (F). In podocytes, EMP2 localized to the sole plate (arrow) and the body (arrowheads) of FPs and to the cytoplasm of podocytes (asterisk, G). In endothelial cells, most of the gold particles (arrowheads) were detected in the nucleus (H). Black scale bars in (F)–(H) represent 1 μm.
(I) Quantification of gold particles in rat glomerular endothelial cells and podocytes. Data represent the mean ± SEM. EMP2 antibody was highly diluted so that background label was negative.
EMP2 is known to regulate the amount of CAVEOLIN-1,13,15 which is the primary structural component of caveolae, flask-shaped invaginations in the plasma membrane. Caveolae contribute to many cellular functions, including endocytosis, cell signaling, and the transcytosis of cholesterol and albumin.20 CAVEOLIN-1 is highly enriched in podocytes and is partially localized to the slit diaphragm.21 Interestingly, it was reported that the amount of CAVEOLIN-1 was significantly higher in the glomeruli of 99 individuals with glomerular diseases than in those of 50 healthy individuals.22 The accumulation of CAVEOLIN-1 was positively correlated with urinary albumin excretion.22 Previously, EMP2 was also shown to positively regulate the amount of VEGF in retinal pigment epithelial cells.23 Podocyte-specific deletion of VEGF-A leads to glomerular disease in mice.24
In order to gain insight into a potential mechanistic link between EMP2 mutations and NS, we performed lentivirus-mediated knockdown of EMP2 in cultured human podocytes by using small hairpin RNAs (shRNAs) (Table S2). We found that upon knockdown of EMP2 in undifferentiated podocytes, the amount of CAVEOLIN-1 was increased (as reported in other cell types13,15), whereas that of VEGFA did not change (Figure 4A). To further define this relationship in podocytes, we performed real-time PCR of CAV1 (MIM 601047) and VEGFA (MIM 192240) mRNA. The amount of CAV1 mRNA was higher in undifferentiated podocytes stably transfected with EMP2 shRNAs than in podocytes transfected with scrambled shRNA (Figure 4B). However, there was no change in the amount of VEGFA mRNA upon EMP2 knockdown (Figure 4C). These results were also confirmed in differentiated podocytes cultured at 37°C for 14 days (Figure S2). EMP2 knockdown in differentiated podocytes also resulted in increased amounts of CAV1 mRNA (Figure S2A), but no change in VEGFA mRNA (Figure S2B). To further test the pathogenicity of the identified mutations, we transfected EMP2 cDNA resulting from the identified mutations (Figure 4D). When transfected, WT EMP2 decreased the amount of CAVEOLIN-1, but it did not change the amount of VEGFA. Like WT EMP2, EMP2 proteins harboring p.Phe7Leu or p.Ala10Thr decreased the amount of CAVEOLIN-1; however, the truncated protein (p.Gln62∗) failed to decrease the amount of CAVEOLIN-1, indicating that the homozygous truncating mutation might at least in part induce loss of function by a CAV1-mediated mechanism. None of the proteins resulting from missense or nonsense mutations affected the amount of VEGFA.
Figure 4.
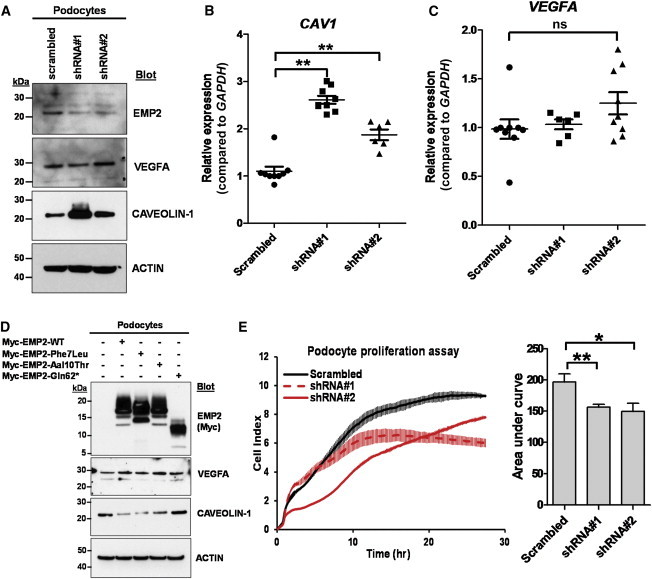
Knockdown of EMP2 in Cultured Human Podocytes Increases the Amount of CAVEOLIN-1 and Decreases Cell Proliferation
(A) shRNA-mediated knockdown of EMP2 resulted in increased amounts of CAVEOLIN-1 without changing the amount of VEGFA.
(B and C) mRNA levels of CAV1 and VEGFA were measured by real-time PCR using TaqMan probes for CAV1 (Hs00971716_m1), VEGFA (Hs00900055_m1), and GAPDH (Hs02758991_g1, control). Knockdown of EMP2 increased CAV1 expression at the transcriptional level (B), whereas the amount of VEGFA mRNA did not change (C). Data represent the mean ± SEM, denoted by three horizontal lines. ∗∗p < 0.01; ns, not significant (two-tailed Student’s t test).
(D) Transfection of cDNAs encoding WT EMP2 and EMP2 harboring missense changes p.Phe7Leu or p.Ala10Thr decreased the amount of CAVEOLIN-1, whereas transfection of cDNAs encoding the truncated protein (p.Gln62∗) failed to decrease the amount of CAVEOLIN-1.
(E) Compared to podocytes transfected with scrambled shRNA negative control (black line), podocytes transfected with EMP2 shRNAs (red lines) showed reduced cell proliferation. Bar graphs represent the area under curves, and data represent the mean ± SEM. ∗p < 0.05, ∗∗p < 0.01.
Finally, we investigated the effect of EMP2 knockdown on cultured human podocyte proliferation by using the xCELLigence system (ACEA Biosciences). We performed the proliferation assay at 33°C to maintain podocytes in the undifferentiated state. Compared to podocytes transfected with scrambled shRNA, podocytes stably transfected with EMP2 shRNAs exhibited reduced cell proliferation (Figure 4E). Because EMP2 is also localized in glomerular cell types other than podocytes (Figure 3B), we also investigated the effect of EMP2 knockdown on human umbilical vein endothelial cells (HUVECs). The knockdown of EMP2 in HUVECs via lentivirus-mediated shRNAs resulted in increased amounts of CAVEOLIN-1 (Figure S3A) and CAV1 mRNA (Figure S3B), consistent with our findings in podocytes (Figures 4A and 4B). Similar to podocytes (Figure 4E), HUVECs exhibited decreased proliferation upon knockdown of EMP2 (Figure S3C). Therefore, EMP2 knockdown causes an increased amount of CAVEOLIN-1 and reduced cell proliferation in both podocytes and HUVECs. However, the pathogenic link among EMP2, CAVEOLIN-1, and NS will require further investigation.
Taken together, our data show that mutations in EMP2 cause an autosomal-recessive form of SSNS. EMP2 localizes to glomeruli in the kidney and regulates the amount of CAVEOLIN-1, and its depletion causes decreased cell proliferation. Knockdown of the zebrafish ortholog, emp2, recapitulated the human nephrosis phenotype. It will be of interest in the future to investigate the exact mechanism through which defects in EMP2 cause NS.
Acknowledgments
The authors thank the families who contributed to this study. We thank David M. Briscoe (Boston Children’s Hospital) for materials and helpful discussion. This research was supported by NIH grants to F.H. (DK076683 and DK086542), E.A.O. (DK090917), W.Z. (DK091405), and D.S.W. (EY07042) and by a Nephcure Foundation grant (to F.H.). F.O. was supported by the European Commission Seventh Framework Programme (EURenOmics, 2012-305608), the Scientific and Technological Research Council of Turkey (TUBITAK 108S417), and the Hacettepe University Infrastructure Project (06A101008 and 011A101003). H.Y.G. is a research fellow of the American Society of Nephrology. W.Z. is a Carl W. Gottschalk Scholar. D.S.W is a Jules and Doris Stein Research to Prevent Blindness Professor. F.H. is an investigator of the Howard Hughes Medical Institute, and Warren E. Grupe is a professor of pediatrics at the Harvard Medical School.
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
Ensembl Genome Browser, http://www.ensembl.org/
HomozygosityMapper, http://www.homozygositymapper.org/HomozygosityMapper/
MutationTaster, http://www.mutationtaster.org/
NHLBI Exome Sequencing Project (ESP) Exome Variant Server, http://evs.gs.washington.edu/EVS/
Online Mendelian Inheritance in Man (OMIM), http://www.omim.org/
PolyPhen-2, http://genetics.bwh.harvard.edu/pph2/
SIFT, http://sift.jcvi.org/
UCSC Genome Browser, http://genome.ucsc.edu/
ZFIN, http://zfin.org
References
- 1.International Study on Kidney Disease in Children Nephrotic syndrome in children: prediction of histopathology from clinical and laboratory characteristics at time of diagnosis. A report of the International Study of Kidney Disease in Children. Kidney Int. 1978;13:159–165. doi: 10.1038/ki.1978.23. [DOI] [PubMed] [Google Scholar]
- 2.Benoit G., Machuca E., Antignac C. Hereditary nephrotic syndrome: a systematic approach for genetic testing and a review of associated podocyte gene mutations. Pediatr. Nephrol. 2010;25:1621–1632. doi: 10.1007/s00467-010-1495-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hildebrandt F. Genetic kidney diseases. Lancet. 2010;375:1287–1295. doi: 10.1016/S0140-6736(10)60236-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Somlo S., Mundel P. Getting a foothold in nephrotic syndrome. Nat. Genet. 2000;24:333–335. doi: 10.1038/74139. [DOI] [PubMed] [Google Scholar]
- 5.Greka A., Mundel P. Cell biology and pathology of podocytes. Annu. Rev. Physiol. 2012;74:299–323. doi: 10.1146/annurev-physiol-020911-153238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gee H.Y., Saisawat P., Ashraf S., Hurd T.W., Vega-Warner V., Fang H., Beck B.B., Gribouval O., Zhou W., Diaz K.A. ARHGDIA mutations cause nephrotic syndrome via defective RHO GTPase signaling. J. Clin. Invest. 2013;123:3243–3253. doi: 10.1172/JCI69134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.McCarthy H.J., Bierzynska A., Wherlock M., Ognjanovic M., Kerecuk L., Hegde S., Feather S., Gilbert R.D., Krischock L., Jones C., RADAR the UK SRNS Study Group Simultaneous sequencing of 24 genes associated with steroid-resistant nephrotic syndrome. Clin. J. Am. Soc. Nephrol. 2013;8:637–648. doi: 10.2215/CJN.07200712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hinkes B.G., Mucha B., Vlangos C.N., Gbadegesin R., Liu J., Hasselbacher K., Hangan D., Ozaltin F., Zenker M., Hildebrandt F., Arbeitsgemeinschaft für Paediatrische Nephrologie Study Group Nephrotic syndrome in the first year of life: two thirds of cases are caused by mutations in 4 genes (NPHS1, NPHS2, WT1, and LAMB2) Pediatrics. 2007;119:e907–e919. doi: 10.1542/peds.2006-2164. [DOI] [PubMed] [Google Scholar]
- 9.Hildebrandt F., Heeringa S.F., Rüschendorf F., Attanasio M., Nürnberg G., Becker C., Seelow D., Huebner N., Chernin G., Vlangos C.N. A systematic approach to mapping recessive disease genes in individuals from outbred populations. PLoS Genet. 2009;5:e1000353. doi: 10.1371/journal.pgen.1000353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Halbritter J., Diaz K., Chaki M., Porath J.D., Tarrier B., Fu C., Innis J.L., Allen S.J., Lyons R.H., Stefanidis C.J. High-throughput mutation analysis in patients with a nephronophthisis-associated ciliopathy applying multiplexed barcoded array-based PCR amplification and next-generation sequencing. J. Med. Genet. 2012;49:756–767. doi: 10.1136/jmedgenet-2012-100973. [DOI] [PubMed] [Google Scholar]
- 11.Taylor V., Suter U. Epithelial membrane protein-2 and epithelial membrane protein-3: two novel members of the peripheral myelin protein 22 gene family. Gene. 1996;175:115–120. doi: 10.1016/0378-1119(96)00134-5. [DOI] [PubMed] [Google Scholar]
- 12.Takemoto M., He L., Norlin J., Patrakka J., Xiao Z., Petrova T., Bondjers C., Asp J., Wallgard E., Sun Y. Large-scale identification of genes implicated in kidney glomerulus development and function. EMBO J. 2006;25:1160–1174. doi: 10.1038/sj.emboj.7601014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Forbes A., Wadehra M., Mareninov S., Morales S., Shimazaki K., Gordon L.K., Braun J. The tetraspan protein EMP2 regulates expression of caveolin-1. J. Biol. Chem. 2007;282:26542–26551. doi: 10.1074/jbc.M702117200. [DOI] [PubMed] [Google Scholar]
- 14.Wadehra M., Forbes A., Pushkarna N., Goodglick L., Gordon L.K., Williams C.J., Braun J. Epithelial membrane protein-2 regulates surface expression of alphavbeta3 integrin in the endometrium. Dev. Biol. 2005;287:336–345. doi: 10.1016/j.ydbio.2005.09.003. [DOI] [PubMed] [Google Scholar]
- 15.Wadehra M., Goodglick L., Braun J. The tetraspan protein EMP2 modulates the surface expression of caveolins and glycosylphosphatidyl inositol-linked proteins. Mol. Biol. Cell. 2004;15:2073–2083. doi: 10.1091/mbc.E03-07-0488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Drummond I.A., Majumdar A., Hentschel H., Elger M., Solnica-Krezel L., Schier A.F., Neuhauss S.C., Stemple D.L., Zwartkruis F., Rangini Z. Early development of the zebrafish pronephros and analysis of mutations affecting pronephric function. Development. 1998;125:4655–4667. doi: 10.1242/dev.125.23.4655. [DOI] [PubMed] [Google Scholar]
- 17.Robu M.E., Larson J.D., Nasevicius A., Beiraghi S., Brenner C., Farber S.A., Ekker S.C. p53 activation by knockdown technologies. PLoS Genet. 2007;3:e78. doi: 10.1371/journal.pgen.0030078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kramer-Zucker A.G., Wiessner S., Jensen A.M., Drummond I.A. Organization of the pronephric filtration apparatus in zebrafish requires Nephrin, Podocin and the FERM domain protein Mosaic eyes. Dev. Biol. 2005;285:316–329. doi: 10.1016/j.ydbio.2005.06.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ebarasi L., He L., Hultenby K., Takemoto M., Betsholtz C., Tryggvason K., Majumdar A. A reverse genetic screen in the zebrafish identifies crb2b as a regulator of the glomerular filtration barrier. Dev. Biol. 2009;334:1–9. doi: 10.1016/j.ydbio.2009.04.017. [DOI] [PubMed] [Google Scholar]
- 20.Parton R.G., del Pozo M.A. Caveolae as plasma membrane sensors, protectors and organizers. Nat. Rev. Mol. Cell Biol. 2013;14:98–112. doi: 10.1038/nrm3512. [DOI] [PubMed] [Google Scholar]
- 21.Sörensson J., Fierlbeck W., Heider T., Schwarz K., Park D.S., Mundel P., Lisanti M., Ballermann B.J. Glomerular endothelial fenestrae in vivo are not formed from caveolae. J. Am. Soc. Nephrol. 2002;13:2639–2647. doi: 10.1097/01.asn.0000033277.32822.23. [DOI] [PubMed] [Google Scholar]
- 22.Moriyama T., Tsuruta Y., Shimizu A., Itabashi M., Takei T., Horita S., Uchida K., Nitta K. The significance of caveolae in the glomeruli in glomerular disease. J. Clin. Pathol. 2011;64:504–509. doi: 10.1136/jcp.2010.087023. [DOI] [PubMed] [Google Scholar]
- 23.Morales S.A., Telander D.G., Leon D., Forward K., Braun J., Wadehra M., Gordon L.K. Epithelial membrane protein 2 controls VEGF expression in ARPE-19 cells. Invest. Ophthalmol. Vis. Sci. 2013;54:2367–2372. doi: 10.1167/iovs.12-11013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Eremina V., Sood M., Haigh J., Nagy A., Lajoie G., Ferrara N., Gerber H.P., Kikkawa Y., Miner J.H., Quaggin S.E. Glomerular-specific alterations of VEGF-A expression lead to distinct congenital and acquired renal diseases. J. Clin. Invest. 2003;111:707–716. doi: 10.1172/JCI17423. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.