

Abstract
QT interval variation is assumed to arise from variation in repolarization as evidenced from rare Na- and K-channel mutations in Mendelian QT prolongation syndromes. However, in the general population, common noncoding variants at a chromosome 1q locus are the most common genetic regulators of QT interval variation. In this study, we use multiple human genetic, molecular genetic, and cellular assays to identify a functional variant underlying trait association: a noncoding polymorphism (rs7539120) that maps within an enhancer of NOS1AP and affects cardiac function by increasing NOS1AP transcript expression. We further localized NOS1AP to cardiomyocyte intercalated discs (IDs) and demonstrate that overexpression of NOS1AP in cardiomyocytes leads to altered cellular electrophysiology. We advance the hypothesis that NOS1AP affects cardiac electrical conductance and coupling and thereby regulates the QT interval through propagation defects. As further evidence of an important role for propagation variation affecting QT interval in humans, we show that common polymorphisms mapping near a specific set of 170 genes encoding ID proteins are significantly enriched for association with the QT interval, as compared to genome-wide markers. These results suggest that focused studies of proteins within the cardiomyocyte ID are likely to provide insights into QT prolongation and its associated disorders.
Introduction
The electrocardiographic QT interval (MIM 610141), an index of ventricular repolarization, is a moderately heritable quantitative trait that has major medical significance because prolongation or shortening of the QT interval is associated with an increased risk of cardiovascular morbidity and mortality.1,2 Extremes of the QT interval are known to trigger ventricular tachycardia and ventricular fibrillation, which can lead to sudden cardiac death (SCD).3 Genetic variation is one major source of QT interval variation and genome-wide association studies (GWASs) have identified at least 35 loci known to create interindividual QT interval variability in individuals of European ancestry (EA) (D.E.A., unpublished data).4–6
The locus with the largest contribution to cardiac repolarization variability, ∼1% of population trait variation, is one on chromosome 1q that contains the gene NOS1AP4 (MIM 605551), although its functional role in cardiac repolarization has not been proven. In neuronal tissues, NOS1AP acts as a C-terminal PDZ domain ligand to neuronal nitric oxide synthase (nNOS), probably regulating translocation of nNOS between synaptic and postsynaptic structures.7 Although the biochemical function of NOS1AP in cardiac tissue remains unknown, the genetic association between NOS1AP and the QT interval, replicated in several studies,8–11 emphasizes its influence on myocardial function. Importantly, the same QT-interval-associated sequence variants at the NOS1AP locus are also associated with ∼30% increased risk of SCD in the general population10,12 and are common genetic modifiers of cardiac events in individuals with long QT syndrome (LQTS)13,14 (MIM 192500). Taken together, these studies implicate NOS1AP as the major genetic locus regulating QT interval in the general population and as a susceptibility factor for cardiac arrhythmias and SCD.
Like all GWASs,15 the identity of the specific gene within this locus, whether it is NOS1AP or not, and the specific variant(s) that modulate the QT interval through this gene remain unknown. Consequently, there is a major gap between genetic findings and their molecular mechanisms, resolution of which can illuminate a novel aspect of cardiac biology. In this paper, we demonstrate, via a variety of contemporary approaches, that the major gene is indeed NOS1AP, that a functional variant lies within an enhancer active in cardiac tissues, and that variable expression of NOS1AP in human heart depends on polymorphism within this enhancer. In addition, we physically localized NOS1AP to the cardiomyocyte intercalated discs (IDs), thereby explaining its role in cardiac biology probably through effects on propagation rather than repolarization. We further show that compared to genome-wide polymorphisms, variants mapping near a specific set of 170 annotated genes encoding proteins that localize to IDs are enriched for association with the QT interval, supporting our hypothesis that population variability of QT interval is significantly regulated by the ID and that genetic variation in its components can lead to interindividual variation in the risk of cardiac arrhythmias and SCD. Detailed studies of ID functions are now necessary to understand how arrhythmias arise, sudden death onsets, and the molecular bases of these complex diseases.
Material and Methods
Samples, Sequencing, and Variant Identification
A total of 46 subjects were selected for targeted sequencing from a population-based survey of volunteers in the KORA cohort,16 in which the original GWAS was performed.4 Table S1 (available online) provides the heart rate-, age-, and sex-corrected QT interval measurements from these subjects.16 Eight CEU (NA06985, NA06993, NA10839, NA10847, NA10859, NA11994, NA12003, NA12006) and eight YRI (NA18486, NA18489, NA18502, NA18505, NA18507, NA18517, NA18522, NA18523) HapMap reference individuals17 were randomly selected for targeted sequencing.
The target locus on human chromosome 1: 161,998,642–162,139,683 (NCBI37/hg19)18 was 141,042 base pairs long and encompassed exon 1, exon 2, intron 1, part of intron 2, and the 5′ upstream region of NOS1AP (Gene ID: 9722; RefSeq accession number NM_014697.2). The target region was recovered from individual samples in 17 overlapping PCR amplicons. Primers for amplifications were designed with Primer319 (Table S1). PCR for individual amplicons in each sample was performed on genomic DNA by TaKaRa LA Taq (Clontech) according to manufacturer’s recommendations. PCR products were gel-purified with QIAquick and QIAEX II gel extraction kits (QIAGEN) and pooled in equimolar amounts per sample. Preparation of short insert paired-end libraries from amplicon pools and subsequent sequencing were performed as described.20
Sequence reads were aligned to the target NOS1AP region by cross_match21 and alignments were then converted to the BAM format, a binary version of the Sequence Alignment/Map.22 For single-nucleotide variant (SNV) and small insertion-deletion (INDEL) genotyping, the Bayesian genotyper “bam2mpg”23 was used to call genotypes at each site for which there was at least one read aligned; only genotypes with a MPG (most probable genotype) score ≥10 were retained. To assess the level of coverage required to discover all observed variants, we performed genotype calling on reduced coverage subsets of the CEU samples’ sequencing data by random sampling of reads. A total of 108 small INDELs were identified and all of these INDELs mapped to homopolymer runs or simple nucleotide repeats; the vast majority of them were observed as singleton alleles (data not shown). Given the short sequence read data and limitations of mapping and accurately genotyping repetitive sequences, we did not use this type of sequence variation in our subsequent studies.
We calculated allele frequencies for observed variants in the KORA samples, but because of the relatively small sample size and selection from extremes of QT interval we do not expect these to be representative for the general population. However, we do expect to find all common variants (≥5% allele frequency) with ≥99% certainty in sequencing of the 54 (108 alleles) CEU and KORA samples.
To assess specificity of this sequencing-based approach, we compared the sequencing-based genotype calls generated in the eight CEU and eight YRI samples with the genotype calls from HapMap17 (HapMap Genome Browser release #28). To assess sensitivity, we calculated the proportion of HapMap polymorphic sites within each sample that were identified by sequencing17 (HapMap Genome Browser release #28).
Imputation and Association Analysis to Identify Candidate Causal Variants
Imputation and association analysis was performed in 9,055 individuals of EA from the ARIC (Atherosclerosis Risk in Communities) study,24 with genotypes from Genome-Wide Human SNP Array 6.0 (Affymetrix) (henceforth labeled as Affymetrix 6.0 chip) and QT interval data.24 We used the BEAGLE genetic analysis software package25 for imputation of reference variants discovered in this study by sequencing of KORA and CEU samples and in the 1000 Genomes Project (1000GP)26 by sequencing of CEU samples, based on 80 genotyped SNVs in the NOS1AP region in ARIC24 samples. These 80 variants were supplemented by first imputing an additional 872 variants identified in this study through de novo sequencing in 46 KORA and 8 CEU HapMap samples (total in both, 897; monomorphic, 25) and then by an additional 39 variants reported only in the 1000GP_CEU pilot data26 catalog (1000GP Pilot data July 2010 release) but not discovered in KORA (total 439 variants in a distributed haplotype file; 63 variants were filtered out by the 1000GP before haplotyping). Out of 911 genotyped and imputed variants in 9,055 ARIC samples, 396 variants were monomorphic after imputation and were subsequently removed before the association analysis. In addition, 3 more variants were removed as a result of having a low imputation score (R2 < 0.8). A total of 512 variants passed quality control checks and were used in association analysis that was conducted in R (The R Project for Statistical Computing) by regressing heart rate-, age-, and sex-corrected QT interval residuals on variant genotypes. To test whether rare variants were collectively associated with QT interval variation, we performed a multiple regression with 92 “rare” variants and tested their association by a global F-test. Here, “rare” was defined as observing no variant-homozygous genotypes in the ARIC samples. The p value for this overall test was 0.12 showing the lack of association, although statistical power to test this hypothesis is probably low.
Intersection of Variants with ENCODE Annotation
SNVs identified in the NOS1AP target region were uploaded into the UCSC genome browser27 as custom tracks in BED file format and intersected with ENCODE tracks28 via the UCSC table browser tool.29 We specifically analyzed sequence variants for overlap with DNaseI hypersensitive sites (DHSs) identified across cardiac cell lines/tissues in the ENCODE project.28
Luciferase Reporter Assays
To assess whether QT-associated variants influence cis-regulatory elements (enhancers or silencers), variant alleles at selected markers were subjected to luciferase reporter assays. The genomic region flanking the variant site was amplified from the genomic DNA of homozygous reference HapMap individuals and cloned upstream of the SV40 promoter driving a firefly luciferase gene in the pGL3-Promoter vector (Promega). The length of the genomic region cloned, flanking the variant site, varied from 423 to 1,160 base pairs (Table S3) and was based on the extent of conservation, presence of annotated functional elements from ENCODE,28 and presence of repetitive sequences. Site-directed mutagenesis (Stratagene) was used to generate the constructs with alternate alleles. The identity of each construct clone was verified by DNA sequencing.
For fine mapping of the cis-regulatory element encompassing the 959 base pair long rs7539120_rs2010491 amplicon, several different amplicons were subcloned by PCR and included the following: (1) 742 bp long rs7539120_rs2010491 amplicon (rs7539120_rs2010491_742), (2) 492 bp long rs7539120_rs2010491 amplicon (rs7539120_rs2010491_492), (3) 709 bp long rs7539120_rs2010491 amplicon with an internal deletion of 250 bases (rs7539120_rs2010491_Δ250), and (4) 731 bp long rs2010491 amplicon with deletion of rs7539120 (±5 bases) (Δrs7539120_rs2010491_742). For each of these amplicons, except (4), constructs carrying both alternate alleles were generated. Variant rs12143842 was re-evaluated in luciferase reporter assays by the cloning of smaller inserts flanking the variant site that included 623 and 325 bp amplicons (rs12143842_623 and rs12143842_325) (Table S3).
The firefly luciferase constructs were transfected into HL1 cells30 and HEK293T cells (ATCC),31 grown in 24-well plates at ∼90% confluency by Lipofectamine-2000 (Invitrogen) according to manufacturer’s protocols. pRLSV40 (Promega), expressing Renilla luciferase, was cotransfected to normalize for transfection efficiency. At 24 hr after transfection, cells were harvested and lysed and firefly and Renilla luciferase activities were measured on a VICTOR2 1420 Multilabel Counter (Wallac) with Dual-Luciferase Reporter Assay System (Promega) according to manufacturer’s protocols. Relative firefly luciferase activities were compared between alternate alleles of a variant and to the empty vector pGL3-Promoter-transfected cells to assess enhancer or silencer function. Luciferase activity from each construct was measured in four biological replicates, and each such experiment was repeated twice (a total of eight biological replicates for each construct in each cell line). Luciferase assays using the 959 bp long rs7539120_rs2010491 haplotype constructs were additionally performed in HL1 cells in eight biological replicates.
In Vivo Enhancer Assays in Zebrafish
All protocols for zebrafish care and use were reviewed and approved by the Institutional Animal Care and Use Committee of Johns Hopkins University and zebrafish were cared for by standard methods.32 The 742 base pair long risk haplotype (rs7539120_rs2010491_742_H2), nonrisk haplotype (rs7539120_rs2010491_742_H1), and the rs7539120 deletion (±5 bases) risk haplotype (Δrs7539120_rs2010491_742_H2) inserts were cloned upstream of a mouse cFos minimal promoter driving enhanced-GFP (eGFP) in a tol2 transposon based reporter vector. Transposase mRNA was in vitro transcribed with plasmid pDB600 (gift from S. Mathavan, Genome Institute of Singapore, Singapore) as template and mMessage mMachine T3 kit (Ambion). For injections, 300 ng of reporter plasmid, 250 ng of transposase mRNA (∼50–100 pg/embryo), and phenol red (0.1% final concentration) were combined in a 10 μl volume and injected into wild-type AB strain embryos at the 1–2 cell stage by a pico-injector (Harvard Instruments). The empty reporter vector injections resulted in no eGFP expression (data not shown). At 24 hr postfertilization (hpf), eggs were moved to egg water containing phenylthiourea to reduce pigmentation and allow better visualization of the eGFP signal.
nos1apa Expression in Zebrafish Embryos
At 24 hpf, 100 zebrafish embryos were collected, 50 of which were used for RNA isolation from entire embryos and 50 of which were used for RNA isolation from dissected heads versus the remainder. Total RNA was isolated with TRIzol reagent (Invitrogen) according to manufacturer’s instructions. DNase treatment and RNA clean-up was performed with RNeasy Mini kit and RNase-Free DNase set (QIAGEN), according to manufacturer’s instructions. cDNA was prepared by oligo-dT primer reverse transcription from 200 ng of total RNA by SuperScript III First-Strand Synthesis System (Invitrogen) according to the manufacturer’s instructions. One-tenth of the cDNA product was used as a template in PCR using nos1apa exon 10/exon 11 spanning primers (nos1apa_exon10F: 5′-aagctggctggacaaggctct-3′ and nos1apa_exon11R: 5′-ccaggcagaaagcgaaaacact-3′) and actb1 exon 5/exon 6 spanning primers (actb1_exon5F: 5′-cggtatccacgagaccaccttc-3′ and actb1_exon6R: 5′-agacggagtatttgcgctcagg-3′). PCR products were resolved by 2% agarose-TAE buffer gel electrophoresis.
Evaluation of rs7539120 as a Cardiac Expression Quantitative Trait Locus
Samples of cardiac tissue were acquired from subjects from the MAGNet consortium. All samples were collected with IRB-approved protocols. Left ventricular free-wall tissue was harvested at the time of cardiac surgery from white subjects with heart failure undergoing transplantation. The heart was perfused with cold cardioplegia prior to cardiectomy to arrest contraction and prevent ischemic damage. Tissue specimens were then obtained and frozen in liquid nitrogen. Genomic DNA from left ventricle was extracted with the Gentra Puregene Tissue Kit (QIAGEN) according to manufacturer’s instruction. Genotypes at rs7539120 were obtained by direct Sanger sequencing of PCR amplicons (959 bp) (Table S3). Total RNA was extracted from left ventricle with the miRNeasy Kit (QIAGEN) including DNase treatment on column. RNA concentration and quality was determined with the NanoVue Plus spectrophotometer (GE Healthcare) and the Agilent 2100 RNA Nano Chip (Agilent). cDNA synthesis by reverse transcription on total RNA was performed as described above. NOS1AP expression was assessed by real-time quantitative PCR (qPCR) with human-specific TaqMan Gene Expression assays (Applied Biosystems) for NOS1AP (Hs00928956_m1) and compared to RPL5 (Hs03044958_g1) (MIM 603634) as a housekeeping control. qPCR was carried on 7900HT Fast Real-Time PCR System (Applied Biosystems) and analyzed with Sequence Detection System Software v.2.1 (Applied Biosystems). All samples were run in duplicate and the −ΔCT (negative delta cycle threshold) was used as the measure of expression. For associations analysis, the −ΔCT expression value of 131 individuals with heart failure, as the dependent variable, was regressed on rs7539120 genotypes (coded as 0, 1, 2). Conventional linear regression analysis was performed with adjustment for age, sex, and study site.
Cardiac eQTL analysis, as described above, was also performed with the MAGNet consortium-generated microarray gene expression data for all protein-coding genes (12) mapping ±500 kb of rs7539120. Robust Multi-array Average expression values for probes specific to the candidate genes and rs7539120 genotypes from 89 individual samples were used for this analysis.
Electrophoretic Mobility Shift Assays
Complementary oligonucleotides encompassing rs7539120 alleles and carrying deletion of rs7539120 (±5 bases) were synthesized (Integrated DNA Technologies), based on the genomic sequence flanking the SNP (UCSC Genome Browser) (see Table S3 for EMSA oligo sequences). For radiolabeled probes, complementary oligonucleotides were annealed, and 5 pmol of double-stranded DNA was labeled with 15 pmol of γ-[32P]ATP (Perkin Elmer) with 20 U of T4 Polynucleotide Kinase (NEB). Unlabeled complementary oligonucleotides were annealed to create competitor probes. Nuclear extract from HL1 cells was prepared with NE-PER Nuclear and Cytoplasmic Extraction Kits (Pierce) according to manufacturer’s instructions. Radiolabeled probes (8,000 cpm) were incubated with 4 μl of HL1 nuclear extract at RT for 20 min in the presence of 10 mM Tris (pH 7.5), 50 mM KCl, 1 mM DTT, 1 μg poly(dI.dC), 2.5% glycerol, 0.05% NP-40, and 5 mM MgCl2 in a 20 μl reaction. Competitive binding assays were performed under the same conditions, with the addition of 10 pmol of unlabeled probe prior to the addition of the labeled probe. Protein-DNA complexes were resolved on 8% native polyacrylamide gels for 2 hr at 200 V in 0.5 × TBE buffer, dried on a sheet of Whatman 3 paper (Whatman), and visualized by autoradiography.
Production of Lentiviruses for NOS1AP Overexpression
Third generation lentiviral vectors were used for cellular NOS1AP overexpression. Coding sequences of human NOS1AP long isoform (RefSeq NM_014697) and short isoform (RefSeq NM_001126060) were cloned as AgeI-SalI fragments into the transfer plasmid pRRLsin18.cPPT.CMV.eGFP.Wpre vector (Inder Verma, Salk Institute, San Diego; hereafter designated as LV-eGFP), after the removal of eGFP, and the resulting plasmids were designated as LV-NOS1AP-Long and LV-NOS1AP-Short. Three third generation vectors, namely pMDLg/pRRE, pRSV-Rev, and pMD2.VSV.G (Trono Didier), served as packaging plasmids for lentivirus production. LVs were produced by transient cotransfection of HEK293T cells as described.33
Cell Culture
HEK293T cells were maintained in DMEM culture medium (Invitrogen) supplemented with 10% fetal bovine serum (FBS) (Invitrogen), 100 U/ml penicillin (Invitrogen), and 100 μg/ml streptomycin (Invitrogen). HL1 cells (gift from W. Claycomb, Louisiana State University) were maintained in Claycomb medium (Sigma) as described.30 Neonatal rat ventricular myocytes (NRVMs) were enzymatically disassociated from the ventricles of 2-day-old Sprague-Dawley rats (Harlan) via trypsin (Amersham Biosciences) and collagenase (Worthington Biochemical). Freshly isolated NRVMs were cultured as described.33 Two 90 min preplatings were performed to reduce fibroblast counts and enrich for cardiac myocyte content in the culture. The final cell suspension was collected, counted for NRVMs, and plated at a concentration of 0.5 million cells per 1 ml of medium. For mapping experiments, 1 million cells were plated on 21 mm plastic coverslips precoated with fibronectin (25 μg/ml in water) at RT for 2 hr (Sigma). After 24 hr, coverslips were washed with warm phosphate-buffered saline (PBS), and fresh medium with 10% FBS was added. After 48 hr, the culture medium was replaced with medium containing 2% FBS, and the cells were maintained in this 2% FBS-containing medium, with medium changes every second day. All protocols for rat care, use, and euthanasia were reviewed and approved by the Institutional Animal Care and Use Committee of Johns Hopkins University and were in accordance with Association for Assessment and Accreditation of Laboratory Animal Care (AAALAC) guidelines.
Optical Mapping in NRVMs
Optical mapping studies were performed on NRVM monolayers 6–7 days posttransduction as described.33 To determine the action potential duration (APD) at 80% repolarization (APD80) and conduction velocity (CV), cells were stimulated with monophasic, 10 ms pulses at 2 Hz delivered by the stimulus electrode at twice the diastolic threshold. A 2 s recording was taken after a 10-beat drive train. Action potentials were recorded from 253 sites by a custom-built contact fluorescence imaging system.33,34 With multisite optical recordings of transmembrane potential, sequential maps of the activation patterns of NRVM cultures were obtained, and normal and arrhythmic electrophysiological behavior were monitored. Data were analyzed in MATLAB (The MathWorks) with customized scripts.
NOS1AP Transcript Expression Profiling in Human and Mouse Tissues
Real-time quantitative expression profiling of NOS1AP in 24 human and 20 mouse tissues was done with Multiple Tissue cDNA (MTC) panels (Clontech) and TaqMan Gene Expression assays (Applied Biosystems). cDNAs from Human MTC Panel I, Human MTC Panel II, Human Fetal MTC Panel, Mouse MTC Panel I, and Mouse MTC Panel III were assessed for NOS1AP expression with human and mouse gene-specific TaqMan Gene Expression assays (Human NOS1AP: Hs00928956_m1, Mouse Nos1ap: Mm01290688_m1) according to manufacturer’s protocols. qPCR was carried on as described above. Expression for each assay-tissue combination was measured in technical triplicates and the averages of the threshold cycle (CT) values were used for analysis. Because the purchased MTC Panels have been normalized to several different housekeeping genes, the average CT values were compared across tissues without any normalization against a housekeeping gene’s expression level.
Anti-NOS1AP Antibody Generation
The production of a polyclonal antibody was contracted to the Custom Immunology Services at Covance (Covance). Using a peptide mapping to amino acid residues 486–503 of human NOS1AP (NP_055512) as an immunogen, an affinity-purified rabbit polyclonal antibody was generated after a 118-day protocol.
Immunoblotting in Mouse Tissue Lysates
Nos1ap expression in various mouse tissues was evaluated by immunoblotting with the rabbit polyclonal antibody we generated. The tissues were dissected from an adult FVB mouse, snap-chilled in liquid nitrogen, and stored at −80°C until used. All protocols for mouse care, use, and euthanasia were reviewed and approved by the Institutional Animal Care and Use Committee of Johns Hopkins University and were in accordance with AAALAC guidelines. Whole-tissue protein extracts from various mouse tissue samples were prepared by homogenization of ∼20 mg of tissue with a handheld homogenizer (Kinematica) in modified RIPA buffer supplemented with protease inhibitor cocktail (Roche). Tissue and cell debris were removed by centrifugation, and the protein concentration was determined by Bio-Rad DC Protein assay (Bio-Rad). Samples (200 μg) were denatured and analyzed by immunoblotting according to standard methods.35
Immunohistochemistry in Formalin-Fixed Paraffin-Embedded Human Heart Sections
FFPE sections of human heart tissue (6 μm) were single stained for NOS1AP and double stained for NOS1AP and Connexin43, N-Cadherin, or Plakoglobin by standard methods. Tissue sections were incubated with rabbit anti-NOS1AP polyclonal antibody alone and simultaneously in combination with commercially available mouse monoclonal antibodies against Connexin43 (Sigma), N-Cadherin (Sigma), or Plakoglobin (Sigma). Anti-rabbit IgG-HRP polymer conjugate was used for single-staining experiments. For double-staining experiments, anti-mouse IgG-HRP and anti-rabbit IgG-AP (alkaline phosphatase) polymer conjugates from PicTure Double staining kit (Invitrogen) were applied simultaneously to the slides. ImmPact DAB (Vector Labs) and Fast Red (Invitrogen) were used as chromogenic substrates for HRP and AP, respectively. Tissue sections were counterstained with Harris Hematoxylin (Sigma) and dehydrated and coverslips were mounted with Cytoseal 60 Mounting Medium (Electron Microscopy Sciences). Because of the presence of substantial auto-fluorescence in FFPE heart sections, we did not use fluorophores for colocalizations (data not shown).
Association between QT Interval and Variants Mapping at Genes Encoding Proteins Localized at Cardiomyocyte Intercalated Disc
The specific gene set, containing 170 autosomal genes whose protein products are localized at cardiomyocycte ID, was selected from a systematic review of cardiac immunohistochemistry data in the Human Protein Atlas (HPA) (with antibodies against more than 5,000 human proteins) and the review on ID proteins by Estigoy and colleagues.36 More than 27,000 polymorphisms mapping in and around (±10 kb) these genes were analyzed for association with QT interval using data from a large GWAS (D.E.A., unpublished data) in >70,000 subjects of EA. A control gene set of 166 autosomal genes, whose protein products are observed in human heart but not localized to ID, were selected by mining the HPA and more than 11,000 variants in and around (±10 kb) these genes were similarly analyzed for association with QT interval. Quantile-quantile plots of association with QT interval were generated and compared against (1) variants at the specific ID gene set, (2) variants at the control heart expressed gene set, and (3) genome-wide variants.
Results
Saturation Sequencing of the NOS1AP Target Interval
Genetic associations are detected by the principle of linkage disequilibrium (LD) across a genomic segment37 so that further analyses are required to identify causal variants. At a ∼141 kb long genomic region flanking the mapped NOS1AP locus,4 we attempted to create a near-complete catalog of common variants by sequencing in eight CEU and eight YRI HapMap17 reference samples and in 46 subjects from the KORA cohort16 where we conducted the original GWAS4 (Table S1). The target region was selected as the genomic segment within which all genotyped markers had p values above the significance threshold in the original QT interval GWAS.4 DNA from the target locus was recovered by long-range PCR and pooling of 17 overlapping amplicons (Table S1) and direct sequencing of the amplicon pool by Illumina technology.20 In all samples, we obtained ∼500-fold or greater coverage across the target locus (Table S1, Figures S1A and S1B); Figure S1C shows the number of variants detected with increasing depth of sequence coverage. In total, 1,063 SNVs were identified, all of which were noncoding (Table S1). Table 1 shows a summary of the sequence variation identified in each of the three groups examined and Table S1 shows the minor allele frequency distribution for the observed variation in KORA samples. No coding variation at the target locus was observed in the samples we sequenced, reflecting the high conservation of the NOS1AP coding sequence, and the variants were randomly distributed with 319, 3, and 741 being located in the upstream, 5′ UTR, and introns 1–2 regions of NOS1AP, respectively (Table S1).
Table 1.
Sequence Variation at the NOS1AP Locus in CEU, YRI, and KORA Samples
| CEU (n = 8) | YRI (n = 8) | KORA (n = 46) | |
|---|---|---|---|
| Total # variants | 474 | 614 | 880 |
| In dbSNP Build 129 | 401 (85%) | 408 (66%) | 456 (52%) |
| Not in dbSNP Build 129 | 73 (15%) | 206 (34%) | 424 (48%) |
| In dbSNP Build 135 | 457 (96%) | 576 (94%) | 705 (80%) |
| Not in dbSNP Build 135 | 17 (4%) | 38 (6%) | 175 (20%) |
| Exonica | 0 | 1 | 3 |
| Coding | 0 | 0 | 0 |
All exonic variants map to the 5′ UTR of NOS1AP.
We assessed the efficiency of de novo sequencing in comparison to variants available in existing databases, an important consideration if we are to census the causal candidate sequence variants within this target locus. We compared variants in the 46 KORA individuals with those reported in the 85 1000GP CEU samples26 (1000GP May 2011 release): in brief, 682 SNVs were observed in the 1000GP_CEU versus 880 in KORA with an overlap of 566 variants, with 314 and 116 variants being unique to KORA and 1000GP_CEU, respectively. The excess of unique variants in KORA samples is most probably improved detection resulting from the higher depth of sequencing at a single ∼141 kb locus (500-fold coverage) in contrast to the low pass (4-fold coverage) across the genome in the 1000 Genomes samples. Among the 314 variants unique to KORA samples, 175 variants were novel (not reported in dbSNP Build 135, Table 1) and of which 19 had >10% allele frequency and so were common in our study sample (Table S1); among the 116 variants unique to 1000GP_CEU, only 4 variants had an allele frequency >10%. Thus, de novo sequencing was necessary to identify potential causal variants (Table 1). We next estimated the specificity and sensitivity of de novo sequencing (Table S1). More than 99.3% and 98.3% concordances for sequencing-based genotype calls were observed in CEU and YRI samples, respectively, when compared to existing HapMap genotypes and more than 98.5% and 97.7% variant detection sensitivity was observed in CEU and YRI samples, respectively (Table S1).
Associations between NOS1AP Candidate Variants and QT Interval
For association tests of variants with the QT interval phenotype, we used 9,055 individuals of EA from the population-based ARIC cohort24 in whom genome-wide polymorphism data (Affymetrix 6.0 SNP array) were available.24 The NOS1AP target locus had 80 Affymetrix 6.0 SNPs (Figure S2A), which were used to impute and test a total of 512 variants. We performed association analysis with standard methods by regressing heart rate-, age-, and sex-corrected QT interval residuals on variant genotypes (Figure 1 and Table S2). Of the total 512 variants assessed, 210 variants were strongly associated with QT interval and reached genome-wide significance (p < 5 × 10−8) and rs12143842 was the most significant (p = 7.19 × 10−22) (Figure 1 and Table S2). This locus could be subdivided into three broad regions based on patterns of LD (LD blocks) (Figure S2B): rs12143842 is located within the first LD block and has a frequency of ∼0.25. At least seven more variants with similar frequencies (range: 0.221–0.251) and statistically significant p values (<4.09 × 10−19) are in high LD (r2 > 0.832) with the peak variant, based on analyses within ARIC. To assess the specificity of the signal, we performed a conditional association analysis by adjusting for the effect of the most significant variant (Figure S2C). A significant but relatively weaker association signal remained at rs75642230 (p = 8.7 × 10−6) in the first LD block, which based on LD is independent of the sentinel variant and not in high LD with variants in the second or the third LD blocks (Figure S2C). The conditional analysis therefore revealed a major independent signal in the NOS1AP region we targeted, confirming the results of our original GWAS,4 with rs75642230 capturing a secondary, relatively weaker signal at the same locus; however, existence of other minor variants cannot be excluded from these analyses. Additional associated polymorphisms do exist within the larger ∼300 kb NOS1AP locus6 but are not considered here because they map outside the peak association region of ∼141 kb studied here; this strategy was adopted because all QT interval association studies so far show that rs12143842 is the most significant signal.
Figure 1.

Regional Association between Variants at the NOS1AP Locus and QT Interval in ARIC
The x axis shows the genomic interval annotated with NOS1AP and OLFML2B transcripts, the left y axis shows the statistical significance of association as negative log10 of p values, and the right y axis shows the human recombination map based on HapMap samples. The most significant SNP, rs12143842, is shown as a purple diamond, the functional SNP, rs7539120 (see Figure 2, below), is shown as a red diamond, and the remainder are shown as gray circles. The genomic interval corresponding to the first major LD region (see Figure S2B) is highlighted in light gray on the x axis. The plot was generated with LocusZoom.
Annotation of Polymorphisms for Functional Analyses
All of the 210 common variants associated with QT interval at the NOS1AP locus were noncoding, indicating the plausibility that variation within a cis-regulatory functional element regulates gene expression and underlies QT interval variation, although other mechanisms (e.g., miRNA, ncRNA, etc.) underlying noncoding variation function are possible. Among the various functional annotations of the human genome,28 we focused on DNaseI hypersensitivity (DH) because it is an in vivo indicator of enhanced chromatin accessibility and is a universal hallmark of active cis-regulatory sequences.38 Because NOS1AP is widely expressed in human tissues (see below), we restricted our analysis to DH maps generated in the following human cardiac tissues and cell lines: human heart (Heart_OC; frozen tissues retrieved from autopsy and surgery) and three cardiac primary cell lines, namely HCF (human cardiac fibroblasts), HCFaa (human cardiac fibroblast, adult atrial), and HCM (human cardiac myocytes).39,40 In the NOS1AP target interval, there are 5, 10, 10, and 18 DHSs41,42 covering 1.68 kb, 1.61 kb, 1.69 kb, and 2.91 kb in Heart_OC, HCF, HCFaa, and HCM samples, respectively (Figure S2D). The combination of these four maps led to identification of 21 cardiac DHSs covering 4.6 kb of the target interval. Among the 210 QT-interval-associated variants, 5 sequence variants map to these cardiac DHSs and were selected for functional assessment (Figure S2D). To broaden our search, we also evaluated genome-wide binding profiles of enhancer-associated coactivator protein p300 and the closely related CBP coactivator protein43 in human heart tissue.44 However, there were no p300/CBP-bound regions in the NOS1AP target interval.
Enhancer/Suppressor Analysis of Selected Variants Identifies a Functional Variant Underlying QT Interval Association
We used two criteria for selecting 12 QT-interval-associated variants for functional analyses: (1) rs12143842 (the sentinel variant) and all other variants in high LD with it; and (2) all variants mapping to cardiac DHSs defined above (Table S3). At each variant, alternate alleles were cloned into luciferase reporter constructs and evaluated for differential enhancer/suppressor activity via transient assays in the mouse cardiomyocyte cell line, HL1.30 We also used the human embryonic kidney cell line HEK293T31 as a noncardiac cell line for these reporter assays. Three variants (rs12123681, rs7366599, and rs12123710), because of their close proximity, were amplified and tested together (referred to as the rs12123710 construct hereafter); however, variant rs7366599 within a homopolymer stretch of adenine bases was not recovered during amplification and not tested in reporter assays. Genomic regions flanking rs12123710, rs2010491, and rs945708 cloned for reporter assays included additional common variants and were tested as QT-prolonging (risk) and QT-reducing (nonrisk) allele-carrying haplotypes. The insert for rs12123710 included rs1415268, the insert for rs2010491 included rs7539120, and the insert for rs945708 included rs10918762. These three additional variants were also genome-wide significant for QT interval association (Table S3).
Across all variants tested in both cell lines, only the rs7539120_rs2010491 haplotype constructs showed a small (1.19-fold) but consistent (8 biological replicates) and significant (p = 3 × 10−5) differential allelic activity in HL1 cells (Figures 2A and S3A). On repeating reporter assays for the rs7539120_rs2010491 haplotype constructs in HL1 cells, in another set of 8 biological replicates, we obtained similar results (1.19-fold, p = 9.5 × 10−4) (Figure S3B), indicating that the differential allelic activity is consistent. The risk allele haplotype rs7539120T_rs2010491G (haplotype 2; H2) had increased reporter expression as compared to the nonrisk allele haplotype rs7539120A_rs2010491A (haplotype 1; H1), so that we needed to test whether one or the other or both variants were responsible for the differential transcriptional activity. The insert size used for the rs7539120_rs2010491 haplotype construct was 959 bp. To refine the functional element driving enhancer activity, we created a series of deletion constructs and evaluated them for reporter expression in HL1 cells (Figure 2B and Table S3). A 217 base long deletion from the 5′ end (rs7539120_rs2010491_742) led to increased differential enhancer activity (1.5-fold; p = 6.7 × 10−6). Deletion of rs7539120 (±5 bases) in the risk allele haplotype (Δrs7539120_rs2010491_742) reduced the expression to levels similar to the nonrisk allele haplotype, indicating that rs7539120 is the functional variant driving enhancer activity in the rs7539120_rs2010491 haplotype construct. A longer 467 base deletion from the 5′ end (rs7539120_rs2010491_492) led to increased reporter expression from both haplotypes but with no significant allelic difference. These experiments indicated that (1) the bases 218–467 (250 bases) of the 959 bp construct carry an enhancer element that increases expression from the rs7539120 risk allele only, (2) the bases 1–217 of the 959 bp construct carry a repressive element, and (3) the internal 250 base enhancer element is necessary to drive enhanced expression from the rs7539120 risk allele. Consequent to this prediction, we can indeed demonstrate that internal deletion of bases 218–467 from the 959 bp construct (rs7539120_rs2010491_Δ250) leads to similar reporter expression from both haplotypes. We also evaluated the sentinel variant rs12143842 in reporter assays by using smaller inserts (623 and 325 bp, Table S3) to identify any insert-size-dependent allelic difference in reporter expression; unlike the rs7539120 variant, no significant allelic expression difference could be observed (Figure S3C). Together, these results show that despite the peak association being at rs12143842, the functional variant is probably rs7539120 whose in vitro enhancer activity is flanking sequence dependent (Figure 2B).
Figure 2.
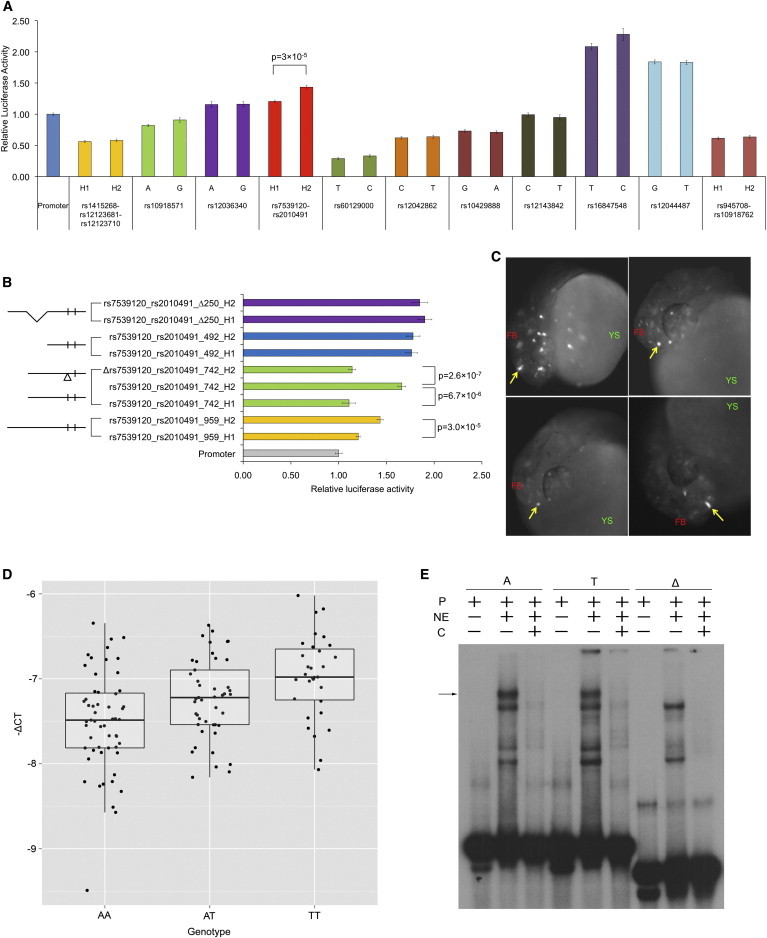
rs7539120 Is a Functional Variant Underlying QT Interval Association at the NOS1AP Locus
(A) Firefly luciferase reporter enhancer/silencer assays in HL1 cells using alternate alleles/haplotypes for selected QT-interval-associated NOS1AP variants. Firefly luciferase expression is plotted relative to Renilla luciferase expression, normalized to the expression from empty vector (Promoter). Mean luciferase expression between alternate alleles/haplotypes constructs was compared with the t test and found to be significantly different between rs2010491 haplotypes, which also include rs7539120 alleles (p = 3 × 10−5). Error bars indicate SEM (n = 8).
(B) Enhancer activity in rs7539120_rs2010491 haplotype is driven by rs7539120 and is dependent on the flanking sequence. Firefly luciferase reporter enhancer/silencer assays in HL1 cells using a deletion series derived from the 959 bp rs7539120_rs2010491 construct. In the construct design on left, small vertical lines represent the rs7539120 and rs2010491 SNPs, Δ represents the 11-base deletion encompassing rs7539120, 5′ deletions are represented by shorter lengths of the horizontal line, and an internal 250 bp deletion is represented by sloping lines. Error bars indicate SEM (n = 8).
(C) rs7539120 acts as an in vivo enhancer. The rs7539120 risk haplotype construct injected (rs7539120_rs2010491_742_H2) at the 1–2 cell stage in developing zebrafish embryos drives transient reporter expression (enhanced-GFP; eGFP) in forebrain 24 hr postfertilization in ∼33% of injected embryos. Representative images from four different embryos with forebrain eGFP expression (white spots, strong expression indicated by yellow arrows) are shown. No eGFP expression was observed from the rs7539120 deletion (±5 bases) constructs (Δrs7539120_rs2010491_742_H2) (see Figure S4). Abbreviations are as follows: FB, forebrain; YS, yolk sac.
(D) rs7539120 acts as a cardiac expression quantitative trait locus (eQTL). Box-whisker plots of NOS1AP mRNA expression in human left ventricle tissue from 131 heart failure subjects genotyped at rs7539120. The y axis shows –ΔCT used as a measure of expression. The risk allele T is associated with higher expression of NOS1AP (p = 4.72 × 10−5).
(E) rs7539120 is bound by an uncharacterized protein(s) from HL1 cell nuclear extract. EMSAs using radiolabeled probes (P) containing nonrisk allele (A), risk allele (T), and deletion (Δ) of rs7539120 in presence or absence of HL1 cells nuclear extract (NE) and excess unlabeled competing probe (C). The black arrow indicates DNA-protein complex formed with both the A and T alleles containing probes, but lacking with the deletion probe. + indicates addition; - indicates absence.
A direct hypothesis from the above results is that rs7539120 is located within an enhancer element. First, we directly evaluated the potential of the rs7539120 variant to act as an enhancer in vivo by using transient developmental enhancer assays in zebrafish embryos. For this, the 742 bp long risk (rs7539120_rs2010491_742_H2) and nonrisk (rs7539120_rs2010491_742_H1) haplotypes and the rs7539120 deletion (±5 bases) risk haplotype (Δrs7539120_rs2010491_742_H2) inserts used in in vitro assays (above) were cloned into minimal promoter driven eGFP reporter constructs and injected into developing zebrafish embryos at 1–2 cells stage and observed for eGFP expression 24 hr postfertilization. The risk allele haplotype drove eGFP expression in forebrain from ∼33% of injected embryos (Figure 2C and Table S3). The nonrisk allele haplotype also drove eGFP expression in forebrain from ∼25% of injected embryos (Figure S4A and Table S3). As expected in transient expression assays, low levels of mosaic expression was also observed. No eGFP expression was observed from the rs7539120 deletion construct (Figure S4A), thus indicating that the rs7539120 variant site and the flanking sequence is necessary for in vivo enhancer activity. We also compared the observed in vivo transient enhancer activity of the rs7539120_rs2010491_742 construct with nos1apa endogenous expression in wild-type zebrafish embryos 24 hpf. We could not detect nos1apa expression by RNA in situ hybridization. However, by RT-PCR, nos1apa expression was observed in the head region and in the remainder of the body from zebrafish embryos as well as in whole embryos (Figure S4B), indicating that nos1apa is expressed at rather low levels not detectable by in situ. Thus, the in vivo enhancer activity observed for rs7539120 construct in transient zebrafish assays overlaps nos1apa expression domains.
Based on the transient in vivo enhancer activity of rs7539120 we observed, the alternate alleles at rs7539120 are expected to demonstrate expression differences in the human heart. Consequently, we measured NOS1AP mRNA levels in human left-ventricular heart tissue obtained from 131 EA subjects with heart failure whom we genotyped for rs7539120. There is no a priori evidence for the NOS1AP locus influencing the risk for heart failure, so the disease state of study subjects is not expected to influence NOS1AP expression. NOS1AP mRNA expression levels were measured by reverse transcriptase-quantitative PCR and rs7539120 was genotyped by direct Sanger sequencing. As predicted, a significant, albeit small, increase in NOS1AP mRNA expression was observed with each copy of the risk allele T at rs7539120 (p = 4.72 × 10−5) (Figure 2D). The same effect of enhanced expression with the risk allele T at rs7539120 was observed in in vitro luciferase reporter assays (above) and therefore shows consistency between the in vitro and in vivo effects.
We also evaluated rs7539120 as a cardiac eQTL for expression of other protein-coding genes nearby. Besides NOS1AP, there are 11 protein-coding genes within ±500 kb of rs7539120. With the MAGNet-consortium-generated cardiac microarray gene expression data from 89 EA samples for which we also had rs7539120 genotypes, eQTL analysis ruled out all but NOS1AP as candidate genes at this locus. Although, based on microarray gene expression data, 8 had cardiac expression, NOS1AP was the only gene whose cardiac gene expression was significantly (p = 1.4 × 10−3) influenced by the rs7539120 genotype (Table S4).
These data support our hypothesis that rs7539120 lies within a cardiac enhancer element. As further evidence we performed EMSAs for both the rs7539120_T risk allele and rs7539120_A nonrisk allele carrying short duplexes (43 bp). These were found to be bound by an uncharacterized protein(s) from HL1 cell nuclear extract, although there is only a small difference in allele-specific binding/shift, a feature common to many, but not all, such allele-specific EMSA assays. However, an 11 base deletion encompassing the variant site abrogated this DNA-protein complex, indicating that the variant base and the flanking sequence act as the binding site (Figure 2E). Consequently, although support for the enhancer hypothesis is strong, the exact nature of the bound cardiac protein is currently unknown.
NOS1AP Regulates Cardiac Cellular Electrophysiology
The genetic results described above, together with expression differences at NOS1AP, suggest that NOS1AP is indeed the basis for QT interval association. To prove its further functional involvement, we next sought direct evidence by altering NOS1AP expression and demonstrating cellular electrophysiological differences in a surrogate system. Two previous studies, prompted by our original genetic screen,4 provide some evidence for a possible role of NOS1AP in regulating cardiac electrophysiology. In a prior collaborative study we demonstrated that morpholino-based knockdown of zebrafish nos1ap leads to shortened APD in excised hearts from developing embryos.45 Second, overexpression of guinea pig Nos1ap in guinea pig ventricular myocytes by in vivo gene transfer leads to shortened APD mediated by inhibition of L-type calcium current.46 These two studies, although performed in different model systems, indicate seemingly opposite directions of effect. Consequently, we evaluated the role of NOS1AP in cardiac cellular electrophysiology with NRVMs as a model system.47 Overexpression of both long and short isoforms of human NOS1AP led to a significant decrease in APD (Figure 3A) and a significant increase in CV measured across the monolayer of cultured NRVMs (Figure 3B), as compared to nontransduced cells. These results are consistent with the findings of Chang et al.46 in other mammalian cardiac cells and show that NOS1AP expression level directly affects cellular electrical properties.
Figure 3.

Expression Levels of NOS1AP Influences Cardiac Electrophysiology
(A) Left: Representative APD (in milliseconds, ms) trace at 80% repolarization (APD80) in NRVMs nontransduced (top) and transduced with human NOS1AP short (middle) and long (bottom) isoforms, respectively. Overexpression of both isoforms of NOS1AP in NRVMs lead to significantly reduced APD80, as compared to nontransduced cells. Right: Bar plots showing mean APD80 from four replicates. Error bars indicate standard deviation and asterisks indicate p < 0.05.
(B) Representative isochronal maps for CV (in cm/s) in monolayer of NRVMs nontransduced (top left) and transduced with human NOS1AP short (top right) and long (bottom left) isoform, respectively. Overexpression of both isoforms of NOS1AP in NRVMs leads to significantly increased CV, as compared to nontransduced cells. Bottom right: Bar plots showing mean CV from four replicates. Error bars indicate standard deviation and asterisks indicate p < 0.05.
NOS1AP Localizes to Intercalated Discs in Cardiac Muscle
NOS1AP transcript and protein are widely expressed as determined from TaqMan-based quantitative PCR (Figures S5A and S5B) and immunoblotting (Figure S5C). Thus, as a clue to its cellular function, we assessed cellular localization of NOS1AP by immunohistochemical staining of formalin-fixed paraffin-embedded (FFPE) sections of human hearts with a custom rabbit polyclonal NOS1AP antibody. Strikingly, and as opposed to the published literature based on commercial antibodies, NOS1AP was mostly localized to ID joining cardiomyocytes (Figure 4A). To confirm this localization, we performed dual immunohistochemical staining for NOS1AP and three well-localized cardiac ID marker proteins, representing each of the three well-recognized structural zones within the ID, namely gap junctions, desmosomes, and fascia adherens.36 By light microscopy, NOS1AP was found to colocalize with all three ID marker proteins, namely Connexin43 (Gap junctions), N-Cadherin (Fascia adherens), and Plakoglobin (Desmosomes) (Figure 4B), pointing to its broader distribution throughout the ID and probably a broader functional role.
Figure 4.

NOS1AP Localizes to ID in Cardiac Muscle
(A) Immunohistochemical staining of FFPE section of human heart (left ventricle free wall) with NOS1AP antibody. Intense staining (dark brown) was observed at ID joining adjacent cardiomyocytes.
(B) NOS1AP colocalizes with proteins marking the three well-recognized structural zones within the ID: desmosomes, gap junctions, and fascia adherens. Immunohistochemical staining of FFPE section of human heart with an antibody against an ID marker protein (Plakoglobin at desmosomes, top; Connexin-43 at gap junctions, middle; and N-cadherin at fascia adherens, bottom) alone (left column) and in combination with NOS1AP antibody (right column).
Intercalated Disc Proteins Play a Significant Role in QT Interval Regulation
The localization of NOS1AP, the major genetic regulator of QT interval trait variation, at cardiomyocyte ID presents a very attractive and testable functional hypothesis for QT interval variation. It is possible that, as currently believed, most QT interval variation arises from variation in repolarization components (Na channels, K channels, and proteins that regulate such channels directly) and that NOS1AP is the exception being localized at the ID. Alternatively, it’s likely that the ID plays a major role in QT interval variation but through variation in propagation. We, therefore, assessed whether sequence variation at genes whose protein products are localized to the ID contributes to QT interval variation. To do so, a specific ID gene list of 170 genes (Table S5) was selected from a systematic review36 based on cardiac immunohistochemical data in the HPA; a control-heart-expressed gene list of 166 genes, whose protein products are observed in the heart but not localized at the ID, was selected by mining HPA (Table S5). We compared both against the results from a recent large GWAS for QT interval in >70,000 EA subjects performed by the QT Interval-International GWAS Consortium (QTIGC) (D.E.A., unpublished data). As Figure 5 clearly shows, using quantile-quantile (QQ) plots, the combined effect of variants in and around the 170 ID genes show a considerably higher enrichment for association than the genome-wide comparison. The parsimonious reason for this finding is the relatively higher proportion of QT-interval-affecting genes in the ID than the whole genome. This enrichment remained even after removing the known and major effect of NOS1AP and this specific effect was not observed for variants in and around the control-heart-expressed genes (Figure 5). Thus, the combined effect of ID genes, implicated by the finding of NOS1AP, is highly significant and shows the important role of the cardiomyocyte ID in the population variability of the QT interval. This suggests that propagation defects, like repolarization defects, are an important source of QT interval prolongation and sudden cardiac death.
Figure 5.
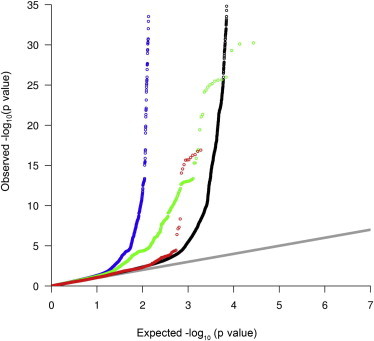
Genes Encoding Proteins Localized at Cardiomyocytes ID Play a Significant Role in Interindividual QT Interval Variation
QQ plots of QT interval GWAS, and association studies of QT interval using a specific set 170 ID genes and NOS1AP, and a control set of 166 heart-expressed genes not localized at ID. The black, blue, green, and red curves show the QQ plots for all GWASs, ID genes including NOS1AP, ID genes minus NOS1AP, and the control heart-expressed genes. The gray line shows the expected values from a theoretical χ2-distribution. For each gene we selected variants in and around (±10 kb) the gene from the GWAS. The y axis has been truncated at 35.
Discussion
GWASs have been an effective screen for common polymorphic markers that underlie genetic variation of quantitative traits and complex diseases.48 Nevertheless, at least so far, they have been ineffective in directly leading to a deeper understanding of either quantitative trait genetic architecture or the pathophysiology of common and chronic human disease. Except for a few examples where association mapping has led to the tacit implication of a nearby gene as a novel disease hypothesis, such as dysregulated complement function in age-related macular degeneration49 (MIM 603075) or autophagy in Crohn disease50 (MIM 266600), in the vast majority of cases it is not clear which of the many genes close to the associated polymorphism(s) is the functional molecule nor which of the many common variants is (are) the causal factors nor the molecular basis of the observed association. Although GWAS results are presented with a nearby gene as the candidate pro tem, this choice is fictive and requires functional data demonstrating its role in that trait or disease.51 In this study, we use a variety of functional analyses to not only implicate a gene but also demonstrate how its cellular localization both suggests a pathophysiological hypothesis and pinpoints functional candidates not known previously.
In this study, we have undertaken a genetic dissection of a primary marker of cardiac repolarization, the QT interval. Our original GWAS identified NOS1AP as the locus with the single largest genetic contribution to interindividual trait variation4–6 and, subsequently, we demonstrated that these same polymorphisms significantly increased the risk for SCD in the general population10,12 and that they also acted as potent genetic modifiers of the LQTS phenotype in individuals with known Na and K channel mutations.13,14 We report here the identification of rs7539120 as one of the functional noncoding variants affecting QT interval variation and provide evidence that NOS1AP is indeed the underlying gene at the associated locus whose expression levels influence cellular electrophysiology. And, based on NOS1AP localization at the ID in human cardiac muscle, we hypothesize that NOS1AP affects cardiac electrical conductance and coupling and thus regulates the QT interval. Identification of NOS1AP as a major genetic regulator of QT interval variation, a gene not previously known to influence cardiac repolarization, thus leads us to a new perspective of QT interval biology.
The localization of NOS1AP to ID opens the way to assess its detailed cellular function, how it regulates cardiac repolarization, and its role in sudden death and other conduction disorders. Interestingly, (1) these functions are not solely attributable to NOS1AP—our QT interval GWAS identifies other loci as well (D.E.A., unpublished data),5,6 (2) the LQTS genetic modification suggests interaction with cardiac Na and K channels,13,14 and, most crucially, (3) the ID harbors many other proteins whose genetic variation also affects the QT interval (Figure 5). Consequently, we advance the hypothesis that one or more proteins at the ID, including NOS1AP, regulate ion flow through the cardiomyocyte gap junctions. A consequence of this hypothesis is that many of these ID proteins can now be directly tested and implicated both in modulating population-level QT interval variation and the risk of SCD likely through propagation defects. This implication, as shown in Figure 5, would be impossible without GWAS data and it is this use of testing functional hypotheses directly that will make the expanding catalogs of GWASs valuable. By restricting attention to variants that map near a functionally enriched set of genes encoding proteins that localize to ID, we were able to identify associations with the QT interval that were missed in the GWAS. Specifically, at false discovery rates of 5% and 1%, we identified 27 and 14 ID genes, respectively, which is a large yield of novel functional candidates. Although these genes need to be further investigated, two exemplars are PTK2 (MIM 600758) and SIPA1L1. PTK2 (protein tyrosine kinase 2) encodes focal adhesion kinase (FAK), a broadly expressed nonreceptor protein-tyrosine kinase, which is concentrated at focal adhesions and regulates many cellular processes in development and disease.52 Specifically, in cardiomyocytes, sarcomeric myosin interacts with FAK to regulate FAK activity and has an impact on the control of cardiomyocyte growth in response to hypertrophic stimuli.53 SIPA1L1 (signal-associated proliferation-associated 1 like 1) encodes a Rap GTPase activating protein that plays a role in noncanonical Wnt signaling and contributes to development.54 Interestingly, common variants at SIPA1L1 locus have been reported to be associated with QRS interval duration, an indicator of ventricular depolarization, in a large GWAS of more than 40,000 subjects.55
The major role of NOS1AP as a QT interval regulator together with the GWAS of other electrocardiographic (ECG) traits, including QT interval, have clearly highlighted the role of non-ion-channel proteins in cardiac electrophysiology. Biochemically, Na channels, K channels, and Ca-handling proteins have been known as the major mediators of cardiac de- and repolarization, but genetic studies in contrast have shown that variation in these channel proteins explain only a minor portion of the trait variance, probably owing to their highly conserved structures and functions. Indeed, the major sources of genetic variation in ECG traits are non-channel proteins that probably regulate cardiac electrical cycle indirectly by influencing ion channel expression, localization, and function. Or, they probably affect propagation as opposed to de- or repolarization. Therefore, the role of the ID may not simply be structural, as usually assumed, but as a rich signaling domain in cardiomyocytes that influences the expression of structural proteins and proteins involved in metabolism as well as electrical excitability.
Our study also has implications for the nature of GWAS variants. The QT-interval-associated functional variant, rs7539120, is a noncoding variant and has an allele frequency of 0.42 (risk allele; T) in directly genotyped EA samples (n = 273) from the MAGNet consortium. The functional enhancer activity of this variant is dependent on the flanking sequence, and deletion experiments show that this variant site demonstrates both consistent and necessary enhancer activity in vitro and in vivo. Also, this variant site is bound by a nuclear protein from HL1 cells, yet uncharacterized, which will provide further clues to NOS1AP function. Surprisingly, the enhancer variant shows only moderate LD (r2 = 0.44) with the original discovery sentinel variant rs12143842, which has a significantly lower allele frequency of 0.26 (risk allele; T) in directly genotyped EA samples (n = 273). These observations are explained by the monophyletic origin of rs12143842 but the polyphyletic origin of rs7539120, the latter occurring on multiple haplotypes, only some of which contain the sentinel variant. This genetic outcome is not unexpected because the functional variant rs7539120 maps to a simple sequence repeat (SSR) (TA)n that we hypothesize leads to increased hypermutability, its multiple origins, and only moderate LD with the sentinel variant. It is important to note that there is growing evidence supporting the role of SSRs as a source of qualitative and quantitative genetic variation and that SSRs coevolve with the genes they are associated with to influence gene regulation, transcription, and protein function.56,57 Given the widespread occurrence of such repetitive sequences in the human genome, this type of enhancer is unlikely to be an isolated example. This implies that contemporary databases of noncoding functional elements (ENCODE28) that are deficient in repeated sequences may miss these elements.
The existence of multiple independent association signals of varying significance at any given locus is not uncommon and has been reported in several GWASs. In our study, association analysis conditional on the sentinel variant rs12143842 revealed the presence of a secondary, relatively weaker association signal captured by rs75642230 (p = 8.7 × 10−6) (Figures S2C and S6A). Association analysis conditional on both the functional variant rs7539120 and the sentinel variant rs12143842 reveals the weaker secondary signal at rs75642230 (p = 1 × 10−5) (Figure S6B); conditioning on these three variants completely removes the association signal at this locus (Figure S6C). These analyses indicate that rs12143842 is the major signal at the QT-interval-associated NOS1AP locus and that rs75642230 captures an independent, relatively weaker signal at the same locus.
Because rs7539120 is located in a repeat region and was imputed for association analysis, we evaluated the accuracy of imputation by directly genotyping rs7539120 by PCR followed by Sanger sequencing in 367 ARIC samples, which were a subset of 9,055 ARIC samples in which imputation and association was performed. The overall match with imputation calls was 85.6%: these match frequencies were 98.5%, 81.3%, and 65.3% for the AA, AT, and TT genotypes, respectively. This would indicate that imputation, which is based on 1000 Genomes reference haplotypes, is undercalling the risk allele T, and thus the mismatch rate is highest for TT genotypes; in other words, the “reference” genotypes are not highly accurate. This makes it likely that the 1000 Genomes reference haplotypes have an underrepresentation of T (risk) allele-carrying haplotypes. Based on these observations, we would conclude that the true association of rs7539120 with the QT interval is higher than what is observed based on imputation in ARIC samples. This is another reason for the apparent lower association of the functional SNP in contrast to the sentinel marker. We also genotyped rs12143842, the sentinel QT-interval-associated SNP, by TaqMan assay in 278 ARIC samples and found the overall match with imputation calls to be 98.9%, indicating that low imputation accuracy for rs7539120 is most likely due to it being in a repeat region.
Our QT interval study demonstrates that replicated genetic associations are almost invariably noncoding, even for loci where the functional candidate gene is clear such as those for the known LQTS Na and K channel genes or the Ca pump regulator phospholamban (PLN). Because the cardiac phenotypes of disruptive coding mutations in these genes are well described,3,58 the overall evidence (hypothesis) for population QT interval variation is that it arises from variation in levels of wild-type (not mutant) protein. Consequently, we propose it is likely that QT interval variation arises from variation in the stoichiometry of specific regulatory proteins—but which ones? QT interval GWASs have implicated at least 35 loci in our recent studies (D.E.A., unpublished data), but which specific genes and variants do they represent? Functional analyses such as the ones we report here will be necessary and can be performed in higher throughput by using parallel cellular electrophysiology studies of gene knockdown or overexpression in NRVMs (Figure 3), sequence variant catalogs,26 and new high-throughput sequencing-based in vitro and in vivo assays of noncoding regulatory function.59,60 As an alternative approach, we could have used DNA sequence conservation across multiple species to prioritize variants.61–63 However, conservation of regulatory function is not always reflected in sequence conservation.28,64 Moreover, many cis-regulatory elements, especially enhancers, are not only species specific but also cell-type specific.65 Finally, loss of highly conserved elements does not always lead to major phenotypic changes because “shadow” enhancers might compensate for these functions.66,67 Therefore, phenotype-driven approaches, such as QT interval GWASs, are important for understanding cardiac regulatory function.
Acknowledgments
We are grateful to William C. Claycomb (Louisiana State University, New Orleans) for providing HL1 cells, Norman Barker (Johns Hopkins University, Baltimore) for help with imaging, and Akhilesh Pandey and Andrew S. McCallion (Johns Hopkins University, Baltimore) for critical discussions. We also thank Ashley O’Connor and Maria X. Sosa (Johns Hopkins University, Baltimore) for technical assistance, Paula Kokko-Gonzales, Louise Fraser, Niall Gormley, and Terena James (Illumina) for sequencing, and Ankit Rakha (Johns Hopkins University, Baltimore), Keira Cheetham, and Lisa Murray (Illumina) for computational support. This work was supported by NIH grants RO1HL086694 and RO1HL105993 and funds from the Donald W. Reynolds Foundation. A.C. is on the Scientific Advisory Board of Biogen Idec and this potential competing interest is managed by the policies of the Johns Hopkins University, School of Medicine. M.R. and D.B. are employees of Illumina, Inc., a public company that develops and markets systems for genetic analysis.
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
1000GP Pilot data July 2010 release, ftp://ftp-trace.ncbi.nih.gov/1000genomes/ftp/pilot_data/release/2010_07/low_coverage/snps/
1000GP May 2011 release, ftp://ftp-trace.ncbi.nih.gov/1000genomes/ftp/release/20110521/
International HapMap Project, release #28 (http://hapmap.ncbi.nlm.nih.gov/cgi-perl/gbrowse/hapmap28_B36/)
LocusZoom, http://csg.sph.umich.edu/locuszoom/
MAGNet, http://www.med.upenn.edu/magnet/
Online Mendelian Inheritance in Man (OMIM), http://www.omim.org/
R statistical software, http://www.r-project.org/
The Human Protein Atlas, http://www.proteinatlas.org/
UCSC Genome Browser, http://genome.ucsc.edu
References
- 1.Dekker J.M., Crow R.S., Hannan P.J., Schouten E.G., Folsom A.R., ARIC Study Heart rate-corrected QT interval prolongation predicts risk of coronary heart disease in black and white middle-aged men and women: the ARIC study. J. Am. Coll. Cardiol. 2004;43:565–571. doi: 10.1016/j.jacc.2003.09.040. [DOI] [PubMed] [Google Scholar]
- 2.Newton-Cheh C., Larson M.G., Corey D.C., Benjamin E.J., Herbert A.G., Levy D., D’Agostino R.B., O’Donnell C.J. QT interval is a heritable quantitative trait with evidence of linkage to chromosome 3 in a genome-wide linkage analysis: The Framingham Heart Study. Heart Rhythm. 2005;2:277–284. doi: 10.1016/j.hrthm.2004.11.009. [DOI] [PubMed] [Google Scholar]
- 3.Priori S.G., Napolitano C. Genetics of cardiac arrhythmias and sudden cardiac death. Ann. N Y Acad. Sci. 2004;1015:96–110. doi: 10.1196/annals.1302.008. [DOI] [PubMed] [Google Scholar]
- 4.Arking D.E., Pfeufer A., Post W., Kao W.H., Newton-Cheh C., Ikeda M., West K., Kashuk C., Akyol M., Perz S. A common genetic variant in the NOS1 regulator NOS1AP modulates cardiac repolarization. Nat. Genet. 2006;38:644–651. doi: 10.1038/ng1790. [DOI] [PubMed] [Google Scholar]
- 5.Newton-Cheh C., Eijgelsheim M., Rice K.M., de Bakker P.I., Yin X., Estrada K., Bis J.C., Marciante K., Rivadeneira F., Noseworthy P.A. Common variants at ten loci influence QT interval duration in the QTGEN Study. Nat. Genet. 2009;41:399–406. doi: 10.1038/ng.364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pfeufer A., Sanna S., Arking D.E., Müller M., Gateva V., Fuchsberger C., Ehret G.B., Orrú M., Pattaro C., Köttgen A. Common variants at ten loci modulate the QT interval duration in the QTSCD Study. Nat. Genet. 2009;41:407–414. doi: 10.1038/ng.362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jaffrey S.R., Snowman A.M., Eliasson M.J., Cohen N.A., Snyder S.H. CAPON: a protein associated with neuronal nitric oxide synthase that regulates its interactions with PSD95. Neuron. 1998;20:115–124. doi: 10.1016/s0896-6273(00)80439-0. [DOI] [PubMed] [Google Scholar]
- 8.Aarnoudse A.J., Newton-Cheh C., de Bakker P.I., Straus S.M., Kors J.A., Hofman A., Uitterlinden A.G., Witteman J.C., Stricker B.H. Common NOS1AP variants are associated with a prolonged QTc interval in the Rotterdam Study. Circulation. 2007;116:10–16. doi: 10.1161/CIRCULATIONAHA.106.676783. [DOI] [PubMed] [Google Scholar]
- 9.Post W., Shen H., Damcott C., Arking D.E., Kao W.H., Sack P.A., Ryan K.A., Chakravarti A., Mitchell B.D., Shuldiner A.R. Associations between genetic variants in the NOS1AP (CAPON) gene and cardiac repolarization in the old order Amish. Hum. Hered. 2007;64:214–219. doi: 10.1159/000103630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kao W.H., Arking D.E., Post W., Rea T.D., Sotoodehnia N., Prineas R.J., Bishe B., Doan B.Q., Boerwinkle E., Psaty B.M. Genetic variations in nitric oxide synthase 1 adaptor protein are associated with sudden cardiac death in US white community-based populations. Circulation. 2009;119:940–951. doi: 10.1161/CIRCULATIONAHA.108.791723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Arking D.E., Khera A., Xing C., Kao W.H., Post W., Boerwinkle E., Chakravarti A. Multiple independent genetic factors at NOS1AP modulate the QT interval in a multi-ethnic population. PLoS ONE. 2009;4:e4333. doi: 10.1371/journal.pone.0004333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Eijgelsheim M., Newton-Cheh C., Aarnoudse A.L., van Noord C., Witteman J.C., Hofman A., Uitterlinden A.G., Stricker B.H. Genetic variation in NOS1AP is associated with sudden cardiac death: evidence from the Rotterdam Study. Hum. Mol. Genet. 2009;18:4213–4218. doi: 10.1093/hmg/ddp356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Crotti L., Monti M.C., Insolia R., Peljto A., Goosen A., Brink P.A., Greenberg D.A., Schwartz P.J., George A.L., Jr. NOS1AP is a genetic modifier of the long-QT syndrome. Circulation. 2009;120:1657–1663. doi: 10.1161/CIRCULATIONAHA.109.879643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tomás M., Napolitano C., De Giuli L., Bloise R., Subirana I., Malovini A., Bellazzi R., Arking D.E., Marban E., Chakravarti A. Polymorphisms in the NOS1AP gene modulate QT interval duration and risk of arrhythmias in the long QT syndrome. J. Am. Coll. Cardiol. 2010;55:2745–2752. doi: 10.1016/j.jacc.2009.12.065. [DOI] [PubMed] [Google Scholar]
- 15.Manolio T.A., Collins F.S., Cox N.J., Goldstein D.B., Hindorff L.A., Hunter D.J., McCarthy M.I., Ramos E.M., Cardon L.R., Chakravarti A. Finding the missing heritability of complex diseases. Nature. 2009;461:747–753. doi: 10.1038/nature08494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pfeufer A., Jalilzadeh S., Perz S., Mueller J.C., Hinterseer M., Illig T., Akyol M., Huth C., Schöpfer-Wendels A., Kuch B. Common variants in myocardial ion channel genes modify the QT interval in the general population: results from the KORA study. Circ. Res. 2005;96:693–701. doi: 10.1161/01.RES.0000161077.53751.e6. [DOI] [PubMed] [Google Scholar]
- 17.International HapMap Consortium A haplotype map of the human genome. Nature. 2005;437:1299–1320. doi: 10.1038/nature04226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lander E.S., Linton L.M., Birren B., Nusbaum C., Zody M.C., Baldwin J., Devon K., Dewar K., Doyle M., FitzHugh W., International Human Genome Sequencing Consortium Initial sequencing and analysis of the human genome. Nature. 2001;409:860–921. doi: 10.1038/35057062. [DOI] [PubMed] [Google Scholar]
- 19.Rozen S., Skaletsky H. Primer3 on the WWW for general users and for biologist programmers. Methods Mol. Biol. 2000;132:365–386. doi: 10.1385/1-59259-192-2:365. [DOI] [PubMed] [Google Scholar]
- 20.Bentley D.R., Balasubramanian S., Swerdlow H.P., Smith G.P., Milton J., Brown C.G., Hall K.P., Evers D.J., Barnes C.L., Bignell H.R. Accurate whole human genome sequencing using reversible terminator chemistry. Nature. 2008;456:53–59. doi: 10.1038/nature07517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gordon D., Abajian C., Green P. Consed: a graphical tool for sequence finishing. Genome Res. 1998;8:195–202. doi: 10.1101/gr.8.3.195. [DOI] [PubMed] [Google Scholar]
- 22.Li H., Handsaker B., Wysoker A., Fennell T., Ruan J., Homer N., Marth G., Abecasis G., Durbin R., 1000 Genome Project Data Processing Subgroup The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2009;25:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Teer J.K., Bonnycastle L.L., Chines P.S., Hansen N.F., Aoyama N., Swift A.J., Abaan H.O., Albert T.J., Margulies E.H., Green E.D., NISC Comparative Sequencing Program Systematic comparison of three genomic enrichment methods for massively parallel DNA sequencing. Genome Res. 2010;20:1420–1431. doi: 10.1101/gr.106716.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.The ARIC Investigators The Atherosclerosis Risk in Communities (ARIC) Study: design and objectives. The ARIC investigators. Am. J. Epidemiol. 1989;129:687–702. [PubMed] [Google Scholar]
- 25.Browning B.L., Browning S.R. A unified approach to genotype imputation and haplotype-phase inference for large data sets of trios and unrelated individuals. Am. J. Hum. Genet. 2009;84:210–223. doi: 10.1016/j.ajhg.2009.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Abecasis G.R., Altshuler D., Auton A., Brooks L.D., Durbin R.M., Gibbs R.A., Hurles M.E., McVean G.A., 1000 Genomes Project Consortium A map of human genome variation from population-scale sequencing. Nature. 2010;467:1061–1073. doi: 10.1038/nature09534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kent W.J., Sugnet C.W., Furey T.S., Roskin K.M., Pringle T.H., Zahler A.M., Haussler D. The human genome browser at UCSC. Genome Res. 2002;12:996–1006. doi: 10.1101/gr.229102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Birney E., Stamatoyannopoulos J.A., Dutta A., Guigó R., Gingeras T.R., Margulies E.H., Weng Z., Snyder M., Dermitzakis E.T., Thurman R.E., ENCODE Project Consortium. NISC Comparative Sequencing Program. Baylor College of Medicine Human Genome Sequencing Center. Washington University Genome Sequencing Center. Broad Institute. Children’s Hospital Oakland Research Institute Identification and analysis of functional elements in 1% of the human genome by the ENCODE pilot project. Nature. 2007;447:799–816. doi: 10.1038/nature05874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Karolchik D., Hinrichs A.S., Furey T.S., Roskin K.M., Sugnet C.W., Haussler D., Kent W.J. The UCSC Table Browser data retrieval tool. Nucleic Acids Res. 2004;32(Database issue):D493–D496. doi: 10.1093/nar/gkh103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Claycomb W.C., Lanson N.A., Jr., Stallworth B.S., Egeland D.B., Delcarpio J.B., Bahinski A., Izzo N.J., Jr. HL-1 cells: a cardiac muscle cell line that contracts and retains phenotypic characteristics of the adult cardiomyocyte. Proc. Natl. Acad. Sci. USA. 1998;95:2979–2984. doi: 10.1073/pnas.95.6.2979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.DuBridge R.B., Tang P., Hsia H.C., Leong P.M., Miller J.H., Calos M.P. Analysis of mutation in human cells by using an Epstein-Barr virus shuttle system. Mol. Cell. Biol. 1987;7:379–387. doi: 10.1128/mcb.7.1.379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Westerfield M. University of Oregon Press; Eugene: 2000. The Zebrafish Book. A Guide for the Laboratory use of Zebrafish (Danio Rerio) [Google Scholar]
- 33.Sekar R.B., Kizana E., Smith R.R., Barth A.S., Zhang Y., Marbán E., Tung L. Lentiviral vector-mediated expression of GFP or Kir2.1 alters the electrophysiology of neonatal rat ventricular myocytes without inducing cytotoxicity. Am. J. Physiol. Heart Circ. Physiol. 2007;293:H2757–H2770. doi: 10.1152/ajpheart.00477.2007. [DOI] [PubMed] [Google Scholar]
- 34.Lim Z.Y., Maskara B., Aguel F., Emokpae R., Jr., Tung L. Spiral wave attachment to millimeter-sized obstacles. Circulation. 2006;114:2113–2121. doi: 10.1161/CIRCULATIONAHA.105.598631. [DOI] [PubMed] [Google Scholar]
- 35.Laemmli U.K. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature. 1970;227:680–685. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- 36.Estigoy C.B., Ponten F., Odeberg J., Herbert B., Guilhaus M., Charleston M., Ho J.W.K., Cameron D., dos Remedios C.G. Intercalated discs: multiple proteins perform multiple functions in non-failing and failing human hearts. Biophys Rev. 2009;1:43–49. doi: 10.1007/s12551-008-0007-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kerem B., Rommens J.M., Buchanan J.A., Markiewicz D., Cox T.K., Chakravarti A., Buchwald M., Tsui L.C. Identification of the cystic fibrosis gene: genetic analysis. Science. 1989;245:1073–1080. doi: 10.1126/science.2570460. [DOI] [PubMed] [Google Scholar]
- 38.Gross D.S., Garrard W.T. Nuclease hypersensitive sites in chromatin. Annu. Rev. Biochem. 1988;57:159–197. doi: 10.1146/annurev.bi.57.070188.001111. [DOI] [PubMed] [Google Scholar]
- 39.Boyle A.P., Guinney J., Crawford G.E., Furey T.S. F-Seq: a feature density estimator for high-throughput sequence tags. Bioinformatics. 2008;24:2537–2538. doi: 10.1093/bioinformatics/btn480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Thurman R.E., Rynes E., Humbert R., Vierstra J., Maurano M.T., Haugen E., Sheffield N.C., Stergachis A.B., Wang H., Vernot B. The accessible chromatin landscape of the human genome. Nature. 2012;489:75–82. doi: 10.1038/nature11232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Boyle A.P., Davis S., Shulha H.P., Meltzer P., Margulies E.H., Weng Z., Furey T.S., Crawford G.E. High-resolution mapping and characterization of open chromatin across the genome. Cell. 2008;132:311–322. doi: 10.1016/j.cell.2007.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.John S., Sabo P.J., Thurman R.E., Sung M.H., Biddie S.C., Johnson T.A., Hager G.L., Stamatoyannopoulos J.A. Chromatin accessibility pre-determines glucocorticoid receptor binding patterns. Nat. Genet. 2011;43:264–268. doi: 10.1038/ng.759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Arany Z., Sellers W.R., Livingston D.M., Eckner R. E1A-associated p300 and CREB-associated CBP belong to a conserved family of coactivators. Cell. 1994;77:799–800. doi: 10.1016/0092-8674(94)90127-9. [DOI] [PubMed] [Google Scholar]
- 44.May D., Blow M.J., Kaplan T., McCulley D.J., Jensen B.C., Akiyama J.A., Holt A., Plajzer-Frick I., Shoukry M., Wright C. Large-scale discovery of enhancers from human heart tissue. Nat. Genet. 2012;44:89–93. doi: 10.1038/ng.1006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Milan D.J., Kim A.M., Winterfield J.R., Jones I.L., Pfeufer A., Sanna S., Arking D.E., Amsterdam A.H., Sabeh K.M., Mably J.D. Drug-sensitized zebrafish screen identifies multiple genes, including GINS3, as regulators of myocardial repolarization. Circulation. 2009;120:553–559. doi: 10.1161/CIRCULATIONAHA.108.821082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Chang K.C., Barth A.S., Sasano T., Kizana E., Kashiwakura Y., Zhang Y., Foster D.B., Marbán E. CAPON modulates cardiac repolarization via neuronal nitric oxide synthase signaling in the heart. Proc. Natl. Acad. Sci. USA. 2008;105:4477–4482. doi: 10.1073/pnas.0709118105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Tung L., Zhang Y. Optical imaging of arrhythmias in tissue culture. J. Electrocardiol. 2006;39(Suppl):S2–S6. doi: 10.1016/j.jelectrocard.2006.04.010. [DOI] [PubMed] [Google Scholar]
- 48.Hirschhorn J.N. Genomewide association studies—illuminating biologic pathways. N. Engl. J. Med. 2009;360:1699–1701. doi: 10.1056/NEJMp0808934. [DOI] [PubMed] [Google Scholar]
- 49.Klein R.J., Zeiss C., Chew E.Y., Tsai J.Y., Sackler R.S., Haynes C., Henning A.K., SanGiovanni J.P., Mane S.M., Mayne S.T. Complement factor H polymorphism in age-related macular degeneration. Science. 2005;308:385–389. doi: 10.1126/science.1109557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Rioux J.D., Xavier R.J., Taylor K.D., Silverberg M.S., Goyette P., Huett A., Green T., Kuballa P., Barmada M.M., Datta L.W. Genome-wide association study identifies new susceptibility loci for Crohn disease and implicates autophagy in disease pathogenesis. Nat. Genet. 2007;39:596–604. doi: 10.1038/ng2032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Musunuru K., Strong A., Frank-Kamenetsky M., Lee N.E., Ahfeldt T., Sachs K.V., Li X., Li H., Kuperwasser N., Ruda V.M. From noncoding variant to phenotype via SORT1 at the 1p13 cholesterol locus. Nature. 2010;466:714–719. doi: 10.1038/nature09266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Schaller M.D. Cellular functions of FAK kinases: insight into molecular mechanisms and novel functions. J. Cell Sci. 2010;123:1007–1013. doi: 10.1242/jcs.045112. [DOI] [PubMed] [Google Scholar]
- 53.Santos A.M., Schechtman D., Cardoso A.C., Clemente C.F., Silva J.C., Fioramonte M., Pereira M.B., Marin T.M., Oliveira P.S., Figueira A.C. FERM domain interaction with myosin negatively regulates FAK in cardiomyocyte hypertrophy. Nat. Chem. Biol. 2012;8:102–110. doi: 10.1038/nchembio.717. [DOI] [PubMed] [Google Scholar]
- 54.Tsai I.C., Amack J.D., Gao Z.H., Band V., Yost H.J., Virshup D.M. A Wnt-CKIvarepsilon-Rap1 pathway regulates gastrulation by modulating SIPA1L1, a Rap GTPase activating protein. Dev. Cell. 2007;12:335–347. doi: 10.1016/j.devcel.2007.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Sotoodehnia N., Isaacs A., de Bakker P.I., Dörr M., Newton-Cheh C., Nolte I.M., van der Harst P., Müller M., Eijgelsheim M., Alonso A. Common variants in 22 loci are associated with QRS duration and cardiac ventricular conduction. Nat. Genet. 2010;42:1068–1076. doi: 10.1038/ng.716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kashi Y., King D., Soller M. Simple sequence repeats as a source of quantitative genetic variation. Trends Genet. 1997;13:74–78. doi: 10.1016/s0168-9525(97)01008-1. [DOI] [PubMed] [Google Scholar]
- 57.Kashi Y., King D.G. Simple sequence repeats as advantageous mutators in evolution. Trends Genet. 2006;22:253–259. doi: 10.1016/j.tig.2006.03.005. [DOI] [PubMed] [Google Scholar]
- 58.Hedley P.L., Jørgensen P., Schlamowitz S., Wangari R., Moolman-Smook J., Brink P.A., Kanters J.K., Corfield V.A., Christiansen M. The genetic basis of long QT and short QT syndromes: a mutation update. Hum. Mutat. 2009;30:1486–1511. doi: 10.1002/humu.21106. [DOI] [PubMed] [Google Scholar]
- 59.Melnikov A., Murugan A., Zhang X., Tesileanu T., Wang L., Rogov P., Feizi S., Gnirke A., Callan C.G., Jr., Kinney J.B. Systematic dissection and optimization of inducible enhancers in human cells using a massively parallel reporter assay. Nat. Biotechnol. 2012;30:271–277. doi: 10.1038/nbt.2137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Patwardhan R.P., Hiatt J.B., Witten D.M., Kim M.J., Smith R.P., May D., Lee C., Andrie J.M., Lee S.I., Cooper G.M. Massively parallel functional dissection of mammalian enhancers in vivo. Nat. Biotechnol. 2012;30:265–270. doi: 10.1038/nbt.2136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Emison E.S., McCallion A.S., Kashuk C.S., Bush R.T., Grice E., Lin S., Portnoy M.E., Cutler D.J., Green E.D., Chakravarti A. A common sex-dependent mutation in a RET enhancer underlies Hirschsprung disease risk. Nature. 2005;434:857–863. doi: 10.1038/nature03467. [DOI] [PubMed] [Google Scholar]
- 62.Drake J.A., Bird C., Nemesh J., Thomas D.J., Newton-Cheh C., Reymond A., Excoffier L., Attar H., Antonarakis S.E., Dermitzakis E.T., Hirschhorn J.N. Conserved noncoding sequences are selectively constrained and not mutation cold spots. Nat. Genet. 2006;38:223–227. doi: 10.1038/ng1710. [DOI] [PubMed] [Google Scholar]
- 63.Emison E.S., Garcia-Barcelo M., Grice E.A., Lantieri F., Amiel J., Burzynski G., Fernandez R.M., Hao L., Kashuk C., West K. Differential contributions of rare and common, coding and noncoding Ret mutations to multifactorial Hirschsprung disease liability. Am. J. Hum. Genet. 2010;87:60–74. doi: 10.1016/j.ajhg.2010.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Fisher S., Grice E.A., Vinton R.M., Bessling S.L., McCallion A.S. Conservation of RET regulatory function from human to zebrafish without sequence similarity. Science. 2006;312:276–279. doi: 10.1126/science.1124070. [DOI] [PubMed] [Google Scholar]
- 65.Bulger M., Groudine M. Functional and mechanistic diversity of distal transcription enhancers. Cell. 2011;144:327–339. doi: 10.1016/j.cell.2011.01.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Ahituv N., Zhu Y., Visel A., Holt A., Afzal V., Pennacchio L.A., Rubin E.M. Deletion of ultraconserved elements yields viable mice. PLoS Biol. 2007;5:e234. doi: 10.1371/journal.pbio.0050234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Hong J.W., Hendrix D.A., Levine M.S. Shadow enhancers as a source of evolutionary novelty. Science. 2008;321:1314. doi: 10.1126/science.1160631. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.