

Abstract
We identified four different missense mutations in the single-exon gene MAB21L2 in eight individuals with bilateral eye malformations from five unrelated families via three independent exome sequencing projects. Three mutational events altered the same amino acid (Arg51), and two were identical de novo mutations (c.151C>T [p.Arg51Cys]) in unrelated children with bilateral anophthalmia, intellectual disability, and rhizomelic skeletal dysplasia. c.152G>A (p.Arg51His) segregated with autosomal-dominant bilateral colobomatous microphthalmia in a large multiplex family. The fourth heterozygous mutation (c.145G>A [p.Glu49Lys]) affected an amino acid within two residues of Arg51 in an adult male with bilateral colobomata. In a fifth family, a homozygous mutation (c.740G>A [p.Arg247Gln]) altering a different region of the protein was identified in two male siblings with bilateral retinal colobomata. In mouse embryos, Mab21l2 showed strong expression in the developing eye, pharyngeal arches, and limb bud. As predicted by structural homology, wild-type MAB21L2 bound single-stranded RNA, whereas this activity was lost in all altered forms of the protein. MAB21L2 had no detectable nucleotidyltransferase activity in vitro, and its function remains unknown. Induced expression of wild-type MAB21L2 in human embryonic kidney 293 cells increased phospho-ERK (pERK1/2) signaling. Compared to the wild-type and p.Arg247Gln proteins, the proteins with the Glu49 and Arg51 variants had increased stability. Abnormal persistence of pERK1/2 signaling in MAB21L2-expressing cells during development is a plausible pathogenic mechanism for the heterozygous mutations. The phenotype associated with the homozygous mutation might be a consequence of complete loss of MAB21L2 RNA binding, although the cellular function of this interaction remains unknown.
Main Text
Structural eye malformations are an important cause of congenital visual impairment.1,2 The terms anophthalmia and microphthalmia are used to indicate the absence or marked reduction in size, respectively, of an eye. Ocular coloboma (MIM 216820) describes the spectrum of eye malformations, including microphthalmia, resulting from failure of optic fissure closure during embryogenesis. These malformations show marked phenotypic and etiological heterogeneity. The most common identifiable genetic causes of structural eye malformations are those involving dosage-sensitive transcription factors (encoded by SOX2 [MIM 184429],3,4 OTX2 [MIM 600037],5 and PAX6 [MIM 607108]6) and retinoic acid metabolism or transport (regulated by STRA6 [MIM 610745],7 ALDH1A3 [MIM 600463],8 RARB [MIM 180220]9). The cause in a significant proportion of individuals with major eye malformations, particularly in those with microphthalmia and coloboma,10,11 remains unknown.
To further elucidate the genetic architecture of ocular coloboma, we performed exome sequencing on genomic DNA from an affected uncle and nephew (individuals II.6 and III.1) in a large family (family 1463) in which apparently isolated bilateral coloboma segregates in a pattern consistent with autosomal-dominant inheritance (Figure 1; Figure S3 and Table S2, available online). These were two of the 99 exome sequences (75 individuals with coloboma and 24 unaffected relatives from 58 different families) that comprised the coloboma contribution to the rare-diseases component of the UK10K project.12 This study was approved by the UK Multiregional Ethics Committee (reference 06/MRE00/76), and informed consent was obtained from all participating families. Exome sequencing was performed as previously described.13 Sequences were aligned with the Burrows-Wheeler Aligner v.0.5.9, duplicates were marked with Picard v.1.43, realignment around indels and base quality scores were recalibrated with the Genome Analysis Toolkit (GATK) v.1.0.5506, and variants were called only with GATK Unified Genotyper. The coverage and depth metrics for these exomes and for each of the other exome analyses mentioned below are provided in Table S4. A total of 27 shared heterozygous, rare (maximum allele frequency < 0.005 and mutation count in UK10K coloboma exomes < 3) variants were identified (Table S1). Two frameshift and one in-frame deletion were called in IFT122 (MIM 606045) but were the result of misalignment of a single IFT122 heterozygous frameshift mutation causing autosomal-recessive cranioectodermal dysplasia (MIM 218330). All the remaining missense mutations or in-frame deletions affected different genes. Only one mutation (c.152G>A [p.Arg51His]; chr4: g.151504333G>A) was found to alter a gene (MAB21L2 [MIM 604357]) on our previously compiled list of 38 candidate genes for eye malformations (Table S3). This mutation is not reported in public databases, including the 1000 Genomes Project, the NHLBI Exome Sequencing Project (ESP) Exome Variant Server, and the Medical Research Council Human Genetics Unit in-house database of variants derived from ∼2,200 exomes. The RefSeq accession numbers NM_006439.4 and NP_006430.1 were used for naming this and all subsequent MAB21L2 variants at cDNA and protein levels, respectively. The entire UK10K coloboma exome data set is available from the European Genome-phenome Archive under a data-access agreement as study number EGAS00001000127.
Figure 1.
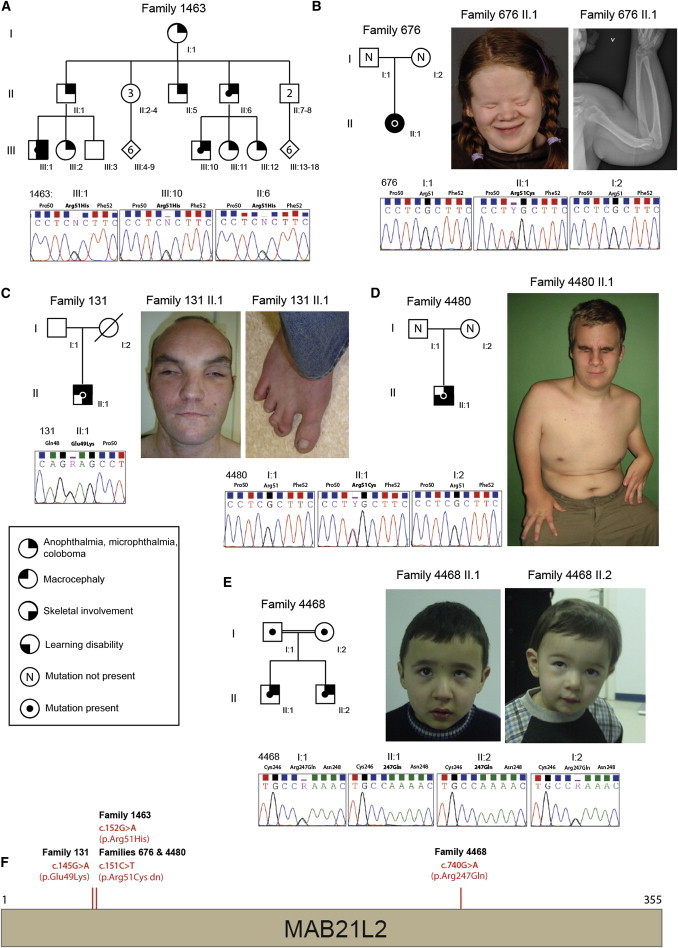
Family Structures and MAB21L2 Mutations
(A–E) Diagrammatic representation of the structure of the five families—1463 (A), 676 (B), 131 (C), 4480 (D), and 4468 (E)—in whom mutations were identified in MAB21L2. The family number is given above each pedigree, and the sequencing chromatograms of the mutated base are given below each pedigree. Clinical images associated with each of the probands are located on the right-hand side of each cognate pedigree.
(F) The location of each missense mutation is provided on a schematic representation of MAB21L2.
Independently, trio whole-exome sequencing of an affected Norwegian female (II.1 in family 676 [Figure 1]) with bilateral anophthalmia, macrocephaly, moderate intellectual disability, and generalized skeletal dysplasia (Table S2) and her parents was performed as previously described14 as part of a study approved by the Regional Committee for Medical and Health Research Ethics in western Norway (institutional review board [IRB] 00001872; written informed consent was obtained from the family). A total of 217 rare variants (with a maximum allele frequency < 0.005 in 1000 Genomes and not present in 80 in-house-generated Norwegian exome samples from the same pipeline) were detected in the proband and filtered against parental exome data in a search for putative de novo variants. Using this approach, we detected four variants, of which only one (c.151C>T [p.Arg51Cys]; chr4: g.151504332C>T; in MAB21L2) was confirmed by Sanger sequencing. This de novo missense mutation was found to alter the same amino acid (Arg51) as that in family 1463. The substitution changes a strictly conserved residue, and both p.Arg51Cys and p.Arg51His are predicted to be deleterious by SIFT, PolyPhen2, and AlignGVGD. Subsequently, one of the authors identified an unrelated male individual (II.1 in family 4480) with more severe rhizomelic skeletal dysplasia associated with bilateral anophthalmia (Figure 1; Table S2). Analysis of DNA samples from this individual and his parents was performed as part of the study approved by the UK Multiregional Ethics Committee (reference 06/MRE00/76; informed consent was obtained from the family). Exactly the same mutation (c.151C>T [p.Arg51Cys]), which had also occurred de novo, was identified in the affected child. Microsatellite analysis of the DNA samples from each family was performed to confirm biological relationships and to exclude sample mix up. The de novo mutation c.151C>T (p.Arg51Cys) thus has a clinically recognizable phenotype.
Resequencing of MAB21L2 was performed in 336 unrelated individuals with major eye malformations (and with no overlap with those who were exome sequenced) as part of the study approved by the UK Multiregional Ethics Committee (reference 06/MRE00/76; informed consent was obtained from all participating families). This analysis revealed one different ultra-rare (not present in the NHLBI ESP Exome Variant Server, 1000 Genomes, or UK10K variant databases) heterozygous missense mutation (Figure 1) in the simplex case of an adult male with bilateral colobomatous microphthalmia (individual II.1 in family 131 [Figure 1; Table S2]). This mutation (c.145G>A [p.Glu49Lys]; chr4: g.151504326G>A) affects the codon encoding a residue two amino acids N-terminal to the substitutions identified above (p.Arg51His and p.Arg51Cys). This man had a history of reasonably good vision until the age of 11 years, after which he became blind over a period of 2 years. He had no evidence of retinal detachment at the age of 30 years, and no retinal electrophysiology was available. He was of normal intelligence and had only minor skeletal dysmorphisms, recurrent dislocation of the patellae, and soft-tissue syndactyly of the third and fourth digits of his hands and of the second and third digits of both feet (Table S2). His mother was deceased, and therefore we were unable to confirm whether this mutation had occurred de novo in this man.
A third independent exome sequencing study of distinct clinical phenotypes in children of consanguineous parents was carried out at the Baylor-Johns Hopkins Center for Mendelian Genomics under ethical approval from the Baylor College of Medicine (BCM) IRB (informed consent was obtained from all participating families). Two male siblings born to first-degree cousins were referred for clinical genetic assessment as a result of eye abnormalities. With the exception of subtle facial dysmorphic features and the eye findings, both boys had normal development. The elder boy (II.1 in family 4468) had left-eye esotropia in addition to a prominent forehead, periorbital fullness, long eyelashes, epicanthus, and a long and prominent philtrum (Table S2). Ophthalmologic examination revealed retinal coloboma including the optic disc and macula in the right eye, whereas there was sparing of the optic disc and macula in the left eye. Refractions were +2.0/+3.0 × 170 and −5.25/−3.25 × 115 in the right and left eyes, respectively. His younger brother (II.2 in family 4468) had right-eye exotropia and microphthalmia in addition to similar facial dysmorphic features. Ophthalmologic examination revealed bilateral retinal coloboma involving the optic disc in the right, but not the left, eye. Refractions were −2.00/−2.00 × 90 and +0.25/−1.50 × 175 in the right and left eyes, respectively. The parents of these siblings had normal vision and had no evidence of an asymptomatic structural eye malformation on ophthalmological examination.
Whole-exome sequencing in both affected siblings identified a homozygous nonsynonymous substitution (c.740G>A [p.Arg247Gln]; chr4: g.151504921G>A, hg19; RefSeq NM_006439) in MAB21L2 in both brothers. This mutation has not been reported in public databases, including the 1000 Genomes Project, the NHLBI ESP Exome Variant Server, and the Atherosclerosis Risk in Communities Study database. In addition, this p.Arg247Gln substitution was not identified in an in-house-generated exome variant database from ∼2,500 individuals at the BCM Human Genome Sequencing Center and BCM Whole Genome Laboratory Database, which includes anonymized data from over 1,000 individuals tested for diagnostic purposes. Sanger sequencing was performed for segregation analysis, and the parents were found to be heterozygous carriers, consistent with Mendelian expectation. All experiments and analyses were performed according to previously described methods.15
The human gene is named after the ortholog in C. elegans. Mutations in Mab-21 cause posterior-to-anterior homeotic transformation of sensory ray 6 in the male tail in this worm.16 In normal development, Mab-21 has been shown to interact with Sin-3, a key component of a histone-deacetylase-containing transcriptional regulatory complex,17 and to be negatively regulated by CET-1 (whose human paralogs are BMP2, BMP4, and BMP7) signaling. After the identification of Mab-21 in C. elegans, multiple orthologous proteins were identified in human18 and mouse.19 In zebrafish, expression of mab21l2 in the eye field is rx3 dependent. Morpholino knockdown of mab21l2 has been shown to produce a proliferation defect within the retinal progenitor cell population, resulting in small but structurally normal eyes.20 Analysis of the cis-regulatory elements surrounding mab21l2 has identified functionally significant subpopulations of cells within the developing eye,21 although the role of the gene product in the formation or maintenance of these cells is not yet clear. Homozygous targeted inactivation of Mab21l2 in mouse embryos causes defects of the ventral body wall, severe eye malformations, and death in midgestation, whereas heterozygous null animals are apparently normal.22 Homozygous null embryos show failure of lens induction and aplasia of the retinal pigment epithelium as a result of a proliferation defect within the optic vesicle. Given the severity of the phenotype observed in the Mab21l2-null mouse embryos and the relatively mild phenotype in the siblings homozygous for c.740G>A (p.Arg247Gln), it seems likely that the human mutation does not result in complete loss of function.
Although developmental expression of Mab21l2 in mouse embryos has been previously reported,23 we wished to examine the expression in the developing eye in more detail. A digoxigenin-labeled antisense riboprobe targeted to the 5′ UTR of Mab21l2 (chr3: 86,547,729–86,548,237, mm10) was used for whole-mount in situ hybridization of 9.5, 10.5, 11.5, and 12.5 day postcoitum (dpc) mouse embryos (Figure S3). In addition to bright-field imaging, optical projection tomography (OPT) was also used for visualizing 10.5 dpc embryos as previously described.12 We chose 10.5 dpc for full descriptive analysis because this time point is prior to optic fissure closure but has a well-formed optic cup. Strong expression was evident in the rostral and distal regions of the developing neural retina (Figure 2A), and there was no expression immediately adjacent to the closing optic fissure (Figure 2B). Expression was also observed in the dorsal and ventral aspects of the developing forelimb bud and in the developing pharyngeal arches. The site- and stage-specific developmental expression pattern of Mab21l2 is thus compatible with the eye and limb phenotypic effects associated with the mutations we identified above. No Mab21l2 expression was observed in the brain at 10.5 dpc on OPT. However, imaging of more intensely stained embryos showed striking midbrain expression of Mab21l2 at 9.5 and 10.5 dpc (Figure S3). This might be important in view of the neurodevelopmental problems reported in the individuals (676 II.1 and 4480 II.1) carrying the substitution p.Arg51Cys.
Figure 2.
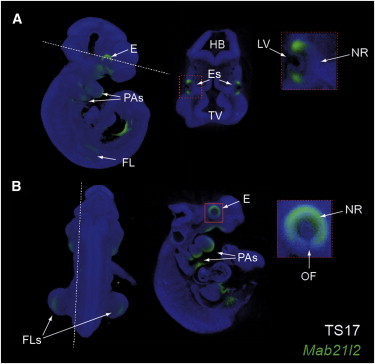
Mab21l2 Expression during Mouse Eye Development
OPT images of Mab21l2 expression at mouse Theiller stage 17 (TS17; 10.5 dpc). Hatched lines indicate the digital sections presented.
(A) Lateral 3D OPT projection showing Mab21l2 expression in the eye (E), pharyngeal arches (PAs), and forelimbs (FLs). The transverse digital section presented alongside shows specific expression in the eyes, and an enlarged image (box) illustrates Mab21l2 expression at the distal regions of the neural retina (NR), but not in the lens vesicle (LV).
(B) Posterior 3D OPT view illustrating specific Mab21l2 expression in the FLs. The sagittal digital section presented alongside and the enlarged box illustrate that Mab21l2 expression was highest dorsally but continued ventrally into the margins of the optic fissure (OF).
Further abbreviations are as follows: HB, hindbrain; and TV, telencephalic vesicle.
The highly localized distribution of the heterozygous missense mutations suggests a mutational mechanism that might not be simple loss of function. The biochemical function of MAB21L2 is not known, but the residues at which each of the amino acid substitutions occurred (Glu49, Arg51, and Arg247) in the human protein are completely conserved in mouse, zebrafish, and C. elegans (Figure S1). The family of 12 human Mab-21 paralogs adopt a nucleotidyltransferase fold24 and include a cyclic GMP-AMP synthase (cGAS), which generates cyclic GMP-AMP in the cytoplasm of cells exposed to DNA.25 Detailed examination of the structure of both cGAS (Protein Data Bank [PDB] accession number 4K9B) and another family member, RNA-activated antiviral protein 2′-5′-oligoadenylate synthetase (OAS26 [PDB 1PX5]), indicated conservation of the active site in MAB21L2 (Figure 3A). However, a sensitive colorimetric assay using purified MAB21L2 (from E. coli or human embryonic kidney 293 [HEK293] cells) with ATP as a substrate (Figure 3B) for analysis of pyrophosphate release27 detected no nucleotidyltransferase activity, whereas OAS purified by the same methods resulted in strong activity. This analysis was repeated with a mixture of all nucleoside triphosphates (Figure S2) with different plausible activator molecules (double-stranded RNA or DNA and single-stranded RNA [ssRNA] or DNA [ssDNA]), but no enzymatic activity was detectable for MAB21L2.
Figure 3.
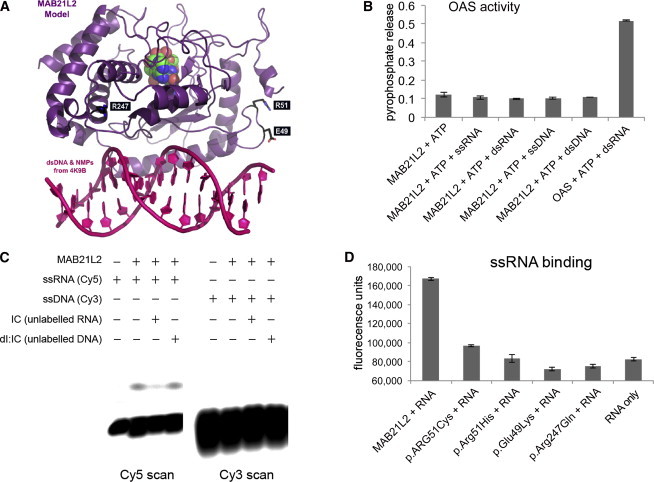
Structural Modeling of MAB21L2 and Prediction of Nucleotidyltransferase Activity
(A) A model of MAB21L2 was generated with PDB 4K9B as a template and is shown in purple; the nucleotide monophosphates are shown in green, blue, and red. This analysis suggests that MAB21L2 has both a nucleotidyltransferase active site and a DNA- and/or RNA-binding domain (double-stranded DNA is shown in pink in the foreground). The position of the residues that were altered in the affected individuals is shown in white text in black boxes. The arginine residues (Arg51 [R51] and Arg247 [R247]) are highlighted in blue, and the glutamic acid residue (Glu49 [E49]) is shown in orange.
(B) A graph showing the absence of OAS-like activity in purified MAB21L2. When OAS protein purified in the same way as MAB21L2 was incubated with ATP and double-stranded RNA (dsRNA), significant pyrophosphate release was detected, indicating nucleotidyltransferase activity. MAB21L2 showed no activity above background with ATP (or other nucleoside triphosphates [Figure S2]) using dsRNA, double-stranded DNA, ssRNA, or ssDNA as an activator.
(C) An electromobility shift assay (EMSA) using fluorescently labeled I:C oligonucleotides shows binding of wild-type MAB21L2 to ssRNA, but not ssDNA. The ssRNA binding could be completed efficiently with unlabeled ssRNA, but not ssDNA.
(D) Solution-based assay showing that wild-type MAB21L2 could efficiently bind a digoxigenin-labeled ssRNA molecule (this was an antisense riboprobe against FZD5, but all probes tested behaved in an identical fashion). None of the altered proteins could bind the ssRNA probe at levels above background.
The error bars in (B) and (D) represent SE. Each experiment represents readings from two biological replicates, and all experiments were repeated twice.
Enzymatic activation of OAS and cGAS occurs via conformational changes induced by binding with RNA and DNA, respectively.28 The structural comparisons suggested that a ∼35 Å long RNA- or DNA-binding groove also exists in MAB21L2 (Figure 3A; Figure S1). A fluorescent electromobility shift assay using Cy5- or Cy3-labeled ssRNA and ssDNA oligonucleotides and bacterially expressed protein showed binding of MAB21L2 to ssRNA, but not ssDNA (Figure 3C). To investigate the effect on ssRNA binding in each of the mutants (Figure 3D), we incubated a digoxigenin-labeled ssRNA (500 nt in length) in solution with sepharose-bead-bound wild-type or altered protein. After extensive washing, the binding of ssRNA to MAB21L2 was determined by fluorometry with an anti-digoxigenin-fluorescein antibody (Roche). Each of the four mutations, including the recessive mutation, resulted in loss of ssRNA-binding activity, consistent with the predicted locations of the affected residues close to the OAS RNA-binding cleft (Figure 3A). Complete loss of RNA binding in association with each of the mutations was remarkable but clearly cannot explain why the c.740G>A (p.Arg247Gln) variant is recessive and the other variants are dominant. We also had no knowledge of what the functional consequence of RNA binding was in the wild-type protein.
We therefore created multiple independent stable tetracycline-inducible HEK293 cell lines expressing the wild-type MAB21L2 and each of the altered forms as full-length GFP-fusion proteins. We used this system first to accurately determine the stability of the induced proteins by using a timed pulse of tetracycline. These analyses showed that all three of the monoallelic mutations resulted in significant stabilization of the protein in comparison to either the wild-type protein or the p.Arg247Gln variant (Figures 4A and 4B). Similar stabilization of altered protein has been reported in the recurrent de novo PACS1 mutations, associated with characteristic facial dysmorphisms and significant intellectual disability.29 In this study, they observed cytoplasmic aggregates of altered GFP-tagged protein, but we could identify no obvious aggregation or localization differences in the MAB21L2 variants (Figure S4). No predicted ubiquitination site could be located in the vicinity of the MAB21L2 substitutions. However, failure of the altered proteins to be recognized by the ubiquitin-mediated degradation system is the most likely mechanism for this observation.30
Figure 4.
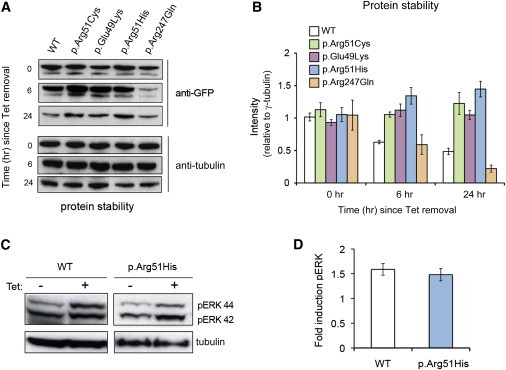
Protein Stability Estimations and Induction of ERK Signaling by MAB21L2
(A) Time-course analysis of protein stability with the use of anti-GFP immunoblotting of MAB21L2 at various time points since tetracycline (Tet) removal. Data presented are representative of five independent replicates.
(B) Quantification of immunoblots indicates higher protein stability for the proteins with substitutions at Glu49 or Arg51 than for the WT, whereas p.Arg247Gln displayed a pattern of protein stability similar to that of the WT. Error bars represent 95% confidence intervals.
(C) Increase in the level of the 44 kDa phospho-ERK band after 5 hr of Tet induction of WT and p.Arg51His MAB21L2 in inducible HEK293 cells.
(D) Graph representing quantification of this induction. Error bars represent 95% confidence intervals.
Given the above-mentioned data from C. elegans, we used the inducible cell system to identify any MAB21L2-dependent alteration in SMAD family member 1, 5, and 8 (SMAD1/5/8) signaling or extracellular-signal-regulated kinase 1 and 2 (ERK1/2) signaling. Immunoblots using anti-phospho-SMAD1/5/8 antibodies detected no alteration in canonical BMP signaling between cells expressing tagged MAB21L2 and the same cells cultured without tetracycline (data not shown). However, induction of wild-type MAB21L2 consistently resulted in an ∼1.5-fold increase in 44 kDa phospho-ERK1 detected on immunoblot (Figures 4C and 4D). A similar level of induction was noted with the p.Arg51His substitution (Figures 4C and 4D). In mouse models of Noonan syndrome, an activating mutation in Ptpn11 has been expressed as a transgene with the use of different tissue-specific promoters. Expression in the developing heart31 and craniofacial region32 produced ERK upregulation associated with specific developmental defects. These malformations disappeared if the transgene was expressed in an ERK1/2-null background31 or when ERK1/2 signaling was chemically ablated.32 The pattern of malformations associated with ERK-activating mutations thus probably reflects the developmental expression of the mutated gene. ERK1/2-mediated signaling is active during eye and skeletal development and is almost entirely dependent on FGF receptor in the early mouse embryo.33 The combination of protein stabilization and phospho-ERK (pERK1/2) induction suggests that the mutational mechanism in the monoallelic mutations affecting MAB21L2 might be activating mutations. Overactive pERK1/2 signaling is generally considered oncogenic, but a paradoxical growth inhibitory effect in chondrocytes has been recently proposed to explain how activating mutations in FGFR3 (MIM 134934) can cause achondroplasia (MIM 100800) and thanatophoric dysplasia (MIM 187600).34 In this model, pERK1/2 overactivity induces cellular defense mechanisms, which are potent inhibitors of growth. Such a mechanism could explain the rhizomelic skeletal dysplasia that is seen to be associated with MAB21L2 mutations. However, given that the wild-type protein and the p.Arg51His substitution result in similar levels of ERK1/2 induction (1.5-fold; Figure 4), the important aspect might be the inability to control the precise timing of the pathway activity during critical developmental processes rather than the absolute level of signaling. The timing of ERK1/2 signaling is known to be crucial for the oscillatory expression of cyclic genes during somitogenesis,35 but it is not yet clear which processes during ocular and skeletal development, if any, are timing critical. ERK1/2 signaling has not been examined in either mouse or zebrafish models for Mab21l2 or mab21l2 loss of function, respectively. However, “knocking in” these monoallelic mutations will be the most effective method of answering the precise relationship among these variants, ERK1/2 signaling, and the developmental pathology.
This report provides compelling human genomic and genetic evidence that mutations in MAB21L2 cause major eye malformations. The combination of dominant and recessive mutations is intriguing, particularly given that the carriers of homozygous mutations are the least severely affected. The restricted repertoire of mutations in the monoallelic cases strongly suggests an unusual genetic mechanism. A similar restricted pattern is seen in disorders caused by the activation of signaling pathways, such as RASopathy disorders36 and Myhre syndrome (MIM 139210).37 It is possible that the monoallelic mutations act as dominant negative. The association between the monoallelic mutations and increased protein stability and the association between MAB21L2 and the induction of pERK signaling raise the possibility that aberrant persistence of a developmental signal might be the mechanism operating in those cases. It could be that complete loss of MAB21L2 RNA-binding activity in association with the recessive Arg247Gln variant might be producing the eye phenotype via a different mechanism, and as such, identifying the in vivo role of the RNA-binding activity is a priority for future work. An understanding of the cellular and developmental function of wild-type MAB21L2 will enable adequate interpretation of mutations at this locus in a clinical setting. Finally, this study illustrates the cumulative value of the active sharing of DNA variation observed in individual patients in order to aggregate sufficient evidence to support specific biological hypotheses.
Consortia
The members of the UK10K Rare Diseases Working Group are Matthew Hurles, David R. FitzPatrick, Saeed Al-Turki, Carl Anderson, Inês Barroso, Philip Beales, Jamie Bentham, Shoumo Bhattacharya, Keren Carss, Krishna Chatterjee, Sebhattin Cirak, Catherine Cosgrove, Allan Daly, Jamie Floyd, Chris Franklin, Marta Futema, Steve Humphries, Shane McCarthy, Hannah Mitchison, Francesco Muntoni, Alexandros Onoufriadis, Victoria Parker, Felicity Payne, Vincent Plagnol, Lucy Raymond, David Savage, Peter Scambler, Miriam Schmidts, Robert Semple, Eva Serra, Jim Stalker, Margriet van Kogelenberg, Parthiban Vijayarangakannan, Klaudia Walter, and Gretta Wood.
Acknowledgments
J.R., H.B., K.A.W., A.M., J.K.R., M.S.T., V.v.H., and D.R.F. are all supported by Medical Research Council (MRC) program grants awarded to the MRC Human Genetics Unit. Funding for UK10K was provided by the Wellcome Trust under award WT091310. Support for the Baylor-Hopkins Center for Mendelian Genomics was provided by the NIH National Human Genome Research Institute (U54 HG006542). G.H. is supported by HelseVest grant 911744. Thanks are given to Inge Jonassen and Kjell Petersen (Computational Biology Unit, Department of Informatics, University of Bergen, Norway) for providing IT infrastructure for the Norwegian next-generation sequencing data through the Elixir.no project and to the HudsonAlpha Institute for Biotechnology (Huntsville) for performing whole-exome sequencing in family 676.
Contributor Information
David R. FitzPatrick, Email: david.fitzpatrick@igmm.ed.ac.uk.
UK10K:
Matthew Hurles, David R. FitzPatrick, Saeed Al-Turki, Carl Anderson, Inês Barroso, Philip Beales, Jamie Bentham, Shoumo Bhattacharya, Keren Carss, Krishna Chatterjee, Sebhattin Cirak, Catherine Cosgrove, Allan Daly, Jamie Floyd, Chris Franklin, Marta Futema, Steve Humphries, Shane McCarthy, Hannah Mitchison, Francesco Muntoni, Alexandros Onoufriadis, Victoria Parker, Felicity Payne, Vincent Plagnol, Lucy Raymond, David Savage, Peter Scambler, Miriam Schmidts, Robert Semple, Eva Serra, Jim Stalker, Margriet van Kogelenberg, Parthiban Vijayarangakannan, Klaudia Walter, and Gretta Wood
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
1000 Genomes, http://www.1000genomes.org/
European Genome-phenome Archive, https://www.ebi.ac.uk/ega/
FANTOM4, http://fantom.gsc.riken.jp/4/
NHLBI Exome Sequencing Project (ESP) Exome Variant Server, http://evs.gs.washington.edu/EVS/
Online Mendelian Inheritance in Man (OMIM), http://www.omim.org/
Picard, http://picard.sourceforge.net
UCSC Genome Browser, http://genome.ucsc.edu/
UK10K project, http://www.uk10k.org/
References
- 1.Haddad M.A., Sei M., Sampaio M.W., Kara-José N. Causes of visual impairment in children: a study of 3,210 cases. J. Pediatr. Ophthalmol. Strabismus. 2007;44:232–240. doi: 10.3928/01913913-20070701-04. [DOI] [PubMed] [Google Scholar]
- 2.Rudanko S.L., Laatikainen L. Visual impairment in children born at full term from 1972 through 1989 in Finland. Ophthalmology. 2004;111:2307–2312. doi: 10.1016/j.ophtha.2004.05.033. [DOI] [PubMed] [Google Scholar]
- 3.Fantes J., Ragge N.K., Lynch S.A., McGill N.I., Collin J.R., Howard-Peebles P.N., Hayward C., Vivian A.J., Williamson K., van Heyningen V., FitzPatrick D.R. Mutations in SOX2 cause anophthalmia. Nat. Genet. 2003;33:461–463. doi: 10.1038/ng1120. [DOI] [PubMed] [Google Scholar]
- 4.Ragge N.K., Lorenz B., Schneider A., Bushby K., de Sanctis L., de Sanctis U., Salt A., Collin J.R., Vivian A.J., Free S.L. SOX2 anophthalmia syndrome. Am. J. Med. Genet. A. 2005;135:1–7. doi: 10.1002/ajmg.a.30642. discussion 8. [DOI] [PubMed] [Google Scholar]
- 5.Ragge N.K., Brown A.G., Poloschek C.M., Lorenz B., Henderson R.A., Clarke M.P., Russell-Eggitt I., Fielder A., Gerrelli D., Martinez-Barbera J.P. Heterozygous mutations of OTX2 cause severe ocular malformations. Am. J. Hum. Genet. 2005;76:1008–1022. doi: 10.1086/430721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Glaser T., Jepeal L., Edwards J.G., Young S.R., Favor J., Maas R.L. PAX6 gene dosage effect in a family with congenital cataracts, aniridia, anophthalmia and central nervous system defects. Nat. Genet. 1994;7:463–471. doi: 10.1038/ng0894-463. [DOI] [PubMed] [Google Scholar]
- 7.Pasutto F., Sticht H., Hammersen G., Gillessen-Kaesbach G., Fitzpatrick D.R., Nürnberg G., Brasch F., Schirmer-Zimmermann H., Tolmie J.L., Chitayat D. Mutations in STRA6 cause a broad spectrum of malformations including anophthalmia, congenital heart defects, diaphragmatic hernia, alveolar capillary dysplasia, lung hypoplasia, and mental retardation. Am. J. Hum. Genet. 2007;80:550–560. doi: 10.1086/512203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Fares-Taie L., Gerber S., Chassaing N., Clayton-Smith J., Hanein S., Silva E., Serey M., Serre V., Gérard X., Baumann C. ALDH1A3 mutations cause recessive anophthalmia and microphthalmia. Am. J. Hum. Genet. 2013;92:265–270. doi: 10.1016/j.ajhg.2012.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Srour M., Chitayat D., Caron V., Chassaing N., Bitoun P., Patry L., Cordier M.P., Capo-Chichi J.M., Francannet C., Calvas P. Recessive and dominant mutations in retinoic acid receptor beta in cases with microphthalmia and diaphragmatic hernia. Am. J. Hum. Genet. 2013;93:765–772. doi: 10.1016/j.ajhg.2013.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gerth-Kahlert C., Williamson K., Ansari M., Rainger J.K., Hingst V., Zimmermann T., Tech S., Guthoff R.F., van Heyningen V., Fitzpatrick D.R. Clinical and mutation analysis of 51 probands with anophthalmia and/or severe microphthalmia from a single center. Mol. Genet. Genomic Med. 2013;1:15–31. doi: 10.1002/mgg3.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chassaing N., Causse A., Vigouroux A., Delahaye A., Alessandri J.L., Boespflug-Tanguy O., Boute-Benejean O., Dollfus H., Duban-Bedu B., Gilbert-Dussardier B. Molecular findings and clinical data in a cohort of 150 patients with anophthalmia/microphthalmia. Clin. Genet. 2013 doi: 10.1111/cge.12275. Published online September 10, 2013. [DOI] [PubMed] [Google Scholar]
- 12.Williamson K.A., Rainger J., Floyd J.A., Ansari M., Meynert A., Aldridge K.V., Rainger J.K., Anderson C.A., Moore A.T., Hurles M.E., UK10K Consortium Heterozygous loss-of-function mutations in YAP1 cause both isolated and syndromic optic fissure closure defects. Am. J. Hum. Genet. 2014;94:295–302. doi: 10.1016/j.ajhg.2014.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Olbrich H., Schmidts M., Werner C., Onoufriadis A., Loges N.T., Raidt J., Banki N.F., Shoemark A., Burgoyne T., Al Turki S., UK10K Consortium Recessive HYDIN mutations cause primary ciliary dyskinesia without randomization of left-right body asymmetry. Am. J. Hum. Genet. 2012;91:672–684. doi: 10.1016/j.ajhg.2012.08.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Haugarvoll K., Johansson S., Tzoulis C., Haukanes B.I., Bredrup C., Neckelmann G., Boman H., Knappskog P.M., Bindoff L.A. MRI characterisation of adult onset alpha-methylacyl-coA racemase deficiency diagnosed by exome sequencing. Orphanet J. Rare Dis. 2013;8:1. doi: 10.1186/1750-1172-8-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bainbridge M.N., Hu H., Muzny D.M., Musante L., Lupski J.R., Graham B.H., Chen W., Gripp K.W., Jenny K., Wienker T.F. De novo truncating mutations in ASXL3 are associated with a novel clinical phenotype with similarities to Bohring-Opitz syndrome. Genome Med. 2013;5:11. doi: 10.1186/gm415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chow K.L., Hall D.H., Emmons S.W. The mab-21 gene of Caenorhabditis elegans encodes a novel protein required for choice of alternate cell fates. Development. 1995;121:3615–3626. doi: 10.1242/dev.121.11.3615. [DOI] [PubMed] [Google Scholar]
- 17.Choy S.W., Wong Y.M., Ho S.H., Chow K.L. C. elegans SIN-3 and its associated HDAC corepressor complex act as mediators of male sensory ray development. Biochem. Biophys. Res. Commun. 2007;358:802–807. doi: 10.1016/j.bbrc.2007.04.194. [DOI] [PubMed] [Google Scholar]
- 18.Margolis R.L., Stine O.C., McInnis M.G., Ranen N.G., Rubinsztein D.C., Leggo J., Brando L.V., Kidwai A.S., Loev S.J., Breschel T.S. cDNA cloning of a human homologue of the Caenorhabditis elegans cell fate-determining gene mab-21: expression, chromosomal localization and analysis of a highly polymorphic (CAG)n trinucleotide repeat. Hum. Mol. Genet. 1996;5:607–616. doi: 10.1093/hmg/5.5.607. [DOI] [PubMed] [Google Scholar]
- 19.Mariani M., Corradi A., Baldessari D., Malgaretti N., Pozzoli O., Fesce R., Martinez S., Boncinelli E., Consalez G.G. Mab21, the mouse homolog of a C. elegans cell-fate specification gene, participates in cerebellar, midbrain and eye development. Mech. Dev. 1998;79:131–135. doi: 10.1016/s0925-4773(98)00180-4. [DOI] [PubMed] [Google Scholar]
- 20.Kennedy B.N., Stearns G.W., Smyth V.A., Ramamurthy V., van Eeden F., Ankoudinova I., Raible D., Hurley J.B., Brockerhoff S.E. Zebrafish rx3 and mab21l2 are required during eye morphogenesis. Dev. Biol. 2004;270:336–349. doi: 10.1016/j.ydbio.2004.02.026. [DOI] [PubMed] [Google Scholar]
- 21.Cederlund M.L., Vendrell V., Morrissey M.E., Yin J., Gaora P.Ó., Smyth V.A., Higgins D.G., Kennedy B.N. mab21l2 transgenics reveal novel expression patterns of mab21l1 and mab21l2, and conserved promoter regulation without sequence conservation. Dev. Dyn. 2011;240:745–754. doi: 10.1002/dvdy.22573. [DOI] [PubMed] [Google Scholar]
- 22.Yamada R., Mizutani-Koseki Y., Koseki H., Takahashi N. Requirement for Mab21l2 during development of murine retina and ventral body wall. Dev. Biol. 2004;274:295–307. doi: 10.1016/j.ydbio.2004.07.016. [DOI] [PubMed] [Google Scholar]
- 23.Wong R.L., Chan K.K., Chow K.L. Developmental expression of Mab21l2 during mouse embryogenesis. Mech. Dev. 1999;87:185–188. doi: 10.1016/s0925-4773(99)00127-6. [DOI] [PubMed] [Google Scholar]
- 24.Kuchta K., Knizewski L., Wyrwicz L.S., Rychlewski L., Ginalski K. Comprehensive classification of nucleotidyltransferase fold proteins: identification of novel families and their representatives in human. Nucleic Acids Res. 2009;37:7701–7714. doi: 10.1093/nar/gkp854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Xiao T.S., Fitzgerald K.A. The cGAS-STING pathway for DNA sensing. Mol. Cell. 2013;51:135–139. doi: 10.1016/j.molcel.2013.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hartmann R., Justesen J., Sarkar S.N., Sen G.C., Yee V.C. Crystal structure of the 2′-specific and double-stranded RNA-activated interferon-induced antiviral protein 2′-5′-oligoadenylate synthetase. Mol. Cell. 2003;12:1173–1185. doi: 10.1016/s1097-2765(03)00433-7. [DOI] [PubMed] [Google Scholar]
- 27.Meng H., Deo S., Xiong S., Dzananovic E., Donald L.J., van Dijk C.W., McKenna S.A. Regulation of the interferon-inducible 2′-5′-oligoadenylate synthetases by adenovirus VA(I) RNA. J. Mol. Biol. 2012;422:635–649. doi: 10.1016/j.jmb.2012.06.017. [DOI] [PubMed] [Google Scholar]
- 28.Hartmann R., Norby P.L., Martensen P.M., Jorgensen P., James M.C., Jacobsen C., Moestrup S.K., Clemens M.J., Justesen J. Activation of 2′-5′ oligoadenylate synthetase by single-stranded and double-stranded RNA aptamers. J. Biol. Chem. 1998;273:3236–3246. doi: 10.1074/jbc.273.6.3236. [DOI] [PubMed] [Google Scholar]
- 29.Schuurs-Hoeijmakers J.H., Oh E.C., Vissers L.E., Swinkels M.E., Gilissen C., Willemsen M.A., Holvoet M., Steehouwer M., Veltman J.A., de Vries B.B. Recurrent de novo mutations in PACS1 cause defective cranial-neural-crest migration and define a recognizable intellectual-disability syndrome. Am. J. Hum. Genet. 2012;91:1122–1127. doi: 10.1016/j.ajhg.2012.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bashir T., Pagano M. Aberrant ubiquitin-mediated proteolysis of cell cycle regulatory proteins and oncogenesis. Adv. Cancer Res. 2003;88:101–144. doi: 10.1016/s0065-230x(03)88305-7. [DOI] [PubMed] [Google Scholar]
- 31.Nakamura T., Colbert M., Krenz M., Molkentin J.D., Hahn H.S., Dorn G.W., 2nd, Robbins J. Mediating ERK 1/2 signaling rescues congenital heart defects in a mouse model of Noonan syndrome. J. Clin. Invest. 2007;117:2123–2132. doi: 10.1172/JCI30756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Nakamura T., Gulick J., Pratt R., Robbins J. Noonan syndrome is associated with enhanced pERK activity, the repression of which can prevent craniofacial malformations. Proc. Natl. Acad. Sci. USA. 2009;106:15436–15441. doi: 10.1073/pnas.0903302106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Corson L.B., Yamanaka Y., Lai K.M., Rossant J. Spatial and temporal patterns of ERK signaling during mouse embryogenesis. Development. 2003;130:4527–4537. doi: 10.1242/dev.00669. [DOI] [PubMed] [Google Scholar]
- 34.Krejci P. The paradox of FGFR3 signaling in skeletal dysplasia: why chondrocytes growth arrest while other cells over proliferate. Mutat. Res. 2014;759:40–48. doi: 10.1016/j.mrrev.2013.11.001. [DOI] [PubMed] [Google Scholar]
- 35.Harima Y., Kageyama R. Oscillatory links of Fgf signaling and Hes7 in the segmentation clock. Curr. Opin. Genet. Dev. 2013;23:484–490. doi: 10.1016/j.gde.2013.02.005. [DOI] [PubMed] [Google Scholar]
- 36.Schubbert S., Bollag G., Lyubynska N., Nguyen H., Kratz C.P., Zenker M., Niemeyer C.M., Molven A., Shannon K. Biochemical and functional characterization of germ line KRAS mutations. Mol. Cell. Biol. 2007;27:7765–7770. doi: 10.1128/MCB.00965-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Le Goff C., Cormier-Daire V. From tall to short: the role of TGFβ signaling in growth and its disorders. Am. J. Med. Genet. C. Semin. Med. Genet. 2012;160C:145–153. doi: 10.1002/ajmg.c.31337. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.