

Abstract
Neu-Laxova syndrome (NLS) is a rare autosomal-recessive disorder characterized by severe fetal growth restriction, microcephaly, a distinct facial appearance, ichthyosis, skeletal anomalies, and perinatal lethality. The pathogenesis of NLS remains unclear despite extensive clinical and pathological phenotyping of the >70 affected individuals reported to date, emphasizing the need to identify the underlying genetic etiology, which remains unknown. In order to identify the cause of NLS, we conducted a positional-mapping study combining autozygosity mapping and whole-exome sequencing in three consanguineous families affected by NLS. Surprisingly, the NLS-associated locus identified in this study was solved at the gene level to reveal mutations in PHGDH, which is known to be mutated in individuals with microcephaly and developmental delay. PHGDH encodes the first enzyme in the phosphorylated pathway of de novo serine synthesis, and complete deficiency of its mouse ortholog recapitulates many of the key features of NLS. This study shows that NLS represents the extreme end of a known inborn error of serine metabolism and highlights the power of genomic sequencing in revealing the unsuspected allelic nature of apparently distinct clinical entities.
Main Text
Neu-Laxova syndrome (NLS [MIM 256520]) is a term coined by Lazjuk in 1979 to unify the independent reports by Neu and Laxova on a lethal multiple-congenital-anomaly syndrome.1–3 The main features of NLS involve defective somatic growth and CNS and skin development, in addition to many other anomalies that might represent primary malformations or a malformation sequence. Growth restriction is a constant feature that is usually apparent in the second trimester, and it could be argued that NLS could be classified as a primordial dwarfism disorder, especially given that frank skeletal dysplasia is uncommon in this condition.4 Another constant feature is abnormal brain development, most commonly in the form of profound microcephaly; a head circumference of 20 cm at term has been reported.5,6 Most of the case reports that describe detailed brain examination by imaging or on autopsy converge on a highly characteristic phenotype of the brain. In addition to being extremely small in volume, the brain has a distinctive pattern of lissencephaly, which some label as lissencephaly type III to differentiate it from type I, seen in Miller-Dieker syndrome (MIM 247200), and the cobblestone type II, seen in Walker-Warburg syndrome (MIM 236670) and related disorders of glycosylation. The cerebellum is often hypoplastic, and complete absence of the vermis has frequently been reported. In addition, the corticomedullary tracts are often small or absent in the brainstem and spinal cord.5 It has been proposed that in view of the severity of CNS involvement, the skeletal manifestations in the form of contractures and syndactyly represent a sequence that is initiated by the brain malformation labeled as cerebroarthrodigital (CAD) sequence.7 Contractures, which are common in NLS and sometimes accompanied by pterygium formation, are associated with hypoplasia of the skeletal muscles. Syndactyly of the hands and feet often takes an unusual form of severe swelling and rudimentary digits that are sometimes undiscernible.8 The skin is usually ichthyotic with marked hyperkeratosis and can resemble the colloidon membrane appearance of other ichthyotic disorders.9 The face is highly characteristic with proptotic eyes, ectropion, eclabion, and a severely hypoplastic nose. Most affected children die shortly after birth, although survival beyond 10 months has been reported and presumed to represent a milder phenotype.10
The nature of this extremely severe multiple-congenital-anomaly syndrome has been debated for decades. In addition to the CAD sequence theory above, another proposal is that NLS might represent an inborn error of fat metabolism.8 The latter was prompted by the often reported finding of significant accumulation of fat and myxoedematous material in the dermis throughout the body; this accumulation gives the typical edematous appearance of affected fetuses, who are sometimes referred to as being “hydropic.”11 In this study, we took advantage of the powerful tools of autozygosity mapping and whole-exome sequencing to show that NLS is in fact an inborn error of serine metabolism and that a mouse model recapitulating key neurological and other features of this disorder can provide insight into its molecular pathogenesis.
Three affected individuals from three families were enrolled under a protocol approved by the institutional review board at King Faisal Specialist Hospital and Research Center (research approval committee 2080006) after signing written informed consent. Blood samples were collected from the affected individuals, their parents, and unaffected siblings when applicable. Family 1 consists of double-first-cousin Saudi parents with one healthy daughter, two spontaneous abortions, and one daughter who died immediately after birth and is said to have had the same phenotype as the index individual. This index individual (II:3 in family 1, Figure 1), a full-term female born vaginally, died immediately after birth. She was very small for her gestational age and had severe microcephaly (records of actual growth parameters at birth could not be retrieved), micrognathia, bulging eyes with absent eyelids, severe ichthyosis of the skin, cleft lip and palate on the right side, a very flat nose, a very short neck, and generalized edema. She also had extremely abnormal limbs with hypoplastic forearms and no discernible digits in the upper or lower limbs (Figure S1A, available online).
Figure 1.
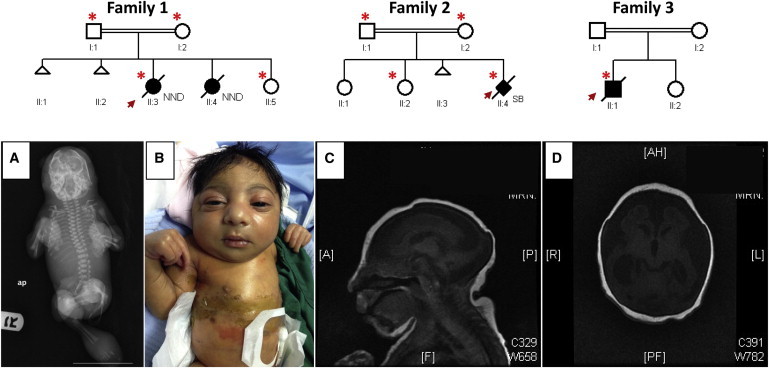
Pedigrees and Clinical Images of the Study Families
The index individual is indicated in each pedigree by an arrow, and asterisks denote individuals whose DNA was available for analysis. Abbreviations are as follows: NND, neonatal death; and SB, stillbirth.
(A) A babygram imaging of the index individual from family 2 shows small distorted calvarial bones without gross vertebral or tubular bone deformity.
(B) A photograph of the index individual from family 3 shows microcephaly, generalized colloidon-like ichthyosis, a sloping forehead, a broad nose, large ears, a short neck, spastic long fingers, and fixed contractures of the extremities.
(C and D) Axial (C) and sagittal (D) MRI of the index individual from family 3 shows a markedly atrophic and small brain with significant ventriculomegaly but normal appearance of the brainstem.
Family 2 consists of first-cousin parents and two healthy daughters. The index individual (II:4 in family 2, Figure 1) was born preterm at 29 weeks after an uneventful vaginal delivery. Antenatal ultrasounds at both 19 and 24 weeks of gestation showed polyhydramnios, curved vertebrae, protruded eyes, an open mouth, low-set ears, a short and broad neck, microcephaly, generalized skin edema (especially of the trunk and scalp), abnormally flexed hands, extended crossed feet with a rocker-bottom appearance, and fetal akinesia. Postnatal screening for congenital infection was inconclusive. Postnatal examination revealed massive body swelling and marked disfigurement of the face and limbs, which appeared engulfed by a thin and shiny membrane (Figure S1B). The eyes were small, fixed, and widely spaced and showed supraorbital massive cystic swelling bilaterally. The nose was completely flat and obliterated, and the mouth was large and fixed open with massively swollen lips. The neck was extremely short. The ear lobules were edematous with tight overlying skin. The trunk was short and shiny with visible veins. The baby exhibited a fixed-flexion appearance with generalized contractures. The massively edematous hands and feet had no discernible digits. A skeletal survey showed defaced and overlapping cranial bones with severe soft-tissue edema. Thoracic, vertebral, pelvic, and other tubular bones had no major skeletal defects (Figure 1).
Family 3 consists of first-cousin parents with one healthy daughter and one affected boy (index) who died at the age of 1 month. The index individual (II:1 in family 3, Figure 1) was delivered vaginally at term with thick meconium-stained liquor. Birth weight was 2.24 kg. The baby was evidently jaundiced with generalized colloidon-like ichthyosis. He was microcephalic (head circumference of 27.5 cm) with a sloping forehead, a broad nose, large ears, a short neck, spastic long fingers, and fixed contractures of the extremities (Figure 1). He succumbed to pneumonia and pseudomonas sepsis and died at 1 month of age.
Autozygome analysis was performed on all affected individuals with the Axiom SNP platform (Affymetrix) and was followed by AutoSNPa genome-wide determination of runs of homozygosity (ROHs) as surrogates of autozygosity essentially as described before.12,13 Autozygomes of the index from the three families overlapped on a single chromosome 1 locus corresponding to the genomic region chr1: 117,058,389–159,754,449 bp (hg19 assembly, UCSC Genome Browser; Figure 2 A). Linkage analysis revealed a single peak (LOD of ∼3.9) corresponding to the same critical ROH highlighted by autozygome analysis (Figure 2B). Within the shared ROH, a region with an identical haplotype (chr1: 119,695,451–120,492,822, spanning 11 RefSeq genes) between the two index individuals in families 1 and 2 was identified, further narrowing the critical interval (Figure 2B). Exome sequencing of the index individual in family 1 revealed a missense variant (c.418G>A [p.Gly140Arg], RefSeq NM_006623.3) in PHGDH as the only homozygous coding or splicing variant absent from public databases within that critical interval. Consistent with their shared haplotype, targeted Sanger sequencing uncovered the same variant in the index individual in family 2 (Figure 2C). We then fully sequenced PHGDH in the index individual (II:1) in family 3 and identified another homozygous missense mutation (c.488G>A [p.Arg163Gln], RefSeq NM_006623.3) (Figure 2C). Both variants were absent in 450 Saudi exomes, the 1000 Genomes Project, and the NHLBI Exome Sequencing Project Exome Variant Server. In addition, 3D modeling of the two variants revealed their localization within the NAD (P) binding domain and specifically at the PHGDH dimer interface, which is important for the optimal function of PHGDH (Figures 3A–3C).14 Furthermore, the two missense variants affect two absolutely conserved residues down to plant, and both SIFT and PolyPhen predict them to be highly pathogenic (PolyPhen: probably damaging [1]; SIFT: deleterious [0]) (Figure 3E). Finally, a dried blood spot from an affected individual in family 3 was analyzed by tandem mass spectrometry,15 and the amino acid profile was significant for low concentrations of serine (50 nmol/ml; reference range 74–448) and glycine (105 nmol/ml; reference range 180–709). These findings are also consistent with a biochemical diagnosis of PHGDH deficiency. These data strongly suggest that each of the two alleles in PHGDH is disease causing in the respective families in the homozygous state.
Figure 2.
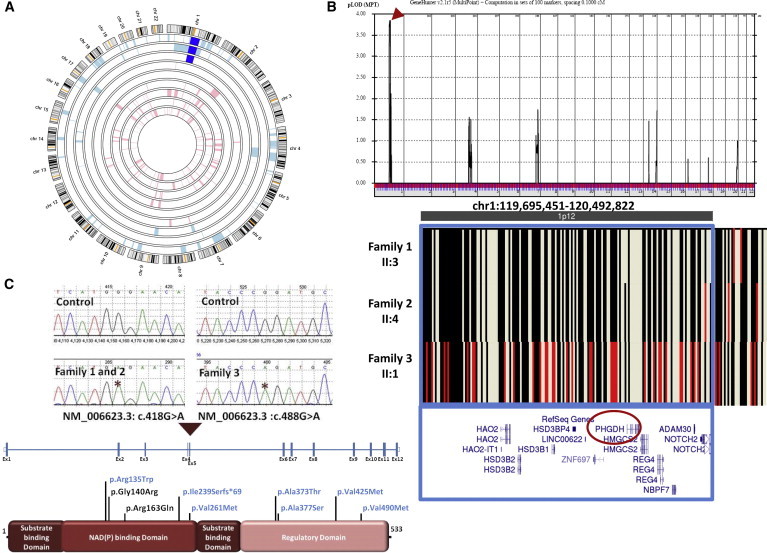
Identification of a NLS-Associated Locus on Chromosome 1
(A) AgileMultiIdeogram showing the chromosome 1 ROH (dark blue) shared among the index individuals from each of the three families.
(B) Combined genome-wide linkage analysis of the three families revealed a single maximal peak (LOD score = ∼3.9) on chromosome 1, and AutoSNPa output shows the critical interval at chr1: 119,695,451–120,492,822 (boxed in blue) within the shared ROH. The identical haplotype between individual II:1 in family 1 and individual II:4 in family 2 is denoted by black lines, whereas the red lines in individual II:1 from family 3 denote divergence in haplotype.
(C) Upper panel: sequence chromatogram of the two missense mutations (control tracing is shown for comparison, and the location of each mutation is denoted by an asterisk). Lower Panel: schematic of PHGDH and the locations of the two homozygous missense substitutions identified in the three families (previously reported substitutions in individuals with PHGDH deficiency are shown in blue for comparison).
Figure 3.
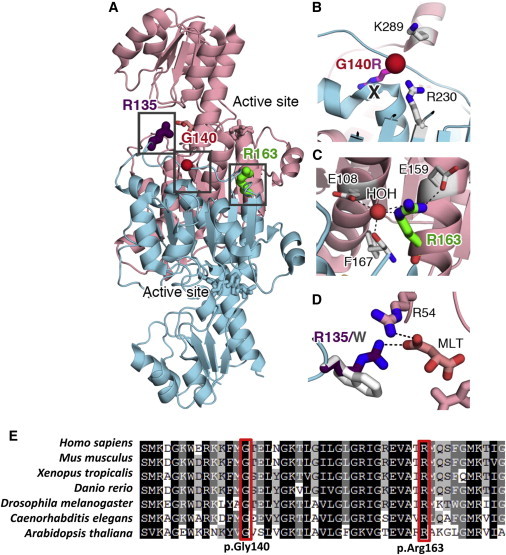
The Two Substituted Amino Acid Residues Are Located in an Important Region of PHGDH
(A) Gly140 (G140, red sphere) and Arg163 (R163, green stick) are located at the PHGDH dimer interface (molecule 1 in salmon, molecule 2 in cyan, two substrates in sticks). The dimer is important for the optimal function of PHGDH. Details of the boxed regions are shown in (B) and (C).
(B) The p.Gly140Arg (Gly140 in red, Arg in purple) substitution would cause steric clash (marked by a “X”) and introduce extra positive charge at the dimer interface, which would most likely weaken the dimerization by steric hindrance and electrostatic repulsion from two nearby positively charged residues (Lys289 [K289] and Arg230 [R230] in sticks).
(C) Arg163 (R163) participates in a water (HOH)-coordinated hydrogen-bonding and salt bridge network at the dimerization interface (Glu108 [E108], Glu159 [E159], and Phe167 [F167] are shown as sticks, and the interaction network is shown as black dashed lines). The p.Arg163Gln substitution might be detrimental to the integrity of this network and weaken dimerization. Also, the loss of the positive charge by the Arg-to-Gln substitution would perturb the surface charge distribution.
(D) Arg135 (R135, purple; involved in the previously reported p.Arg135Trp substitution in PHGDH deficiency) and Arg54 (R54, pink sticks) interact with the tail of the substrate malate (MLT, pink sticks) by providing two salt bridges (black dashed lines). The p.Arg135Trp substitution (gray sticks) would eliminate one of the salt bridges and weaken the overall electrostatic attraction. Some activity presumably remains given that Arg54 might still hold the MLT substrate in position. Furthermore, Arg135 does not interact with other protein side chains, and there is enough space to accommodate a Trp substitution, so the p.Arg135Trp substitution might not perturb the overall protein structure and function significantly.
(E) Multisequence-alignment orthologs of the two missense substitutions. The affected glycine and arginine residues (boxed in red) are absolutely conserved across species down to C. elegans and plant.
The realization that the distinction between metabolism and dysmorphology is artificial is not new, and mucopolysaccharidoses (MIM 252700), Smith-Lemli-Opitz syndrome (MIM 270400), and peroxisomal biogenesis disorder (MIM 214100) are among the earliest examples of metabolic disorders with a significant dysmorphology profile.16 However, the historical branching of clinical genetics into inborn errors of metabolism and dysmorphology continues to hamper the exchange of phenotyping expertise, which in turn can delay the proper molecular classification of disorders, as has been shown recently in the case of congenital disorder of glycosylation type IIa (MIM 212066).17,18 Fortunately, the recent availability of genomic tools that are largely unaffected by this problem of phenotyping bias has ushered in a new era of molecular diagnostics and revealed many surprises as a result. Interesting in this regard is the fact that the first Mendelian disorder to be solved by whole-exome sequencing—Miller syndrome—is a classic dysmorphology syndrome that was found to be an inborn error of pyrimidine metabolism.19 Thus, our surprising finding that NLS is an inborn error of metabolism should be viewed in the context of a trend where whole-exome sequencing is demolishing arbitrary distinctions. In addition, we note that whole-exome sequencing has confirmed allelism between NLS and PHGDH deficiency, a surprising finding that could not have been predicted on clinical grounds.
PHGDH deficiency, an inborn error of serine metabolism, was first described by Jaeken et al. in 1996.20 Although serine can be synthesized with alternative pathways, reports of PHGDH deficiency were viewed as evidence that PHGDH and the pathway in which it serves are the major source of this nonessential amino acid because affected individuals have low serine levels in plasma and CSF despite the intact nature of those alternative pathways. These individuals are almost uniformly microcephalic and delayed in their development, and epilepsy and intrauterine growth retardation are common features.21
Our finding that NLS and PHGDH deficiency are allelic disorders raises an obvious question: why do the phenotypes of these two disorders differ significantly? There are several possible explanations for this discrepancy. First, ascertainment bias makes it possible that only individuals at the mild end of the PHGDH spectrum present to metabolic specialists with the common referring diagnosis of nonspecific intellectual disability and epilepsy, whereas those with more severe deficiency present with structural anomalies and are referred instead to dysmorphologists, who are less likely to conduct a detailed metabolic assessment. We are not aware of any previously published NLS report that explicitly describes results of plasma amino acid measurements. Second, a closer look at the literature suggests that a spectrum does indeed exist such that it is conceivable to view NLS and PHGDH deficiency as part of a spectrum, as illustrated by the previously reported 10-month-old infant with a mild form of NLS. Third, it is possible that the mutations that cause NLS exert a more detrimental effect on the protein function than do those previously reported in PHGDH deficiency. For example, 3D modeling suggests that the previously reported p.Arg135Trp (c.403C>T) (RefSeq NM_006623.3) substitution in PHGDH deficiency is likely to affect the protein more mildly than the adjacent p.Gly140Arg, which we report here (Figure 3). Fourth, mice that completely lack the murine ortholog of PHGDH display a universally lethal phenotype that recapitulates key pathogenic features of NLS.22 The embryos do not usually survive beyond 13.5 days postcoitus, are extremely small in size, and have a limb phenotype (where the terminal limb bud is swollen and fails to digitize) that is remarkably similar to that of NLS. Moreover, the brains of these embryos are small and show evidence of abnormal development, as seen in NLS individuals.
Given the above features, Phgdh−/− mice make a valuable NLS disease model and provide insights into the molecular pathogenesis of NLS. Serine is required for the synthesis of key brain lipids, including sphingolipids and gangliosides, and these are indeed markedly deficient in the brains of Phgdh−/− embryos.22 These mice have also been shown to have reduced aminophospholipids (e.g., phosphatidylserine and phosphatidylethanolamine) in addition to a documented repression of protein translation initiation.23 Interestingly, PHGDH is highly expressed in neuroprogenitor cells during embryogenesis and is expressed in glial cells of postnatal brains.24,25 Indeed, complete deficiency of PHGDH is associated with a marked proliferation defect of neuroprogenitors of Phgdh−/− brains, which can explain the severe microlissencephaly, a phenotype that has been previously shown to result from a mitosis defect of neuroprogenitors.26 On the other hand, heterozygous Phgdh+/− mice are completely normal, so it is conceivable that residual activity of PHGDH in previously published individuals with PHGDH deficiency (range 12%–25%) might have provided sufficient serine to overcome a particular threshold below which the major developmental defects of NLS take place. It is worth mentioning here that another serine-metabolism-related knockout mouse (Psid−/−) in which phosphatidylserine decarboxylase deficiency results in decreased synthesis of phosphatidylethanolamine is also characterized by embryonic lethality.27
Our direct observation of serine depletion in NLS and the encouraging results of serine supplementation to treat individuals with PHGDH deficiency raise interesting possibilities about the potential benefit of serine supplementation during pregnancy to treat NLS or at least mitigate the severity of its associated developmental defects.28 The practicality of such an approach will most likely be limited by the inability to identify this very rare disorder sufficiently early in pregnancy, but it can theoretically be attempted in families with high recurrence risk, such as the families studied here.
In summary, we report on the identification of homozygous mutations in PHGDH and serine deficiency in individuals with NLS. This disorder thus seems to be an extremely severe expression of PHGDH deficiency. Our finding that NLS is an allelic disorder of the much milder PHGDH-deficiency phenotype highlights the additional layer of complexity in linking genes to human diseases in that although genes are limited in number, their pathogenic alleles are not, and each of these alleles can have its own phenotypic consequences. Genomic sequencing is a powerful tool in tackling this challenge.
Acknowledgments
We thank the affected individuals and their families for their enthusiastic participation. We thank Mais Hashem, Niema Ibrahim, and Firdous Mohammed for their help as clinical research coordinators. We also thank the Genotyping and Sequencing Core Facilities at King Faisal Specialist Hospital and Research Center for their technical help. Thanks are also due to Dietrich Matern and Piero Rinaldo of the Biochemical Genetics Laboratory, Mayo Clinic, for the amino acid analysis in a dried blood spot from the index individual in family 3. This work was supported by a collaborative research grant from the Dubai Harvard Foundation for Medical Research (to F.S.A.).
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
AgileMultiIdeogram, http://dna.leeds.ac.uk/agile/AgileMultiIdeogram/
NHLBI Exome Sequencing Project (ESP) Exome Variant Server, http://evs.gs.washington.edu/EVS/
Online Mendelian Inheritance in Man (OMIM), http://www.omim.org/
PolyPhen, www.genetics.bwh.harvard.edu/pph2/
SIFT, www.sift.jcvi.org/
UCSC Genome Browser, http://genome.ucsc.edu
References
- 1.Neu R.L., Kajii T., Gardner L.I., Nagyfy S.F. A lethal syndrome of microcephaly with multiple congenital anomalies in three siblings. Pediatrics. 1971;47:610–612. [PubMed] [Google Scholar]
- 2.Laxova R., Ohara P.T., Timothy J.A. A further example of a lethal autosomal recessive condition in sibs. J. Ment. Defic. Res. 1972;16:139–143. doi: 10.1111/j.1365-2788.1972.tb01585.x. [DOI] [PubMed] [Google Scholar]
- 3.Lazjuk G.I., Lurie I.W., Ostrowskaja T.I., Cherstvoy E.D., Kirillova I.A., Nedzved M.K., Usoev S.S. Brief clinical observations: the Neu-Laxova syndrome—a distinct entity. Am. J. Med. Genet. 1979;3:261–267. doi: 10.1002/ajmg.1320030304. [DOI] [PubMed] [Google Scholar]
- 4.Shaheen R., Faqeih E., Ansari S., Abdel-Salam G., Al-Hassnan Z.N., Al-Shidi T., Alomar R., Sogaty S., Alkuraya F.S. Genomic analysis of primordial dwarfism reveals novel disease genes. Genome Res. 2014;24:291–299. doi: 10.1101/gr.160572.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ostrovskaya T.I., Lazjuk G.I. Cerebral abnormalities in the Neu-Laxova syndrome. Am. J. Med. Genet. 1988;30:747–756. doi: 10.1002/ajmg.1320300308. [DOI] [PubMed] [Google Scholar]
- 6.Mueller R.F., Winter R.M., Naylor C.P. Neu-Laxova syndrome: two further case reports and comments on proposed subclassification. Am. J. Med. Genet. 1983;16:645–649. doi: 10.1002/ajmg.1320160424. [DOI] [PubMed] [Google Scholar]
- 7.Scott C.I., Louro J.M., Laurence K.M., Tolarová M., Hall J.G., Reed S., Curry C.J. Comments on the Neu-Laxova syndrome and CAD complex. Am. J. Med. Genet. 1981;9:165–175. doi: 10.1002/ajmg.1320090211. [DOI] [PubMed] [Google Scholar]
- 8.Shved I.A., Lazjuk G.I., Cherstvoy E.D. Elaboration of the phenotypic changes of the upper limbs in the Neu-Laxova syndrome. Am. J. Med. Genet. 1985;20:1–11. doi: 10.1002/ajmg.1320200102. [DOI] [PubMed] [Google Scholar]
- 9.Carder K.R., Fitzpatrick J.E., Weston W.L. What syndrome is this? Neu-Laxova syndrome. Pediatr. Dermatol. 2003;20:78–80. doi: 10.1046/j.1525-1470.2003.03017.x. [DOI] [PubMed] [Google Scholar]
- 10.Horn D., Müller D., Thiele H., Kunze J. Extreme microcephaly, severe growth and mental retardation, flexion contractures, and ichthyotic skin in two brothers: a new syndrome or mild form of Neu-Laxova syndrome? Clin. Dysmorphol. 1997;6:323–328. doi: 10.1097/00019605-199710000-00004. [DOI] [PubMed] [Google Scholar]
- 11.Karimi-Nejad M.H., Khajavi H., Gharavi M.J., Karimi-Nejad R. Neu-Laxova syndrome: report of a case and comments. Am. J. Med. Genet. 1987;28:17–23. doi: 10.1002/ajmg.1320280104. [DOI] [PubMed] [Google Scholar]
- 12.Alkuraya F.S. Discovery of rare homozygous mutations from studies of consanguineous pedigrees. Curr. Protoc. Hum. Genet. 2012;Chapter 6:12. doi: 10.1002/0471142905.hg0612s75. [DOI] [PubMed] [Google Scholar]
- 13.Alkuraya F.S. Autozygome decoded. Genet. Med. 2010;12:765–771. doi: 10.1097/GIM.0b013e3181fbfcc4. [DOI] [PubMed] [Google Scholar]
- 14.Mishra V., Kumar A., Ali V., Nozaki T., Zhang K.Y., Bhakuni V. Glu-108 is essential for subunit assembly and dimer stability of D-phosphoglycerate dehydrogenase from Entamoeba histolytica. Mol. Biochem. Parasitol. 2012;181:117–124. doi: 10.1016/j.molbiopara.2011.10.008. [DOI] [PubMed] [Google Scholar]
- 15.Turgeon C., Magera M.J., Allard P., Tortorelli S., Gavrilov D., Oglesbee D., Raymond K., Rinaldo P., Matern D. Combined newborn screening for succinylacetone, amino acids, and acylcarnitines in dried blood spots. Clin. Chem. 2008;54:657–664. doi: 10.1373/clinchem.2007.101949. [DOI] [PubMed] [Google Scholar]
- 16.Hunter A.G.W. Medical genetics: 2. The diagnostic approach to the child with dysmorphic signs. CMAJ. 2002;167:367–372. [PMC free article] [PubMed] [Google Scholar]
- 17.Alkuraya F.S. Mental retardation, growth retardation, unusual nose, and open mouth: an autosomal recessive entity. Am. J. Med. Genet. A. 2010;152A:2160–2163. doi: 10.1002/ajmg.a.33575. [DOI] [PubMed] [Google Scholar]
- 18.Alazami A.M., Monies D., Meyer B.F., Alzahrani F., Hashem M., Salih M.A., Alkuraya F.S. Congenital disorder of glycosylation IIa: the trouble with diagnosing a dysmorphic inborn error of metabolism. Am. J. Med. Genet. A. 2012;158A:245–246. doi: 10.1002/ajmg.a.34347. [DOI] [PubMed] [Google Scholar]
- 19.Ng S.B., Buckingham K.J., Lee C., Bigham A.W., Tabor H.K., Dent K.M., Huff C.D., Shannon P.T., Jabs E.W., Nickerson D.A. Exome sequencing identifies the cause of a mendelian disorder. Nat. Genet. 2010;42:30–35. doi: 10.1038/ng.499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jaeken J., Detheux M., Van Maldergem L., Foulon M., Carchon H., Van Schaftingen E. 3-Phosphoglycerate dehydrogenase deficiency: an inborn error of serine biosynthesis. Arch. Dis. Child. 1996;74:542–545. doi: 10.1136/adc.74.6.542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.van der Crabben S.N., Verhoeven-Duif N.M., Brilstra E.H., Van Maldergem L., Coskun T., Rubio-Gozalbo E., Berger R., de Koning T.J. An update on serine deficiency disorders. J. Inherit. Metab. Dis. 2013;36:613–619. doi: 10.1007/s10545-013-9592-4. [DOI] [PubMed] [Google Scholar]
- 22.Yoshida K., Furuya S., Osuka S., Mitoma J., Shinoda Y., Watanabe M., Azuma N., Tanaka H., Hashikawa T., Itohara S., Hirabayashi Y. Targeted disruption of the mouse 3-phosphoglycerate dehydrogenase gene causes severe neurodevelopmental defects and results in embryonic lethality. J. Biol. Chem. 2004;279:3573–3577. doi: 10.1074/jbc.C300507200. [DOI] [PubMed] [Google Scholar]
- 23.Sayano T., Kawakami Y., Kusada W., Suzuki T., Kawano Y., Watanabe A., Takashima K., Arimoto Y., Esaki K., Wada A. L-serine deficiency caused by genetic Phgdh deletion leads to robust induction of 4E-BP1 and subsequent repression of translation initiation in the developing central nervous system. FEBS J. 2013;280:1502–1517. doi: 10.1111/febs.12146. [DOI] [PubMed] [Google Scholar]
- 24.Furuya S., Tabata T., Mitoma J., Yamada K., Yamasaki M., Makino A., Yamamoto T., Watanabe M., Kano M., Hirabayashi Y. L-serine and glycine serve as major astroglia-derived trophic factors for cerebellar Purkinje neurons. Proc. Natl. Acad. Sci. USA. 2000;97:11528–11533. doi: 10.1073/pnas.200364497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yamasaki M., Yamada K., Furuya S., Mitoma J., Hirabayashi Y., Watanabe M. 3-Phosphoglycerate dehydrogenase, a key enzyme for l-serine biosynthesis, is preferentially expressed in the radial glia/astrocyte lineage and olfactory ensheathing glia in the mouse brain. J. Neurosci. 2001;21:7691–7704. doi: 10.1523/JNEUROSCI.21-19-07691.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Alkuraya F.S., Cai X., Emery C., Mochida G.H., Al-Dosari M.S., Felie J.M., Hill R.S., Barry B.J., Partlow J.N., Gascon G.G. Human mutations in NDE1 cause extreme microcephaly with lissencephaly [corrected] Am. J. Hum. Genet. 2011;88:536–547. doi: 10.1016/j.ajhg.2011.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Steenbergen R., Nanowski T.S., Beigneux A., Kulinski A., Young S.G., Vance J.E. Disruption of the phosphatidylserine decarboxylase gene in mice causes embryonic lethality and mitochondrial defects. J. Biol. Chem. 2005;280:40032–40040. doi: 10.1074/jbc.M506510200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.de Koning T.J., Klomp L.W., van Oppen A.C., Beemer F.A., Dorland L., van den Berg I., Berger R. Prenatal and early postnatal treatment in 3-phosphoglycerate-dehydrogenase deficiency. Lancet. 2004;364:2221–2222. doi: 10.1016/S0140-6736(04)17596-X. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.