

Abstract
Diets high in sugar and saturated fat (Western diet) contribute to obesity and pathophysiology of metabolic syndrome. A common physiological response to obesity is hypertension, which induces cardiac remodeling and hypertrophy. Hypertrophy is regulated at the level of chromatin by repressor element 1-silencing transcription factor (REST), and pathological hypertrophy is associated with reexpression of a fetal cardiac gene program. Reactivation of fetal genes is commonly observed in hypertension-induced hypertrophy; however, this response is blunted in diabetic hearts, partially due to upregulation of the posttranslational modification O-linked-β-N-acetylglucosamine (O-GlcNAc) to proteins by O-GlcNAc transferase (OGT). OGT and O-GlcNAc are found in chromatin-modifying complexes, but it is unknown whether they play a role in Western diet-induced hypertrophic remodeling. Therefore, we investigated the interactions between O-GlcNAc, OGT, and the fetal gene-regulating transcription factor complex REST/mammalian switch-independent 3A/histone deacetylase (HDAC). Five-week-old male C57BL/6 mice were fed a Western (n = 12) or control diet (n = 12) for 2 wk to examine the early hypertrophic response. Western diet-fed mice exhibited fasting hyperglycemia and increased body weight (P < 0.05). As expected for this short duration of feeding, cardiac hypertrophy was not yet evident. We found that REST is O-GlcNAcylated and physically interacts with OGT in mouse hearts. Western blot analysis showed that HDAC protein levels were not different between groups; however, relative to controls, Western diet hearts showed increased REST and decreased ANP and skeletal α-actin. Transcript levels of HDAC2 and cardiac α-actin were decreased in Western diet hearts. These data suggest that REST coordinates regulation of diet-induced hypertrophy at the level of chromatin.
Keywords: cardiac hypertrophy, chromatin, O-GlcNAc, fetal genes
diets high in sugar and saturated fat (Western diet) contribute to the development of obesity, metabolic syndrome, and Type 2 diabetes, and complications arising from these conditions can lead to cardiovascular disease (3, 7, 14, 23). Abnormal or pathological hypertrophic remodeling is a normal, adaptive response to cardiac stress that is a precursor to cardiomyopathy and heart failure. However, the derangement of the hypertrophic response in the diabetic heart may underlie the more rapid degeneration to heart failure observed relative to nondiabetic heart disease (5). Pathological cardiac signaling in hypertrophy involves epigenetic changes at the chromatin level (1). Diet- and diabetes-related pathological hypertrophies are characterized by cardiac dysfunction, but with a less pronounced increase in heart size than that seen in pressure-overloaded hearts; additionally, the genetic and cellular regulation of these types of hypertrophic signaling is poorly understood (5). It is not known whether diet alone can induce changes in transcriptional control of the early stages of hypertrophy.
Hypertrophy in response to pressure overload or myocardial stretch activates a fetal profile of genes that are typically silenced in the adult heart, and are reactivated under pathological hypertrophic stimuli (11). Components of the fetal gene program include atrial natriuretic peptide (ANP), brain natriuretic peptide (BNP), and skeletal α-actin. Skeletal α-actin is the fetal cardiac muscle isoform; this dominates over the adult cardiac α-actin muscle isoform in pathological remodeling. Activation of the fetal gene program is controlled by a protein complex of the DNA-binding protein repressor element 1-silencing transcription factor (REST), mammalian switch-independent 3A (mSin3A), and multiple histone deacetylase (HDAC) proteins (10, 26). REST suppresses the fetal gene program by binding to repressor element-1 in the 3′ untranslated region and the 5′ promoter regions of ANP and BNP, respectively (2, 9, 11). mSin3A colocalizes with REST and recruits HDAC proteins, which regulate transcription through removal of histone acetyl groups (6). The outcome of this regulation is partly determined by the HDAC classes that are associated with mSin3A: class I HDACs include HDACs 1–3 and 8, and they promote hypertrophy, while class II HDACs include 4–7 and 9, and inhibit hypertrophy (4, 8, 25).
The posttranslational attachment of O-linked β-N-acetylglucosamine (O-GlcNAc) to serine/threonine residues of cardiac proteins is increased following chronic consumption of a Western diet (17) and also in the Type 2 diabetic heart (7, 16). We recently demonstrated that pharmacological upregulation of O-GlcNAc in the presence of euglycemia mimics the blunted hypertrophic response observed in diabetic cardiomyocytes (15). A single enzyme, O-GlcNAc transferase (OGT), catalyzes the attachment of O-GlcNAc to proteins while O-GlcNAcase (OGA) removes the moiety. In mammalian cells, OGT is a single protein with three splice variants, although only two of the variants have known functions (13). The longest isoform is ∼116 kDa and is localized to both the nucleus and cytoplasm, the 103-kDa OGT isoform, mitochondrial, contains a mitochondrial targeting sequence at its NH2 terminus, while the shortest OGT isoform at 78 kDa is nuclear and cytoplasmic but has not been further classified, and its specific function remains unknown. O-GlcNAc modifies histone tails and directly impacts gene transcription (20). However, it is unknown whether this occurs in the heart or if this is a mechanism by which O-GlcNAc is involved with hypertrophic gene expression.
OGT complexes with mSin3A and HDAC1 in vhepg2 liver carcinoma cells (24). We recently showed that OGT also complexes with REST and that HDACs 1,2,4,5 are O-GlcNAc modified in cardiac tissue (18). These data showed that OGT is present in a chromatin-remodeling complex that directly controls fetal gene expression, and suggest that the interaction of OGT and O-GlcNAc with REST/mSin3A/HDAC may control the expression of fetal genes in the adult heart. Because the expression of these genes underlies the hypertrophic response to hyperglycemia and obesity, we hypothesized that a short duration of consumption of a Western diet would stimulate hypertrophic signaling cascades. We further hypothesized that this would be associated with changes in interactions between O-GlcNAc, OGT, and components of the REST/mSin3A/HDAC complex, and chose a 2-wk time point to investigate the early signaling mechanisms in this process.
METHODS
Ethical approval.
This protocol was approved by the Institutional Animal Care and Use Committee at Washington State University and conformed to the Care and Use of Laboratory Animals published by the National Institutes of Health (NIH publication number 85-23, 1996).
Study protocol.
Five-week-old male C57BL/6 mice were housed three per cage, maintained on a 12:12-h light-dark cycle, and provided with food and tap water ad libitum. Animals were randomized to receive control (12% kcal fat/16% kcal protein/72% kcal carbohydrate; 3.84 kcal/g; n = 12) or Western (40% kcal fat/16% kcal protein/44% carbohydrate; 4.55 kcal/g; n = 12) diets for 2 wk; diets were prepared and analyzed by TestDiet (Richmond, IN). It should be noted that the diets are not isocaloric by weight; saturated fat content was higher in Western diet (12%) compared with control (1.65%), and the contribution of simple sugars to total carbohydrates was also different (34% in Western compared with 11% in control).
Tissue collection.
Mice were fasted overnight and then quickly anesthetized using 2–4% isoflurane in 100% oxygen and immediately decapitated. Fasting blood glucose was measured in neck blood. Whole hearts were rapidly excised, atria were removed, and remaining ventricular tissue was wet weighed. Heart samples were snap frozen in liquid nitrogen and stored at −80°C until use. Tibia length was measured from the tibial plateau to the lateral malleolus on each animal.
Cell culture.
H9c2 cardiomyoblasts were maintained in high-glucose (25 mM) complete growth medium [American Type Culture Collection-formulated (ATCC) DMEM; 30–2002; ATCC; Manassas, VA] and 10% FBS (30–2020; ATCC) and were passaged at 50–70% confluence. Twenty-four hours prior to individual cell treatments, all cells were supplemented with growth medium containing low (5.6 mM) glucose (Sigma; DMEM; D5546) and 1% FBS for the remainder of the study. To increase GlcNAc, cells were treated with high glucose (25 mM), glucosamine (5 mM), or Thiamet-G (10 nM) for 24 h; mannitol was used as an osmotic control. Cells were then harvested with ice-cold RIPA buffer containing 0.2% (vol/vol) PUGNac and 3% (vol/vol) each of protease inhibitor, phosphatase inhibitor 2, and phosphatase inhibitor 3 (Sigma). Lysates were sonicated for five pulses of 2 s each and centrifuged; the supernatants were stored at −80°C until use for Western blot analysis.
Western blot analysis.
Frozen heart tissue was ground into a fine powder under liquid nitrogen and fractionated into nuclear and cytosolic components using a commercially available kit (Thermo Fisher Scientific, Rockford, IL) in the presence of O-(2-acetamido-2-deoxy-d-glucopyranosylidene)amino-N-phenylcarbamate (PUGNAc; Sigma-Aldrich, St. Louis, MO) to inhibit OGA, and a protease inhibitor cocktail (Sigma-Aldrich). Protein concentrations were determined using the Bio-Rad protein assay kit (Bio-Rad, Hercules, CA). Lysates were prepared in Laemmli buffer (Bio-Rad), and 25 μg protein/sample were separated using SDS-PAGE and transferred to either nitrocellulose (for O-GlcNAc immunoblots only) or PVDF membranes at a constant voltage of 100 V for 75 min for PVDF or constant amperage of 0.2A for 85 min. Equal protein loading was initially confirmed by Ponceau-S staining, and immunoblots were then probed for O-GlcNAc (CTD110.6, 1:5,000; Epitope Recognition and Immunodetection Facility, University of Alabama at Birmingham, Birmingham, AL; or RL-2, 1:2,000; ab2739; Abcam, Cambridge, MA), OGT (1:1,500; O6139; Sigma-Aldrich), Anti-NCOAT/OGA (1:1,000; sc66612; Santa Cruz Biotechnology, Santa Cruz, CA), skeletal α-actin (1:2,000; ab28052; Abcam), ANP (1:500; ab91250; Abcam), HDACs 1–5 (1:1,000; HDAC1: 5356; HDAC2: 5113; HDAC3: 3949; HDAC4: 5392; HDAC5: 2082; Cell Signaling, Danvers, MA), mSin3A (1:2,000; ab3479; Abcam), or REST (1:1,000; 07–579; Millipore, Billerica, MA). Blots were then stripped using 1 M NaOH and reprobed with GAPDH (1:1,500; ab9484; Abcam) and TATA binding protein antibodies (1:1,000; ab51841; Abcam) as purity and loading controls for the cytoplasmic and nuclear fractions, respectively, or calsequestrin (1:4,000; ab3516; Abcam) as the protein loading control for whole cell lysates. For loading controls on anti-O-GlcNAc blots, nitrocellulose membranes were stripped with 100 mM glycine/HCl (pH 2.5) and reprobed with GAPDH or TATA binding protein. After incubation with appropriate secondary antibodies, blots were washed with SuperSignal West Pico or Femto chemiluminescent substrate (Thermo Fisher Scientific) and either exposed to film or visualized using the Chemidoc XRS imager (Bio-Rad). Densitometric analysis was performed using ImageJ software (National Institutes of Health, Bethesda, MD).
Coimmunoprecipitation.
Left ventricular (LV) tissue was homogenized in 10× wt/vol T-PER buffer (Thermo Fisher Scientific) containing 1% protease inhibitor cocktail (Sigma-Aldrich), 1% each phosphatase inhibitor cocktail 2 and 3 (Sigma-Aldrich), and 0.1% PUGNac (20 mM stock; Sigma-Aldrich) to prevent the removal of O-GlcNAc, as previously described (12) and incubated 45 min on ice. Tissue debris was pelleted, and the supernatants were stored at −80°C. Lysates were precleared over protein A/G resin beads (Thermo Fisher Scientific) for 4 h at 4°C. Precleared supernatants were then incubated with antibody-coated beads [anti-OGT (ab0271) or anti-O-GlcNAc (RL-2, ab2739), Abcam] overnight with slow rotation at 4°C. Immunoprecipitates (IP) were eluted by boiling in Laemmli buffer, and subjected to Western blot analysis, as described above. LV tissue lysate diluted to the IP concentration was used for input; precleared lysates immunoprecipitated without antibody were used as IgG controls. Analysis for co-IP was performed by dividing the quantity of coimmunoprecipitated protein by the quantity of IP target protein.
Immunohistochemistry.
To determine myocyte cross-sectional area, hearts from a small cohort of mice (n = 3 per diet) were harvested. Horizontal short-axis sections through the middle LV were fixed in 4% paraformaldehyde, transferred to 70% ethanol until being paraffin embedded, sectioned at 5 μm, and mounted on slides. Slides were then deparaffinized in xylene, rehydrated in ethanol, and blocked with 5% goat serum in 1% BSA for 1 h at room temperature. Sections were incubated with a primary antibody against laminin (ab11575, Abcam; 1:100); diluted in 5% goat serum in 1% BSA overnight at 4°C; secondary antibody, which was conjugated to Alexa Fluor 594 (red) (A11012; Invitrogen, Carlsbad, CA), was used to visualize the cellular membrane with 4,6-diamidino-2-phenylindole (DAPI; blue) to identify nuclei. Images of tissue in cross-sectional orientation from the anterior, posterior, and free walls of the LV endocardium were acquired (63× objective), total field size was measured, and myocytes were counted within the field to determine average area. Eight fields were analyzed per heart with an average of 17 cardiomyocytes per field. Image acquisition and analysis were performed on a Zeiss Axioplan 2 epifluorescence microscope with an AxioCam MRm cooled charge-coupled device camera and AxioVision software (Carl Zeiss Microimaging, Thornwood, NY).
Real-time quantitative PCR.
Total RNA was extracted from snap-frozen whole hearts with the RNeasy mini kit (Qiagen, Valencia, CA) and converted to cDNA using the Verso cDNA kit (Thermo Fisher Scientific), according to the respective manufacturer's instructions. Real-time quantitative PCR was performed on a MyiQ real-time PCR detection system (Bio-Rad) with RT2 FAST SYBR Green quantitative PCR kit (Qiagen) combined with 2 μM of each primer (Table 1) and 50 ng cDNA to a final volume of 25 μl. The quantitative PCR cycles were as follows: 95°C for 2 min, 35 cycles of 95°C for 30 s followed by 60°C for 30 s, and 72°C for 5 min. Fluorescent signals were monitored sequentially at the end of each elongation step. Specificity of RT-PCR products was confirmed by melting curve analysis and omission of template cDNA. All samples were run in triplicate and normalized to GAPDH mRNA using the ΔΔCt (cycle threshold) method, as described by Pfaffl, with primer efficiency set at 2 (19).
Table 1.
Primer sequences for qRT-PCR analysis
| Gene | Sequence |
|---|---|
| OGT | |
| Forward | 5′-TGCTCTCAAAGAGAAGGGCAGTGT-3′ |
| Reverse | 5′-TGTTCCCGTTTGATGTTGGCAAGG-3′ |
| REST | |
| Forward | 5′-AGGGTTGGCTGGTAAAGTGACTGA-3′ |
| Reverse | 5′-CAAGTGGCGATTGAGGTGTTTGCT-3′ |
| ANP | |
| Forward | 5′-TTCCGGTACCGAAGATAACAGCCA-3′ |
| Reverse | 5′-TGACACACCACAAGGGCTTAGGAT-3′ |
| BNP | |
| Forward | 5′-AGACAAGGGAGAACACGGCATCAT-3′ |
| Reverse | 5′-ACAGAATCATCTGGGACAGCACCT-3′ |
| α-MHC | |
| Forward | 5′-GGCAAAGTCACTGCGGAAACTGAA-3′ |
| Reverse | 5′-TCTTGTCGAACTTGGGTGGGTTCT-3′ |
| β-MHC | |
| Forward | 5′-TGGCTGGTGAGGTCATTGACAGAA-3′ |
| Reverse | 5′-TGGCTGGTGAGGTCATTGACAGAA-3′ |
| α-actin (Skeletal) | |
| Forward | 5′-TTGTGCACCGCAAATGCTTCTAGG-3′ |
| Reverse | 5′-GCAACCACAGCACGATTGTCGATT-3′ |
| α-actin (Cardiac) | |
| Forward | 5′-TGTAGGTGATGAAGCCCAGAGCAA-3′ |
| Reverse | 5′-TGGTGCCAGATCTTCTCCATGTCA-3′ |
| HDAC 1 | |
| Forward | 5′-TTCCTGCGTTCTATTCGCCCAGAT-3′ |
| Reverse | 5′-AACAAGCCATCAAACACCGGACAG-3′ |
| HDAC 2 | |
| Forward | 5′-TACAACAGATCGCGTGATGACCGT-3′ |
| Reverse | 5′-TCCCTTTCCAGCACCAATATCCCT-3′ |
| HDAC 4 | |
| Forward | 5′-AAGGAGAAGGGCAAAGAGAGTGCT-3′ |
| Reverse | 5′-GCTGGAAATGCAGTGGTTCAGGTT-3′ |
| GAPDH | |
| Forward | 5′-TGTGATGGGTGTGAACCACGAGAA-3′ |
| Reverse | 5′-CATGAGCCCTTCCACAATGCCAAA-3′ |
Statistical analysis.
Data were analyzed using SigmaPlot (Systat Software, San Jose, CA) with two-tailed unpaired Student's t-tests, two-way ANOVA, or Kruskal-Wallis ANOVA on ranks, where appropriate. Interactions for ANOVA and Kruskal-Wallis ANOVA were investigated using Bonferroni and Dunn's post hoc analyses, respectively. Values are presented as means ± SE, and significance between groups was established at P < 0.05.
RESULTS
Two weeks of Western diet induces severe fasting hyperglycemia and weight gain.
Fasting blood glucose and body weight were significantly elevated in Western diet compared with control (P < 0.05). Neither skeleton size, as assessed by tibia length, nor heart wet weight, were different between groups (Table 2). Cardiac hypertrophy was not different between diets, either at the whole heart level, as denoted by heart wet-weight relative to tibia length (HW/TL), or at the cellular level with cardiomyocyte cross-sectional area (CSA) (Fig. 1A).
Table 2.
Animal Characteristics
| Control Diet | Western Diet | |
|---|---|---|
| Week 0 body weight, g | 18.00 ± 0.30 | 17.53 ± 0.39 |
| Week 2 body weight, g | 19.50 ± 0.26 | 21.89 ± 0.43* |
| Weight gain, g | 1.49 ± 0.29 | 4.36 ± 0.25* |
| Fasting blood glucose, mmol/l | 5.86 ± 0.36 | 10.49 ± 0.57* |
| Heart wet weight, g | 0.010 ± 0.003 | 0.011 ± 0.003 |
| Tibia Length, cm | 2.25 ± 0.01 | 2.24 ± 0.01 |
Values are expressed as means ± SE; n = 12 per group.
P < 0.05 vs. control diet.
Fig. 1.
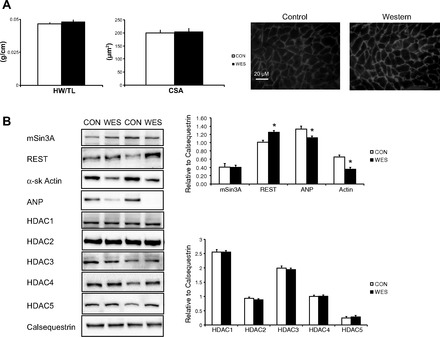
A: effects of the Western (WES) diet on markers of cardiac hypertrophy compared with control (CON) hearts. Heart weight to tibia length (HW/TL), cardiomyocyte cross-sectional area (CSA), and representative images of cardiomyocyte size. Scale bar: 20 μm. Background and contrast were adjusted using ImageJ software (National Institutes of Health, Bethesda, MD). B: left ventricular homogenates from WES-fed mice exhibited higher protein levels of repressor element 1-silencing transcription factor (REST), α-sk Actin, and ANP vs. CON. Calsequestrin was the internal loading control. n = 6 per group except for CSA; n = 3 per group, *P < 0.05 vs. CON.
Increased REST suppressed the fetal gene program with Western diet consumption.
Ventricular tissue from Western diet-fed mice showed significantly higher levels of REST and lower skeletal α-actin and ANP compared with control (P < 0.05; Fig. 1B). There were no changes in protein levels of any HDAC proteins. Despite the increase in blood glucose with the Western diet, total protein O-GlcNAcylation was not different between diets (Fig. 2). However, since densitometry of the entire sample lane is dominated by the intensely immunoreactive bands at ∼39 and ∼85 kDa, the analysis was also performed over the high-, middle-, and low-molecular-weight ranges; this revealed that O-GlcNAcylation of high-molecular weight proteins was higher in nuclear fractions (P < 0.05). As expected, we found compartment-specific localization of both OGT and OGA, although there were no diet-specific differences between these proteins in the heart (Fig. 2).
Fig. 2.
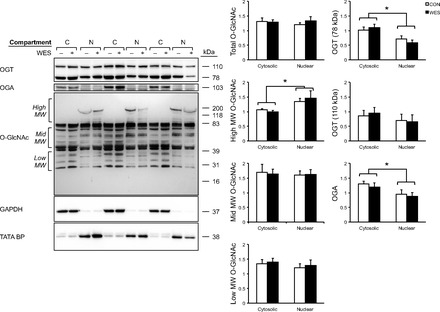
Nuclear and cytosolic distribution of O-GlcNAc transferase (OGT), O-GlcNAcase (OGA), and O-GlcNAc. Densitometric analysis of cytosolic and nuclear proteins are presented as relative to GAPDH and TATA-BP, respectively. n = 6 per group. *P < 0.05.
mRNA expression levels of α-cardiac-actin and HDAC2 are decreased with the Western diet.
Two weeks of Western diet consumption decreased mRNA expression of cardiac α-actin and HDAC2 in mouse hearts. However, in contrast to the protein levels shown in Fig. 1, diet did not alter transcript levels of any other genes of interest (Fig. 3).
Fig. 3.
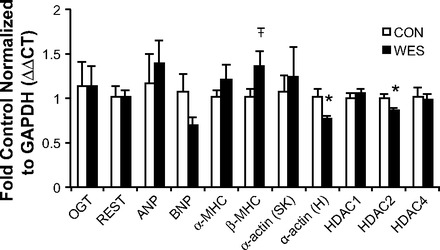
qRT-PCR showing that the Western diet induced increased mRNA levels of cardiac α-actin and histone deacetylase 2 (HDAC2) but no other genes within the O-GlcNAc pathway or REST complex. n = 5 per group, *P < 0.05;  = 0.08.
= 0.08.
REST, HDAC4, and HDAC5 are O-GlcNAc targets in mouse hearts.
To investigate the interaction between the O-GlcNAc pathway and components of the REST complex, we coimmunoprecipitated O-GlcNAc, OGT, REST, and mSin3A. Immunoblotting showed that total amounts of capture of these proteins were not different between groups. OGT, HDACs 1,2,4, and 5, mSin3A, and REST were O-GlcNAc modified, but this was not different between diets (Fig. 4A). We were unable to identify O-GlcNAcylation of HDAC3 through immunoprecipitation due to its molecular weight, as the HDAC3 signal overlaps with the signal of the IgG band. We found that OGT exists in complex with mSin3A, REST, and HDACs 1, 2, 4, and 5 in the heart, but that this is not affected by consumption of a Western diet (Fig. 4B). mSin3A is weakly O-GlcNAcylated and is also weakly associated with OGT and REST in the mouse heart (Fig. 5A); this was not different between diet groups. We did not find that REST had appreciable interactions with any proteins aside from OGT (Fig. 5B).
Fig. 4.
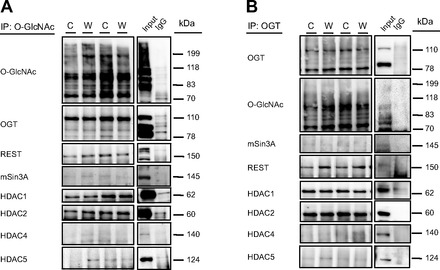
Coimmunoprecipitation (IP) of O-GlcNAc (A) and OGT (B) followed by immunoblots targeting protein involved in the O-GlcNAc pathway and REST complex. The immunoblots presented are representative images from a total of n = 6 per group. C, Control; W, Western.
Fig. 5.
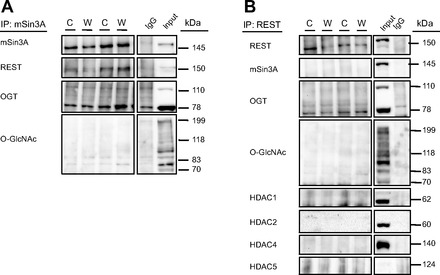
Coimmunoprecipitation of mSin3A (A) and REST (B), immunoblotted for mSin3A, REST, OGT, O-GlcNAc, HDAC1, HDAC2, HDAC4, and HDAC5. The position from which additional lanes were removed is indicated by the lined box and white spaces; immunoblots presented are representative images from a total of n = 6 per group.
O-GlcNAc is not associated with fetal gene program signaling in cardiomyoblasts.
In an attempt to determine whether the fetal gene program changes were a result of hyperglycemia and/or O-GlcNAc, we performed a series of cell culture experiments in which substrates for O-GlcNAc were increased either alone or in combination with a potent OGA inhibitor, Thiamet-G (Fig. 6). Protein O-GlcNAcylation was increased with Thiamet-G treatments, although not with any other individual substrates. Skeletal α-actin protein levels were increased in response to 25 mM glucose. Global fetal gene program signaling was not altered, regardless of the treatment.
Fig. 6.
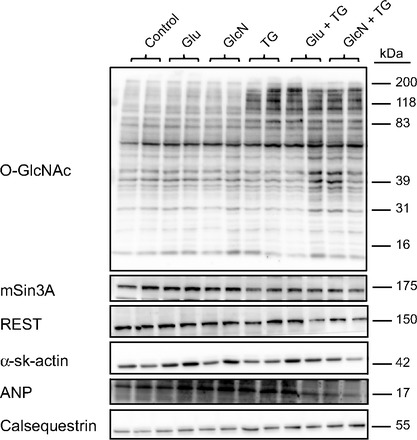
H9c2 cardiomyoblasts were subjected to treatments that increase protein O-GlcNAcylation. Glu, glucose; GlcN, glucosamine; TG, Thiamet-G. n = 3 per group.
DISCUSSION
Chronic Western diet consumption causes increased levels of cardiac protein O-GlcNAcylation (17); however, little is known about how pathological signaling in cardiac hypertrophy is regulated with this diet. We recently demonstrated that pharmacological elevation of O-GlcNAc in primary cardiomyocytes mimics the blunted hypertrophic response observed in diabetic cardiomyocytes (15). In light of these findings, we sought to identify a relationship between O-GlcNAc and the chromatin-modifying proteins that regulate pathological hypertrophy. We specifically chose to study the REST/mSin3A/HDAC complex due to its regulation of the fetal gene program, as well as our recent finding that OGT and REST interact in the heart (18). To our knowledge, early cardiac hypertrophic signaling in response to Western diet consumption has previously not been examined; therefore, we fed mice a Western diet for 2 wk to observe early changes in the regulation of fetal gene expression.
In this study, 2 wk of Western diet consumption significantly increased body weight and fasting blood glucose prior to any evidence of cardiac hypertrophy. As tibia length was the same between diets, any increases in body weight were due to diet-induced obesity and not a differential effect of skeletal (linear) growth caused by overnutrition with the Western diet.
We then investigated fetal gene expression as an early indicator of diet-induced hypertrophy. REST suppresses fetal cardiac gene components such as skeletal α-actin and ANP by recruiting mSin3A and HDACs 1 and 2 to the NH2 and/or COOH terminal regions of REST under basal conditions (2). In early pathology, REST continues to inhibit the heart's response to pathological stimuli by maintaining suppression of these genes (2, 11). However, once hypertrophic stimuli are present in the heart for an extended period, REST no longer represses transcription of proteins that are part of the fetal cardiac gene program (11). This occurs via recruitment of various corepressors and transcription factors, such as HDACs, to both the NH2 and COOH terminal regions of REST. Although the process remains poorly understood, our observations of elevated REST in combination with decreased ANP and α-actin indicate preemptive REST-mediated suppression of the fetal gene program before overt hypertrophy was evident. Suppression of the fetal gene program is consistent with previous reports, and these are the first data to demonstrate an effect of diet on cardiac REST. Interestingly, we also found that HDAC2 and cardiac α-actin mRNA levels were reduced in Western diet-fed animals compared with control mice. HDAC2 is specifically associated with cardiac hypertrophy (22); therefore, this may indicate early suppression of prohypertrophic transcription factors after Western diet consumption. The only effect observed in vitro was a decrease in skeletal α-actin when O-GlcNAc was increased through glucose administration (Fig. 6). However, when additional substrates were added along with Thiamet-G, no fetal gene program alterations were observed. Although this rules out hyperglycemia as an independent effector of the altered signaling observed following Western diet consumption, it does not preclude the involvement of O-GlcNAc because the level of O-GlcNAcylation is not dependent on the availability of glucose substrate (21). We also cannot discount the potential contributions of obesity and/or systemic metabolic disorder on suppression of the fetal gene program with a Western diet, particularly given that O-GlcNAc levels are often increased in various tissues with these pathologies. Given that both obesity and metabolic disorder exert numerous direct and indirect effects on the heart and that O-GlcNAcylation can be increased by stress independent of substrate levels, investigation of the effects of these conditions is beyond the scope of the current study. However, we are currently exploring these pathologies in other experimental settings to elucidate potential mechanisms.
We also examined O-GlcNAcylation of key transcription factors that regulate fetal genes. In this study, we have confirmed our previous findings that OGT and HDACs 1,2,4, and 5 are O-GlcNAc modified in the heart (18), and we have additionally shown that mSin3a and REST are O-GlcNAc modified in the mouse heart. Interestingly, we were only able to visualize slight levels of mSin3A with both our O-GlcNAc and OGT co-IPs. This is contrary to the original findings both by Yang et al. (24), which clearly show that mSin3A is O-GlcNAcylated in HepG2 liver carcinoma cells. Therefore, O-GlcNAcylation of mSin3A may be a tissue-specific effect; alternatively, it is possible that the RL-2 antibody does not capture O-GlcNAcylated mSin3A due to differences in epitope targeting. If this is the case, then using the alternative anti-O-GlcNAc antibody CTD 110.6 may demonstrate O-GlcNAcylation of mSin3A. However, we chose to perform immunoprecipitation with the RL-2 antibody due to its much higher affinity for protein A/G-coated resin.
Although it is well established that OGT is O-GlcNAc modified, we showed here that the 110-kDa catalytic isoform of OGT is more O-GlcNAcylated than the 78-kDa regulatory isoform in the mouse heart. Additionally, although the OGT antibody selects for the 110-kDa isoform, IP of both REST and mSin3A coimmunoprecipitated with primarily the 78-kDa regulatory isoform. Because of the unclear function of this short 78-kDa isoform of OGT, this is an important finding that we are currently exploring further. OGT is observed here to form a complex with REST that appears to be independent of mSin3A, as mSin3A did not immunoprecipitate with either OGT (Fig. 4B) or REST (Fig. 5B). However, these complex formations were not influenced by Western diet consumption.
Conclusion.
In this study, we have demonstrated that the Western diet induces obesity and fasting hyperglycemia in mice in as little as 2 wk. In Western diet-fed mice, protein levels of REST were increased, and ANP and skeletal α-actin were decreased. This suggests that a short period of feeding induces suppression of these fetal genes in the heart and that this is an early event in the hypertrophic signaling cascade that occurs prior to the appearance of hypertrophy at the organ or cellular levels. We have also demonstrated that high-molecular-weight cardiac proteins are more O-GlcNAc modified in the nuclear fraction; this may reflect preferential O-GlcNAcylation of high-molecular-weight transcription factors such as HDAC4, HDAC5, REST, and mSin3A. Collectively, these data show that OGT and O-GlcNAc interact with transcription factors that regulate fetal genes in the heart and suggest that 2 wk of Western-diet feeding suppresses fetal genes that are associated with cardiac hypertrophy; this is consistent with the blunted fetal gene response in hypertrophy of diabetic hearts. Therefore, we propose that Western diet-induced suppression of the fetal gene program is consistent with the deleterious cardiac remodeling and pathology seen in Type 2 diabetic hearts and that this may be mediated by either O-GlcNAcylation of components of the REST complex or the interaction of OGT with these proteins.
Perspectives and Significance
Because of the lack of overall changes to the fetal gene program in vitro, we propose that there is a systemic effect of Western diet consumption that induces deleterious cardiac signaling after as little as two weeks of consumption. The fact that REST and its corepressors interact directly with DNA to alter signaling in the heart after such a short intervention is an important finding and indicates a potential for irreversible damage. We are exploring the role of OGT in this process and propose that this complex could be a potential therapeutic target for treatment of cardiac disease.
GRANTS
This work was supported by the National Institutes of Health (HL-104549 to S. A. Marsh) and Washington State University College of Pharmacy. H. M. Medford is supported by a William Townsend Porter Predoctoral Fellowship from the American Physiological Society.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
AUTHOR CONTRIBUTIONS
Author contributions: H.M.M. and S.A.M. conception and design of research; H.M.M., E.J.C., and L.E.M. performed experiments; H.M.M., E.J.C., and L.E.M. analyzed data; H.M.M., E.J.C., L.E.M., and S.A.M. interpreted results of experiments; H.M.M., E.J.C., and L.E.M. prepared figures; H.M.M. drafted manuscript; H.M.M., E.J.C., L.E.M., and S.A.M. edited and revised manuscript; H.M.M., E.J.C., L.E.M., and S.A.M. approved final version of manuscript.
ACKNOWLEDGMENTS
We thank Mary Ann Accavitti from the University of Alabama at Birmingham for her generous gift of the anti-O-GlcNAc antibody CTD110.6 and Brad P. Dieter for his technical assistance.
REFERENCES
- 1.Barry SP, Davidson SM, Townsend PA. Molecular regulation of cardiac hypertrophy. Int J Biochem Cell Biol 40: 2023–2039, 2008 [DOI] [PubMed] [Google Scholar]
- 2.Bingham AJ, Ooi L, Kozera L, White E, Wood IC. The repressor element 1-silencing transcription factor regulates heart-specific gene expression using multiple chromatin-modifying complexes. Mol Cell Biol 27: 4082–4092, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Brownlee M. Biochemistry and molecular cell biology of diabetic complications. Nature 414: 813–820, 2001 [DOI] [PubMed] [Google Scholar]
- 4.Cavasin MA, Demos-Davies K, Horn TR, Walker LA, Lemon DD, Birdsey N, Weiser-Evans MC, Harral J, Irwin DC, Anwar A, Yeager ME, Li M, Watson PA, Nemenoff RA, Buttrick PM, Stenmark KR, McKinsey TA. Selective class I histone deacetylase inhibition suppresses hypoxia-induced cardiopulmonary remodeling through an antiproliferative mechanism. Circ Res 110: 739–748, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chavali V, Tyagi SC, Mishra PK. Predictors and prevention of diabetic cardiomyopathy. Diabetes Metab Syndr Obes 6: 151–160, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dannenberg JH, David G, Zhong S, van der Torre J, Wong WH, Depinho RA. mSin3A corepressor regulates diverse transcriptional networks governing normal and neoplastic growth and survival. Genes Dev 19: 1581–1595, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Fulop N, Mason MM, Dutta K, Wang P, Davidoff AJ, Marchase RB, Chatham JC. Impact of Type 2 diabetes and aging on cardiomyocyte function and O-linked N-acetylglucosamine levels in the heart. Am J Physiol Cell Physiol 292: C1370–C1378, 2007 [DOI] [PubMed] [Google Scholar]
- 8.Grozinger CM, Hassig CA, Schreiber SL. Three proteins define a class of human histone deacetylases related to yeast Hda1p. Proc Natl Acad Sci USA 96: 4868–4873, 1999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kuwahara K, Nishikimi T, Nakao K. Transcriptional regulation of the fetal cardiac gene program. J Pharm Sci 119: 198–203, 2012 [DOI] [PubMed] [Google Scholar]
- 10.Kuwahara K, Saito Y, Ogawa E, Takahashi N, Nakagawa Y, Naruse Y, Harada M, Hamanaka I, Izumi T, Miyamoto Y, Kishimoto I, Kawakami R, Nakanishi M, Mori N, Nakao K. The neuron-restrictive silencer element-neuron-restrictive silencer factor system regulates basal and endothelin 1-inducible atrial natriuretic peptide gene expression in ventricular myocytes. Mol Cell Biol 21: 2085–2097, 2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kuwahara K, Saito Y, Takano M, Arai Y, Yasuno S, Nakagawa Y, Takahashi N, Adachi Y, Takemura G, Horie M, Miyamoto Y, Morisaki T, Kuratomi S, Noma A, Fujiwara H, Yoshimasa Y, Kinoshita H, Kawakami R, Kishimoto I, Nakanishi M, Usami S, Harada M, Nakao K. NRSF regulates the fetal cardiac gene program and maintains normal cardiac structure and function. EMBO J 22: 6310–6321, 2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Laczy B, Marsh SA, Brocks CA, Wittmann I, Chatham JC. Inhibition of O-GlcNAcase in perfused rat hearts by NAG-thiazolines at the time of reperfusion is cardioprotective in an O-GlcNAc-dependent manner. Am J Physiol Heart Circ Physiol 299: H1715–H1727, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lazarus BD, Love DC, Hanover JA. Recombinant O-GlcNAc transferase isoforms: identification of O-GlcNAcase, yes tyrosine kinase, and tau as isoform-specific substrates. Glycobiology 16: 415–421, 2006 [DOI] [PubMed] [Google Scholar]
- 14.Malik VS, Fung TT, van Dam RM, Rimm EB, Rosner B, Hu FB. Dietary patterns during adolescence and risk of type 2 diabetes in middle-aged women. Diabetes Care 35: 12–18, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Marsh SA, Dell'Italia LJ, Chatham JC. Activation of the hexosamine biosynthesis pathway and protein O-GlcNAcylation modulate hypertrophic and cell signaling pathways in cardiomyocytes from diabetic mice. Amino Acids 40: 819–828, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Marsh SA, Powell PC, Dell'italia LJ, Chatham JC. Cardiac O-GlcNAcylation blunts autophagic signaling in the diabetic heart. Life Sci 92: 648–656, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Medford HM, Chatham JC, Marsh SA. Chronic ingestion of a Western diet increases O-linked-β-N-acetylglucosamine (O-GlcNAc) protein modification in the rat heart. Life Sci 90: 883–888, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Medford HM, Porter K, Marsh SA. Immediate effects of a single exercise bout on protein O-GlcNAcylation and chromatin regulation of cardiac hypertrophy. Am J Physiol Heart Circ Physiol 305: H114–H123, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pfaffl MW. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res 29: e45, 2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sakabe K, Wang Z, Hart GW. Beta-N-acetylglucosamine (O-GlcNAc) is part of the histone code. Proc Natl Acad Sci USA 107: 19915–19920, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Taylor RP, Parker GJ, Hazel MW, Soesanto Y, Fuller W, Yazzie MJ, McClain DA. Glucose deprivation stimulates O-GlcNAc modification of proteins through up-regulation of O-linked N-acetylglucosaminyltransferase. J Biol Chem 283: 6050–6057, 2008 [DOI] [PubMed] [Google Scholar]
- 22.Trivedi CM, Luo Y, Yin Z, Zhang M, Zhu W, Wang T, Floss T, Goettlicher M, Noppinger PR, Wurst W, Ferrari VA, Abrams CS, Gruber PJ, Epstein JA. Hdac2 regulates the cardiac hypertrophic response by modulating Gsk3 β activity. Nat Med 13: 324–331, 2007 [DOI] [PubMed] [Google Scholar]
- 23.Wilson CR, Tran MK, Salazar KL, Young ME, Taegtmeyer H. Western diet, but not high fat diet, causes derangements of fatty acid metabolism and contractile dysfunction in the heart of Wistar rats. Biochem J 406: 457–467, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yang X, Zhang F, Kudlow JE. Recruitment of O-GlcNAc transferase to promoters by corepressor mSin3A: coupling protein O-GlcNAcylation to transcriptional repression. Cell 110: 69–80, 2002 [DOI] [PubMed] [Google Scholar]
- 25.Zhang CL, McKinsey TA, Chang S, Antos CL, Hill JA, Olson EN. Class II histone deacetylases act as signal-responsive repressors of cardiac hypertrophy. Cell 110: 479–488, 2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zheng D, Zhao K, Mehler MF. Profiling RE1/REST-mediated histone modifications in the human genome. Genome Biol 10: R9, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]