

Abstract
Purpose
We provide a comprehensive review of adaptive phase I clinical trials in oncology that used a statistical model to guide dose escalation to identify the maximum-tolerated dose (MTD). We describe the clinical setting, practical implications, and safety of such applications, with the aim of understanding how these designs work in practice.
Methods
We identified 53 phase I trials published between January 2003 and September 2013 that used the continual reassessment method (CRM), CRM using escalation with overdose control, or time-to-event CRM for late-onset toxicities. Study characteristics, design parameters, dose-limiting toxicity (DLT) definition, DLT rate, patient-dose allocation, overdose, underdose, sample size, and trial duration were abstracted from each study. In addition, we examined all studies in terms of safety, and we outlined the reasons why escalations occur and under what circumstances.
Results
On average, trials accrued 25 to 35 patients over a 2-year period and tested five dose levels. The average DLT rate was 18%, which is lower than in previous reports, whereas all levels above the MTD had an average DLT rate of 36%. On average, 39% of patients were treated at the MTD, and 74% were treated at either the MTD or an adjacent level (one level above or below).
Conclusion
This review of completed phase I studies confirms the safety and generalizability of model-guided, adaptive dose-escalation designs, and it provides an approach for using, interpreting, and understanding such designs to guide dose escalation in phase I trials.
INTRODUCTION
There has been an ongoing debate1,2 regarding the use of new dose-escalation methods in oncology.3 The increased accuracy of novel designs in estimating the maximum-tolerated dose (MTD), along with the fact that they can reach the MTD faster, makes them appealing.4–6 The greater flexibility of adaptive approaches allows investigators to address more involved clinical issues, such as patient heterogeneity, treatment scheduling, and combination therapies, in a single study. However, model-based adaptive designs are viewed as complex and difficult to implement in real time and are not easily understood by clinicians. It is not easy to anticipate ongoing dose recommendations, and the approach requires input from a multidisciplinary team. It has been argued that in certain cases, the recommendation of such designs might violate our clinical intuition. In this article, we address the question of how adaptive designs work in practice by providing a review of phase I trials that have used model-based algorithms to guide dose escalation and find the MTD.
The performance of model-based designs has been evaluated in the literature through simulated studies.3 For example, given hypothetical data, we can evaluate when the method escalates, de-escalates, or stays at the same level; where patients are likely to be treated; and how often the true MTD or a dose close to the MTD is selected.7 Several authors have shown through simulations and theoretical derivations that model-based designs are more accurate and efficient than the 3 + 3 design, because they reach the MTD faster and treat most patients at or near the MTD.1,5,7–9 Although simulations enable us to assess to what extent such behavior is desirable, simulations are not convincing to clinicians. We recognize it is challenging to conceptualize the implications of a large number of similar hypothetical studies. For this reason, we illustrate the behavior and safety of such designs in the clinical setting through a review of completed case studies. A review of the safety of these designs based on real trials is lacking in the literature, and we aim to provide this in our review.
We summarize a number of key questions (Table 1), such as whether adaptive dose-finding designs are safe, and whether they treat patients at a therapeutic dose interval; whether they tend to overdose or underdose; whether they take too long to complete because of nonstaggered accrual; and how many patients they require for completion. Our review assesses the flexibility of these designs to deal with different treatment schedules, heterogeneity of patient populations in terms of disease type or prior treatment, drug combinations, and accommodation of different disease settings. Finally, we discuss specific examples of clinical trials and summarize the current state of phase I designs.
Table 1.
Key Questions Review Aims to Address Regarding Model-Based Designs
| Question | Outcome |
|---|---|
| Are model-based dose-escalation designs safe? | DLT rate and toxicity rate above MTD |
| Are model-based designs treating patients at subtherapeutic levels? | Underdosing and overdosing; percentage of patients treated at MTD or within MTD |
| Do model-based phase I trials result in longer trial duration? | Trial duration |
| Is sample size too large? | Sample size |
| Are these designs flexible enough to deal with different schedules, patient populations, or drug combinations? | Specific parameters such as acceptable toxicity rate, No. of groups, model assumptions, and DLT window of observation |
| Do designs recommend counterintuitive escalations? | Specific examples |
Abbreviations: DLT, dose-limiting toxicity; MTD, maximum-tolerated dose.
METHODS
Search Strategy
In September 2013, we conducted a search of trials and abstracts in the PubMed database that were published between January 2003 and September 2013. The search was limited to phase I or dose-finding trials in humans that used the continual reassessment method (CRM), CRM designs using escalation with overdose control (EWOC-CRM),10 and CRM designs that allow for time to event or late-onset toxicities (TITE-CRM).11 We searched PubMed for articles with the search terms listed in the Appendix (online only).
Outcome Measures
The following parameters describing the study design and disease setting, safety, and optimal treatment allocation were extracted from each article:
Disease setting, treatment type (agent type, single- or multiple-agent regimen), dose-limiting toxicity (DLT) definition, and timeframe of DLT observation.
Patient population, MTD for ≥ one group of patients, and evaluation of safety for single or multiple schedules.
Overall DLT rate, primary outcome (total number of DLTs divided by number of evaluable patients), and toxicity above the MTD (proportion of DLTs observed among patients treated above MTD; key question 1).
Overdose, underdose, and treatment at the MTD (proportion of patients treated above, below, and at MTD, respectively); the proportion of patients treated within the MTD plus or minus one level is reported (key question 2).
Trial duration and sample size (key question 3).
Design parameters and model specifications: acceptable toxicity rate, cohort size, planned sample size, and number of dose levels (key question 4).
RESULTS
We identified 64 potentially eligible articles (Fig 1) published between January 2003 and September 2013 through our search, as well as 39 articles through other sources (Pediatric Brain Tumor Consortium, articles that cited EWOC-CRM or CRM or similar). Forty-nine articles were excluded primarily because they described methodology without a completed phase I trial. After these exclusions, 53 phase I trials in patients with cancer were included in the final synthesis.
Fig 1.
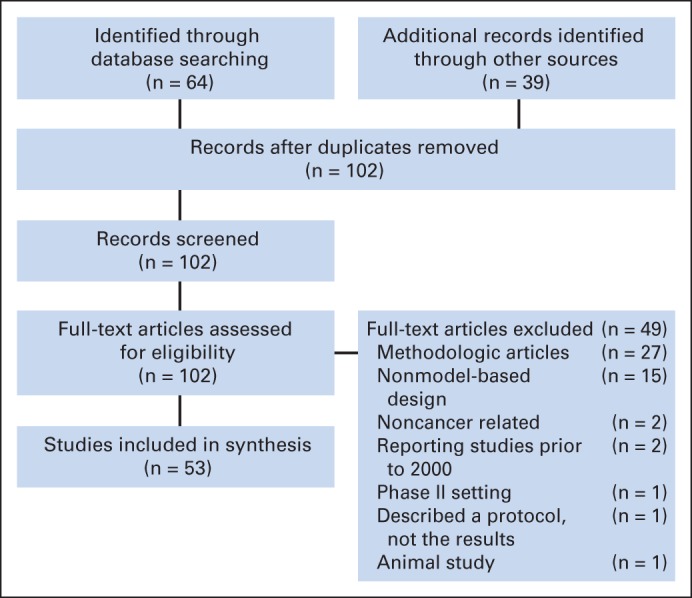
Trials included in this review; trial search and selection PRISMA (preferred reporting items for systematic reviews and meta-analyses) diagram.
Design Setup and Trial Flexibility
The Data Supplement lists protocol-defined clinical parameters for each trial, such as the aim of the trial, disease setting, agent or treatment type, DLT definition and DLT observation timeframe (cycles), design and model parameters, and other relevant details. Through these details, we aim to describe each specific trial and provide a qualitative review and understanding of how the trial design was set up. On average, these trials enrolled 35 patients, with 25 patients evaluable for DLT; they were completed within approximately 25 months, tested five dose levels, and targeted an acceptable toxicity rate of 26% (range, 10% to 33%; Table 2). Fifty-four percent of trials (28 of 52) tested a single-agent regimen, and 46% (24 of 52) tested a combination regimen. The average DLT timeframe of observation was 38 days (median, 28 days), corresponding to approximately two 21-day cycles. Excluding radiation therapy trials, the average DLT window of observation was 28 days. There was one early-phase chemoprevention trial in patients with a history of breast carcinoma that aimed to find the MTD.42 The safety profile of such a trial and the patient population (without disease at study entry) could have skewed our results. To account for this, we calculated all summary measures with and without the trial by Crew et al,42 and the average DLT rate remained unchanged: 18.31% with and 18.33% without. In general, model-based designs can be used in settings where the acceptable DLT rate or threshold varies, because this is a design-tuning parameter, and it can be set specific to each protocol or type of drug. This is evident from the fact that the acceptable DLT rate established at the outset in different trials in this review varied from 10% to 33%.
Table 2.
Results for Each Study: DLT Rate and Treatment Allocation by Trial
| Study | No. of Groups | No. of Levels | No. of Evaluable Patients | Target | DLT Rate |
Toxicity Above MTD |
Patients Treated Below MTD |
Patients Treated at MTD |
Patients Treated Above MTD (%) | Patients Treated Within MTD (%) | ||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| No. | % | No.* | % | No. | % | No. | % | |||||||
| Thornton et al12 | 1 | 5 | 15 | 0.20 | NR | NR | NR | NR | NR | NR | 9 of 15 | 0.60 | NR | NR |
| Harvey et al13 | 1 | 7 | 30 | 0.33 | NR | NR | NR | NR | 8 of 30 | 0.27 | 3 of 30 | 0.10 | NA | 0.53 |
| Ben-Josef et al14 | 1 | 6 | 51 | 0.25 | 11 of 51 | 0.22 | 3 of 9 | 0.33 | 22 of 51 | 0.43 | 20 of 51 | 0.39 | 0.18 | 0.88 |
| Tsien et al15 | 1 | 5 | 38 | 0.25 | 7 of 38 | 0.18 | 5 of 16 | 0.31 | 13 of 38 | 0.34 | 9 of 38 | 0.24 | 0.42 | 0.74 |
| Schneider et al16 | 1 | 4 | 12 | 0.30 | 5 of 12 | 0.42 | NA | NA | NA | NA | NA | NA | NA | NA |
| Reardon et al17 | 1 | 2 | 13 | 0.20-0.35 | 4 of 13 | 0.31 | NA | NA | NA | NA | NA | NA | NA | NA |
| Sinha et al18 | 1 | 3 | 19 | 0.3 | 5 of 19 | 0.26 | 3 of 12 | 0.25 | 1 of 19 | 0.05 | 6 of 19 | 0.32 | 0.63 | 1.00 |
| Mehnert et al19 | 1 | 5 | 16 | 0.25 | 0 of 16 | 0.00 | 0 | 0.00 | 13 of 16 | 0.81 | 3 of 16 | 0.19 | 0.00 | 0.38 |
| Lonial et al20 | 2 | 3 | 19 | 0.33 | 1 of 19 | 0.05 | 0 | 0.00 | NA | NA | NA | NA | NA | NR |
| 3 | 20 | 0.33 | 1 of 20 | 0.05 | 0 | 0.00 | NA | NA | NA | NA | NA | NR | ||
| Guillot et al21 | 1 | 4 | 31 | 0.33 | 8 of 31 | 0.26 | 4 of 11 | 0.36 | 2 of 31 | 0.06 | 18 of 31 | 0.58 | 0.35 | 0.97 |
| Morita et al22 | 1 | 3 | 13 | 0.20 | 4 of 13 | 0.31 | 2 of 5 | 0.40 | 0 | 0.00 | 8 of 13 | 0.62 | 0.38 | 1.00 |
| Broniscer et al23 | 2 | 5 | 27 | 0.25 | 6 of 27 | 0.22 | 6 of 14 | 0.43 | 6 of 27 | 0.22 | 7 of 27 | 0.26 | 0.52 | 0.74 |
| 3 | 15 | 0.25 | 3 of 15 | 0.20 | 3 of 6 | 0.50 | 3 of 15 | 0.20 | 6 of 15 | 0.40 | 0.40 | 1.00 | ||
| Saji et al24 | 1 | 5 | 16 | 0.33 | 3 of 16 | 0.19 | 0 | 0.00 | 10 of 16 | 0.63 | 6 of 16 | 0.38 | 0.00 | 0.56 |
| Freedman et al25 | 1 | 1 | 8 | 0.2 | 3 of 8 | 0.38 | NA | NA | NA | NA | NA | NA | NA | NA |
| Mackler et al26 | 1 | 5 | 31 | 0.30 | 4 of 31 | 0.13 | 0 | 0.00 | 14 of 31 | 0.45 | 17 of 31 | 0.55 | 0.00 | 0.84 |
| Geoerger et al27 | 2 | 3 | 25 | 0.20 | 7 of 25 | 0.28 | 3 of 8 | 0.38 | 6 of 25 | 0.24 | 11 of 25 | 0.44 | 0.32 | 1.00 |
| 3 | 13 | 0.20 | 3 of 13 | 0.23 | 1 of 2 | 0.50 | 0 | 0.00 | 11 of 13 | 0.85 | 0.15 | 1.00 | ||
| Khuri et al28 | 1 | 5 | 21 | 0.20 | 7 of 21 | 0.33 | 4 of 9 | 0.44 | 3 of 21 | 0.14 | 9 of 21 | 0.43 | 0.43 | 0.81 |
| Eder et al29 | 1 | 6 | 33 | 0.30 | 2 of 33 | 0.06 | 2 of 9 | 0.22 | 20 of 33 | 0.61 | 4 of 33 | 0.12 | 0.27 | 0.58 |
| Muler et al30 | 1 | 4 | 18 | 0.20 | 4 of 18 | 0.22 | 4 of 8 | 0.50 | 5 of 18 | 0.28 | 5 of 18 | 0.28 | 0.44 | 1.00 |
| Geoerger et al31 | 2 | 4 | 24 | 0.20 | 4 of 24 | 0.17 | 2 of 4 | 0.50 | 6 of 24 | 0.25 | 14 of 24 | 0.58 | 0.17 | 0.88 |
| 3 | 20 | 0.20 | 2 of 20 | 0.10 | 0 | 0.00 | 12 of 20 | 0.60 | 8 of 20 | 0.40 | 0.00 | 0.70 | ||
| Rathkopf et al32 | 2 | 1 | 8 | 0.25 | 1 of 8 | 0.13 | NA | NA | NA | NA | NA | NA | NA | NA |
| 1 | 8 | 0.25 | 6 of 8 | 0.75 | NA | NA | NA | NA | NA | NA | NA | NA | ||
| Bailey et al33 | 2 | 5 | 50 | 0.20-0.35 | 9 of 50 | 0.18 | 4 of 5 | 0.80 | 7 of 34 | 0.21 | 16 of 34 | 0.48 | 0.15 | 0.79 |
| Borghaei et al34 | Many | Continuous | 52 | 0.10 | 6 of 39 | 0.15 | NA | NA | NA | NA | NA | NA | NA | NA |
| 1 of 13 | 0.08 | NA | NA | NA | NA | NA | NA | NA | NA | |||||
| Mathew et al35 | 1 | 6 | 22 | 0.30 | 11 of 22 | 0.50 | 8 of 10 | 0.80 | 0 | 0.00 | 12 of 22 | 0.55 | 0.45 | 0.82 |
| Angevin et al36 | 1 | 2 | 19 | NR | 3 of 19 | 0.16 | 2 of 4 | 0.50 | 0 | 0.00 | 15 of 19 | 0.79 | 0.21 | 1.00 |
| Sharma et al37 | 2 | 3 | 20 | 0.20-0.35 | 1 of 20 | 0.05 | 0 | 0.00 | 13 of 20 | 0.65 | 7 of 20 | 0.35 | 0.00 | 0.70 |
| 2 | 8 | 0.20-0.35 | 2 of 8 | 0.25 | 1 of 2 | 0.50 | 0 | 0.00 | 6 of 8 | 0.75 | 0.25 | 1.00 | ||
| Sessa et al38 | 1 | 9 | 93 | 0.16-0.33 | 8 of 93 | 0.09 | 0 | 0.00 | 69 of 93 | 0.74 | 24 of 93 | 0.26 | 0.00 | 0.45 |
| Bendell et al39 | 1 | 6 | 30 | 0.33 | 6 of 30 | 0.20 | 1 of 3 | 0.33 | 11 of 30 | 0.37 | 16 of 30 | 0.53 | 0.10 | 0.83 |
| Markman et al40 | 1 | 8 | 57 | 0.33 | 3 of 57 | 0.05 | 0 | 0.00 | 46 of 57 | 0.81 | 11 of 57 | 0.19 | 0.00 | 0.42 |
| Roberts et al41 | 2 | 4 | 15 | 0.30 | 3 of 15 | 0.20 | 1 of 3 | 0.33 | 9 of 15 | 0.60 | 3 of 15 | 0.20 | 0.20 | 0.80 |
| 4 | 14 | 0.30 | 3 of 14 | 0.21 | 1 of 4 | 0.25 | 7 of 14 | 0.50 | 3 of 14 | 0.21 | 0.29 | 0.79 | ||
| Crew et al42 | 1 | 3 | 30 | 0.25 | 5 of 30 | 0.17 | 1 of 3 | 0.33 | 16 of 30 | 0.53 | 11 of 30 | 0.37 | 0.10 | 1.00 |
| Feng et al43 | 2 | 8 | 13 | 0.15 | 2 of 13 | 0.15 | 0 | 0.00 | 11 of 13 | 0.85 | 2 of 13 | 0.15 | 0.00 | 0.62 |
| 9 | 10 | 2 of 10 | 0.20 | NA | NA | NA | NA | NA | NA | NA | NA | |||
| Tevaarwerk et al44 | 1 | 8 | 24 | 0.33 | 1 of 24 | 0.04 | 0 | 0.00 | 19 of 24 | 0.79 | 5 of 24 | 0.21 | 0.00 | 0.46 |
| Fouladi et al45 | 1 | 3 | 21 | 0.25 | 3 of 21 | 0.14 | 0 | 0.00 | 3 of 21 | 0.14 | 18 of 21 | 0.86 | 0.00 | 1.00 |
| Satoh et al46 | 3 | 1 | 40 | 0.30 | NR | NR | NR | NR | NR | NR | NR | NR | NR | NR |
| 3 | 20 | 0.30 | 0 of 20 | 0.00 | 0 | 0.00 | 4 of 20 | 0.20 | 16 of 20 | 0.80 | 0.00 | 0.90 | ||
| 4 | 19 | 0.30 | 6 of 19 | 0.32 | 0 | 0.00 | 4 of 19 | 0.21 | 15 of 19 | 0.79 | 0.00 | 0.84 | ||
| Warren et al47 | 1 | 10 | 44 | 0.25 | 2 of 44 | 0.05 | 0 | 0.00 | NA | NA | NA | NA | NA | NA |
| Fouladi et al48 | 2 | 6 | 29 | 0.25 | 3 of 29 | 0.10 | 3 of 3 | 1.00 | 20 of 29 | 0.69 | 6 of 29 | 0.21 | 0.10 | 0.55 |
| 5 | 21 | 0.25 | 2 of 21 | 0.10 | 0 | 0.00 | 15 of 21 | 0.71 | 6 of 21 | 0.29 | 0.00 | 0.67 | ||
| Peereboom et al49 | 2 | 16 | 0.33 | 1 of 16 | 0.06 | 0 | 0.00 | 11 of 16 | 0.69 | 2 of 16 | 0.13 | 0.00 | 0.31 | |
| 16 | 0.33 | 4 of 16 | 0.25 | 3 of 4 | 0.75 | 9 of 16 | 0.56 | 3 of 16 | 0.19 | 0.25 | 0.56 | |||
| Loeb et al50 | 1 | 6 | 12 | 0.30 | 4 of 12 | 0.33 | 3 of 6 | 0.50 | 4 of 12 | 0.33 | 2 of 12 | 0.17 | 0.33 | 0.42 |
| de Bono et al51 | 1 | 7 | 26 | 0.33 | 4 of 26 | 0.15 | 1 of 1 | 1.00 | 19 of 26 | 0.73 | 2 of 26 | 0.23 | 0.04 | 0.42 |
| Grossman et al52 | 2 | 4 | 31 | 0.33 | 4 of 31 | 0.13 | 2 of 6 | 0.33 | 13 of 31 | 0.42 | 12 of 31 | 0.39 | 0.19 | 0.81 |
| 3 | 17 | 0.33 | 1 of 17 | 0.06 | 0 | 0.00 | NA | NA | NA | NA | NA | NA | ||
| Gururangan et al53 | 2 | 3 | 10 | 0.25 | 3 of 10 | 0.30 | 3 of 4 | 0.75 | 0 | 0 | 6 of 10 | 0.60 | 0.40 | 0.80 |
| 3 | 11 | 0.25 | 3 of 11 | 0.27 | 3 of 4 | 0.75 | 2 of 11 | 0.18 | 5 of 11 | 0.45 | 0.36 | 1.00 | ||
| MacDonald et al54 | 1 | 7 | 31 | 0.25 | 0 of 31 | 0.00 | NA | NA | NA | NA | NA | NA | NA | NA |
| Neuenschwander et al55 | 1 | 15 | 27 | 0.30 | 4 of 27 | 0.15 | 2 of 2 | 1.00 | 16 of 27 | 0.59 | 9 of 27 | 0.33 | 0.07 | 0.56 |
| Kieran et al56 | 1 | 5 | 32 | 0.20 | 9 of 32 | 0.28 | 4 of 9 | 0.44 | 18 of 32 | 0.56 | 5 of 32 | 0.16 | 0.28 | 0.59 |
| Pollack et al57 | 3 | 4 | 23 | 0.20 | 6 of 23 | 0.26 | 2 of 6 | 0.33 | 6 of 23 | 0.26 | 11 of 23 | 0.48 | 0.26 | 0.87 |
| 3 | 20 | 0.20 | 2 of 20 | 0.10 | 2 of 8 | 0.25 | 3 of 20 | 0.15 | 9 of 20 | 0.45 | 0.40 | 1.00 | ||
| 4 | 12 | 0.20 | 0 of 12 | 0.00 | NA | NA | NA | NA | NA | NA | NA | NA | ||
| Grossman et al58 | 2 | 3 | 12 | 0.33 | 2 of 12 | 0.17 | 2 of 3 | 0.67 | 3 of 12 | 0.25 | 6 of 12 | 0.50 | 0.25 | 1.00 |
| 5 | 18 | 0.33 | 2 of 18 | 0.11 | 1 of 3 | 0.33 | 12 of 18 | 0.67 | 6 of 18 | 0.33 | 0.00 | 0.33 | ||
| Gururangan et al59 | 1 | 5 | 23 | 0.20 | 3 of 23 | 0.13 | 2 of 4 | 0.50 | 12 of 23 | 0.52 | 7 of 23 | 0.30 | 0.17 | 0.61 |
| Cheng et al60 | Many | Continuous | 78 | 0.1 | 8 of 78 | 0.10 | NA | NA | NA | NA | NA | NA | NA | NA |
| Schöffski et al61 | 1 | 14 | 34 | 0.20 | 4 of 34 | 0.12 | 2 of 5 | 0.40 | 22 of 34 | 0.65 | 7 of 34 | 0.21 | 0.15 | 0.65 |
| Garrison et al62 | 2 | 3 | 31 | 0.20 | 9 of 31 | 0.29 | 6 of 9 | 0.67 | 6 of 31 | 0.19 | 16 of 31 | 0.52 | 0.29 | 0.74 |
| 4 | ||||||||||||||
| Gilbert et al63 | 2 | 9 | 30 | 0.30 | 2 of 30 | 0.07 | 2 of 3 | 0.67 | 21 of 30 | 0.70 | 6 of 30 | 0.20 | 0.10 | 0.40 |
| 3 | 9 | 0.30 | 3 of 9 | 0.33 | 3 of 3 | 1.00 | 3 of 9 | 0.33 | 3 of 9 | 0.33 | 0.33 | 1.00 | ||
| Rowinsky et al64 | 2 | 7 | 36 | 0.20 | 5 of 36 | 0.14 | 2 of 3 | 0.67 | 20 of 36 | 0.56 | 13 of 36 | 0.36 | 0.08 | 0.69 |
| Summary | ||||||||||||||
| Mean | 5 | 24 | 0.26 | 0.18 | 0.37 | 0.39 | 0.39 | 0.19 | 0.74 | |||||
Abbreviations: DLT, dose-limiting toxicity; MTD, maximum-tolerated dose; NA, not applicable; NR, not reported.
Indicates No. of DLTs among patients treated above MTD.
To account for late-onset adverse events (AEs) as DLTs without slowing accrual, TITE-CRM was used in eight of 53 trials; CRM with continuous dosing was used in nine of 53 trials; 12 trials used CRM-EWOC; and one trial used lower-grade toxicities. The remaining 23 (43%) of 53 trials used the original CRM, as introduced by O'Quigley et al.3 These trials included two-stage designs, where the model guided escalation only in the second stage after the occurrence of the first DLT, and/or used a design with varying cohort sizes.
Safety, Therapeutic Treatment, and Efficiency
One of the most important questions in a phase I trial is safety. The observed DLT rate in these trials was 18% (range, 0% to 75%), much lower than the acceptable DLT rate of 26% (Table 2). The acceptable DLT rate is the threshold we plug into the design and call the model to find the dose level that is associated with that rate. In these trials, investigators defined 10%, 20%, 25%, 30%, or 33% as an acceptable rate in different trials. The trials that had a high observed DLT rate (≥ 50%) are described in detail. Among patients enrolled in dose levels above the MTD, 36% of patients had DLTs. This is also consistent with the 3 + 3 design, which has a DLT rate > 33% in doses above the MTD. Nineteen percent of patients were treated at levels above the MTD, and given a sample size of 25 patients, on average, we can expect 4.7 patients treated above the MTD. Thirty-nine percent of patients were treated at the MTD, 39% of patients were treated at levels below the MTD, and 74% of patients were treated at levels within one level from the MTD. These results confirm previous studies5,7 showing that on average, ≥ 10 patients are treated at the MTD when using model-based designs. All of these results are consistent with the statistical literature that has shown identical summary measures through simulations.1,3,5–8 Appendix Table A1 (online only) lists additional summary measures, such as trial duration and accrual rates. Next, we address whether these designs provide recommendations that may be counterintuitive or clinically not acceptable in the context of specific trials.
A review of Table 2 shows that there are unique trials with a high DLT rate above the MTD. The choice of the starting dose level and dose range in general is a common problem in phase I clinical trials, because preclinical models are not necessarily informative to human studies.65 Often, the selection of the starting level or the actual dose increments between consecutive levels are based on preclinical models with notable uncertainty. Whether we start experimenting near the MTD or potentially too far below the MTD is typically not known in practice.16 For this reason, treating patients in cohorts of six without updating the model is not advised with these adaptive methods, because they lose their advantages of acting on the observed toxicities and reaching the MTD faster.35 Another important question is what to do while waiting for the DLT results of already included patients who have been observed for less than the DLT observation period when a new patient presents for enrollment. In the trial by Muler et al,30 for example, 19 patients were enrolled in 15 months, and the observation timeframe for the DLT was 9 weeks. In the setting of late-onset AEs, designs can either be aggressive and move through the levels quickly without waiting for the full observation/follow-up of previous patients, or be more cautious by waiting for late toxicities before enrolling new cohorts of patients. Model-based designs can be updated at any time, and the conflict of fast accrual and late-onset toxicities is relevant in any design and has been discussed in previous articles.66,67 More details about these two trials and a third trial are presented in the Appendix (Appendix Table A2; Appendix Figs A1 and A2, online only).
DISCUSSION
This review confirms that adaptive phase I designs pose no safety concerns. We show that on average, these trials require 25 patients to test five to six levels. The average observed DLT rate of these studies was 18%, which is lower than that in previous reports.30,35 These results are in agreement with reports from the statistical literature that were based on simulated trials.4 The toxicity rate above the MTD was 35%; 19% or, on average, four to five patients per trial were treated above the MTD, illustrating these designs are aligned with clinical expectations in real trials. These designs are efficient in terms of sample size and short trial duration, and they locate the MTD rapidly and accurately, with most patients treated at or near the MTD. This review also illustrates that model-based designs are flexible in addressing multiple objectives in a single trial. For example, a study of combination agents where two schedules are being evaluated,20 studies with drug combinations where both agents are being escalated,24,27 and studies aiming to find different MTDs for different patient groups (for example, based on the use of enzyme-inducing antiepileptic drug use or metastatic v primary disease) were also presented here.49 Typically, investigators carry out separate phase I trials to establish different MTDs in heterogenous patient groups or schedules, which leads to increased use of resources.
Another important finding of this review is that the designs are flexible through the model parameters. This is important in early-phase clinical trials or first-in-man trials, where it is quite reasonable to include other auxiliary information not included in the model (pharmacokinetics, results from ongoing phase 0 studies, type, onset and resolution of AEs) in the decision to escalate. We suggest that the clinical team be able to override the recommendation of the model when dictated by clinical reasoning. A discrepancy of one level for some patients, but certainly a minority, would not pose a serious risk. However, it is not advisable to override the dose recommendations of the model consistently to the point where the model is disregarded completely.
Other authors have reviewed the acceptability of model-based designs,2 provided methodologic advancements, or examined the relationship of dose and benefit.68 Rogatko et al2 reported how often phase I trials follow a statistical design and, if they do, the time lag associated with their use. Our review is different, in that we provide extensive details on the design and conduct of these trials so that investigators have the necessary tools to evaluate their utility based on safety data from real trials. We limit our review to studies using the CRM and present them in detail. One of the limitations of this review is that we obtained all reported information from published articles; thus, not all of the AE information (attribution, onset, patient characteristics) and parameters that the investigators used when they carried out these trials were reported. Few trials (two of 53) reported on the practical difficulties encountered or how the designs were carried out between the clinical team and statisticians in real time.
Another limitation is that we could not confirm from published reports how many trials required mandatory or optional biopsies at baseline and whether this affected accrual rates or enrollment. Phase I studies have increasingly evaluated targeted therapy in specific patient populations where efficacy and safety are important criteria before moving forward to new studies. Whether certain biomarkers relate to clinical response is also a question these studies aim to answer with the use of biopsies from archived or fresh tumor. In our review, 14 of 53 studies reported that they had collected biopsies (in one, biopsies were mandatory; in 13, they were optional), and 12 of 14 trials reported specific results based on biopsies, mostly exploring biomarkers and their relationship to clinical response. These results do not preclude other exploratory analyses from tumor biopsies that were not reported in these articles. In addition, dose-expansion cohorts are often added once the MTD is found, with the aim of further evaluating safety and obtaining preliminary evidence of efficacy in disease- or histology-specific patient populations. We believe design considerations of dose-expansion cohorts are important, and more research is needed to understand how to best match the objective of each study and the mechanism of action of a specific drug (cytotoxic, molecular, biologic, immunotherapeutic, and so on) with the appropriate dose-escalation design.69
The dose-escalation designs we present aim to find the MTD, defined as a dose with an acceptable DLT rate (eg, 30%), so there is a uniform definition of MTD, consistent with the objective of the design. There are phase I studies that aim to find the minimum effective dose or biologically effective dose and designs that take into account antitumor activity or other response end points in addition to safety to decide the recommended phase II dose. If the dose-toxicity curve plateaus and a secondary criterion, such as immune response or T-cell response, are used to select the minimum effective dose in settings where immunotherapy is being evaluated, model-based designs can easily be modified to address this objective. Ongoing research is being performed to modify current designs to answer these objectives.70
Phase I studies that aim to find the MTD of a single agent in a homogeneous group of patients pose no difficulties when the MTD happens to be close to the starting dose.71,72 Challenges arise in studies in which a heterogeneous patient population is involved, combination therapies or various treatment schedules are evaluated, toxicity attribution is not clear, and the definition of drug- versus disease-related toxicity is ambiguous. All of these challenges can be handled within the framework of model-based adaptive designs.73–75 Information obtained from preclinical data typically informs us regarding the range of tested dose levels, but it can never be accurate as to the safety profile of a drug in humans. This is the reason more efficient and accurate designs should be used in phase I trials.
Supplementary Material
Appendix
Search Terms
We searched PubMed for articles with the following terms: (Clinical Trials, Phase I [Mesh] OR “Phase I” OR “Phase 1”) AND (Models, Statistical[Mesh] OR “Continual Reassessment Method”[All Fields] OR “CRM”[All Fields]) AND (“Maximum Tolerated Dose”[All Fields] OR MTD[All Fields]) AND (“dose escalation”[All Fields] OR escalation[All Fields] OR “drug overdose”[MeSH Terms] OR “drug overdose”[All Fields] OR “overdose”[All Fields] OR EWOC[All Fields] OR “time to event”[All Fields] OR TITE[All Fields] OR “late onset toxicity”[All Fields] OR “late onset toxicities”[All Fields] OR “dose-response relationship, Drug”[MeSH Terms] OR “drug overdose”[MeSH Terms]) AND (“neoplasms”[MeSH Terms] OR “neoplasms”[All Fields] OR “cancers”[All Fields] OR “neoplasms”[MeSH Terms] OR “neoplasms”[All Fields] OR “neoplasm”[All Fields] OR “neoplasms”[MeSH Terms] OR “neoplasms”[All Fields] OR “neoplasms”[MeSH Terms] OR “carcinoma”[MeSH Terms] OR “carcinoma”[All Fields] OR “carcinoma”[MeSH Terms] OR “carcinoma”[All Fields] OR “carcinomas”[All Fields] OR “tumour”[All Fields] OR “neoplasms”[MeSH Terms] OR “neoplasms”[All Fields] OR “tumor”[All Fields] OR “tumours”[All Fields] OR “neoplasms”[MeSH Terms] OR “neoplasms”[All Fields] OR “tumors”[All Fields] OR “tumour”[All Fields] OR “neoplasms”[MeSH Terms] OR “neoplasms”[All Fields] OR “tumor”[All Fields] OR “tumours”[All Fields] OR “neoplasms”[MeSH Terms] OR “neoplasms”[All Fields] OR “tumors”[All Fields] OR Lesion[All Fields] OR Lesions[All Fields] OR “neoplasms”[MeSH Terms] OR “neoplasms”[All Fields] OR “neoplasia”[All Fields]). To capture articles that failed to mention the dose-finding algorithm in the abstract, title, or MESH term, we also searched Web of Science (version 5.11) for articles that cited one of these methods (continual reassessment method [CRM] or CRM designs with escalation with overdose control). Trials reported in abstracts from conference proceedings that were not published as articles in a peer-reviewed journal were not included. Studies that were classified as commentaries, reviews, editorials, or reported methodologic methods without a completed phase I or dose-finding trial, and studies conducted in a nononcology setting, were excluded from the analysis. Authors of the included trials were contacted and asked to provide data on the primary outcome (dose-limiting toxicity rate) if those data had not been reported in the article itself.
Specific Case Studies
Trial one: phase I trial of platelet-derived growth factor receptor inhibitor imatinib mesylate and docetaxel in patients with prostate cancer.
The study35 consisted of a lead-in period where patients were treated with imatinib mesylate at 600 mg/m2 daily in single or divided doses for 30 days and a combination-therapy phase. Patients who received ≥ 80% of the prescribed drug were eligible for combination therapy, where they received intravenous docetaxel at one of the six tested dose levels for 4 consecutive weeks (days 1, 8, 15, and 22). Details of the statistical design are listed in the Data Supplement. Dose escalation for docetaxel with imatinib at a fixed dose was guided by CRM, starting from 30 mg/m2 as the initial dose level and treating patients in cohorts of six. The maximum-tolerated dose (MTD) was defined as the dose of docetaxel in combination with imatinib at 600 mg that achieved a dose-limiting toxicity (DLT) rate closest to 30%. The study enrolled 28 patients; 22 were evaluable for toxicity assessment within cycle one. Zero DLTs were observed among the first six patients treated at dose 30 mg/m2, and CRM recommended escalation to 45 mg/m2, where three of four patients experienced a DLT (Appendix Fig A1). Note that the decision to enroll four patients at dose 45 mg/m2 was a result of the cohort size, because the design called for assignments of cohorts of six. However, because of excessive toxicity, this cohort stopped early, and on the basis of the data from 10 patients, the model was reevaluated. CRM correctly de-escalated to dose 35 mg/m2, and a cohort of six patients was assigned to that level. This dose was still too high in terms of toxicity, because five of six patients experienced DLTs. CRM recommended another de-escalation to dose 30 mg/m2, where three of six patients experienced DLTs, at which point the trial was terminated. The MTD for docetaxel based on the updated model was 30 mg/m2 in combination with 600 mg/m2 imatinib in a 6-week cycle, with an observed DLT rate of three (25%) of 12.
The choice of the starting dose level and dose range in general is a common problem in phase I clinical trials, because preclinical models are not necessarily informative to human studies.65 Overexposing patients to excessive risk because the starting dose is, in retrospect, too toxic is not specific to the dose-escalation design. However, we can minimize this risk by enrolling fewer patients in each cohort and by avoiding skipping dose levels. In this trial, had the cohort size been three, CRM would have recommended de-escalation of two levels after three patients were treated at dose 45 mg/m2 to dose 30 mg/m2. Similarly, if three instead of six patients were treated at dose 35 mg/m2, a further de-escalation would have been recommended to level 30 mg/m2, thus exposing fewer patients to excessive risk that resulted in the observed number of DLTs (five of six) at 35 mg/m2. The last cohort based on the model should have been treated one level lower, at dose 25 mg/m2, based on the data from 16 patients. On the basis of the data from all 22 patients, the model recommendation is level two—25 mg/m2—with a predicted DLT rate of 28% (model-predicted DLT rates for all six levels: 16%, 28%, 43%, 53%, 58%, and 64%, respectively). However, the recommended phase II dose was level three—30 mg/m2—with an estimated DLT rate of 43%, which investigators considered sufficiently safe in this setting, because only three (25%) of 12 patients experienced DLTs.
Trial two: phase I trial of cisplatin with gemcitabine in pancreatic cancer.
The aim of the study30 was to find the MTD of cisplatin that could be added to full-dose gemcitabine and radiation therapy (RT) in patients with pancreatic cancer. Treatment consisted of two 28-day cycles of chemotherapy, with RT administered during the first cycle of chemotherapy. Radiation and gemcitabine doses were held constant, while the dose level of cisplatin was assigned based on CRM designs that allow for time to event or late-onset toxicities (TITE-CRM). RT was administered daily in 2.4-Gy fractions five times weekly, for a total of 36 Gy, beginning on day 1 of the first cycle. Gemcitabine 1,000 mg/m2 was administered over 30 minutes on days 1, 8, and 15 of a 28-day cycle. Cisplatin was administered intravenously 1 to 2 hours after gemcitabine in 250 mL of 0.9% saline over 30 minutes on days 1 and 15 of a chemotherapy cycle. After combination treatment with chemotherapy and RT, a second cycle of gemcitabine and cisplatin was administered. The initial dose of cisplatin was 30 mg/m2 and escalated to a target dose of 50 mg/m2. The protocol implemented a restriction that at least two patients must have completed therapy of at least 9 weeks at the lower level before the first patient could be assigned at a higher level. As a result of this restriction, four patients were treated at 30 mg/m2 and four evaluable patients at 40 mg/m2, although the model indicated a higher MTD (Appendix Fig A2). After zero DLTs in eight patients, there was escalation to dose 50 mg/m2. Seven patients were treated at dose 50mg/m2, with two of seven experiencing DLTs, because the recommendation was to remain at the same dose level. However, patient No. 16 was de-escalated to a dose of 40 mg/m2 because of concerns about a potential DLT for patient No. 15, and patient No. 17 was treated at a level of 50 mg/m2 because the DLT of patient No. 15 had not yet been established. After patient No. 17 had a DLT and previously enrolled patients No. 12 and 15 developed late-onset toxicities that counted as DLTs, patient No. 18 was treated at dose 30 mg/m2. The model recommended level 40 mg/m2 as the final MTD (estimated DLT rate, 0.20; observed DLT rate, zero of five; acceptable toxicity rate, 0.20). The criticism is that at the level above the MTD, four (50%) of eight patients had been exposed to DLTs. As Appendix Figure A2 shows, this trial experimented at three levels, two of which had no DLTs. The dose-toxicity curve in this trial is too steep, jumping from 0% to 50% between doses of 40 and 50 mg/m2; hence, the model and the investigators recommended 40 mg/m2, with an observed DLT rate of zero.
In this trial, the accrual rate was much greater than the rate at which the DLTs could be observed, because 19 patients were enrolled in 15 months, and the observation timeframe for DLTs was 9 weeks. This raises the question of what to do while waiting for the DLT results of already included patients who have been observed for < 9 weeks when a new patient presents for enrollment. The investigators chose to use the TITE-CRM as opposed to the standard CRM. The standard CRM, in choosing the best current estimate of the MTD at which to treat, will only make use of patients for whom a DLT assessment can be made definitively. The TITE-CRM uses information on all included patients, and those patients for whom the follow-up time is incomplete are treated as nontoxicities, although with a model weight to scale down their influence proportional to their total follow-up time. Once the follow-up time is complete for any individual, a DLT within that interval counts as a DLT. CRM and TITE-CRM react in the same way to this information. The same is true for a non-DLT if, at interval completion, it is still a non-DLT. However, CRM ignores patients without a DLT and for whom follow-up is < the entire 9-week interval, whereas TITE-CRM counts it toward a non-DLT at that level. Thus, many such patients, by summing, can count as full or even several full non-DLTs at the levels at which they are treated. If ≥ one of these weighted non-DLTs turns out to be a late-onset DLT, the method is behaving in an anticonservative fashion for at least some part of the trial, because an observed DLT has been counted as some percentage of a non-DLT.11 The effect of this is to push toward more aggressive escalation. Late-onset DLTs were being counted for some period of the trial as partial non-DLTs, with escalation proceeding accordingly. Subsequently, the model re-evaluated the recommended level and de-escalated based on the known DLTs when these late-onset DLTs occurred, which is why the final MTD is the correct dose. Given the fast accrual in this trial and the number of nonevaluable patients for DLTs in the entire 9-week window, eight of 18 patients were treated at the level above the MTD, because the model could not react to late toxicities until they actually happened. Alternatively, investigators could have waited for each cohort of three patients to be observed for a full 9-week period (ie, 4.5 months per cohort) and completed the trial in 27 rather than 15 months.
Trial three: dose-escalation cancer trial.
The trial55 was designed with the aim of characterizing the safety, tolerability, and pharmacokinetic profiles of a drug. The details of the drug, patient population, and definition of DLT are not given; only the recommendation to escalate or de-escalate, based on the data, is provided.55 The initial design indicated a total of 15 possible levels ranging from 1 to 250 mg, with the prior distribution indicating dose level 10 (50 mg) to be the likely MTD before seeing any data. However, the starting dose for the trial was level one (1 mg), with the prior centered at a dose 50× higher (50 mg) than the starting dose. After zero DLTs were seen in 16 patients treated at levels one to four, a decision was taken by the investigators to skip dose levels five and six and treat two patients at level seven. At level seven, two DLTs were observed in two patients, and on the basis of these two DLTs among 18 patients, the chosen model, and the prior information, the recommendation was to escalate to level nine, with an estimated DLT rate of 33%. Furthermore, as the authors illustrate,55 the reason the estimated rate at level seven was so low (16%) is because the method relies on two initial parameters: an initial dose-toxicity curve and the prior distribution that controls the uncertainty around the location of the curve. The initial curve was low (shallow), almost zero in low levels, and increased rapidly at the last levels, and the prior also favored high levels heavily (level 10). As a result, an enormous amount of data was required to override these strong initial working assumptions. The investigators correctly identified the initial curve as the problem and illustrated the more sensible choice of a recommendation to stay at the current level (ie, level seven), which is three levels lower than the prior assumption (ie, level 10). The recommendation to stay at the same level is reasonable given the previous data supporting no DLTs at any of the lower levels. Note that the recommendation would be de-escalation to level six had we followed the model from the beginning of the trial, as summarized in Appendix Table A2. Appendix Table A2 shows how the trial would have escalated with cautious dose recommendations if the model had been followed from the beginning of the trial. CRM has been shown to be coherent (ie, it never escalates in presence of DLTs or de-escalates in absence of DLTs), assuming the model has been followed from the trial onset (Cheung YK et al: Biometrics 58:671-674, 2002).
In this trial by Neuenschwander et al,55 the dose-level allocation decisions made for the first 18 patients did not comply with the model-based recommendations. This is understandable, given that the a priori model indicated experimenting at level 10 (50 mg) and higher, whereas clinical investigators had to start experimentation at the lowest dose (1 mg). In general, investigators need to experiment at a level indicated by the prior, or they need to use a prior consistent with where they wish to experiment. Here, the prior was indicating experimentation at level 10, whereas the investigators' allocation was indicating lower levels. In fact, before seeing any data, the prior amounts to two DLTs of 17 observations at level seven support level seven to be a safe, acceptable dose.
There are two possible approaches to a CRM trial: either a fully Bayesian approach,3 in which from the outset we experiment at the level indicated by our prior information or beliefs, or a two-stage design starting out at a low-, or the lowest-, level dose, and we escalate according to any nonstatistical rule until we run into the first observed DLT.8 Once that first DLT has been observed, we fit our model, just like in the Bayesian case, and we then continue in the same way. Although this study seems to be Bayesian, the design forced experimentation far from the recommendation of the Bayesian prior. Starting experimentation at the lowest levels in groups and escalating until the first toxicities made the design seem to be a two-stage design. However, it is not a two-stage design, because of the Bayesian prior, and it is not a correctly structured Bayesian design for the first 18 patients. Choosing either a fully Bayesian design or a two-stage likelihood design with a steadily increasing curve would have avoided the difficulties encountered in this trial (Appendix Table A3). The solution is not to make use of the seemingly more flexible two-parameter logistic model, because a two-parameter CRM based on logistic regression has been studied, and it performed less well than the one-parameter CRM, even when the actual observations were generated by the two parameter model (Shu J et al: Stat Med 27:5345-5353, 2008; discussion 54-55).
Table A1.
No. of Patients Enrolled and Evaluable, Accrual Rate, and Trial Duration
| Study | No. of Groups | No. of Levels | DLT Window (days) | No. of Evaluable Patients | No. of Patients Enrolled | Trial Duration (months) | Accrual Rate* (patients per month) |
|---|---|---|---|---|---|---|---|
| Thornton et al12 | 1 | 5 | 56 | 15 | 15 | NR | |
| Harvey et al13 | 1 | 7 | 42 | 30 | 31 | NR | |
| Ben-Josef et al14 | 1 | 6 | 126 | 51 | 51 | 45 | 1.1 |
| Tsien et al15 | 1 | 5 | 90 | 38 | 42 | 44 | 1.0 |
| Schneider et al16 | 1 | 4 | 42 | 12 | 12 | 14 | 0.9 |
| Reardon et al17 | 1 | 2 | 28 | 13 | 16 | 12 | 1.3 |
| Sinha et al18 | 1 | 3 | 21 | 19 | 19 | 24 | 0.8 |
| Mehnert et al19 | 1 | 5 | 21 | 16 | 16 | 11 | 1.5 |
| Lonial et al20 | 2 | 3 | 100 | 19 | NR | ||
| 3 | 100 | 20 | 39 | ||||
| Guillot et al21 | 1 | 4 | 28 | 31 | 31 | NR | |
| Morita et al22 | 1 | 3 | 42 | 13 | 13 | 27 | 0.5 |
| Broniscer et al23 | 2 | 5 | 28 | 27 | 72 | 32 | 2.3 |
| 3 | 15 | ||||||
| Saji et al24 | 1 | 5 | 42 | 16 | 17 | 26 | 0.7 |
| Freedman et al25 | 1 | 1 | 30 | 8 | 8 | 21 | 0.4 |
| Mackler et al26 | 1 | 5 | 90 | 31 | 33 | 21 | 1.6 |
| Geoerger et al27 | 2 | 3 | 28 | 25 | 39 | 13 | 3.0 |
| 3 | 28 | 13 | |||||
| Khuri et al28 | 1 | 5 | 21 | 21 | 24 | 9 | 2.7 |
| Eder et al29 | 1 | 6 | 28 | 33 | 33 | 12 | 2.8 |
| Muler et al30 | 1 | 4 | 63 | 18 | 19 | 15 | 1.3 |
| Geoerger et al31 | 2 | 4 | 21 | 24 | |||
| 3 | 42 | 20 | 51 | 26 | 2.0 | ||
| Rathkopf et al32 | 2 | 2 | 21 | 16 | 16 | 15 | 1.1 |
| Bailey et al33 | 2 | 5 | 21-28 | 50 | 53 | 11 | 4.8 |
| Bonghaei et al34 | Many | Continuous | 28 | 52 | 52 | 31 | 1.7 |
| Mathew et al35 | 1 | 6 | 42 | 22 | 28 | NR | |
| Angevin et al36 | 1 | 2 | 28 | 19 | 20 | 28 | 0.7 |
| Sharma et al37 | 2 | 3 | 28 | 20 | 24 | NR | NR |
| 2 | 21 | 8 | 9 | ||||
| Sessa et al38 | 1 | 9 | 28 | 93 | 101 | 25 | 4 |
| Bendell et al39 | 1 | 6 | 28 | 30 | 35 | 11 | 3.2 |
| Markman et al40 | 1 | 8 | 28 | 57 | 57 | 26 | 2.2 |
| Roberts et al41 | 2 | 4 | 21 | 15 | |||
| 4 | 21 | 14 | 29 | 23 | 1.3 | ||
| Crew et al42 | 1 | 3 | 180 | 30 | 40 | 25 | 1.6 |
| Feng et al43 | 2 | 8 | 60 | 13 | |||
| 9 | 60 | 10 | 23 | 54 | 0.4 | ||
| Tevaarwerk et al44 | 1 | 8 | 28 | 24 | 24 | 16 | 1.5 |
| Fouladi et al45 | 1 | 3 | 28 | 21 | 23 | NR | |
| Satoh et al46 | 3 | 1 | 14 | 40 | |||
| 3 | 14 | 20 | |||||
| 4 | 14 | 19 | 82 | 23 | 3.6 | ||
| Warren et al47 | 1 | 10 | 28 | 44 | 51 | 36 | 1.4 |
| Fouladi et al48 | 2 | 6 | 28 | 29 | 32 | NR | |
| 5 | 28 | 21 | 27 | ||||
| Peereboom et al49 | 2 | 21 | 16 | ||||
| 21 | 16 | 57 | 37 | 1.5 | |||
| Loeb et al50 | 1 | 6 | 42 | 12 | 13 | NR | |
| de Bono et al51 | 1 | 7 | 21 | 39 | 39 | NR | |
| Grossman et al52 | 2 | 4 | 28 | 31 | 31 | NR | |
| 3 | 28 | 17 | 18 | ||||
| Gururangan et al53 | 2 | 3 | 42 | 10 | 42 | 15 | 2.8 |
| 3 | 42 | 11 | |||||
| MacDonald et al54 | 1 | 7 | 28 | 31 | 35 | 20 | 1.8 |
| Neuenschwander et al55 | 1 | 15 | NR | 27 | 27 | NR | |
| Kieran et al56 | 1 | 5 | 28 | 32 | 55 | NR | |
| Pollack et al57 | 3 | 4 | 56 | 23 | 84 | 36 | 2.3 |
| 3 | 56 | 10 | |||||
| 4 | 56 | 12 | |||||
| Grossman et al58 | 2 | 3 | 21 | 12 | |||
| 5 | 21 | 18 | 32 | 27 | 1.2 | ||
| Gururangan et al59 | 1 | 5 | 14 | 23 | 28 | 34 | 0.8 |
| Cheng et al60 | Many | Continuous | 28 | 78 | 78 | 27 | 2.9 |
| Schöffski et al61 | 1 | 14 | 21 | 34 | 41 | 30 | 1.4 |
| Garrison et al62 | 2 | 3 | 21 | 31 | 31 | NR | |
| 4 | 28 | ||||||
| Gilbert et al63 | 2 | 9 | 28 | 30 | 40 | NR | |
| 3 | 28 | 9 | |||||
| Rowinsky et al64 | 2 | 7 | 21 | 36 | 37 | NR | |
| Summary | |||||||
| Mean | 5 | 38 | 25 | 35 | 25 | 1.79 |
Abbreviations: DLT, dose-limiting toxicity; NR, not reported.
Accrual rate was estimated by dividing No. of enrolled patients by trial duration (months) when duration was reported.
Table A2.
Case Study: Trial Three
| Patient No. | Treatment |
Initial Curve: Actual Recommendation* |
Updated DLT Rate at 50-mg Dose | Correct Curve: Correct Recommendation† |
|||||
|---|---|---|---|---|---|---|---|---|---|
| Dose (mg) | Level | DLT | Dose (mg) | Level | Dose (mg) | Level | DLT | ||
| 1 | 1 | 1 | No | 50 | 10 | 21 | 1 | 1 | No |
| 2 | 1 | 1 | No | 50 | 10 | 17 | 5 | 3 | No |
| 3 | 1 | 1 | No | 50 | 10 | 15 | 15 | 5 | No |
| 4 | 2.5 | 2 | No | 50 | 10 | 13 | 20 | 6 | No |
| 5 | 2.5 | 2 | No | 50 | 10 | 11 | 25 | 7 | Yes |
| 6 | 2.5 | 2 | No | 50 | 10 | 10 | 15 | 5 | No |
| 7 | 2.5 | 2 | No | 50 | 10 | 9 | 20 | 6 | No |
| 8 | 5 | 3 | No | 50 | 10 | 8 | 20 | 6 | No |
| 9 | 5 | 3 | No | 50 | 10 | 8 | 25 | 7 | Yes |
| 10 | 5 | 3 | No | 50 | 10 | 7 | 20 | 6 | No |
| 11 | 5 | 3 | No | 50 | 10 | 7 | 20 | 6 | No |
| 12 | 5 | 3 | No | 50 | 10 | 6 | 25 | 7 | Yes |
| 13 | 10 | 4 | No | 50 | 10 | 6 | 20 | 6 | No |
| 14 | 10 | 4 | No | 50 | 10 | 6 | 20 | 6 | No |
| 15 | 10 | 4 | No | 50 | 10 | 6 | 20 | 6 | No |
| 16 | 10 | 4 | No | 50 | 10 | 5 | 25 | 7 | Yes |
| 17 | 25 | 7 | Yes | 40 | 9 | 38 | 20 | 6 | No |
| 18 | 25 | 7 | Yes | 40 | 9 | 47 | 20 | 6 | No |
| Recommended dose | 40 | 9 | 25 | 7 | |||||
NOTE. Initial curve assigns 30% DLT rate at level 10 (50 mg) and low rates at all remaining levels (rates for each respective dose level: 0.01, 0.015, 0.02, 0.025, 0.03, 0.04, 0.05, 0.1, 0.17, and 0.3). Correct curve assigns 30% initial DLT rate (before seeing data) at dose one so that experimentation starts at dose one. Rates under correct curve for each dose level: 0.30, 0.40, 0.48, 0.56, 0.64, 0.72, 0.80, 0.88, 0.92, and 0.99.
Abbreviation: DLT, dose-limiting toxicity.
Prior/initial values: 50 mg, level 10.
Prior/initial values: 1 mg, level one.
Fig A1.
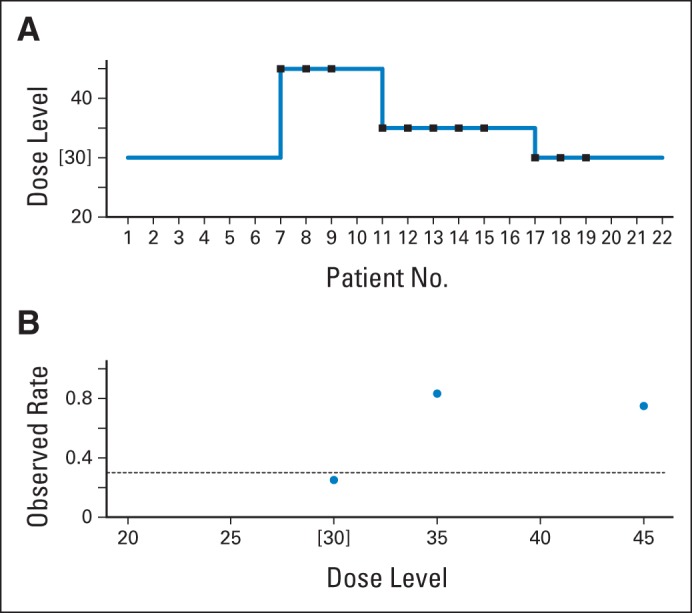
Case study, trial one: phase I trial of platelet-derived growth factor receptor inhibitor imatinib mesylate and docetaxel in patients with prostate cancer. (A) Trial history (dose units are in mg/m2); (B) observed dose-limiting toxicity (DLT) rate at each dose level. Brackets indicate maximum-tolerated dose; squares indicate DLTs; circles indicate observed DLT rate; dashed line indicates acceptable/target DLT rate. Data adapted.35
Fig A2.
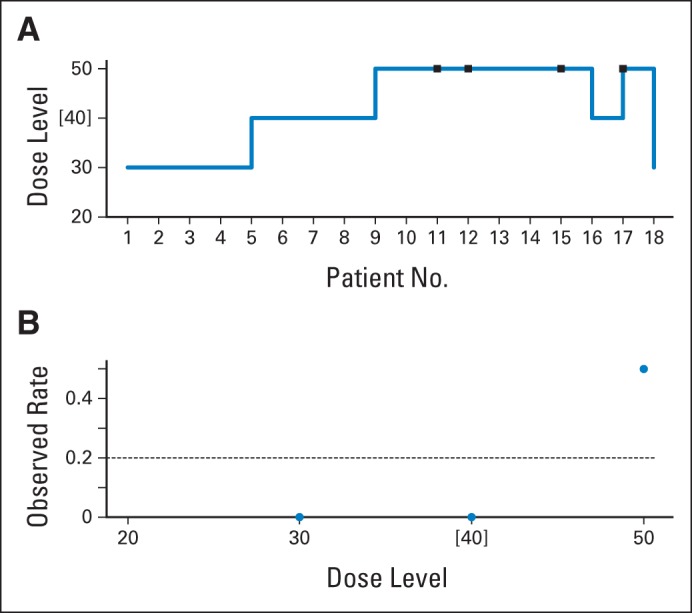
Case study, trial two: phase I trial of cisplatin with gemcitabine in patients with pancreatic cancer. (A) Trial history (dose units are in mg/m2); (B) observed dose-limiting toxicity (DLT) rate at each dose level. Brackets indicate maximum-tolerated dose; squares indicate dose-limiting toxicities; circles indicate observed DLT rate; dashed line indicates acceptable/target DLT rate. Data adapted.30
Footnotes
Supported in part by the National Institutes of Health Grants No. U01 CA069856 and P30-CA008748 (A.I.) and R01-CA142859 (J.O.).
Authors' disclosures of potential conflicts of interest and author contributions are found at the end of this article.
AUTHORS' DISCLOSURES OF POTENTIAL CONFLICTS OF INTEREST
The author(s) indicated no potential conflicts of interest.
AUTHOR CONTRIBUTIONS
Conception and design: All authors
Collection and assembly of data: Alexia Iasonos
Data analysis and interpretation: All authors
Manuscript writing: All authors
Final approval of manuscript: All authors
REFERENCES
- 1.Garrett-Mayer E. The continual reassessment method for dose-finding studies: A tutorial. Clin Trials. 2006;3:57–71. doi: 10.1191/1740774506cn134oa. [DOI] [PubMed] [Google Scholar]
- 2.Rogatko A, Schoeneck D, Jonas W, et al. Translation of innovative designs into phase I trials. J Clin Oncol. 2007;25:4982–4986. doi: 10.1200/JCO.2007.12.1012. [DOI] [PubMed] [Google Scholar]
- 3.O'Quigley J, Pepe M, Fisher L. Continual reassessment method: A practical design for phase 1 clinical trials in cancer. Biometrics. 1990;46:33–48. [PubMed] [Google Scholar]
- 4.Ahn C. An evaluation of phase I cancer clinical trial designs. Stat Med. 1998;17:1537–1549. doi: 10.1002/(sici)1097-0258(19980730)17:14<1537::aid-sim872>3.0.co;2-f. [DOI] [PubMed] [Google Scholar]
- 5.Goodman SN, Zahurak ML, Piantadosi S. Some practical improvements in the continual reassessment method for phase I studies. Stat Med. 1995;14:1149–1161. doi: 10.1002/sim.4780141102. [DOI] [PubMed] [Google Scholar]
- 6.Piantadosi S, Fisher JD, Grossman S. Practical implementation of a modified continual reassessment method for dose finding trials. Cancer Chemother Pharmacol. 1998;41:429–436. doi: 10.1007/s002800050763. [DOI] [PubMed] [Google Scholar]
- 7.Iasonos A, Wilton AS, Riedel ER, et al. A comprehensive comparison of the continual reassessment mehod to the standard 3+3 dose escalation scheme in phase I dose-finding studies. Clin Trials. 2008;5:465–477. doi: 10.1177/1740774508096474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.O'Quigley J, Shen LZ. Continual reassessment method: A likelihood approach. Biometrics. 1996;52:673–684. [PubMed] [Google Scholar]
- 9.Jaki T, Clive S, Weir CJ. Principles of dose finding studies in cancer: A comparison of trial designs. Cancer Chemother Pharmacol. 2013;71:1107–1114. doi: 10.1007/s00280-012-2059-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Babb J, Rogatko A, Zacks S. Cancer phase I clinical trials: Efficient dose escalation with overdose control. Stat Med. 1999;17:1103–1120. doi: 10.1002/(sici)1097-0258(19980530)17:10<1103::aid-sim793>3.0.co;2-9. [DOI] [PubMed] [Google Scholar]
- 11.Cheung YK, Chappell R. Sequential designs for phase I clinical trials with late-onset toxicities. Biometrics. 2000;56:1177–1182. doi: 10.1111/j.0006-341x.2000.01177.x. [DOI] [PubMed] [Google Scholar]
- 12.Thornton KA, Chen AR, Trucco MM, et al. A dose-finding study of temsirolimus and liposomal doxorubicin for patients with recurrent and refractory bone and soft tissue sarcoma. Int J Cancer. 2013;133:997–1005. doi: 10.1002/ijc.28083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Harvey RD, Owonikoko TK, Lewis CM, et al. A phase 1 Bayesian dose selection study of bortezomib and sunitinib in patients with refractory solid tumor malignancies. Br J Cancer. 2013;108:762–765. doi: 10.1038/bjc.2012.604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ben-Josef E, Schipper M, Francis IR, et al. A phase I/II trial of intensity modulated radiation (IMRT) dose escalation with concurrent fixed-dose rate gemcitabine (FDR-G) in patients with unresectable pancreatic cancer. Int J Radiat Oncol Biol Phys. 2012;84:1166–1171. doi: 10.1016/j.ijrobp.2012.02.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tsien CI, Brown D, Normolle D, et al. Concurrent temozolomide and dose-escalated intensity-modulated radiation therapy in newly diagnosed glioblastoma. Clin Cancer Res. 2012;18:273–279. doi: 10.1158/1078-0432.CCR-11-2073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Schneider BJ, Kalemkerian GP, Bradley D, et al. Phase I study of vorinostat (suberoylanilide hydroxamic acid, NSC 701852) in combination with docetaxel in patients with advanced and relapsed solid malignancies. Invest New Drugs. 2012;30:249–257. doi: 10.1007/s10637-010-9503-6. [DOI] [PubMed] [Google Scholar]
- 17.Reardon DA, Cloughesy T, Rich J, et al. Pharmacokinetic drug interaction between AEE788 and RAD001 causing thrombocytopenia in patients with glioblastoma. Cancer Chemother Pharmacol. 2012;69:281–287. doi: 10.1007/s00280-011-1754-1. [DOI] [PubMed] [Google Scholar]
- 18.Sinha R, Kaufman JL, Khoury HJ, Jr, et al. A phase 1 dose escalation study of bortezomib combined with rituximab, cyclophosphamide, doxorubicin, modified vincristine, and prednisone for untreated follicular lymphoma and other low-grade B-cell lymphomas. Cancer. 2012;118:3538–3548. doi: 10.1002/cncr.26660. [DOI] [PubMed] [Google Scholar]
- 19.Mehnert JM, Tan AR, Moss R, et al. Rationally designed treatment for solid tumors with MAPK pathway activation: A phase I study of paclitaxel and bortezomib using an adaptive dose-finding approach. Mol Cancer Ther. 2011;10:1509–1519. doi: 10.1158/1535-7163.MCT-10-0944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lonial S, Kaufman J, Tighiouart M, et al. A phase I/II trial combining high-dose melphalan and autologous transplant with bortezomib for multiple myeloma: A dose- and schedule-finding study. Clin Cancer Res. 2010;16:5079–5086. doi: 10.1158/1078-0432.CCR-10-1662. [DOI] [PubMed] [Google Scholar]
- 21.Guillot B, Khamari A, Cupissol D, et al. Temozolomide associated with PEG-interferon in patients with metastatic melanoma: A multicenter prospective phase I/II study. Melanoma Res. 2008;18:141–146. doi: 10.1097/CMR.0b013e3282f6309c. [DOI] [PubMed] [Google Scholar]
- 22.Morita S, Nakata B, Tsuji A, et al. A phase I study of combination therapy of the oral fluorinated pyrimidine compound S-1 with low-dose cisplatin twice-a-week administration (JFMC27-9902 Step2) in patients with advanced gastric cancer using a continual reassessment method. Jpn J Clin Oncol. 2007;37:924–929. doi: 10.1093/jjco/hym124. [DOI] [PubMed] [Google Scholar]
- 23.Broniscer A, Gururangan S, MacDonald TJ, et al. Phase I trial of single-dose temozolomide and continuous administration of o6-benzylguanine in children with brain tumors: A pediatric brain tumor consortium report. Clin Cancer Res. 2007;13:6712–6718. doi: 10.1158/1078-0432.CCR-07-1016. [DOI] [PubMed] [Google Scholar]
- 24.Saji S, Toi M, Morita S, et al. Dose-finding phase I and pharmacokinetic study of capecitabine (Xeloda) in combination with epirubicin and cyclophosphamide (CEX) in patients with inoperable or metastatic breast cancer. Oncology. 2007;72:330–337. doi: 10.1159/000113062. [DOI] [PubMed] [Google Scholar]
- 25.Freedman GM, Meropol NJ, Sigurdson ER, et al. Phase I trial of preoperative hypofractionated intensity-modulated radiotherapy with incorporated boost and oral capecitabine in locally advanced rectal cancer. Int J Radiat Oncol Biol Phys. 2007;67:1389–1393. doi: 10.1016/j.ijrobp.2006.11.017. [DOI] [PubMed] [Google Scholar]
- 26.Mackler NJ, Dunn RL, Hellerstedt B, et al. Dose escalation of oral vinorelbine in combination with estramustine in hormone-refractory adenocarcinoma of the prostate. Cancer. 2006;106:2617–2623. doi: 10.1002/cncr.21927. [DOI] [PubMed] [Google Scholar]
- 27.Geoerger B, Vassal G, Doz F, et al. Dose finding and O6-alkylguanine-DNA alkyltransferase study of cisplatin combined with temozolomide in paediatric solid malignancies. Br J Cancer. 2005;93:529–537. doi: 10.1038/sj.bjc.6602740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Khuri FR, Glisson BS, Kim ES, et al. Phase I study of the farnesyltransferase inhibitor lonafarnib with paclitaxel in solid tumors. Clin Cancer Res. 2004;10:2968–2976. doi: 10.1158/1078-0432.ccr-03-0412. [DOI] [PubMed] [Google Scholar]
- 29.Eder JP, Jr, Garcia-Carbonero R, Clark JW, et al. A phase I trial of daily oral 4′-N-benzoyl-staurosporine in combination with protracted continuous infusion 5-fluorouracil in patients with advanced solid malignancies. Invest New Drugs. 2004;22:139–150. doi: 10.1023/B:DRUG.0000011790.31292.ef. [DOI] [PubMed] [Google Scholar]
- 30.Muler JH, McGinn CJ, Normolle D, et al. Phase I trial using a time-to-event continual reassessment strategy for dose escalation of cisplatin combined with gemcitabine and radiation therapy in pancreatic cancer. J Clin Oncol. 2004;22:238–243. doi: 10.1200/JCO.2004.03.129. [DOI] [PubMed] [Google Scholar]
- 31.Geoerger B, Hargrave D, Thomas F, et al. Innovative therapies for children with cancer pediatric phase I study of erlotinib in brainstem glioma and relapsing/refractory brain tumors. Neuro Oncol. 2011;13:109–118. doi: 10.1093/neuonc/noq141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rathkopf D, Wong BY, Ross RW, et al. A phase I study of oral panobinostat alone and in combination with docetaxel in patients with castration-resistant prostate cancer. Cancer Chemother Pharmacol. 2010;66:181–189. doi: 10.1007/s00280-010-1289-x. [DOI] [PubMed] [Google Scholar]
- 33.Bailey S, Neuenschwander B, Laird G, et al. A Bayesian case study in oncology Phase I combination dose-finding using logistic regression with covariates. J Biopharm Stat. 2009;19:469–484. doi: 10.1080/10543400902802409. [DOI] [PubMed] [Google Scholar]
- 34.Borghaei H, Alpaugh K, Hedlund G, et al. Phase I dose escalation, pharmacokinetic and pharmacodynamic study of naptumomab estafenatox alone in patients with advanced cancer and with docetaxel in patients with advanced non–small-cell lung cancer. J Clin Oncol. 2009;27:4116–4123. doi: 10.1200/JCO.2008.20.2515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mathew P, Thall PF, Jones D, et al. Platelet-derived growth factor receptor inhibitor imatinib mesylate and docetaxel: A modular phase I trial in androgen-independent prostate cancer. J Clin Oncol. 2004;22:3323–3329. doi: 10.1200/JCO.2004.10.116. [DOI] [PubMed] [Google Scholar]
- 36.Angevin E, Lopez-Martin JA, Lin CC, et al. Phase I study of dovitinib (TKI258), an oral FGFR, VEGFR, and PDGFR inhibitor, in advanced or metastatic renal cell carcinoma. Clin Cancer Res. 2013;19:1257–1268. doi: 10.1158/1078-0432.CCR-12-2885. [DOI] [PubMed] [Google Scholar]
- 37.Sharma S, Beck J, Mita M, et al. A phase I dose-escalation study of intravenous panobinostat in patients with lymphoma and solid tumors. Invest New Drugs. 2013;31:974–985. doi: 10.1007/s10637-013-9930-2. [DOI] [PubMed] [Google Scholar]
- 38.Sessa C, Shapiro GI, Bhalla KN, et al. First-in-human phase I dose-escalation study of the HSP90 inhibitor AUY922 in patients with advanced solid tumors. Clin Cancer Res. 2013;19:3671–3680. doi: 10.1158/1078-0432.CCR-12-3404. [DOI] [PubMed] [Google Scholar]
- 39.Bendell JC, Rodon J, Burris HA, et al. Phase I, dose-escalation study of BKM120, an oral pan–class I PI3K inhibitor, in patients with advanced solid tumors. J Clin Oncol. 2012;30:282–290. doi: 10.1200/JCO.2011.36.1360. [DOI] [PubMed] [Google Scholar]
- 40.Markman B, Tabernero J, Krop I, et al. Phase I safety, pharmacokinetic, and pharmacodynamic study of the oral phosphatidylinositol-3-kinase and mTOR inhibitor BGT226 in patients with advanced solid tumors. Ann Oncol. 2012;23:2399–2408. doi: 10.1093/annonc/mds011. [DOI] [PubMed] [Google Scholar]
- 41.Roberts AW, Seymour JF, Brown JR, et al. Substantial susceptibility of chronic lymphocytic leukemia to BCL2 inhibition: Results of a phase I study of navitoclax in patients with relapsed or refractory disease. J Clin Oncol. 2012;30:488–496. doi: 10.1200/JCO.2011.34.7898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Crew KD, Brown P, Greenlee H, et al. Phase IB randomized, double-blinded, placebo-controlled, dose escalation study of polyphenon E in women with hormone receptor-negative breast cancer. Cancer Prev Res (Phila) 2012;5:1144–1154. doi: 10.1158/1940-6207.CAPR-12-0117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Feng M, Smith DE, Normolle DP, et al. A phase I clinical and pharmacology study using amifostine as a radioprotector in dose-escalated whole liver radiation therapy. Int J Radiat Oncol Biol Phys. 2012;83:1441–1447. doi: 10.1016/j.ijrobp.2011.10.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Tevaarwerk A, Wilding G, Eickhoff J, et al. Phase I study of continuous MKC-1 in patients with advanced or metastatic solid malignancies using the modified time-to-event continual reassessment method (TITE-CRM) dose escalation design. Invest New Drugs. 2012;30:1039–1045. doi: 10.1007/s10637-010-9629-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Fouladi M, Stewart CF, Olson J, et al. Phase I trial of MK-0752 in children with refractory CNS malignancies: A pediatric brain tumor consortium study. J Clin Oncol. 2011;29:3529–3534. doi: 10.1200/JCO.2011.35.7806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Satoh T, Ura T, Yamada Y, et al. Genotype-directed, dose-finding study of irinotecan in cancer patients with UGT1A1*28 and/or UGT1A1*6 polymorphisms. Cancer Sci. 2011;102:1868–1873. doi: 10.1111/j.1349-7006.2011.02030.x. [DOI] [PubMed] [Google Scholar]
- 47.Warren KE, Goldman S, Pollack IF, et al. Phase I trial of lenalidomide in pediatric patients with recurrent, refractory, or progressive primary CNS tumors: Pediatric Brain Tumor Consortium study PBTC-018. J Clin Oncol. 2011;29:324–329. doi: 10.1200/JCO.2010.31.3601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Fouladi M, Stewart CF, Blaney SM, et al. Phase I trial of lapatinib in children with refractory CNS malignancies: A Pediatric Brain Tumor Consortium study. J Clin Oncol. 2010;28:4221–4227. doi: 10.1200/JCO.2010.28.4687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Peereboom DM, Supko JG, Carson KA, et al. A phase I/II trial and pharmacokinetic study of ixabepilone in adult patients with recurrent high-grade gliomas. J Neurooncol. 2010;100:261–268. doi: 10.1007/s11060-010-0190-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Loeb DM, Garrett-Mayer E, Hobbs RF, et al. Dose-finding study of 153Sm-EDTMP in patients with poor-prognosis osteosarcoma. Cancer. 2009;115:2514–2522. doi: 10.1002/cncr.24286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.de Bono JS, Kristeleit R, Tolcher A, et al. Phase I pharmacokinetic and pharmacodynamic study of LAQ824, a hydroxamate histone deacetylase inhibitor with a heat shock protein-90 inhibitory profile, in patients with advanced solid tumors. Clin Cancer Res. 2008;14:6663–6673. doi: 10.1158/1078-0432.CCR-08-0376. [DOI] [PubMed] [Google Scholar]
- 52.Grossman SA, Carson KA, Batchelor TT, et al. The effect of enzyme-inducing antiseizure drugs on the pharmacokinetics and tolerability of procarbazine hydrochloride. Clin Cancer Res. 2006;12:5174–5181. doi: 10.1158/1078-0432.CCR-06-0932. [DOI] [PubMed] [Google Scholar]
- 53.Gururangan S, Turner CD, Stewart CF, et al. Phase I trial of VNP40101M (Cloretazine) in children with recurrent brain tumors: A Pediatric Brain Tumor Consortium study. Clin Cancer Res. 2008;14:1124–1130. doi: 10.1158/1078-0432.CCR-07-4242. [DOI] [PubMed] [Google Scholar]
- 54.MacDonald TJ, Stewart CF, Kocak M, et al. Phase I clinical trial of cilengitide in children with refractory brain tumors: Pediatric Brain Tumor Consortium study PBTC-012. J Clin Oncol. 2008;26:919–924. doi: 10.1200/JCO.2007.14.1812. [DOI] [PubMed] [Google Scholar]
- 55.Neuenschwander B, Branson M, Gsponer T. Critical aspects of the Bayesian approach to phase I cancer trials. Stat Med. 2008;27:2420–2439. doi: 10.1002/sim.3230. [DOI] [PubMed] [Google Scholar]
- 56.Kieran MW, Packer RJ, Onar A, et al. Phase I and pharmacokinetic study of the oral farnesyltransferase inhibitor lonafarnib administered twice daily to pediatric patients with advanced central nervous system tumors using a modified continuous reassessment method: A Pediatric Brain Tumor Consortium study. J Clin Oncol. 2007;25:3137–3143. doi: 10.1200/JCO.2006.09.4243. [DOI] [PubMed] [Google Scholar]
- 57.Pollack IF, Jakacki RI, Blaney SM, et al. Phase I trial of imatinib in children with newly diagnosed brainstem and recurrent malignant gliomas: A Pediatric Brain Tumor Consortium report. Neuro Oncol. 2007;9:145–160. doi: 10.1215/15228517-2006-031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Grossman SA, Carson KA, Phuphanich S, et al. Phase I and pharmacokinetic study of karenitecin in patients with recurrent malignant gliomas. Neuro Oncol. 2008;10:608–616. doi: 10.1215/15228517-2008-030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Gururangan S, Petros WP, Poussaint TY, et al. Phase I trial of intrathecal spartaject busulfan in children with neoplastic meningitis: A Pediatric Brain Tumor Consortium study (PBTC-004) Clin Cancer Res. 2006;12:1540–1546. doi: 10.1158/1078-0432.CCR-05-2094. [DOI] [PubMed] [Google Scholar]
- 60.Cheng JD, Babb JS, Langer C, et al. Individualized patient dosing in phase I clinical trials: The role of escalation with overdose control in PNU-214936. J Clin Oncol. 2004;22:602–609. doi: 10.1200/JCO.2004.12.034. [DOI] [PubMed] [Google Scholar]
- 61.Schöffski P, Riggert S, Fumoleau P, et al. Phase I trial of intravenous aviscumine (rViscumin) in patients with solid tumors: A study of the European Organisation for Research and Treatment of Cancer New Drug Development Group. Ann Oncol. 2004;15:1816–1824. doi: 10.1093/annonc/mdh469. [DOI] [PubMed] [Google Scholar]
- 62.Garrison MA, Hammond LA, Geyer CE, Jr, et al. A phase I and pharmocokinetic study of exatecan mesylate administered as a protracted 21-day infusion in patients with advanced solid malignancies. Clin Cancer Res. 2003;9:2527–2537. [PubMed] [Google Scholar]
- 63.Gilbert MR, Supko JG, Batchelor T, et al. Phase I clinical and pharmacokinetic study of irinotecan in adults with recurrent malignant glioma. Clin Cancer Res. 2003;9:2940–2949. [PubMed] [Google Scholar]
- 64.Rowinsky EK, Rizzo J, Ochoa L, et al. A phase I and pharmacokinetic study of pegylated camptothecin as a 1-hour infusion every 3 weeks in patients with advanced solid malignancies. J Clin Oncol. 2003;21:148–157. doi: 10.1200/JCO.2003.03.143. [DOI] [PubMed] [Google Scholar]
- 65.Le Tourneau C, Stathis A, Vidal L, et al. Choice of starting dose for molecularly targeted agents evaluated in first-in-human phase I cancer clinical trials. J Clin Oncol. 2010;28:1401–1407. doi: 10.1200/JCO.2009.25.9606. [DOI] [PubMed] [Google Scholar]
- 66.Thall PF, Lee JJ, Tseng CH, et al. Accrual strategies for phase I trials with delayed patient outcome. Stat Med. 1999;18:1155–1169. doi: 10.1002/(sici)1097-0258(19990530)18:10<1155::aid-sim114>3.0.co;2-h. [DOI] [PubMed] [Google Scholar]
- 67.Bekele BN, Ji Y, Shen Y, et al. Monitoring late-onset toxicities in phase I trials using predicted risks. Biostatistics. 2008;9:442–457. doi: 10.1093/biostatistics/kxm044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Gupta S, Hunsberger S, Boerner SA, et al. Meta-analysis of the relationship between dose and benefit in phase I targeted agent trials. J Natl Cancer Inst. 2012;104:1860–1866. doi: 10.1093/jnci/djs439. [DOI] [PubMed] [Google Scholar]
- 69.Iasonos A, O'Quigley J. Design considerations for dose-expansion cohorts in phase I trials. J Clin Oncol. 2013;31:4014–4021. doi: 10.1200/JCO.2012.47.9949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Chiuzan C, Garrett-Mayer E. A likehood-based approach to selecting doses with acceptable toxicity in a standard algorithm-based phase I cancer trial. Presented at the 34th Annual Meeting of the Society of Clinical Trials; May 19-22, 2013; Boston, MA. [Google Scholar]
- 71.Iasonos A, O'Quigley J. Continual reassessment and related designs in dose-finding studies. Stat Med. 2011;30:2057–2061. doi: 10.1002/sim.4215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Hamberg P, Ratain MJ, Lesaffre E, et al. Dose escalation models for combination phase I trials. Eur J Cancer. 2010;46:2870–2878. doi: 10.1016/j.ejca.2010.07.002. [DOI] [PubMed] [Google Scholar]
- 73.Iasonos A, Gounder M, Spriggs DR, et al. The impact of non-drug-related toxicities on the estimation of the maximum tolerated dose in phase I trials. Clin Cancer Res. 2012;18:5179–5187. doi: 10.1158/1078-0432.CCR-12-0726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.O'Quigley J, Shen LZ, Gamst A. Two-sample continual reassessment method. J Biopharm Stat. 1999;9:17–44. doi: 10.1081/BIP-100100998. [DOI] [PubMed] [Google Scholar]
- 75.Wages NA, Conaway MR, O'Quigley J. Continual reassessment method for partial ordering. Biometrics. 2011;67:1555–1563. doi: 10.1111/j.1541-0420.2011.01560.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.