

Abstract
Development of higher rates of nondiabetic glomerulosclerosis (GS) in African Americans has been attributed to two coding sequence variants (G1 and G2) in the APOL1 gene. To date, the cellular function and the role of APOL1 variants (Vs) in GS are still unknown. In this study, we examined the effects of overexpressing wild-type (G0) and kidney disease risk variants (G1 and G2) of APOL1 in human podocytes using a lentivirus expression system. Interestingly, G0 inflicted podocyte injury only at a higher concentration; however, G1 and G2 promoted moderate podocyte injury at lower and higher concentrations. APOL1Vs expressing podocytes displayed diffuse distribution of both Lucifer yellow dye and cathepsin L as manifestations of enhanced lysosomal membrane permeability (LMP). Chloroquine attenuated the APOL1Vs-induced increase in podocyte injury, consistent with targeting lysosomes. The chloride channel blocker DIDS prevented APOL1Vs- induced injury, indicating a role for chloride influx in osmotic swelling of lysosomes. Direct exposure of noninfected podocytes with conditioned media from G1- and G2-expressing podocytes also induced injury, suggesting a contributory role of the secreted component of G1 and G2 as well. Adverse host factors (AHFs) such as hydrogen peroxide, hypoxia, TNF-α, and puromycin aminonucleoside augmented APOL1- and APOL1Vs-induced podocyte injury, while the effect of human immunodeficiency virus (HIV) on podocyte injury was overwhelming under conditions of APOLVs expression. We conclude that G0 and G1 and G2 APOL1 variants have the potential to induce podocyte injury in a manner which is further augmented by AHFs, with HIV infection being especially prominent.
Keywords: APOL1 risk variants, kidney disease, podocyte, necrosis, lysosomal membrane permeability
african americans (aas) develop higher rates of progressive nephropathy, including focal segmental glomerulosclerosis (FSGS), human immunodeficiency virus (HIV)-associated nephropathy (HIVAN), and hypertension-associated end-stage kidney disease (ESKD) compared with European Americans (EAs) (25, 37, 43). This disparity is the most pronounced in the case of HIVAN, reaching a disparity which is >10-fold (25). This overwhelming population disparity between AAs and EAs and, greater familial clustering among AAs, point to a prominent contribution of underlying genetic factor(s) (14). Recently, increasing evidence has shown that this major health disparity is strongly associated with two coding sequence variants (G1 and G2) in APOL1 (12, 15, 19, 42).
APOL1 is located at chromosome 22q13.1, and it encodes the minor apolipoprotein L1 component of HDL. This apolipoprotein is expressed in the liver, pancreas, kidney, and brain and in macrophages, endothelial, and several other cell types (9, 10). The G1 variant (rs73885319) is a compound missense mutation (S342G:I384M) encoding two nonsynonymous amino acids. The G2 variant is a 6-bp in-frame deletion which has resulted in the loss of two amino acids (N388 and Y389) at the C-terminal helix of APOL1 (18, 48). These two allelic variants arose on separate human phylogenetic lineages, and they have never been observed together on the same parental chromosome. Approximately 34% of AAs possess one of the two risk variants (APOL1Vs), and ∼13% have both coding variants (18, 25, 48). In the United States, ∼3 million AAs carry both risk alleles. In contrast, APOL1Vs occur infrequently in EAs, ∼0.3% carry G1 and 0.1% G2 alleles. These studies are consistent with African admixture (18, 25, 48). Wild-type (WT) APOL1 circulates in plasma and has the ability to kill the trypanosome (Trypanosome brucei) responsible for causing sleeping sickness in cattle.
The Trypanosoma brucei rhodesiense (Tbr) subspecies is resistant to the trypanolytic effect of APOL1. This is due to the presence of a serum resistance-associated (SRA) factor. SRA allows for Tbr to induce human sleeping sickness (25, 44). The G1 and G2 coding variants confer resistance, in humans, to Tbr. The presence of even a single variant has been shown to confer resistance. Thus these variants have gained prominence in parts of the world with significant rates of sleeping sickness. The risk allele state is now highly associated with increased risk for major forms of nondiabetic kidney disease in descendants, including AAs (18, 19, 25, 42, 48). Recent clinical studies have also implicated the coding variants with renal allograft failure (24, 38). In these studies, kidneys donated from deceased donors with APOL1Vs lasted for shorter periods, independently of recipient ancestry.
Although clinical studies have clearly shown a strong association of APOL1Vs with progressive nondiabetic kidney disease, the cellular function and the role of APOL1Vs in the pathogenesis of kidney injury are still unknown. Localization studies have implicated the podocyte as a target (31). Most proteinuric diseases including glomerular sclerosis and HIVAN are associated with podocytopathy (altered podocyte phenotype, reduction in number, and effacement of foot processes) and tubulopathy (32, 33).
In the present study, we examined the effect of APOL1WT (G0) and APOL1Vs (G1 and G2) on human podocytes. Additionally, we have studied the mechanism involving APOL1Vs-induced podocyte injury.
MATERIALS AND METHODS
Culture of human podocytes.
Human podocytes were cultured as previously reported (27, 40). Briefly, immortalized human podocytes proliferated in the growth medium containing RPMI 1640 supplemented with 10% fetal bovine serum, 1× penicillin-streptomycin, 1 mM l-glutamine, and 1× insulin, transferrin, and selenium (ITS; Invitrogen, Grand Island, NY) at permissive temperature (33°C). When the cells attained ∼80% confluence, they were transferred at 37°C for differentiation in a medium without ITS for 5–7 days.
Production of pseudotyped retroviral supernatant and transduction of podocytes.
Replication-defective viral supernatants were prepared as published previously (21). Briefly, a green fluorescent protein (GFP) reporter gene (from pEGFP-C1; Clontech, Palo Alto, CA) was substituted in place of gag/pol genes in HIV-1 proviral construct pNL4–3. This parental construct (pNL4–3: ΔG/P-GFP) was used to produce VSV.G pseudotyped viruses to provide pleiotropism and high-titer virus stocks. Infectious viral supernatants were produced by transient transfection of 293T cells using Effectene (Qiagen). The HIV-1 gag/pol and VSV.G envelope genes were provided in trans using pCMV R8.91 and pMD2.G plasmids, respectively (gifts of Dr. Didier Trono, Salk Institute, La Jolla, CA). As a negative control, virus was also produced from pHR-CMV-IRES2-GFP-ΔB, which contained HIV-1 LTRs and a GFP empty expression vector. The viral stocks were titrated by infecting 293T cells with 10-fold serial dilution. Viral stocks ranging from 105 to 106 GFP-expressing units (GEU)/ml were obtained. The cells were infected with MOI of 0.5 GEU for 2 h.
Lentivirus preparation.
To amplify the DNA fragments corresponding to the whole APOL1 open reading frames, a forward primer GGATCCATGGAGGGAGCTGCTTTGCTGAGAG (BamHI cleavage site underlined) and reverse primer GTCGACTCACAGTTCTTGGTCCGCCTGCAG (SalI cleavage site underlined) were designed based on the sequences around the initial and stop codons, respectively. The PCR-amplified products were double-digested with BamHI and SalI and subcloned into a LG12 lentiviral vector to obtain a transfer plasmid designated LG12-“APOL1” 5. The transcription of targeting genes was driven by a CMV promoter. psPAX2 was the HIV-derived packaging construct, and the pMD2.G construct expressed the VSV-G envelope protein. The APOL11 lentivirus was produced by transfection of 293T cells by using Effectene Transfection Reagent (Qiagen), and the virus titer was determined by using QuickTiter Lentivirus Titer Kit (Cell Biolabs, San Diego, CA), following the manufacturer's instructions.
Quantitative RT-PCR.
Total RNA was isolated from human podocytes using TRIzol reagent (Invitrogen). Five micrograms of total RNA were reverse transcribed using the first-strand synthesis system (Invitrogen). Quantitative PCR (qPCR) was carried out in an ABI Prism 7900HT sequence detection system. The primer sequences were AGGGAGCTGCTTTGCTGAGAGT (APOL1-FW) and AGTCACCGAGGGGCTTACTTTG (APOL1-RV). GAPDH was used as an internal control, and the primers were GGGAAGCTCACTGGCATGGCCTTCC (GAPDH-FW) and CATGTGGGCCATGAGGTCCACCAC (GAPDH-RV).
Lactate dehydrogenase activity detection.
After infection with the lentivirus, human podocytes were cultured in medium containing 1% serum. At 24 h postinfection (hpi), the medium was harvested for lactate dehydrogenase (LDH) activity detection with a LDH Cytotoxicity Detection Kit (Clontech, Mountain View, CA) following the manufacturer's instructions.
Trypan blue staining.
Differentiated human podocytes were transduced with the lentivirus for 48 h and then rinsed with PBS to remove the floating cells and cell debris. Cells were detached with Accutase (Innovative Cell Technologies, San Diego, CA), subsequently labeled with 0.2% trypan blue, and counted under the light microscope. The unstained cells (white) were considered as live cells, while the blue cells were dead.
Propidium iodide and Hoechst staining.
Proidium iodide (PI) and Hoechst staining was performed as previously reported (46). Briefly, after appropriate treatment, the culture media was removed from the cells and fresh media containing Hoechst 33342 (10 μg/ml) was added. Cells were subsequently incubated for 10 min at 37°C. Then, a PI solution was added and the cell dishes were kept on ice for 7 min. Cell images were taken immediately with a Zeiss microscope (Carl Zeiss MicoImaging, Jena, Germany) equipped with a digital imaging system.
Assessment of lysosomal structural integrity.
Lysosomes were stained with Lucifer yellow (Invitrogen) and LysoTracker Red (Invitrogen) as described previously (6, 7). Briefly, cells were cultured overnight in the medium containing 1 mg/ml Lucifer yellow, and then 500 nM LysoTracker was added followed by an additional 30-min incubation. Then, the medium was removed, and the cells were rinsed three times with PBS. The cells were then fixed with 4% paraformaldehyde (PFA) for 10 min. The lysosomes were visualized, and images were captured with a Zeiss microscope. Cathepsin L activity was detected using a Magic Red Cathepsin L Kit (ImmunoChemistry Technologies, Bloomington, MN) following the manufacturer's instructions. The F-actin cytoskeleton was visualized using Alexa Fluor 594-conjugated phalloidin, as previously reported (28).
Western blotting.
Western blotting was performed as described (26, 27). Briefly, cells were washed with PBS and lysed in RIPA buffer [1× PBS, pH 7.4, 0.1% SDS, 1% NP-40, 0.5% sodium deoxycholate, 1.0 mM sodium orthovanadate, 10 μl of protease inhibitor cocktail (100×, Calbiochem)/1 ml of buffer, and 100 μg/ml PMSF]. Proteins (20–30 μg) were separated by 12% SDS-PAGE and then transferred to an Immuno-Blot polyvinylidene fluoride (PVDF) membrane (Bio-Rad, Hercules, CA). After blocking in PBS/Tween (0.1%) with 5% nonfat milk, the membrane was incubated with primary antibodies overnight at 4°C followed by horseradish peroxidase-conjugated secondary antibodies (1:3,000, Santa Cruz Biotechnology) and then developed using Enhanced Chemiluminescent (ECL) solution (Pierce). The primary antibodies used for immunodetection were goat anti-APOL1 (1:1,000, Santa Cruz Biotechnology) and goat anti-actin (1:3,000, Santa Cruz Biotechnology). For protein expression quantification, the developed films were scanned with a CanonScan 9950F scanner, and the acquired images were then analyzed using the public domain National Institutes of Health image-analysis program (http://rsb.info.nih.gov/nih-image/).
Statistical analyses.
Data are presented as means ± SD unless otherwise noted. All experiments were conducted and repeated at least three times with duplicate or triplicate samples in each assay. All data were evaluated statistically by ANOVA, followed by Newman-Keuls multiple comparison tests using Prism 4.0 (GraphPad Software). In the case of single mean comparison, data were analyzed by Student's t-test. P values <0.05 were considered as statistically significant.
RESULTS
Overexpression of APOL1Vs increases podocyte necrosis.
To investigate the role of APOL1Vs in the induction of podocyte injury, we overexpressed APOL1Vs (G1 and G2) as well as APOL1WT (G0) in human podocytes employing a GFP-containing lentivirus expression system. Human podocytes were transduced with the same quantity of lentivirus (titrated as 0.8 pg p24 protein/cell), harboring APOL1WT or APOL1Vs cDNAs. Transduction efficiency was quantified using GFP expression. It was observed to be 90%. Subsequently, the overexpression of recombinant APOL1 in podocytes was confirmed. For time course experiments, cell lysates were collected for Western blot assay. Podocytes displayed initial APOL1WT and Vs expression within 24 h of infection (Fig. 1, A and B), with increasing overexpression by 48 h (Fig. 1, C and D).
Fig. 1.

Overexpression of APOL1 in human podocytes. Differentiated human podocytes were transduced with a lentivirus [titrated as 0.8 pg human immunodeficiency virus (HIV) p24 protein/cell] for 3 h and were cultured in fresh medium. Cell lysates were collected at 24 (A) and 48 (C) h postinfection (hpi) and were subjected to Western blot assay. B and D: quantification of the expression of APOL1 in A and C, respectively, and the results are means ± SD of 3 independent samples. V, vector alone; W, APOL1, wild-type (WT); G1, APOL1 G1 variant; G2, APOL1 G2 variant. *P < 0.05 compared with control (vector).
To determine the effect of APOL1WT and Vs on podocyte injury, we overexpressed these variants in differentiated human podocytes and analyzed podocyte LDH activity. At 24 h, APOL1WT increased podocyte LDH activity, and podocytes expressing G1/G2 variants exhibited even higher levels of LDH activity compared with APOL1WT-expressing cells (Fig. 2A).
Fig. 2.
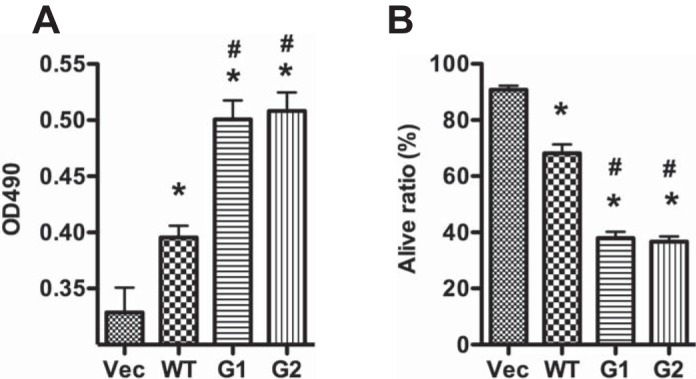
APOL1 risk variants increase podocyte injury. Human podocytes were transduced with the lentivirus (titrated as 0.8 pg HIV p24 protein/cell) for 3 h and were cultured in medium containing 1% serum. A: medium was collected for lactate dehydrogenase (LDH) activity determination at 24 h hpi. B: cells were detached with Accutase and subjected to trypan blue staining at 48 hpi. *P < 0.05 compared with vector. #P < 0.05 compared with APOL1WT.
We then evaluated the effect of APOL1WT (G0) and APOL1Vs (G1 and G2) on podocyte viability using trypan blue staining. Although both APOL1WT and APOL1Vs displayed an increased number of trypan blue-positive podocytes compared with noninfected podocytes, nonetheless podocytes expressing either G1 or G2 constructs displayed a greater number of trypan blue-positive cells compared with G0 (Fig. 2B).
Overexpression of APOL1WT has been reported to induce autophagic cell death in cancer cell lines (47, 49). To determine the type of the cytotoxicity induced by APOL1WT and APOL1Vs, we examined their effect on podocyte morphology under a phase-contrast microscope. Podocytes transduced with G0 or G1- and G2-containing lentiviruses showed a higher percentage of cells expressing a swollen phenotype compared with the vector alone (Fig. 3, A and B). This swollen phenotype precipitated detachment, signifying loss of cell viability. The effect was significantly more pronounced for cells transduced with the G1 and G2 lentivirus.
Fig. 3.

APOL1 risk variants increase podocyte swelling. A–C: human podocytes were transduced with the lentivirus (titrated as 0.8 pg HIV p24 protein/cell) for 3 h and were cultured in the fresh medium for 48 h. Swollen cells (black arrows) were visualized (A) and counted (B) under a contrast-phase microscope. C: swollen cells were stained with propidium iodide (PI) and visualized under a contrast-phase microscope. D and E: human podocytes were infected with half the dose of lentivirus (titrated as 0.4 pg HIV p24 protein/cell), and swollen cells (D) and living cells (E) were counted. Scale bars = 100 and 20 μm for A and C, respectively. *P < 0.05 compared with vector. #P < 0.05 compared with APOL1WT. F: podocytes were incubated in media in the absence or presence of different concentrations of interferon-γ (0, 1, 2.5, 10, and 20 ng/ml) for 48 h. Subsequently, RNA was extracted and cDNA was probed for APOL1 with qPCR. *P < 0.05 compared with 0 interferon-γ.
We then stained podocytes transduced with APOL1G0 and APOL1Vs with PI and Hoechst 33342 and examined these cells under an inverted fluorescence microscope. Interestingly, the swollen cells also contained swollen nuclei (Fig. 3C) and were either necrotic or experiencing necrosis.
Since APOL1WT expression inflicted podocyte injury, we asked whether injury was podocyte APOL1 expression dependent. To determine the effects of lowering expression of APOL1, podocytes were infected with half the dose of lentivirus (titrated as 0.4 pg p24 protein/cell) for 3 h and then incubated in fresh media for 48 h. Live and swollen cells were counted. As shown in Fig. 3, D and E, podocytes expressing the APOL1WT did not display obvious podocyte injury; on the other hand, podocytes expressing G1 or G2 displayed a decreased percentage of viable cells and an increased percentage of swollen cells.
Since higher concentrations of APOL1G0 induced podocyte injury, we asked whether an inflammatory milieu has the potential to enhance podocyte G0 expression. Podocytes were incubated in media containing different concentrations of interferon-γ (0, 1, 2.5, 10, and 20 ng/ml) for 48 h. Subsequently, RNA was extracted and cDNA was probed for APOL1. As shown in Fig. 3F, interferon-γ enhanced podocyte endogenous APOL1 expression in a dose-dependent manner. Thus elevated APOL1 expression in response to an inflammatory milieu could have the potential to contribute to ongoing injury.
To visualize the effect of APOL1Vs on podocyte injury, we performed PI/Hoechst staining after infecting the cells with the lentivirus at 48 hpi. APOL1Vs promoted both primary (PI stained single swollen nuclei) and secondary (PI stained fragmented nuclei) necrosis compared with APOL1G0 (Fig. 4). Taken together, these findings indicate that high levels of APOL1Vs are cytotoxic to human podocytes.
Fig. 4.

APOL1 risk variants increase podocyte swelling. Human podocytes were transduced with the lentivirus (titrated as 0.8 pg HIV p24 protein/cell) for 3 h and were cultured in fresh medium. Cells were subjected to PI/Hoechst staining at 48 hpi. Representative microphotographs (A) were selected to show the apoptotic (green arrow), necrotic (white arrow), and secondary necrotic (yellow arrow) cells, and the ratios of these cells were calculated (B). Scale bars = 50 μm. *P < 0.05 compared with vector, while # P < 0.05 compared with APOL1WT.
Knockdown of APOL1 doesn't increase podocyte injury.
Gene mutation often leads to the loss of functionality of the encoded protein, in turn causing a loss-of-function disease phenotype with recessive inheritance (e.g., sickle cell anemia). To examine whether podocyte loss of function was involved in APOL1-associated nephropathy, APOL1 expression was silenced in human podocytes [small interfering (si) RNA-APOL1/podocytes], and then podocyte phenotype was examined. The siRNA-APOL1/podocytes displayed a ∼60% decrease in protein expression (Fig. 5, A and B), confirming silencing of this gene. However, this was not associated with induction of either LDH activity or cell loss (Fig. 5, C and D). Moreover, siRNA-APOL1/podocytes did not display morphological alterations. These results suggest that APOL1 is not essential for the short-term structural or functional integrity of podocytes. This supports the notion that APOL1Vs-associated nephropathy is not due to the loss of baseline APOL1 functionality in podocytes. It is also important to note that the APOL1 gene exists only in humans and several nonhuman primates and appears to be dispensable in rodents and other mammals (3).
Fig. 5.

Knockdown of APOL1 does not increase podocyte injury. Differentiated human podocytes were transfected with small interfering (si) APOL1 or siCon for 12 h and then were cultured in fresh medium for 48 h. A: cell lysates were collected for Western blotting. B: quantification of the expression of APOL1 in A. Values are means ± SD of 3 independent samples. NS, not statistically different. *P < 0.05 compared with siCon.
Overexpression of APOL1Vs increases podocyte lysosomal membrane permeability.
Pérez-Morga et al. (36, 45) reported that APOL1 promotes trypanosomal lysis by forming pores in lysosomal membranes. We considered whether APOL1 could compromise podocyte integrity through a similar mechanism. To test this, we compared the effects of APOL1G0 and Vs on podocyte lysosomal membrane permeability (LMP), with LysoTracker to label lysosomes. Both G1 and G2 variants drastically decreased the number of punctated structures (normal lysosomes) in podocytes compared with vector control (Fig. 6, A and B). Such a decrease may represent a consequence of the dramatic necrotic response evident in Figs. 2–4. To confirm the specificity of APOL1-induced effects on LMP, corresponding podocytes were stained with Lucifer yellow. Vector-treated podocytes displayed containment of dye within lysosomes; on the other hand, G1 or G2 podocytes displayed diffuse lysosomal leak of dye into the cytosolic and nuclear compartments (Fig. 6, A–C).
Fig. 6.

APOL1 risk variants increase podocyte lysosomal membrane permeability. A–G: human podocytes were transduced with the lentivirus (titrated as 0.8 pg HIV p24 protein/cell) for 3 h and were cultured in fresh medium for 48 h. LysoTrack red (A and B) and Lucifer yellow (A and C) staining was performed. Cathepsin L activity (D and E) was detected with a Magic Red Cathepsin L kit. F-actin (F and G) was detected with Alexa Fluor 594-conjugated phalloidin. Images were captured with a confocal fluorescence microscope, and representative microphotographs were selected (A, D, and F). Scale bars = 10, 20, and 100 μm for A, D, and F, respectively. The average fluorescence intensity was measured per region of interest (ROI) for each group (B, C, E, and G). H and I. human podocytes were transduced with the lentivirus (titrated as 0.4 pg HIV p24 protein/cell) for 3 h and were cultured in 1% serum medium with or without 100 μM DIDS for 72 h. Then, swollen cells (H) and living cells (I) were counted. *P < 0.05 compared with vector. #P < 0.05 compared with control.
Since leakage of lysosomal enzymes such as cathepsin L has been reported to cause podocyte injury (39, 41), we examined the leakage of podocyte lysosomal cathepsin L in response to transduction with the APOL1G0 and APOL1Vs encoding lentivirus constructs. In these studies, cathepsin L labeling was carried out using the Magic Red kit as described in materials and methods. APOL1Vs podocytes displayed diffuse distribution of cathepsin L in the cytosolic compartment; on the contrary, vector-treated podocytes showed containment of cathepsin L within lysosomes (punctate morphology) (Fig. 6, D and E). In turn, a consequence of enhanced LMP and lysosomal cathepsin L leakage is the degradation of the cytoskeletal protein F-actin (41). To compare the effect of APOL1G0 with APOL1Vs on podocyte F-actin, control and experimental podocytes were labeled for F-actin by using Alexa-labeled phalloidin. APOL1Vs podocytes displayed scant actin filaments compared with both vector alone and APOL1G0 (Fig. 6, F and G). These findings suggest that APOL1 variants could be compromising the actin cytoskeleton by enhancing podocyte LMP.
A chloride channel blocker inhibits APOL1Vs-induced podocyte swelling.
Since APOL1 has been demonstrated to induce lysosomal membrane depolarization and continuous influx of chloride, and subsequent osmotic swelling of the lysosomes in trypanosomes (36), we evaluated the effects of chloride channel blockers on APOLVs-induced podocyte swelling. G1 and G2 podocytes were incubated in media containing either buffer or the chloride channel inhibitor DIDS for 48 h. Subsequently, swollen and viable nonswollen podocytes were counted. As shown in Fig. 6, H and I, the chloride channel inhibitor provided protection against G1- and G2-induced podocyte injury.
Blocking membrane traffic attenuates APOL1Vs-induced podocyte injury.
Since APOL1 is also a secreted protein and normally present in the circulation, it could reach the lysosomal compartments via an endosomal trafficking pathway (37). Therefore, we studied the effect of chloroquine (CQ), which blocks this pathway. Chloroquine attenuated the number of swollen and dead cells in APOL1Vs podocytes (Fig. 7, A and B). Similarly, changing the media every 12 h resulted in increased cell viability (Fig. 7C); these findings indicated that uptake of secreted APOL1Vs partially contributed to podocyte injury.
Fig. 7.

Role of secretory APOL1 in podocyte injury. A and B: human podocytes were transduced with the lentivirus for 3 h and were cultured in 1% serum medium with or without 2.5 μM chloroquine (CQ). At 48 hpi, swollen cells (A) and living cells (B) were counted. *P < 0.05 compared with control group without chloroquine. C: human podocytes were transduced with the lentivirus (titrated as 0.8 pg HIV p24 protein/cell) for 3 h and were cultured in fresh medium. After every 12 h, medium was removed and cells were refreshed (washed) with new medium. At 48 hpi, cells were detached with Accutase and were subjected to trypan blue staining *P < 0.05 compared with control (without wash). D and E: human podocytes were transduced with the lentivirus (titrated as 0.8 pg HIV p24 protein per cell) for 3 h, then were washed with PBS and cultured in fresh medium. At 72 hpi, the medium was harvested. After centrifugation, the supernatant was collected as conditioned medium (CM) and was added to new human podocytes. After another 36 h, swollen cells (D) and living cells (E) were counted. *P < 0.05 compared with vector. #P < 0.05 compared with APOL1WT.
To confirm the role of secreted APOL1, podocytes were incubated in media containing conditioned media (CM; collected from vector-, G0-, G1-, and G2-expressing podocytes) for 36 h. Swollen cells and viable cells were quantified. Podocytes treated with CM-G1 and CM-G2 displayed higher numbers of swollen cells and lower numbers of viable cells compared with respective CM-vector and CM-G0 (Fig. 7, D and E). These findings indicate that factor elaborated into the media in the CM-G1 and CM-G2 contributed to podocyte injury.
APOL1Vs make podocytes vulnerable to adverse host factors.
Only a percentage fraction of the population with the APOL1 risk genotype actually develop chronic kidney disease, suggesting the need for a secondary hit factor (42). Accordingly, we examined the effect of several anticipated adverse host factors (AHFs) on the state of podocyte injury in the setting of the WT and APOL1Vs. To determine the effects of such previously well-characterized AHFs, podocytes expressing vector, G0, G1, and G2 were exposed to H2O2, hypoxia, TNF-α, puromycin aminonucleoside, and HIV for different time periods, as indicated in Fig. 8. Subsequently, the percentage of viable cells was counted. AHFs decreased the number of living cells both in human podocytes infected with vector and with APOL1G0. However, the effects of AHFs were greatly augmented in podocytes expressing the APOL1 G1 and G2 risk variants. The most prominent difference was observed in the case of the response to HIV. These findings indicate that podocytes expressing APOL1 are more susceptible to injury in the presence of AHFs. Thus it seems likely that depending on the severity of such secondary hit AHFs, and the degree of APOL1 expression, podocytes may pass a threshold of injury sufficient to cause progressive nephropathy. In this regard, it is noteworthy that some of the AHF's which cause greater injury in podocytes expressing risk versions of APOL1 might themselves also induce the expression of APOL1. This would be the case especially for HIV and possibly other viral pathogens, which elicit an interferon-γ response (17).
Fig. 8.

Adverse host factors accentuate APOL1-induced podocyte injury. Human podocytes were transduced with the lentivirus (titrated as 0.4 pg HIV p24 protein/cell) for 3 h and were cultured in fresh medium. After 24 h, H2O2 (600 μM), TNF-α (100 ng/ml), or puromycin aminonucleoside (30 μM) was added. For hypoxia treatment, cells were moved into a 1% oxygen incubator; for HIV treatment, cells were infected with pseudotype NL4–3 following the APOL1-expressing lentivirus infection. After another 8 (H2O2), 24 (hypoxia, TNF-α, puromycin), or 48 h (HIV), cells were detached with Accutase and were subjected to trypan blue staining. *P < 0.05 compared with control (without wash).
DISCUSSION
There have been many clinical reports suggesting the role of APOL1Vs in the development of APOL1-associated nephropathy. However, to date there is a paucity of information in in vitro studies exploring the mechanism. Accordingly, this study was conducted to test the biological effects of APOL1 and its variants to determine whether there is a pathological counterpart to the differential effect of G0 and its kidney disease risk association variants, in a potentially relevant cell type. We examined several potential mechanisms wherein APOL1Vs could be causative in the greater susceptibility of African Americans to progressive major forms of chronic kidney disease in the category of podocytopathies. Since even APOL1G0 is toxic to cells when markedly overexpressed (47, 49), we titrated lentivirus-expressing APOL1 non-risk and risk variants to achieve levels of expression wherein G0 would have minimal toxicity. At the same doses, G1 and G2 display cell toxicity. These findings provide the first clear-cut biological, functional counterpart to the observed association studies and validate the variants as being causative and not only associated with cellular injury. Podocyte injury was evident as the increased percentages of swollen and necrosed cells. However, higher levels of G0 expression also increased LDH activity, decreased cell viability, and enhanced LMP and necrotic cell death; moreover, comparably high levels of G1 or G2 still further enhanced the manifestation of these phenotypes. These findings suggest that at comparable lower levels of expression, the G1 and G2 variants of APOL1 are more conducive to podocyte cell injury compared with G0 or APOL1WT. This is potentially relevant to the formulation of a threshold effect as an explanation for the recessive or two-risk allele inheritance mode, as has been consistently reported (18).
The absence of a cellular phenotype after knockdown of endogenous APOL1 in human podocytes is also consistent with the observation that APOL1 is dispensable for kidney integrity (22). Interestingly, frequent changes of incubation media decreased APOLVs-induced podocyte injury, while direct exposure of conditioned media of G1 or G2 promoted podocyte injury; these findings indicate that a secretory component of APOL1Vs contributes to podocyte injury. Thus it is premature to dismiss the potential contributory roles of both intracellular and secretory APOL1 gene products in the pathophysiology of kidney injury in different clinical states and perhaps reflecting isoforms of APOL1 with differences in the signal peptide domain (8). Since chloroquine partially attenuated APOL1-induced podocyte injury, a role of endosomal traffic in transport of APOL1 may be inferred. Interestingly, the chloride channel inhibitor DIDS attenuated APOL1-induced podocyte swelling; these findings indicate that secretory effector molecule(s) transported via membrane trafficking may be targeted to lysosomes, in turn contributing to abnormal chloride influx and lysosomal swelling. As a cellular organelle, the lysosome is a membrane-bound vesicle loaded with low pH-dependent hydrolytic enzymes. The integral double-layer membrane around lysosome limits these enzymes within the lumen and protects the cytosolic proteins from degradation. Increasing LMP will lead to the leakage of lysosomal enzymes into the cytosol, resulting in a series of severe cell injuries, including cytoskeleton degradation, apoptosis, and necrosis (1, 4, 5, 20). In the present study, overexpression of APOL1G1 and G2 in podocytes dramatically increased both LMP and the leakage of lysosomal components into the cytosol, which was also accompanied by APOL1-induced F-actin degradation and necrotic cell death in podocytes.
In this study, chloroquine, a known membrane traffic blocker, partially attenuated APOL1Vs-induced podocyte cell injury; moreover, the change of media every 12 h also provided partial protection against APOL1Vs-induced podocyte injury. Direct exposure of the conditioned media of G1 and G2 to podocytes inflicted podocyte injury. All of these findings indicate that uptake of secreted APOL1Vs through endocytosis might have also contributed to podocyte injury and that abnormal chloride flux could be contributing to osmotic lysosomal swelling, overwhelming the capacity to remove damaged lysosomes. Lysosome-mediated cell injury is also consistent with the observations of Perez-Morga et al. (36) displaying the effect of APOL1 on the depolarization of lysosomal membranes and continuous influx of chloride. Since it appears from the current findings that endosomal pathways may be required for APOL1 to target lysosomes, as also suggested by other investigators studying APOL1-induced lysosomal injury (36, 45), modulation of such membrane traffic, in addition to chloride channel blockade, could be used as a potential therapeutic means to ameliorate APOLVs-induced podocyte injury. Nonetheless, it will be important in future studies to delineate the details of the membrane traffic pathways and their role in the induction of podocyte lysosomal injury associated with APOL1Vs.
Genetic epidemiological studies have demonstrated that, while the odds ratios for kidney disease risk are high in association with APOL1Vs, only a small percentage of the population with harboring of APOL1Vs actually develops kidney disease. This suggests that genetic susceptibility is combined with a nongenetic second, which transforms risk to disease causation. This is especially prominent in the case of HIVAN, wherein in the absence of APOL1 genetic susceptibility HIVAN is rare to absent, and in the presence of APOL1 genetic susceptibility, poor control of HIV viral load leads to HIVAN with a very high likelihood (3, 11). To examine the pathobiological underpinnings of this second-hit formulation, we evaluated the effect of G0, G1, and G2 versions of APOL1 in the presence of AHFs such as H2O2, hypoxia, TNF-α, puromycin aminonucleoside, and HIV. AHFs induced only mild podocyte injury in vector- and G0-expressing cells; however, the injury's effect was more pronounced on top of a higher level of baseline injury in G1 and G2 podocytes. Thus it appears that both the expression levels of APOL1 and severity of one or more second hits may determine the sustenance of podocyte injury, past a threshold which may not be altered by compensatory mechanisms utilized for the maintenance of integrity of the glomerular filtration barrier (16).
Consistent with the clinical and epidemiological observations, the second-hit counterpart was the most pronounced in the case of HIV. Among individuals with APOL1Vs genotypic susceptibility, >50% will develop HIVAN, which is about 10 times higher than those without HIV infection. This type of injury can be mitigated by successful viral control (2, 25, 30, 32). These studies indicate that APOL1Vs may cause particularly severe renal injury in the setting of HIV infection. In a previous study, we noted that HIV increased lysosomal enzyme cathepsin L activity, which further compromised the integrity of the podocyte cytoskeleton (7). Taken together, we speculate that APOL1Vs and HIV may act synergistically on such lysosomal damage, leading to enhanced leakage of lysosomal components, exceeding the capacity to deal with damaged lysosomes by normally available pathways, and stronger cytosol cathepsin L activity than in the case of either APOL1Vs or HIV alone. This could be a possible mechanism for the prominence of the association of APOL1Vs with HIVAN. Thus it appears that both the expression levels of APOL1 and the severity of a second hit act in concert to determine the extent of podocyte injury. In patients, there may also be compensatory repair mechanisms utilized for the maintenance of integrity of the glomerular filtration barrier.
Although we observed that APOL1 variants would lead to podocyte loss, the underlying molecular mechanism is still unknown. The T. brucei subspecies rhodesiense and gambiense resist the lytic activity of Apol1WT and can infect humans, causing sleeping sickness (29). In the case of Tbr, resistance, their lysis involves interaction of the SRA protein with the C-terminal helix of APOL1 (14, 19, 25). This observation led us to believe that there may be functional SRA analogs in human serum or on the cell vesicle membranes. We propose that it would be important to examine whether the APOL1 protein may have the capacity to cause lysosomal membrane damage, while this toxic function is blocked in the WT through binding to such a functional SRA homolog at the C terminus. To examine this possibility, we constructed an artificial G1/G2 APOL1 variant containing both mutations of G1 and G2 on the same sequence. We observed that expression of G1/G2 showed far greater toxicity to podocytes compared with G1 or G2 alone. For example, trypan blue staining showed a ratio of dead cells at 48 hpi was close to 50% for either G1 or G2, but 70% for the combined G1/G2 construct (data not shown). These findings are consistent with the gain-of-injury formulation noted and provide a potentially important tool to screen for protective molecules. While the current study focused on the effects of APOL1 on podocytes in culture, this does not exclude additional sites of expression, cellular targets, or mechanisms. We also recognize that cells in culture can amplify and accelerate cellular injury processes, causing them to appear in relatively shorter time periods, compared with the corresponding clinical situations. This may be attributed to lack of compensatory defense and repair mechanisms, which apply in the whole organism. Cell culture systems amplify effects, which take longer or are milder in vivo, and this can be attributed to a hostile cell growth environment with an absence of biological matrices, reduced serum factors, and induction of increased cellular expression of protein. Nevertheless, this platform has enabled the clear-cut discernment of differential biological effects of APOL1 and its variants in a relevant experimental platform that has been used extensively in the past to study mechanisms involved in podocytopathies. By titrating expression of APOL1 G0 to levels which did not induce podocyte injury, but in which corresponding levels of G1 and G2 expression did inflict such injury, together with the differences in degrees of injury at higher concentrations using multiple assays, and the augmented adverse host factor effects, this study provides compelling preliminary evidence that extends beyond genetic association and should enable causative mechanisms to be examined.
In summary, overexpression of APOL1G0, G1, and G2 are toxic to podocytes. However, importantly, the APOL1 G1 and G2 variants induce a greater degree of podocyte injury, which manifests in the form of cellular swelling and necrosis. This effect is accompanied by evidence of enhanced LMP, F-actin disruption, and can be blocked partially by chloroquine and a chloride channel inhibitor. It is very likely that many other mechanisms may be involved, as those cells other than podocytes may be targeted as well. Nevertheless, as far as current literature is concerned, this is the first report of a differential effect of APOL1 risk variants on cellular function in kidney cells and therefore provides a platform for unraveling the molecular interaction that is responsible. On that account, the present study provides insight into the mechanisms involved in APOL1-associated nephropathy and would help to set up high-throughput compound screening and potential prevention and therapy for APOL1-associated nephropathy.
GRANTS
This work was supported by Grants RO1DK 098074, RO1DK084910, and RO1 DK083931 (P. C. Singhal) from the National Institutes of Health (Bethesda, MD). K. Skorecki was supported by grants from the Ernest and Bonnie Beutler Grant Program at Rambam Medical Center, the Binational Science Foundation, and the Israel Science Foundation.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
Author contributions: X.L. and P.C.S. provided conception and design of research; X.L., A.J., and S.A. performed experiments; X.L., H.W., J.M., and A.M. analyzed data; X.L. drafted manuscript; X.L., A.J., K.C., H.W., M.A.S., P.W.M., S.A., A.M., K.L.S., and P.C.S. approved final version of manuscript; K.C., M.A.S., P.W.M., J.M., and K.L.S. interpreted results of experiments; A.M., K.L.S., and P.C.S. edited and revised manuscript.
REFERENCES
- 1.Artal-Sanz M, Samara C, Syntichaki P, Tavernarakis N. Lysosomal biogenesis and function is critical for necrotic cell death in Caenorhabditis elegans. J Cell Biol 173: 231–239, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Atta MG. Diagnosis and natural history of HIV-associated nephropathy. Adv Chronic Kidney Dis 17: 52–58, 2010 [DOI] [PubMed] [Google Scholar]
- 3.Behar DM, Kedem E, Rosset S, Haileselassie Y, Tzur S, Kra-Oz Z, Wasser WG, Shenhar Y, Shahar E, Hassoun G, Maor C, Wolday D, Pollack S, Skorecki K. Absence of APOL1 risk variants protects against HIV-associated nephropathy in the Ethiopian population. Am J Nephrol 34: 452–459, 2011 [DOI] [PubMed] [Google Scholar]
- 4.Boya P, Andreau K, Poncet D, Zamzami N, Perfettini JL, Metivier D, Ojcius DM, Jäättelä M, Kroemer G. Lysosomal membrane permeabilization induces cell death in a mitochondrion-dependent fashion. J Exp Med 197: 1323–1334, 2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Boya P, Kroemer G. Lysosomal membrane permeabilization in cell death. Oncogene 27: 6434–6451, 2008 [DOI] [PubMed] [Google Scholar]
- 6.Castro J, Bittner CX, Humeres A, Montecinos VP, Vera JC, Barros LF. A cytosolic source of calcium unveiled by hydrogen peroxide with relevance for epithelial cell death. Cell Death Differ 11: 468–478, 2004 [DOI] [PubMed] [Google Scholar]
- 7.Chandel N, Sharma B, Husain M, Salhan D, Singh T, Rai P, Mathieson PW, Saleem MA, Malhotra A, Singhal PC. HIV compromises integrity of the podocyte actin cytoskeleton through downregulation of the vitamin D receptor. Am J Physiol Renal Physiol 304: F1347–F1357, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Duchateau PN, Pullinger CR, Cho MH, Eng C, Kane JP. Apolipoprotein L gene family: tissue-specific expression, splicing, promoter regions; discovery of a new gene. J Lipid Res Lipid Res 42: 620–630, 2001 [PubMed] [Google Scholar]
- 9.Duchateau PN, Pullinger CR, Orellana RE, Kunitake ST, Naya-Vigne J, O'Connor PM, Malloy MJ, Kane JP. Apolipoprotein L, a new human high density lipoprotein apolipoprotein expressed by the pancreas. Identification, cloning, characterization, and plasma distribution of apolipoprotein L. J Biol Chem 272: 25576–25582, 1997 [DOI] [PubMed] [Google Scholar]
- 10.Dunham I, Hunt AR, Collins JE, Bruskiewich R, Beare DM, Clamp M, Smink LJ, Ainscough R, Almeida JP, Babbage A, Bagguley C, Bailey J, Barlow K, Bates KN, Beasley O, Bird CP, Blakey S, Bridgeman AM, Buck D, Burges J, Burrill WD, Burton J, Carder C, Carter NP, Che Y, Clark G, Clegg SM, Cobley V, Cole CG, Collier RE, Connor RE, Conroy D, Corby N, Coville GJ, Cox AV, Davis J, Dawson E, Dhami PD, Dockree C, Dodsworth SJ, Durbin RM, Ellington A, Evans KL, Fey JM, Fleming K, French L, Garner AA, Gilbert JGR, Goward ME, Grafham D, Griffiths MN, Hall C, Hall R, Hall-Tamlyn G, Heathcott RW, Ho S, Holmes S, Hunt SE, Jones MC, Kershaw J, Kimberley A, King A, Laird GK, Langford CF, Leversha MA, Lloyd C, Lloyd DM, Martyn ID, Mashreghi-Mohammad M, Matthews L, McCann OT, McClay J, McLaren S, McMurray AA, Milne SA, Mortimore BJ, Odell CN, Pavit R, Pearce AV, Pearson D, Phillimore BJ, Phillips SH, Plumb RW, Ramsay H, Ramsey Y, Rogers L, Ross MT, Scott CE, Sehra HK, Skuce CD, Smalley S, Smith ML, Soderlund C, Spragon L, Steward C, Sulston JE, Swann RM, Vaudin M, Wall M, Wallis JM, Whiteley MN, Willey D, Williams L, Williams S, Williamson H, Wilmer TE, Wilming L, Wright CL, Hubbard T, Bentley DR, Beck S, Rogers J, Shimizu N, Minoshima S, Kawasaki K, Sasaki T, Asakawa S, Kudoh J, Shintan A, Shibuya K, Yoshizaki Y, Aoki N, Mitsuyam S, Roe BA, Chen F, Chu L, Crabtree J, Deschamps S, Do A, Do T, Dorman A, Fang F, Fu Y, Hu P, Hua A, Kenton S, Lai H, Lao HI, Lewis J, Lewis S, Lin SP, Loh P, Malaj E, Nguyen T, Pan H, Phan S, Qi S, Qian Y, Ray L, Ren Shaull S, Sloan D, Song L, Wang Q, Wang Y, Wang Z, White J, Willingham D, Wu H, Yao Z, Zhan M, Zhang G, Chissoe S, Murra J, Miller N, Min P, Fulton R, Johnson D, Bemis G, Bentley D, Bradshaw H, Bourne S, Cordes M, Du Z, Fulto L, Goela D, Graves T, Hawkins J, Hinds K, Kemp K, Latreille P, Layman D, Ozersky P, Rohlfing T, Scheet C, Walke C, Wamsley A, Wohldmann P, Pepin K, Nelson J, Korf I, Bedell JA, Hillier L, Mardis E, Waterston R, Wilson R, Emanuel BS, Shaikh T, Kurahashi H, Saitta S, Budarf ML, McDermid HE, Johnson A, Wong ACC, Morrow BE, Edelmann L, Kim UJ, Shizuy H, Simon MI, Dumanski JP, Peyrard M, Kedra D, Seroussi E, Fransson I, Tapia I, Bruder CE, O'Brien KP. The DNA sequence of human chromosome 22. Nature 402: 489–495, 1999 [DOI] [PubMed] [Google Scholar]
- 11.Fine DM, Wasser WG, Estrella MM, Atta MG, Kuperman M, Shemer R, Rajasekaran A, Tzur S, Racusen LC, Skorecki K. APOL1 risk variants predict histopathology and progression to ESRD in HIV-related kidney disease. J Am Soc Nephrol 23: 343–350, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Foster MC, Coresh J, Fornage M, Astor BC, Grams M, Franceschini N, Boerwinkle E, Parekh RS, Kao WH. APOL1 variants associate with increased risk of CKD among African Americans. J Am Soc Nephrol 24: 1484–1491, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Freedman BI, Langefeld CD, Turner J, Núñez M, High KP, Spainhour M, Hicks PJ, Bowden DW, Reeves-Daniel AM, Murea M, Rocco MV, Divers J. Association of APOL1 variants with mild kidney disease in the first-degree relatives of African American patients with non-diabetic end-stage renal disease. Kidney Int 82: 805–811, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Freedman BI, Soucie JM, Stone SM, Pegram S. Familial clustering of end-stage renal disease in blacks with HIV-associated nephropathy. Am J Kidney Dis 34: 254–258, 1999 [DOI] [PubMed] [Google Scholar]
- 15.Friedman DJ, Kozlitina J, Genovese G, Jog P, Pollak MR. Population-based risk assessment of APOL1 on renal disease. J Am Soc Nephrol 22: 2098–2105, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fukuda A, Wickman LT, Venkatareddy MP, Sato Y, Chowdhury MA, Wang SQ, Shedden KA, Dysko RC, Wiggins JE, Wiggins RC. Angiotensin II-dependent persistent podocyte loss from destabilized glomeruli causes progression of end stage kidney disease. Kidney Int 81: 40–55, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gasser O, Brander C, Wolbers M, Brown NV, Rauch A, Günthard HF, Battegay M, Hess C; Swiss HIV Cohort Study. Expansion of interferon-γ-secreting HIV-specific T cells during successful antiretroviral therapy. HIV Med 14: 241–246, 2013 [DOI] [PubMed] [Google Scholar]
- 18.Genovese G, Friedman DJ, Pollak MR. APOL1 variants and kidney disease in people of recent African ancestry. Nat Rev Nephrol 9: 240–244, 2013 [DOI] [PubMed] [Google Scholar]
- 19.Genovese G, Friedman DJ, Ross MD, Lecordier L, Uzureau P, Freedman BI, Bowden DW, Langefeld CD, Oleksyk TK, Uscinski Knob AL, Bernhardy AJ, Hicks PJ, Nelson GW, Vanhollebeke B, Winkler CA, Kopp JB, Pays E, Pollak MR. Association of trypanolytic ApoL1 variants with kidney disease in African Americans. Science 329: 841–845, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gulbins E, Kolesnick RN. It takes a CAD to kill a tumor cell with a LMP. Cancer Cell 24: 279–281, 2013 [DOI] [PubMed] [Google Scholar]
- 21.Husain M, Meggs LG, Vashistha H, Simoes S, Griffiths KO, Kumar D, Mikulak J, Mathieson PW, Saleem MA, Del Valle L, Pina-Oviedo S, Wang JY, Seshan SV, Malhotra A, Reiss K, Singhal PC. Inhibition of p66ShcA longevity gene rescues podocytes from HIV-1-induced oxidative stress and apoptosis. J Biol Chem 284: 16648–16658, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Johnstone DB, Shegokar V, Nihalani D, Rathore YS, Mallik L, Ashish Zare V, Ikizler HO, Powar R, Holzman LB. APOL1 null alleles from a rural village in India do not correlate with glomerulosclerosis. PLoS One 7: e51546, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kieft R, Capewell P, Turner CM, Veitch NJ, MacLeod A, Hajduk S. Mechanism of Trypanosoma brucei gambiense (group 1) resistance to human trypanosome lytic factor. Proc Natl Acad Sci USA 107: 16137–16141, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kofman T, Audard V, Narjoz C, Gribouval O, Matignon M, Leibler C, Desvaux D, Lang P, Grimbert P. APOL1 polymorphisms and development of CKD in an identical twin donor and recipient pair. Am J Kidney Dis 63: 816–819, 2014 [DOI] [PubMed] [Google Scholar]
- 25.Kopp JB, Nelson GW, Sampath K, Johnson RC, Genovese G, An P, Friedman D, Briggs W, Dart R, Korbet S, Mokrzycki MH, Kimmel PL, Limou S, Ahuja TS, Berns JS, Fryc J, Simon EE, Smith MC, Trachtman H, Michel DM, Schelling JR, Vlahov D, Pollak M, Winkler CA. APOL1 genetic variants in focal segmental glomerulosclerosis and HIV-associated nephropathy. J Am Soc Nephrol, 22: 2129–2137, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lan X, Cheng K, Chandel N, Lederman R, Jhaveri A, Husain M, Malhotra A, Singhal PC. High glucose enhances HIV entry into T cells through upregulation of CXCR4. J Leukoc Biol 94: 769–777, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lan X, Rai P, Chandel N, Cheng K, Lederman R, Saleem MA, Mathieson PW, Husain M, Crosson JT, Gupta K, Malhotra A, Singhal PC. Morphine induces albuminuria by compromising podocyte integrity. PLoS One 8: e55748, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lan X, Xu J, Kiyota T, Peng H, Zheng JC, Ikezu T. HIV-1 reduces Abeta degrading enzymatic activities in primary human mononuclear phagocytes. J Immunol 186: 6925–6932, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lecordier L, Vanhollebeke B, Poelvoorde P, Tebabi P, Paturiaux-Hanocq F, Andris F, Lins L, Pays E. C-terminal mutants of apolipoprotein L1 efficiently kill both Trypanosoma brucei brucei and Trypanosoma brucei rhodesiense. PLoS Pathog 5: e1000685, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lipkowitz MS, Freedman BI, Langefeld CD, Comeau ME, Bowden DW, Kao WH, Astor BC, Bottinger EP, Iyengar SK, Klotman PE, Freedman RG, Zhang W, Parekh RS, Choi MJ, Nelson GW, Winkler CA, Kopp JB SK. Investigators. Apolipoprotein L1 gene variants associate with hypertension-attributed nephropathy and the rate of kidney function decline in African Americans. Kidney Int 83: 114–120, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Madhavan SM, O'Toole JF, Konieczkowski M, Ganesan S, Bruggeman LA, Sedor JR. APOL1 localization in normal kidney and nondiabetic kidney disease. J Am Soc Nephrol 22: 2119–2128, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Medapalli RK, He JC, Klotman PE. HIV-associated nephropathy: pathogenesis. Curr Opin Nephrol Hypertens 20: 306–311, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mundel P, Shankland SJ. Podocyte biology and response to injury. J Am Soc Nephrol 13: 3005–3015, 2002 [DOI] [PubMed] [Google Scholar]
- 34.Oli MW, Cotlin LF, Shiflett AM, Hajduk SL. Serum resistance associated protein blocks lysosomal targeting of trypanosome lytic factor in Trypanosoma brucei. Eukaryotic Cell 5: 132–139, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Pays E, Vanhollebeke B, Vanhamme L, Paturiaux-Hanocq F, Nolan DP, Pérez-Morga D. The trypanolytic factor of human serum. Nat Rev Microbiol 4: 477–486, 2006 [DOI] [PubMed] [Google Scholar]
- 36.Pérez-Morga D, Vanhollebeke B, Paturiaux-Hanocq F, Nolan DP, Lins L, Homblé F, Vanhamme L, Tebabi P, Pays A, Poelvoorde P, Jacquet A, Brasseur R, Pays E. Apolipoprotein LI promotes trypanosome lysis by forming pores in lysosomal membranes. Science 309: 469–472, 2005 [DOI] [PubMed] [Google Scholar]
- 37.Quaggin SE, George AL. Apolipoprotein l1 and the genetic basis for racial disparity in chronic kidney disease. J Am Soc Nephrol 22: 1955–1958, 2011 [DOI] [PubMed] [Google Scholar]
- 38.Reeves-Daniel AM, DePalma JA, Bleyer AJ, Rocco MV, Murea M, Adams PL, Langefeld CD, Bowden DW, Hicks PJ, Stratta RJ, Lin JJ, Kiger DF, Gautreaux MD, Divers J, Freedman BI. The APOL1 gene and allograft survival after kidney transplantation. Am J Transplant 11: 1025–1030, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Reiser J, Oh J, Shirato I, Asanuma K, Hug A, Mundel TM, Honey K, Ishidoh K, Kominami E, Kreidberg JA, Tomino Y, Mundel P. Podocyte migration during nephrotic syndrome requires a coordinated interplay between cathepsin L and alpha3 integrin. J Biol Chem 279: 34827–34832, 2004 [DOI] [PubMed] [Google Scholar]
- 40.Saleem MA, O'Hare MJ, Reiser J, Coward RJ, Inward CD, Farren T, Xing CY, Ni L, Mathieson PW, Mundel P. A conditionally immortalized human podocyte cell line demonstrating nephrin and podocin expression. J Am Soc Nephrol 13: 630–638, 2002 [DOI] [PubMed] [Google Scholar]
- 41.Sever S, Altintas MM, Nankoe SR, Möller CC, Ko D, Wei C, Henderson J, del Re EC, Hsing L, Erickson A, Cohen CD, Kretzler M, Kerjaschki D, Rudensky A, Nikolic B, Reiser J. Proteolytic processing of dynamin by cytoplasmic cathepsin L is a mechanism for proteinuric kidney disease. J Clin Invest 117: 2095–2104, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Tzur S, Rosset S, Shemer R, Yudkovsky G, Selig S, Tarekegn A, Bekele E, Bradman N, Wasser WG, Behar DM, Skorecki K. Missense mutations in the APOL1 gene are highly associated with end stage kidney disease risk previously attributed to the MYH9 gene. Hum Genet 128: 345–350, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.US Renal Data System. USRDS Annual Data Report: Atlas of Chronic Kidney Disease and End-Stage Renal Disease in the United States. Bethesda, MD: National Institutes of Health, National Institute of Diabetes and Digestive and Kidney Diseases, 2009 [Google Scholar]
- 44.Uzureau P, Uzureau S, Lecordier L, Fontaine F, Tebabi P, Homblé F, Grélard A, Zhendre V, Nolan DP, Lins L, Crowet JM, Pays A, Felu C, Poelvoorde P, Vanhollebeke B, Moestrup SK, Lyngsø J, Pedersen JS, Mottram JC, Dufourc EJ, Pérez-Morga D, Pays E. Mechanism of Trypanosoma brucei gambiense resistance to human serum. Nature 501: 430–434, 2013 [DOI] [PubMed] [Google Scholar]
- 45.Vanhollebeke B, Pays E. The trypanolytic factor of human serum: many ways to enter the parasite, a single way to kill. Mol Microbiol 76: 806–814, 2010 [DOI] [PubMed] [Google Scholar]
- 46.Vashistha H, Husain M, Kumar D, Singhal PC. Tubular cell HIV-1 gp120 expression induces caspase 8 activation and apoptosis. Ren Fail 31: 303–312, 2009 [DOI] [PubMed] [Google Scholar]
- 47.Wan G, Zhaorigetu S, Liu Z, Kaini R, Jiang Z, Hu CA. Apolipoprotein L1, a novel Bcl-2 homology domain 3-only lipid-binding protein, induces autophagic cell death. J Biol Chem 283: 21540–21549, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wasser WG, Tzur S, Wolday D, Adu D, Baumstein D, Rosset S, Skorecki K. Population genetics of chronic kidney disease: the evolving story of APOL1. J Nephrol 25: 603–618, 2012 [DOI] [PubMed] [Google Scholar]
- 49.Zhaorigetu S, Wan G, Kaini R, Jiang Z, Chien-an AH. ApoL1, a BH3-only lipid-binding protein, induces autophagic cell death. Autophagy 4: 1079–1082, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]