

Abstract
Activation of histone deacetylases (HDACs) is required for renal epithelial cell proliferation and kidney development. However, their role in renal tubular cell survival and regeneration after acute kidney injury (AKI) remains unclear. In this study, we demonstrated that all class I HDAC isoforms (1, 2, 3, and 8) were expressed in the renal epithelial cells of the mouse kidney. Inhibition of class I HDACs with MS-275, a highly selective inhibitor, resulted in more severe tubular injury in the mouse model of AKI induced by folic acid or rhabdomyolysis, as indicated by worsening renal dysfunction, increased neutrophil gelatinase-associated lipocalin expression, and enhanced apoptosis and caspase-3 activation. Blocking class I HDAC activity also impaired renal regeneration as evidenced by decreased expression of renal Pax-2, vimentin, and proliferating cell nuclear antigen. Injury to the kidney is accompanied by increased phosphorylation of epidermal growth factor receptor (EGFR), signal transducers and activators of transcription 3 (STAT3), and Akt. Inhibition of class I HDACs suppressed EGFR phosphorylation as well as reduced its expression. MS-275 was also effective in inhibiting STAT3 and Akt phosphorylation, but this treatment did not affect their expression levels. Taken together, these data suggest that the class I HDAC activity contributes to renal protection and functional recovery and is required for renal regeneration after AKI. Furthermore, renal EGFR signaling is subject to regulation by this class of HDACs.
Keywords: acute kidney injury, histone deacetylases, dedifferentiation, proliferation, epidermal growth factor receptor
acute kidney injury (AKI) is a common and serious clinical entity associated with high morbidity and mortality. It can arise in multiple causes such as ischemia-reperfusion (I/R), sepsis, rhabdomyolysis, trauma, and nephrotoxin exposure (18, 30). Animal studies have shown that the kidney possesses a remarkable regenerative capacity after AKI. During the regenerative process, surviving tubular cells undergo dedifferentiation, migration, and proliferation that result in morphological and functional recovery of renal epithelium (1). Proliferation of surviving resident tubular epithelial cells has been shown to be the predominant mechanism for renal regeneration after AKI (12). Therefore, investigation of the mechanism that regulates this process after acute injury will help to develop novel therapeutic treatments for accelerating renal regeneration and renal functional recovery.
Currently, the molecular mechanism mediating renal regeneration is incompletely understood. Studies have shown that some molecules such as vimentin, Pax-2, and neural cell adhesion molecule, which are expressed in metanephric mesenchyme but not in mature kidneys, are reexpressed in renal epithelial cells during recovery from acute injury (1, 8). This suggests that renal regeneration following acute injury is similar to early kidney development. Kidney development is a complex process that is subjected to genetic and epigenetic regulation. Epigenetic control is the regulation of gene transcription without any change in the DNA sequence. Among epigenetic events, histone acetylation/deacetylation is one of the most studied genetic modifications, and it is regulated by histone acetyltransferases (HATs) and histone deacetylases (HDACs), respectively. There are 18 HDACs, which are classified into four groups: class I HDACs (HDAC1, 2, 3, and 8), class II HDACs (HDAC 4, 5, 6, 7, 9, and 10), class III HDACs (SIRT1–7), and class IV HDACs (HDAC 11). Of those HDACs, activation of class I HDACs is required for early nephron gene expression, growth, and differentiation during kidney development (3, 17). Moreover, class I HDACs have been implicated in the regulation of cell cycle progression and cell proliferation in various cell types, including renal epithelial cells (14, 16, 24, 26).
Proliferation of renal tubular cells is induced by activation of multiple growth factor receptors. Among them, epidermal growth factor receptor (EGFR) has proven to be critically involved in kidney development and renal regeneration: genetic or pharmacologic reduction of EGFR activity reduces the rates of renal function recovery and inhibits the kidney's regenerative response after AKI (2, 8, 29); and blockage of EGFR activity inhibits dedifferentiation and proliferation of renal tubular cell in the animal model of AKI induced by folic acid (8). EGFR is a tyrosine kinase that initiates activation of several signaling pathways, including the signal transducers and activators of transcription 3 (STAT3) and the phosphoinositide 3-kinase (PI3K)/Akt pathways. Activation of these two pathways is also reported to mediate renal epithelial cell cycle progression and proliferation (2, 8, 26). Recently, we demonstrated that inhibition of HDAC activity can reduce activation of EGFR and STAT3 in the normally cultured renal epithelial cells (26). This suggests a link between epigenetic modification and activation of cellular signaling pathway important for renal cell proliferation.
Although the fundamental functions of HDACs in cancer, cardiac and immune disorders are well-documented, their role in tissue development has just begun to be elucidated (7). It has been reported that pharmacological blockade of class I/II HDACs increases histone acetylation levels and inhibits tail and limb regeneration in the amputated tail of Xenopus laevis larvae (27, 28). Inhibiting HDAC activities also delays liver regeneration and inhibition of hepatocyte proliferation (4). More importantly, selective knockout of HDAC1 or/and HDAC2 genes in hepatocytes led to impaired liver regeneration (31). These studies clearly indicate the importance of HDACs in regulating regeneration of epithelial tissues. However, the role of HDACs in renal regeneration after acute injury remains unclear. In this study, we examined this issue by using MS-275, a selective inhibitor of class I HDAC (9), in a murine model of AKI induced by either folic acid (FA) or rhabdomyolysis (RM).
MATERIALS AND METHODS
Antibodies and reagents.
Antibodies to p-EGFR, p-Akt, Akt, p-STAT3, STAT3, Acetyl-H3, cleaved caspased-3, vimentin were purchased from Cell Signaling Technology (Dancers, MA). Antibody against Pax-2 was purchased from Invitrogen (Grand Island, NY). Antibodies to GAPDH, EGFR, HDAC1, HDAC2, HDAC3, HDAC8, and proliferating cell nuclear antigen (PCNA) were purchased from Santa Cruz Biotechnology (Santa Cruz, CA). Antibodies to neutrophil gelatinase-associated lipocalin (NGAL) were purchased from R&D Systems (Minneapolis, MN). Anti-α-tubulin antibody, secondary antibodies, and all other chemicals were from Sigma (St. Louis, MO).
Animals and treatment.
Male C57/black mice, each weighing 20–25 g (Jackson Laboratory, Bar Harbor, ME), were housed under a 12:12-h light-dark cycle with food and water supplied ad libitum. To establish FA-induced AKI, the animals were injected intraperitoneally with FA at 250 mg/kg body wt. Sodium bicarbonate (0.3 M NaHCO3, the vehicle used for FA administration) alone was used as controls. To establish RM-induced AKI, the animals were injected with 50% glycerol (GL; 10 ml/kg) intramuscularly to the two hind legs or injected with saline as a control. To examine the efficacy of class I HDACs on AKI, MS-275 (20 mg/kg) in 50 μl of DMSO was given intraperitoneally immediately after FA injection or 2 h after GL injection and then administered daily. Animals treated with an equal volume of DMSO were used as controls. At the end of the experiments, animals were killed and the kidneys were removed for protein analysis and histological examination. Six to eight mice were used in each group. Animal protocol was reviewed and approved by the Institutional Animal Care and Use Committee at Tongji University, China.
Measurement of renal function.
Renal function was estimated by serum creatinine and blood urea nitrogen (BUN), measured using a colorimetric kit (Sigma Diagnostics) and enzymatic assay Kit (Sigma Diagnostics), respectively, according to the protocol provided by the manufacturer.
Assessment of tubular injury.
Tubular injury was scored on a scale from 0 to 3, where 0 = normal, 1 = injury <30%, 2 = injury 30–60%, 3 = injury >60%. The TdT-mediated dUTP nick-end labeling (TUNEL) staining was performed according to the protocol provided by Roches Molecular System (Branchburg, NJ).
Immunoblot analysis.
Immunoblot analysis for tissue samples was performed according to our previous protocols (22). The densitometry analysis of immunoblot results was conducted with Image J software (National Institutes of Health, Bethesda, MD).
Immunofluorescent and immunohistochemical staining.
Immunofluorescent staining was carried out according to the procedure described in our previous studies (22). Renal tissues were fixed in 4.5% buffered formalin, dehydrated, and embedded in paraffin. For immunofluorescent staining, the tissue sections were rehydrated and labeled with antibodies, including primary antibodies Acetyla-H3 (1:100), Pax-2 (1:50), HDAC1 (1:100), HDAC2 (1:100), HDAC3 (1:50), HDAC8 (1:50), NGAL (1:250), PCNA (1:50), and then exposed to Texas red-labeled secondary antibodies (Invitrogen). Periodic acid-Schiff staining was performed in the Department of Pathology at Rhode Island Hospital.
Statistical analysis.
All data were presented as means ± SE for each group. Comparisons between intergroups were made by using one-way ANOVA followed by Tukey's test. Statistical significant difference was considered at P < 0.05.
RESULTS
Expression and location of class I HDAC isoforms in the kidney.
Our recent studies showed that all the class I HDAC isoforms are expressed in cultured renal tubular cells (26). To determine their expression and location in the kidney, we conducted immunoblot analysis and immunofluorescence staining. Figure 1A showed that HDAC1, HDAC2, HDAC3, and HDAC8 were abundantly expressed in the kidney. As expected, HDAC1 and HDAC2 were primarily in the nucleus as evidenced by their costaining with DAPI (a nuclear dye) in all tubular cells, and HDAC3 was also partially located in the nucleus (Fig. 1B). However, HDAC8 was mainly expressed in the cytoplasm since it was not colocalized with DAPI (Fig. 1B). These data illustrated the expression of all four members of class I HDAC and their distinct locations in renal tubule cells in vivo.
Fig. 1.

Expression and location of class I histone deacetylases (HDACs) in the kidney. A: tissue lysates from 2 normal kidneys were subjected to immunoblot analysis with specific antibodies against HDAC1, HDAC2, HDAC3, HDAC8, and α-tubulin. B: photomicrographs (×200) illustrate immunofluorescent costaining of HDAC1, HDAC2, HDAC3, or HDAC8 with DAPI in normal kidney sections.
Inhibition of class I HDAC activity by MS-275 aggravates renal dysfunction and kidney damage in FA- or RM-induced AKI.
Recent studies have shown that class I HDAC activity is acquired for kidney development and proliferation of embryonic proximal tubular cells (3). To demonstrate the role of class I HDACs in AKI, murine models of FA- or RM-induced AKI were used and a selective class I HDAC inhibitor, MS-275 (9), was given immediately after FA injection or 2 h after GL injection. As shown in Fig. 2, at 48 h after FA or GL injection, serum creatinine and BUN were significantly increased and kidney damage (tubular dilatation, swelling, necrosis, luminal congestion) was clearly observed. MS-275 significantly increased the level of creatinine and BUN and potentiated the degree of renal tubular damage in mice subjected to FA or GL. Scoring of kidney sections showed more severe tubular damage in the injured kidney after MS-275 administration relative to the injured kidney without MS-275 treatment. In contrast, injection of MS-275 alone neither affected renal function nor caused pathological damage to the kidney. These data illustrate that endogenous class I HDAC activity is required for renal protection in these two models of AKI.
Fig. 2.

Class I HDAC inhibition potentiates renal dysfunction and damage in the murine model of folic acid (FA)- or rhabdomyolysis (RM)-induced acute kidney injury (AKI). After various treatments as indicated, blood was collected and the serum creatinine and blood urea nitrogen (BUN) were measured (A–D). The kidneys underwent periodic acid-Schiff (PAS) staining (E). Morphological changes were scored based on the scale described in materials and methods (F). Data are represented as means ± SE (n = 6). Means with different superscript letters are significantly different from one another (P < 0.05).
MS-275 increases acetyl-histone H3 expression in the kidney of FA- or RM-induced AKI.
Blocking HDACs induces acetylation of histone and nonhistone proteins, and global histone H3 hyperacetylation is frequently used to indicate the effectiveness of HDAC inhibitors. To demonstrate the inhibitory effect of MS-275 on class I HDAC activity in the kidney, we examined the expression of acetyl-histone H3 in the injured kidney with or without MS-275 administration. Immunofluorescence staining revealed very few number of acetyl-histone H3-positive cells in the sham-operated kidney and the kidney injured by injection of FA or GL alone. Treatment with MS-275 significantly increased this population of cells in the injured kidney (Fig. 3, A–B and E–F). To confirm this observation, we also examined expression of acetyl-histone H3 by immunoblot analysis. As shown in Fig. 3, C–D and G–H, the basal level of acetyl-histone H3 was detected in sham-operated kidneys, and injury to the kidney by injection of FA or GL resulted in a slight increase in its expression. MS-275 treatment induced a small enhancement of acetyl-histone H3 in the sham-operated kidney, which reached a level comparable to that in the injured kidney without MS-275 application. However, acetyl-histone H3 expression levels were markedly upregulated in the injured kidney administered by MS-275. Of note, acetyl-histone H3 is primarily located in renal tubular cells (Fig. 3, A and E). These data suggest that normal kidneys maintain a high level of HDAC activity, which is slightly decreased by acute injury, and further repressed by MS-275.
Fig. 3.

Class I HDAC inhibition enhances expression of acetyl-histone H3 in the kidney of FA- or RM-induced AKI. A, E: photomicrographs (×200) illustrate acetyl-histone H3 with immunofluorescent staining of the kidney tissues collected at 48 h after FA or glycerol (GL) injection with or without MS-275 administration in C57/black mice. B, F: tubular cells with positive acetyl-histone H3 staining were counted in 10 high-power fields and expressed as means ± SE. C, G: kidney tissue lysates were subjected to immunoblot analysis with specific antibodies against acetyl-histone H3 and α-tubulin. D, H: expression level of acetyl-histone H3 was quantified by densitometry and normalized with α-tubulin. Data are means ± SE (n = 6). Representative immunoblots are 2 samples from 6 animals in each group. Means with different superscript letters are significantly different from one another (P < 0.05).
Inhibition of class I HDACs activity by MS-275 potentiates tubular damage in FA- or RM-induced AKI.
To determine the role of class I HDACs in FA- and RM-induced AKI, we first examined NGAL expression by immunofluorescence staining. As expected, NGAL expression is limited to renal tubular cells. Whereas the NGAL signal was not observed in the sham-operated kidney with/without MS-275 treatment, it was clearly seen in some tubules in the kidney after FA or GL administration. Treatment with MS-275 potentiated this response (Fig. 4, A and C). In agreement with this observation, NGAL was not detected in sham-operated kidneys by immunoblot analysis, but its expression was induced in the kidney after FA or GL administration and further enhanced by MS-275 (Fig. 4, B and D). These data further indicate that the class I HDACs activity is necessary for protecting the kidney from acute injury.
Fig. 4.
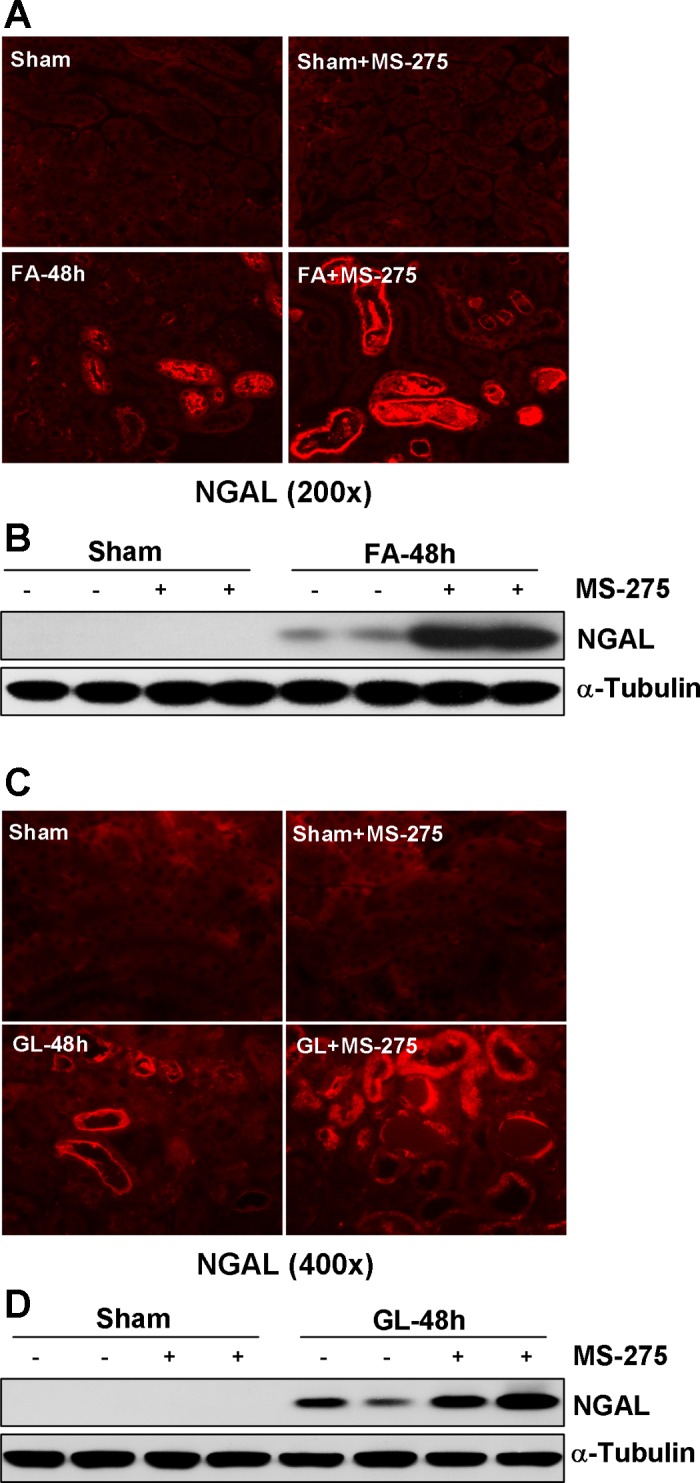
Class I HDAC inhibition enhances expression of neutrophil gelatinase-associated lipocalin (NGAL) in the kidney of FA- or RM-induced AKI. A, C: photomicrographs illustrate immunofluorescent staining of NGAL in kidney tissues collected at 48 h after sham and FA or GL injection with or without MS-275 administration in C57/black mice. B, D: kidney tissue lysates were subjected to immunoblot analysis with specific antibodies against NGAL and α-tubulin. Representative immunoblots are 2 samples from 6 animals in each group.
Inhibition of class I HDACs activity by MS-275 potentiates tubular cell apoptosis in FA- or RM-induced AKI.
As apoptosis of tubular epithelial cells is a hallmark of AKI, we further examined the effect of MS-275 on apoptosis in the kidney by the TUNEL assay. TUNEL-positive cells were not observed in the sham-operated kidney with or without MS-275 treatment, but they were evident in the kidney after FA or GL injection. MS-275 administration significantly increased the number of apoptotic tubular cells in the injured kidney (Fig. 5, A–B and E–F). In addition, we examined expression of cleaved caspase-3 by immunoblot analysis as its activation is central to apoptosis. As shown in Fig. 5, C, D, and G, H, FA or GL injection induced expression of cleaved caspased-3 and its expression was also potentiated by MS-275. It should be noted that MS-275 did not induce cleavage of caspase-3 in the sham-operated kidney. Together, these data further suggest that class I HDAC activation contributes to renal protection in FA- or RM-induced AKI.
Fig. 5.

Class I HDAC inhibition enhances renal tubular cell apoptosis in the kidney of FA- or RM-induced AKI. A, E: photomicrographs illustrate TdT-mediated dUTP nick-end labeling (TUNEL) staining of the kidney tissues collected at 48 h after sham and FA or GL injection with or without MS-275 administration in C57/black mice. B, F: positive TUNEL staining cells were counted in 10 high-power fields and expressed as means ± SE. C, G: kidney tissue lysates were subjected to immunoblot analysis with specific antibodies against cleaved caspased-3 and GAPDH. D, H: expression level of cleaved caspased-3 was quantified by densitometry and normalized with GAPDH. Data are means ± SE (n = 6). Representative immunoblots are 2 samples from 6 animals in each group. Means with different symbols are significantly different from one another (P < 0.05).
Class I HDAC activity is required for renal tubular cell dedifferentiation in FA- or RM-induced AKI.
Fate mapping studies indicated that renal repair after AKI occurs predominantly by proliferation of dedifferentiated intrinsic renal tubular cells (12). To determine whether class I HDAC activation contributes to this process, we examined expression of Pax-2 and vimentin, two markers of renal epithelial cell dedifferentiation, in the kidney after injury with/without administration of MS-275. Immunofluorescence staining showed that Pax-2-positive cells were not seen in the shamed kidney treated with either vehicle or MS-275 (Fig. 6, A, B, and E, F), but this population of cells was increased in the kidney after FA or GL injection. In contrast, inhibition of class I HDACs by MS-275 significantly reduced the number of cells. To confirm these results, we also examined the expression level of vimentin by immunoblot analysis (Fig. 6, C, D, and G, H). Increased expression of vimentin in the injured kidney was observed in FA- or GL-injured kidney and MS-275 administration abolished its expression. Cells expressing Pax-2 and vimentin were not observed in the sham-operated kidney with or without treatment with MS-275.
Fig. 6.

Class I HDAC inhibition suppresses renal tubular cell dedifferentiation in the kidney of FA- or RM-induced AKI. A, E: photomicrographs illustrate Pax-2 staining of the kidney tissue collected at 48 h after sham and FA or GL injection with or without MS-275 administration in C57/black mice. B, F: tubular cells with positive staining of Pax-2 were counted in 10 high-power fields and expressed as means ± SE. C, G: kidney tissue lysates were subjected to immunoblot analysis with specific antibodies against vimentin and GAPDH. D, H: expression level of vimentin was quantified by densitometry and normalized with GAPDH. Data are means ± SE (n = 6). Representative immunoblots are 2 samples from 6 animals in each group. Means with different symbols are significantly different from one another (P < 0.05).
Class I HDAC activity is required for renal tubular cell proliferation in FA- or RM-induced AKI.
We proceed to assess the effect of class I HDAC inhibition on renal tubular cell proliferation using PCNA as a marker. As shown in Fig. 7, A, B, E, and F, very few PCNA-positive cells were seen in the sham-operated kidney, which corresponds to its turnover rate at 1/1,000 under physiological state. After FA or GL injection, the number of PCNA-positive cells was markedly increased, and MS-275 treatment blocked this response. Similar results were observed when PCNA expression was examined by immunoblot analysis (Fig. 7, C, D, and G, H).
Fig. 7.

Class I HDAC inhibition suppresses renal tubular cell proliferation in the kidney of FA- or RM-induced AKI. A, E: photomicrographs illustrate proliferating cell nuclear antigen (PCNA) staining of the kidney tissues collected at 48 h after sham and FA injection with or without MS-275 administration in C57/black mice. B, F: PCNA-positive cells were counted in 10 high-power fields and expressed as means ± SE. C, G: kidney tissue lysates were subjected to immunoblot analysis with specific antibodies against PCNA and α-tubulin. D, H: expression level of PCNA was quantified by densitometry and normalized with α-tubulin. Data are means ± SE (n = 6). Representative immunoblots are 2 samples from 6 animals in each group. Means with different symbols are significantly different from one another (P < 0.05).
These data, together with Fig. 6, suggest that class I HDAC activity is critically involved in the regulation of renal tubular cell dedifferentiation and proliferation after AKI.
Class I HDACs activity is required for EGFR, Akt, and STAT3 phosphorylation in FA- or RM-induced AKI.
Studies have shown that the EGFR signaling plays an important role in the regulation of renal epithelial cell proliferation and dedifferentiation (15, 33). Akt and STAT3 are two signaling molecules that act downstream of EGFR to mediate these biological processes (26, 33). We tested whether class I HDACs would mediate renal regeneration through activation of EGFR signaling in the kidney after acute injury. As shown in Fig. 8, FA or GL injection-induced AKI is accompanied by increased expression of phospho-EGFR (p-EGFR) and total EGFR. However, the upregulation of EGFR phosphorylation was not only due to the increased level of total EGFR because calculation of the ratio of p-EGFR to total EGFR was still increased in the injured kidney. MS-275 treatment blocked expression of both p-EGFR and total EGFR in the kidney injured; the basal level of total EGFR was not significantly different in the sham-operated kidney with or without administration of MS-275. Notably, the ratio of p-EGFR to total EGFR was still decreased in the injured kidney treated with MS-275, compared with injured kidneys without MS-275 treatment. These data indicate that downregulation of EGFR by MS-275 is not only due to its inhibitory effect on total EGFR but that class I HDACs activity is also critically involved in the regulation of EGFR activation.
Fig. 8.

Class I HDAC activity is required for expression and activation of renal epidermal growth factor receptor (EGFR) after FA- or RM-induced AKI. The kidney tissues were collected at 48 h after FA or GL injection. A, D: kidney tissue lysates were subjected to immunoblot analysis with specific antibodies against p-EGFR, EGFR, and GAPDH. B, E: expression levels of p-EGFR, EGFR, and GAPDH were calculated by densitometry and the ratio between p-EGFR and total EGFR was determined. C, F: total EGFR levels were normalized with GAPDH. Data are means ± SE (n = 6). Representative immunoblots are 2 samples from 6 animals in each group. Means with different symbols are significantly different from one another (P < 0.05).
We further examined the effect of class I HDAC inhibition on expression and phosphorylation of Akt and STAT3. After injection of FA increased expression of p-Akt, p-STAT3, total Akt, or STAT3 was observed in the injured kidney; administration of MS-275, however, blocked STAT3 phosphorylation and to a lesser extent, reduced Akt phosphorylation. The total expression levels of these two kinases remain constant in the kidney with/without treatment of MS-275 (Fig. 9, A–E). In the parallel experiments, we also examined the effect of class I HDAC inhibition on the renal expression and phosphorylation of Akt and STAT 3 in the mice after injection of GL, and the similar results as seen in mice with FA administration were obtained (data not shown).
Fig. 9.

Class I HDAC activity is required for activation of renal Akt and STAT3 after FA-induced AKI. Kidney tissues were collected at 48 h after FA injection, with or without MS-275 administration. A: kidney tissue lysates were subjected to immunoblot analysis with specific antibodies against p-Akt, Akt, p-STAT3, STAT3, and GAPDH. B: expression levels of p-STAT3, STAT3, and α-tubulin were calculated by densitometry and the ratio between p-STAT3 and total STAT3 was determined. C: total STAT3 levels were normalized with GAPDH. D: ratio between p-Akt and total Akt was determined. E: total Akt levels were normalized with GAPDH. Data are means ± SE (n = 6). Representative immunoblots are 2 samples from 6 animals in each group. Means with different symbols are significantly different from one another (P < 0.05).
Taken together, these data demonstrate that class I HDAC activity contributes to activation of EGFR and of Akt and STAT3 signaling pathways.
DISCUSSION
The purpose of this study was to examine the role of class I HDAC in AKI. Our results showed that inhibition of class I HDACs by MS-275 resulted in worse renal dysfunction and increased tubular cell damage and apoptosis in a murine model of AKI induced by FA or RM. MS-275 was also effective in decreasing renal epithelial cell dedifferentiation and proliferation in the injured kidney. Furthermore, inactivation of class I HDACs inhibited FA- or RM-induced phosphorylation of EGFR and its downstream signaling molecules STAT3 and Akt. Thus, these results provide strong evidence that class I HDAC activity is required for renal epithelial tubular cell survival and regeneration in the kidney after acute injury. In line with our observations, recent studies have indicated that either employment of suberoylanilide hydroxyamic acid (SAHA), a class HDAC I/II inhibitor, or conditional deletion of some isoforms of class I HDACs such as HDAC1 and HDAC2 from hepatocytes also impairs liver regeneration (10, 31), suggesting the importance of HDAC activity in promoting liver regeneration as well.
Previous studies showed that activation of HDACs either contributes to cell death or survival, depending on cell types and stimuli (3, 6, 24). Our studies clearly indicated that activation of class I HDACs is protective against AKI because blocking class I HDACs with MS-275 induced expression of NGAL and increased apoptotic tubular cells in the kidney following FA or GL injection. Moreover, class I HDAC inhibition enhanced expression of active caspase-3, a key enzyme responsible for execution of apoptosis, in the injured kidney. Notably, treatment with MS-275 did not induce/enhance expression of NGAL, and apoptosis in the sham-operated kidney, suggesting that the inducible/enhancing effect of MS-275 on renal injury was not due to its possible toxic effect. In support of this statement, our recent in vitro studies also indicated that treatment of cultured renal epithelial cells with MS-275 for 48 h resulted in marked suppression of cell proliferation without induction of apoptosis (26). The underlying mechanism by which class I HDACs mediate cell survival is not clear, but may be related to preservation and activation of some survival signaling pathways (i.e., STAT3 and PI3K/Akt). It is well-documented that activation of STAT3 and PI3K/Akt signaling pathways is required for survival of renal epithelial cells (19, 23). And our data showed that acute injury to the kidney induced phosphorylation (activation) of STAT3 and Akt, and MS-275 treatment suppressed this response.
Following acute injury, renal epithelial cells expand by self-duplication of surviving renal tubular cells in the repairing kidney (11). To determine whether activation of class I HDACs is involved in renal tubular cell dedifferentiation and proliferation, we also examined the effect of MS-275 on the expression of Pax-2 and vimentin, two dedifferentiation markers, and that of PCNA, a maker of proliferation, in the kidney. Our results showed that injury to the kidney induced their expression in renal tubular cells and inhibition of class I HDACs by MS-275 suppressed expression of these markers. These data, together with our previous results obtained in in vitro studies that MS-275 was effective in inhibiting proliferation of cultured renal proximal tubular cells (RPTC) (26), suggest that class I HDAC activity is required for renal tubular cell dedifferentiation and proliferation, two key regenerative responses in the process of renal recovery. These results are supported by recent observations that inhibition of HDACs with class I/II inhibitors SAHA or valproic acid, and genetic depletion of HDAC1 or/and HDAC2, also resulted in the impairment of liver regeneration after partial hepatectomy (10, 13, 31).
Currently, the specific mechanism by which class I HDACs regulate renal tubular cell survival and regeneration is not fully understood. It has been reported that blockage of HDACs activity reduces the expression of EGFR in tumor cells (4). Our recent studies also indicated that inhibition of class I HDACs with MS-275 reduced both phosphorylation and expression of EGFR in cultured RPTC (26). In this study, we found that expression and phosphorylation of EGFR were upregulated in the injured kidney and inhibition of class I HDACs also markedly reduced its phosphorylation and expression. Since EGFR is critically involved in nephrogenesis and the renal regenerative process after AKI (25), we suggest that activation of EGFR may be part of signaling mechanism by which HDACs regulate renal survival and regeneration. In addition, because EGFR is an upstream activator of STAT3 and Akt, and because in vitro and in vivo studies have demonstrated that STAT3 and Akt mediate regulation of renal epithelial cell cycle progression and cell proliferation as well as survival (2, 8, 23, 26, 33), it is speculated that class I HDACs regulate renal regeneration through EGFR-mediated activation of these two signaling pathways. In support of this idea, we revealed that MS-275 treatment reduced phosphorylation of STAT3 and Akt in the kidney after acute injury. However, the EGFR/STAT3 or/and Akt pathway may not be the only one that mediates the biological actions of EGFR since HDAC inhibition can also attenuate activation of several other signaling pathways, including β-caternin, hedgehog, PAX2-Six2-GNDF-cRet, and p53 pathways in the developing kidneys (3).
In addition, class I HDACs may also mediate renal regeneration through epigenetic modification of key genes associated with this process. A recent study showed that expression of some key developmental renal regulators is dependent on intact HDAC activity (3). For example, HDAC inhibition is associated with histone hyperacetylation of Pax2, and knockdown of both HDAC1 and HDAC2 reduces PAX2 expression (3). Furthermore, long-term treatment of embryonic kidneys with MS-275 impairs ureteric bud branching morphogenesis program and induces growth arrest and apoptosis (3). As renal regeneration after acute injury is generally considered to be a process similar to renal development (1), the requirement of HDAC activity for expression of renal developmental regulators in the embryonic kidney suggests that HDACs may also be involved in the regulation of genes that contribute to renal regeneration. Additional studies are required to identify the HDAC-regulated genes in the regenerating kidney after acute injury.
Although it was reported that conditional deletion of HDAC1 and HDAC2 from hepatocytes impairs liver regeneration (31), the role that individual HDACs play during renal regeneration in vivo remains unclear. Our in vitro studies have demonstrated that siRNA-mediated silencing of HDAC1, 3, or 8, but not 2, decreased cell proliferation and inhibited EGFR phosphorylation/expression in cultured renal epithelial cells (26). However, since in vitro studies have shown that MS-275 preferentially inhibits HDAC1 and HDAC3 (9) and the specificity of this inhibitor on class II-IV HDACs has not been evaluated, we suggest that HDAC1 and HDAC3 may play a predominant role in renal regeneration after acute injury, but other HDACs may also be involved in this process. It will be interesting to elucidate the role of individual members of class I HDACs in renal regeneration in the kidney using knockout and overexpression approaches.
In contrast to the necessity of the class I HDAC activity in promoting renal and liver regeneration after acute injury, there are also reports showing that HDAC inhibition accelerates renal recovery after injury in murine models of AKI induced by I/R or aristolochic acid (5, 20). These effects were demonstrated by the same group using another HDAC inhibitor, methyl-4-(phenythio) butanoate (m4PTB), and mechanistically linked to acceleration of cell cycle progression and reduction of G2/M arrest of regenerating renal tubular epithelial cells and inhibition of fibrosis (5, 20). Unlike MS-275, the capacity and profile of m4PTB-inactivated HDACs remain unclear. There are no available data showing whether this compound is specific to one or more than one class of HDACs. Given that there are four groups of HDACs that are composed of 18 members, and each isoform of HDACs has multiple functions, it is possible that the profile of m4PTB-mediated inhibition of HDACs is different from that offered by MS-275, and consequently results in different results. Another possibility is that m4PTB exerted its promoting effect on renal functional recovery after AKI through inhibition of renal fibrosis. In this respect, MS-275 and m4PTB may share a common effect (5, 15, 20).
In summary, our studies demonstrated that class I HDAC activity is required for preservation of renoprotective effects and regulation of multiple regenerative processes after AKI. The effects of class I HDACs are associated with positive regulation of renal tubular cell survival, dedifferentiation, and proliferation through a mechanism involved in EGFR signaling. Given the specificity and redundancy of HDACs, additional studies using isotype-specific HDAC inhibitors or genetic approaches will help to further identify the role of individual HDACs in regulation of renal regeneration. Moreover, experiments are also required to examine the effect of various HDAC inhibitors on tubular survival and regenerative responses in animal models of AKI induced by diverse insults.
GRANTS
This work was supported by grants from the National Institutes of Health (DK-085065 to S. Zhuang), the National Natural Science Foundation of China (81270778 to S. Zhuang, 81170638 to H. Yan), and Pudong New District Foundation of China (PWZxk2014-6).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
AUTHOR CONTRIBUTIONS
Author contributions: J.T. and Y.Y. performed experiments; J.T. and S.Z. interpreted results of experiments; J.T. and S.Z. prepared figures; J.T. drafted manuscript; T.C.Z., R.G., H.Y., and S.Z. conception and design of research; G.B. and S.Z. edited and revised manuscript; S.Z. analyzed data; S.Z. approved final version of manuscript.
REFERENCES
- 1.Bonventre JV. Dedifferentiation and proliferation of surviving epithelial cells in acute renal failure. J Am Soc Nephrol 14, Suppl 1: S55–S61, 2003 [DOI] [PubMed] [Google Scholar]
- 2.Chen J, Chen JK, Harris RC. Deletion of the epidermal growth factor receptor in renal proximal tubule epithelial cells delays recovery from acute kidney injury. Kidney Int 82: 45–52, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chen S, Bellew C, Yao X, Stefkova J, Dipp S, Saifudeen Z, Bachvarov D, El-Dahr SS. Histone deacetylase (HDAC) activity is critical for embryonic kidney gene expression, growth, and differentiation. J Biol Chem 286: 32775–32789, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chou CW, Wu MS, Huang WC, Chen CC. HDAC inhibition decreases the expression of EGFR in colorectal cancer cells. PLos One 6: e18087, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cianciolo Cosentino C, Skrypnyk NI, Brilli LL, Chiba T, Novitskaya T, Woods C, West J, Korotchenko VN, McDermott L, Day BW, Davidson AJ, Harris RC, de Caestecker MP, Hukriede NA. Histone deacetylase inhibitor enhances recovery after AKI. J Am Soc Nephrol 24: 943–953, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Glaser KB, Li J, Staver MJ, Wei RQ, Albert DH, Davidsen SK. Role of class I and class II histone deacetylases in carcinoma cells using siRNA. Biochem Biophys Res Commun 310: 529–536, 2003 [DOI] [PubMed] [Google Scholar]
- 7.Haberland M, Montgomery RL, Olson EN. The many roles of histone deacetylases in development and physiology: implications for disease and therapy. Nat Rev Genet 10: 32–42, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.He S, Liu N, Bayliss G, Zhuang S. EGFR activity is required for renal tubular cell dedifferentiation and proliferation in a murine model of folic acid-induced acute kidney injury. Am J Physiol Renal Physiol 304: F356–F366, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hu E, Dul E, Sung CM, Chen Z, Kirkpatrick R, Zhang GF, Johanson K, Liu R, Lago A, Hofmann G, Macarron R, de los Frailes M, Perez P, Krawiec J, Winkler J, Jaye M. Identification of novel isoform-selective inhibitors within class I histone deacetylases. J Pharmacol Exp Ther 307: 720–728, 2003 [DOI] [PubMed] [Google Scholar]
- 10.Huang J, Barr E, Rudnick DA. Characterization of the regulation and function of zinc-dependent histone deacetylases during rodent liver regeneration. Hepatology 57: 1742–1751, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Humphreys BD, Czerniak S, DiRocco DP, Hasnain W, Cheema R, Bonventre JV. Repair of injured proximal tubule does not involve specialized progenitors. Proc Natl Acad Sci USA 108: 9226–9231, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Humphreys BD, Valerius MT, Kobayashi A, Mugford JW, Soeung S, Duffield JS, McMahon AP, Bonventre JV. Intrinsic epithelial cells repair the kidney after injury. Cell Stem Cell 2: 284–291, 2008 [DOI] [PubMed] [Google Scholar]
- 13.Ke Q, Yang RN, Ye F, Wang YJ, Wu Q, Li L, Bu H. Impairment of liver regeneration by the histone deacetylase inhibitor valproic acid in mice. J Zhejiang Univ Sci B 13: 695–706, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lee KW, Kim JH, Park JH, Kim HP, Song SH, Kim SG, Kim TY, Jong HS, Jung KH, Im SA, Kim NK, Bang YJ. Antitumor activity of SK-7041, a novel histone deacetylase inhibitor, in human lung and breast cancer cells. Anticancer Res 26: 3429–3438, 2006 [PubMed] [Google Scholar]
- 15.Liu N, He S, Ma L, Ponnusamy M, Tang J, Tolbert E, Bayliss G, Zhao TC, Yan H, Zhuang S. Blocking the class I histone deacetylase ameliorates renal fibrosis and inhibits renal fibroblast activation via modulating TGF-beta and EGFR signaling. PLos One 8: e54001, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Liu S, Cheng H, Kwan W, Lubieniecka JM, Nielsen TO. Histone deacetylase inhibitors induce growth arrest, apoptosis, and differentiation in clear cell sarcoma models. Mol Cancer Ther 7: 1751–1761, 2008 [DOI] [PubMed] [Google Scholar]
- 17.Majumdar G, Adris P, Bhargava N, Chen H, Raghow R. Pan-histone deacetylase inhibitors regulate signaling pathways involved in proliferative and pro-inflammatory mechanisms in H9c2 cells. BMC Genomics 13: 709, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mehta RL, Kellum JA, Shah SV, Molitoris BA, Ronco C, Warnock DG, Levin A. Acute Kidney Injury Network: report of an initiative to improve outcomes in acute kidney injury. Crit Care 11: R31, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Nishino T, Pusey CD, Domin J. Elevated Akt phosphorylation as an indicator of renal tubular epithelial cell stress. J Biol Chem 277: 33943–33949, 2002 [DOI] [PubMed] [Google Scholar]
- 20.Novitskaya T, McDermott L, Zhang KX, Chiba T, Paueksakon P, Hukriede N, de Caestecker MP. A PTBA class small molecule enhances recovery and reduces postinjury fibrosis after aristolochic acid-induced kidney injury. Am J Physiol Renal Physiol 306: F496–F504, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Oehme I, Deubzer HE, Wegener D, Pickert D, Linke JP, Hero B, Kopp-Schneider A, Westermann F, Ulrich SM, von Deimling A, Fischer M, Witt O. Histone deacetylase 8 in neuroblastoma tumorigenesis. Clin Cancer Res 15: 91–99, 2009 [DOI] [PubMed] [Google Scholar]
- 22.Pang M, Kothapally J, Mao H, Tolbert E, Ponnusamy M, Chin YE, Zhuang S. Inhibition of histone deacetylase activity attenuates renal fibroblast activation and interstitial fibrosis in obstructive nephropathy. Am J Physiol Renal Physiol 297: F996–F1005, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ponnusamy M, Pang M, Annamaraju PK, Zhang Z, Gong R, Chin YE, Zhuang S. Transglutaminase-1 protects renal epithelial cells from hydrogen peroxide-induced apoptosis through activation of STAT3 and AKT signaling pathways. Am J Physiol Renal Physiol 297: F1361–F1370, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sikandar S, Dizon D, Shen X, Li Z, Besterman J, Lipkin SM. The class I HDAC inhibitor MGCD0103 induces cell cycle arrest and apoptosis in colon cancer initiating cells by upregulating Dickkopf-1 and non-canonical Wnt signaling. Oncotarget 1: 596–605, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tang J, Liu N, Zhuang S. Role of epidermal growth factor receptor in acute and chronic kidney injury. Kidney Int 83: 804–810, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tang J, Yan Y, Zhao TC, Bayliss G, Yan H, Zhuang S. Class I histone deacetylase activity is required for proliferation of renal epithelial cells. Am J Physiol Renal Physiol 305: F244–F254, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Taylor AJ, Beck CW. Histone deacetylases are required for amphibian tail and limb regeneration but not development. Mech Dev 129: 208–218, 2012 [DOI] [PubMed] [Google Scholar]
- 28.Tseng AS, Carneiro K, Lemire JM, Levin M. HDAC activity is required during Xenopus tail regeneration. PLos One 6: e26382, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wang Z, Chen JK, Wang SW, Moeckel G, Harris RC. Importance of functional EGF receptors in recovery from acute nephrotoxic injury. J Am Soc Nephrol 14: 3147–3154, 2003 [DOI] [PubMed] [Google Scholar]
- 30.Wen X, Murugan R, Peng Z, Kellum JA. Pathophysiology of acute kidney injury: a new perspective. Contrib Nephrol 165: 39–45, 2010 [DOI] [PubMed] [Google Scholar]
- 31.Xia J, Zhou Y, Ji H, Wang Y, Wu Q, Bao J, Ye F, Shi Y, Bu H. Loss of histone deacetylases 1 and 2 in hepatocytes impairs murine liver regeneration through Ki67 depletion. Hepatology 58: 2089–2098, 2013 [DOI] [PubMed] [Google Scholar]
- 32.Zhu J, Wan H, Xue C, Jiang T, Qian C, Zhang Y. Histone deacetylase 3 implicated in the pathogenesis of children glioma by promoting glioma cell proliferation and migration. Brain Res 1520: 15–22, 2013 [DOI] [PubMed] [Google Scholar]
- 33.Zhuang S, Dang Y, Schnellmann RG. Requirement of the epidermal growth factor receptor in renal epithelial cell proliferation and migration. Am J Physiol Renal Physiol 287: F365–F372, 2004 [DOI] [PubMed] [Google Scholar]