

Abstract
Adipose tissue metabolism is a critical regulator of adiposity and whole body energy expenditure; however, metabolic changes that occur in white adipose tissue (WAT) with obesity remain unclear. The purpose of this study was to understand the metabolic and bioenergetic changes occurring in WAT with obesity. Wild-type (C57BL/6J) mice fed a high-fat diet (HFD) showed significant increases in whole body adiposity, had significantly lower V̇o2, V̇co2, and respiratory exchange ratios, and demonstrated worsened glucose and insulin tolerance compared with low-fat-fed mice. Metabolomic analysis of WAT showed marked changes in lipid, amino acid, carbohydrate, nucleotide, and energy metabolism. Tissue levels of succinate and malate were elevated, and metabolites that could enter the Krebs cycle via anaplerosis were mostly diminished in high-fat-fed mice, suggesting altered mitochondrial metabolism. Despite no change in basal oxygen consumption or mitochondrial DNA abundance, citrate synthase activity was decreased by more than 50%, and responses to FCCP were increased in WAT from mice fed a high-fat diet. Moreover, Pgc1a was downregulated and Cox7a1 upregulated after 6 wk of HFD. After 12 wk of high-fat diet, the abundance of several proteins in the mitochondrial respiratory chain or matrix was diminished. These changes were accompanied by increased Parkin and Pink1, decreased p62 and LC3-I, and ultrastructural changes suggestive of autophagy and mitochondrial remodeling. These studies demonstrate coordinated restructuring of metabolism and autophagy that could contribute to the hypertrophy and whitening of adipose tissue in obesity.
Keywords: metabolomics, insulin resistance, adipocyte, autophagy, mitochondria
the increasing prevalence of obesity is a principal health concern worldwide. In 2008, approximately 1.5 billion adults aged 20 yr or older were overweight, and 10% were obese (2). In the US, more than one-third of the adult population is currently obese (BMI >30), and 68% have a BMI of >25 (17). By 2025, these numbers are expected to increase by more than 50% (70). These statistics are a cause for alarm. Obesity is a powerful predictor of insulin resistance (51) and a major risk factor for several common medical conditions such as type 2 diabetes, cardiovascular disease, nonalcoholic fatty liver disease, gallstones, Alzheimer's disease, and cancers such as breast, colon, endometrial, and esophageal cancer (21).
Although lack of exercise is an undeniable risk factor for weight gain (21, 27, 50), excessive caloric intake appears to be one of the key factors fueling the obesity epidemic. In the past three decades, the average consumption of calories in the US has increased by ≥200 kcal/day per person, which could be partly attributable to an increase in the intake of energy-dense foods (7, 29, 40, 77). Such poor dietary habits negatively affect metabolic homeostasis, which could not only promote obesity but hasten the development of obesity-related comorbidities as well. Despite the simplicity of the apparent remedy (i.e., decreasing caloric intake), treatment of obesity remains a challenging crisis facing the health care system. The efficacy of losing weight via caloric restriction is limited by multiple challenges. These include an evolutionarily engendered guard against starvation and low fat mass (70, 78) and a propensity to increase caloric efficiency during dieting (34, 47). Although drugs with anorectic and antiobesogenic properties are in clinical use, many of these show marginal long-term efficacy or have unacceptable or overtly dangerous side effects. Thus, recent strategies to modulate obesity have begun to target tissues that naturally regulate energy metabolism (70).
Increasing energy expenditure by modulating adipose tissue activity could be an attractive target for therapy. Because adult humans maintain small depots of brown fat that are capable of burning significant amounts of caloric energy (16, 53, 72, 75), multiple studies have focused on the physiological and molecular mechanisms regulating the thermogenic capacity of adipose tissue. These studies have shown that adaptive thermogenesis in brown fat can be a powerful regulator of systemic energy metabolism. However, the relatively small amount of brown adipose (<0.4% of body weight) compared with white adipose tissue (WAT; which can comprise >40% of the body weight of an obese human) suggests that WAT may be a more tangible target. Interestingly, white adipose depots, which typically function to esterify free fatty acids and store excess lipids, have the capacity to develop into brown adipose-like tissue that is capable of modulating systemic metabolism and preventing obesity and insulin resistance (65).
Although the phenomenon of adipose tissue “browning” is an active area of research, there is also considerable interest in understanding the metabolic changes that occur in WAT with obesity. It is becoming increasingly clear that conditions of nutrient excess promote “whitening” of adipose tissue characterized by a decrease in mitochondrial abundance (6, 14, 68). Hence, although promoting browning is one way to positively modulate metabolism, decreasing adipose tissue whitening could be another strategy to prevent dysregulation of systemic metabolism during obesity. Indeed, the beneficial metabolic changes induced by drugs such as rosiglitazone and pioglitazone have been suggested to be due in part to their ability to prevent loss of mitochondria or increase mitochondrial function in WAT (6, 79). Nevertheless, mitochondrial changes in WAT during the development of obesity have not been thoroughly examined, and it remains unclear how changes in adipocyte metabolism affect adipocyte hypertrophy and metabolic homeostasis.
In this study, we examined WAT-specific changes in metabolism in a mouse model of diet-induced obesity. Our data indicate extensive metabolic remodeling of WAT that precedes both the infiltration of inflammatory cells and overt decreases in mitochondrial abundance. This renovation of adipocyte metabolism involved changes in glucose and lipid pathways that appear to favor fat storage and prevent excessive lipid oxidation. The metabolite profile also yields evidence of osmotic stress and adipocyte inflammation. Restructuring of intermediary metabolism was accompanied by biochemical and ultrastructural evidence of autophagy, which may promote degradation of mitochondria. These findings have important implications for understanding the metabolic effects of obesity on adipose tissue metabolism and suggest that targeting metabolic pathways that contribute to adipose tissue whitening or hypertrophy could form the basis for novel therapies to combat metabolic disease.
METHODS
Animal studies.
All procedures were approved by the University of Louisville Institutional Animal Care and Use Committee. C57BL/6J [wild-type (WT)] mice were purchased from The Jackson Laboratory (Bar Harbor, ME). At 8 wk of age, male mice were placed on either a 10% low-fat diet (LFD) (no. D12450B; Research Diets) or a 60% high-fat diet (HFD) (no. D12492; Research Diets) for 6 or 12 wk. Water and diet were provided ad libitum. Body weights were recorded weekly.
Metabolic phenotyping.
Body composition was measured by dual-energy X-ray absorptiometry using a mouse densitometer (PIXImus2; Lunar, Madison, WI), and whole body energy expenditure, respiratory exchange ratio, food consumption, and locomotion, ambulatory, and fine movements were measured using a physiological/metabolic cage system (TSE PhenoMaster System, Bad Homberg, Germany) (54). Glucose and insulin tolerance tests and plasma levels of insulin were measured exactly as described by Sansbury et al. (54).
Adipocyte size measurements.
Adipose tissue was excised at the time of euthanization, and wet weight was recorded. All adipose tissue was either snap-frozen at −80°C or fixed in 10% formalin, paraffin embedded, and sectioned. The sections were stained in hematoxylin and eosin. Adipocyte cross-sectional area and distribution were determined using Nikon Elements software. Adipose tissue sections were assessed for crown-like structures, as described previously (22).
Adipose tissue metabolite profiling.
WAT from the epididymal fat pads of mice fed LFD or HFD for 6 wk was used for these analyses. Prior to tissue collection, the mice were fasted for 16 h. After euthanization, the adipose tissue was removed and immediately snap-frozen in liquid nitrogen. Relative metabolite abundance was then measured by gas chromatography-mass spectrometry or liquid chromatography-mass spectrometry, as described before (54, 55). Metabolites with missing values were imputed by replacing missing values with half of the minimum positive value in the original data. After a generalized logarithm transformation, the data were autoscaled, i.e., mean-centered and divided by the standard deviation of each variable. This step was performed to transform the intensity values so that the distribution was more Gaussian. Univariate (e.g., volcano plots), multivariate [e.g., partial least squares-discriminant analysis (PLS-DA)], and cluster (heatmap and dendogram) analyses were then performed. Most analyses were performed using Metaboanalyst 2.0 software (http://www.metaboanalyst.ca/) (82); z-score plots were constructed in GraphPad 5.0 software.
Adipose tissue bioenergetic measurements.
The oxygen consumption rate (OCR) of intact WAT explants was measured using a Seahorse XF24 analyzer (Seahorse Bioscience, Billerica, MA), as described previously (54). At least two replicates from each animal were used for the assay. After baseline measurements, the maximal OCR was measured by exposing the explants to FCCP (10 μM). The nonmitochondrial OCR was measured following injection of antimycin A (25 μM) and rotenone (5 μM).
Citrate synthase activity assay.
Citrate synthase assay was performed in 100 mM Tris·HCl, pH 8.0, containing 1 mM EDTA, 1 mM 5′,5′-dithiobis 2-nitrobenzoic acid, and 10 mM acetyl-CoA. The reaction was initiated by the addition of 10 mM oxaloacetate. Cuvettes were warmed to 37°C, and upon addition of 10 μg of protein from WAT lysates, absorbance at 420 nm was measured for 10 min. Activity is expressed as nanomoles per minute per microgram of protein.
Expression analyses.
For quantitative RT-PCR, RNA was extracted from tissues using the RNeasy lipid tissue kit (Qiagen), followed by cDNA synthesis. Real-time PCR amplification was performed with SYBR Green quantitative PCR (qPCR) Master Mix (SA Biosciences) using a 7900HT Fast Real-Time PCR System (Applied Biosystems) and primers for Il1, Tnfa, Il6, Arg1, Il10, Ym1, Hif1a, Emr1, Pgc1a, Cytc, Sirt1, Sirt3, Pdk4, Cpt1a, Cpt1b, Cox7a1, Hprt, and Idh3a (IDT Bioscience). Relative expression was determined by the 2−ΔΔCT method. M1 macrophages in WAT were measured by flow cytometry, as described previously (22).
For measuring protein abundance, WAT homogenates were prepared as described in Horrillo et al. (25). Equal amounts of protein were separated by SDS-PAGE, electroblotted to PVDF membranes, and probed using primary antibodies according to the respective manufacturers' protocols. The following antibodies were used: aldehyde dehydrogenase 2 (ALDH2; Abcam), sirtuin 3 (Sirt3; Cell Signaling Technology), MitoProfile Total OXPHOS Rodent WB Antibody Cocktail (Mitosciences), COX4I1 (Cell Signaling Technology), GAPDH (Cell Signaling Technology), Parkin (Abcam), Pink1 (Cell Signaling Technology), p62 (Cell Signaling Technology), LC3 (Cell Signaling Technology), protein-ubiquitin (Cell Signaling Technology), and α-tubulin (Sigma). Fluorescent or horseradish peroxidase-linked secondary antibodies (Invitrogen or Cell Signaling Technology, respectively) were used to detect and visualize the protein bands with a Typhoon 9400 variable mode imager (GE Healthcare). Band intensity was quantified using Image Quant TL software.
Relative mitochondrial DNA measurements.
Mitochondrial abundance in adipose tissue was estimated by measuring mitochondrial DNA (mtDNA) abundance relative to nuclear DNA (nDNA) (73). Total DNA was isolated from WAT using a QIAamp DNA Mini Kit (Qiagen). A 25-mg aliquot of the tissue was homogenized, followed by overnight digestion in proteinase K at 55°C. Following isolation, relative amounts of mtDNA and nDNA were compared using quantitative real-time PCR, using 2 ng of the isolated DNA. Primers for cytochrome b (mtDNA) and β-actin (nDNA) were used; the sequences are cytochrome b, 5′-TTGGGTTGTTTGATCCTGTTTCG-3′ and 5′-CTTCGCTTTCCACTTCATCTTACC-3′; and β-actin, 5′-CAGGATGCCTCTCTTGCTCT-3′ and 5′-CGTCTTCCCCTCCATCGT-3′.
Electron microscopy.
Adipose tissues were fixed with 3% glutaraldehyde in 0.1 M sodium phosphate buffer (pH 7.4) for 4 h at room temperature (25°C). The tissues were then postfixed with 1% osmium tetroxide for 1 h, dehydrated, and embedded in Embed-812 plastic (Electron Microscopy Sciences). Ultrathin sections were stained with uranyl acetate and Reynolds lead citrate, and electron micrographs were taken using a Philips CM10 transmission electron microscope operating at 80 kV.
Statistical analyses.
Data are means ± SE. Unpaired Student's t-test was used for direct comparisons. Statistical analyses for metabolomic data sets were performed using Metaboanalyst 2.0 software. A P value of <0.05 was considered significant.
RESULTS
HFD increases adiposity and alters systemic metabolism.
WT C57BL/6J mice were placed on a LFD or HFD for 6 wk. Significant weight gain occurred as early as 1 wk on HFD, and the change in total body mass was nearly 10 g by 6 wk on the diet (Fig. 1A). Food and water intake were not significantly different between groups (Fig. 1, B and C). Dual-energy X-ray absorptiometry scan analysis showed a twofold increase in percent fat mass and a concomitant decrease in percent lean mass in HFD-fed mice (Fig. 1, D and E). These results are typical of this commonly utilized model of diet-induced obesity (30, 54).
Fig. 1.

Effects of high-fat diet (HFD) on weight gain, adiposity, and systemic metabolism. Male wild-type (WT) C57BL/6J mice were fed a low-fat diet (LFD; 10% kcal fat) or HFD (60% kcal fat) for 6 wk, and the following measurements were recorded: mouse weights during 6 wk of feeding (n = 20/group; A); food intake (n = 7/group; B); water intake (n = 7/group; C); representative dual-energy X-ray absorptiometry scan images (D); %lean mass and %body fat (n = 10/group; E); and average oxygen consumption (V̇o2; F), average carbon dioxide production (V̇co2; G), respiratory exchange ratio (RER; H); total activity level (I), ambulatory counts (J), and fine movements (n = 7/group; K). *P < 0.05 vs. LFD.
To determine how diet affects systemic metabolism, mice fed either LFD or HFD for 6 wk were placed in metabolic chambers, and their oxygen consumption (V̇o2), carbon dioxide production (V̇co2), and physical activity were measured. As shown in Fig. 1, F and G, average V̇o2 and V̇co2 values decreased in HFD-fed mice compared with mice fed a LFD. The respiratory exchange ratio (RER) was also decreased in HFD mice vs. mice fed LFD (Fig. 1H). Physical activity, measured by total beam breaks (Fig. 1I), ambulatory counts (Fig. 1J), and fine movements (Fig. 1K), was not significantly different between groups, although the group fed a HFD appeared to show a trend toward decreased activity. Consistent with our previous results (54), mice fed a HFD also demonstrated worsened glucose and insulin tolerance (Fig. 2) as well as a significant increase in plasma insulin levels (WT LFD 151 ± 71 pg/ml vs. WT HFD 2,690 ± 593 pg/ml; n = 3/group, P < 0.05).
Fig. 2.
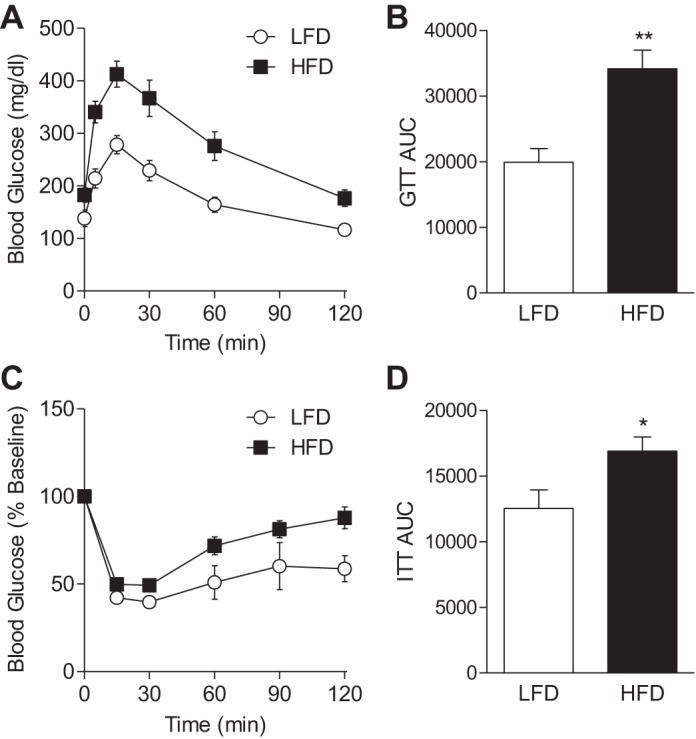
Glucose and insulin tolerance in mice fed LFD or HFD. After 6 wk of a LFD or HFD, glucose tolerance and insulin sensitivity were examined: glucose tolerance test (GTT; A), GTT area under the curve (AUC; B), insulin tolerance test (ITT) shown as %baseline (C), and ITT AUC (D); n = 7/group. *P < 0.05 and **P < 0.01 vs. LFD.
Robust macrophage infiltration does not occur with 6 wk of HFD.
To examine the effects of HFD on macrophage infiltration, we placed mice on LFD or HFD for 6 or 12 wk and measured adipocyte size, crown-like structures that are indicative of macrophage infiltration, and the expression of inflammatory genes. As shown in Fig. 3, A–C, mice fed a HFD for 6 and 12 wk showed a three- to fourfold increase in adipocyte size when compared with LFD controls. Whereas sections of WAT derived from mice fed a HFD for 12 wk showed obvious increases in crown-like structures, WAT from mice fed HFD for 6 wk showed minimal increases in such structures. These observations suggest that with 6 wk of HFD there is minimal macrophage accumulation. Indeed, the expression of Emr1, a marker of macrophages, as well as that of other inflammatory genes was not changed with 6 wk of HFD (Fig. 3D). Moreover, the abundance of M1 macrophages in adipose tissue stromal vascular fractions was not different between mice fed different diets (F4/80+/CD11c+/CD301− cells as %F4/80+ cells: LFD 37.4 ± 3.5, HFD 45.9 ± 1.8; n = 9–10/group, P > 0.05). Most likely, the modest, insignificant increase in Tnfa is due to adipocytes, which have been shown to be capable of producing TNFα (26, 60). In addition, no increase in plasma levels of inflammatory mediators such as IL-6 were identified at 6 wk of HFD [IL-6 (pg/ml): LFD, 23.6 ± 7.5; HFD, 18.8 ± 4.2]. Collectively, these data show that adipocyte size was increased after 6 wk of HFD, without significant changes in infiltrating inflammatory cells.
Fig. 3.

Effect of HFD on adipose tissue expansion and inflammation. Morphological and molecular changes in adipose tissues. A: representative hematoxylin and eosin stains of epididymal adipose tissue from mice fed a LFD or HFD for 6 or 12 wk; inset shows a crown-like structure (CLS) in the 12-wk HFD group. B: average area of adipocytes. C: adipocyte size distribution. D: quantitative RT-PCR analyses of markers of inflammation in adipose tissues from mice fed a LFD or HFD for 6 wk; n = 4–5/group. *P < 0.05 vs. indicated groups.
Obesity alters the metabolite profile of adipose tissue.
To examine the effect of obesity on adipose tissue metabolism, epididymal WAT from mice fed a LFD or HFD for 6 wk was subjected to unbiased metabolomic analysis. The relative concentration of adipose metabolites was measured by mass spectrometry and queried against the Metabolon reference library. It is important to note that the composition of adipose tissue derived from mice fed a LFD differed from mice fed a HFD. Compared with the LFD group, the protein yield per milligram wet weight was 43% lower in the adipose tissue of the HFD group (μg protein/mg wet wt: 6 wk LFD, 10.86 ± 1.70; 6 wk HFD, 6.21 ± 1.28; n = 10–12/group). Hence, for metabolomic analyses, we corrected for this difference in protein content. Partial least squares-discriminant analysis (PLS-DA) with the corrected data showed that the LFD samples separate clearly from HFD samples (Fig. 4A), and cluster analysis showed that the abundance of several metabolites was associated with diet (Fig. 4B). Of the 191 metabolites measured, 49 were found to be significantly different (P < 0.05) in the WAT of mice fed a HFD compared with the adipose tissue from mice fed a LFD. Volcano plot analysis, using data corrected for protein content, showed significantly higher levels of 34 metabolites and significantly lower levels of 15 metabolites in the adipose tissue of HFD-fed mice (Fig. 4C and Table 1). Volcano plot analysis based on uncorrected raw data showed a large skew toward decreased abundance of most metabolites (Fig. 4C, inset). To visualize the data in the biological context of metabolic pathways, metabolites that were statistically different in each group were analyzed using the MetPA tool of Metaboanalyst 2.0 software. The pathways were calculated as the sum of the importance measures of the matched metabolites normalized by the sum of the importance measures of all metabolites in each pathway (55, 81). As shown in Fig. 4D, the highest pathway impact values were related to linoleic acid metabolism and Phe, Tyr, and Trp metabolism. Branched-chain amino acids (BCAA), i.e., Val, Leu, and Ile, and pathways related to taurine metabolism, glycerolipid metabolism, Ala, Asp, and Glu metabolism, carbohydrate metabolism, and pyrimidine metabolism also showed relatively high pathway impact and significant values.
Fig. 4.

Metabolomic analyses of adipose tissue. Analyses of epididymal adipose tissue metabolites from WT mice fed a LFD or HFD for 6 wk. A: multivariate analysis; partial least squares-discriminant analysis (PLS-DA). Inset shows PLS-DA analysis from the uncorrected data set. B: hierarchial clustering; heat map and dendogram. C: univariate analysis; volcano plot of metabolites. Those metabolites that were significantly higher in abundance are in the quadrant shaded red, and those that were significantly lower are shaded green (P < 0.05, t-test). The identity of these metabolites is found in Table 1. The inset shows volcano plot analysis using data that were not corrected for protein content. D: metabolites found to be significantly different were subjected to pathway impact analysis using Metaboanalyst MetPA and the Mus musculus pathway library. Fisher's exact test was used for overrepresentation analysis, and relative betweenness centrality was used for pathway topology analysis; n = 14 animals (7 WT LFD and 7 WT HFD).
Table 1.
List of adipose tissue metabolites that changed significantly in high-fat-fed mice
| Metabolite | Fold Change | P Value | FDR |
|---|---|---|---|
| Stearoyl sphingomyelin* | 2.8 | 0.000542 | 0.010414 |
| Glycerophosphoethanolamine* | 2.7 | 0.000000 | 0.000014 |
| Succinate* | 2.34 | 0.000139 | 0.006385 |
| Inosine 5′-monophosphate | 2.27 | 0.016131 | 0.088492 |
| Uridine monophosphate (5′ or 3′) | 2.26 | 0.009442 | 0.064744 |
| Dihomo-linoleate (20:2n6) | 2.03 | 0.011868 | 0.071209 |
| Pelargonate (9:0) | 2.02 | 0.030984 | 0.14203 |
| Cytidine 5′-monophosphate* | 1.95 | 0.000214 | 0.006839 |
| Isobar: fructose 1,6-diphosphate, glucose 1,6-diphosphate, myo-inositol 1,4 or 1,3-diphosphate* | 1.87 | 0.000166 | 0.006385 |
| Maltose | 1.86 | 0.03107 | 0.14203 |
| Docosapentaenoate (22:5n3) | 1.83 | 0.033432 | 0.14928 |
| Glutamate* | 1.81 | 0.000111 | 0.006385 |
| Glucose | 1.8 | 0.005764 | 0.046111 |
| Azelate (nonanedioate) | 1.75 | 0.001215 | 0.01795 |
| Dihomo-linolenate (20:3n3 or n6) | 1.75 | 0.006864 | 0.052713 |
| Stearate (18:0) | 1.74 | 0.003972 | 0.038131 |
| Arachidonate (20:4n6) | 1.68 | 0.001536 | 0.021059 |
| Deoxycarnitine | 1.64 | 0.000539 | 0.010414 |
| Glycerophosphorylcholine | 1.64 | 0.005243 | 0.045087 |
| Hypotaurine | 1.62 | 0.024921 | 0.12592 |
| 12-Hydroxyeicosatetraenoic acid | 1.61 | 0.036945 | 0.16121 |
| Acetylcarnitine | 1.54 | 0.000362 | 0.008923 |
| Margarate (17:0) | 1.51 | 0.046138 | 0.18455 |
| Aspartate | 1.47 | 0.003862 | 0.038131 |
| Creatine | 1.46 | 0.000666 | 0.01162 |
| Malate | 1.45 | 0.008981 | 0.063867 |
| Linoleate (18:2n6) | 1.42 | 0.026561 | 0.13076 |
| Palmitoyl ethanolamide | 1.38 | 0.02444 | 0.12592 |
| Propionylcarnitine | 1.37 | 0.048868 | 0.18902 |
| Taurine | 1.33 | 0.001968 | 0.024159 |
| Glutamine | 1.32 | 0.000372 | 0.008923 |
| Choline phosphate | 1.32 | 0.011199 | 0.06936 |
| Citrulline | 1.29 | 0.002367 | 0.025244 |
| Cholesterol | 1.28 | 0.002251 | 0.025244 |
| Leucine* | 0.79 | 0.005401 | 0.045087 |
| Tyrosine* | 0.78 | 0.042828 | 0.18273 |
| Pantothenate | 0.73 | 0.049223 | 0.18902 |
| Mead acid (20:3n9) | 0.66 | 0.028737 | 0.13794 |
| Phenylalanine | 0.62 | 0.011086 | 0.06936 |
| Valine | 0.6 | 0.00483 | 0.044159 |
| 3-Dehydrocarnitine | 0.58 | 0.013332 | 0.077568 |
| 1,5-Anhydroglucitol | 0.56 | 0.010344 | 0.068482 |
| Glucose 6-phosphate | 0.51 | 0.045306 | 0.18455 |
| Histamine | 0.46 | 0.017878 | 0.095349 |
| Ergothioneine | 0.45 | 0.000163 | 0.006385 |
| N-acetylmethionine* | 0.35 | 0.002013 | 0.024159 |
| Mannose 6-phosphate* | 0.33 | 0.013998 | 0.079048 |
| Glycerate* | 0.07 | 0.008323 | 0.061462 |
| Glycylleucine* | 0.05 | 0.000986 | 0.015774 |
FDR, false discovery rate. Wild-type mice were fed a low-fat (LFD) or high-fat diet (HFD) for 6 wk. Epididymal adipose tissue was then subjected to liquid chromatography or gas chromatography mass spectrometric analysis. Raw spectral values from white adipose tissue (WAT) of HFD-fed mice were corrected on the basis of mean difference in protein content (per mg wet wt) compared with WAT from LFD-fed mice. Adipose tissue metabolites in HFD-fed mice that were ≥25% changed in abundance and significantly different (P < 0.05) compared with LFD-fed mice are shown; n = 7 mice/group.
Metabolites that were significant even when ±2 SE was applied in protein correction.
To delineate changes in metabolites further, metabolites were categorized based on superfamily class, and z-score plots were constructed. As shown in Fig. 5, the abundance of multiple members of the lipid superfamily, including those involved in glycerolipid metabolism and linoleic acid metabolism, was higher. Several carnitine derivatives, with the sole exception of 3-dehydrocarnitine, were higher in abundance as well. In the amino acid superfamily, the levels of nonessential amino acids Glu, Gln, and Asp were elevated, and those of essential amino acids Phe, Leu, and Val were lower. Notably, the organic osmolytes taurine, hypotaurine, and creatine were more abundant. In the HFD group, levels of the Krebs cycle intermediate succinate were higher; levels of malate were higher as well, although pantothenate, a precursor to CoA, was less abundant. Although both glucose and maltose levels were higher, phosphorylated sugars were diminished. Plasma 1,5-anhydroglucitol (1,5-AG), which is present in most organs and tissues and has been shown to decrease with loss of glycemic control (67, 83), was diminished in abundance, which is in agreement with insulin resistance in skeletal muscle and liver occurring at 6 wk of HFD (30). Interestingly, the pyrimidine monophosphates cytidine-5′-monophosphate (CMP) and uridine monophosphate (UMP), as well as the purine inosine-5′-monophosphate (IMP), were elevated in WAT from HFD-fed mice. A graphic illustration of these changes, placed into context of known pathways of intermediary metabolism, is shown in Fig. 6.
Fig. 5.

Z-score plot analysis of metabolite changes in adipose tissue from HFD-fed mice. Data are shown as standard deviations from the mean of LFD. Z-scores of some metabolites were not plotted due to lack of detectable signal in either the LFD or HFD group, e.g., glycylleucine, glycerate. Each point represents 1 metabolite in 1 sample; n = 7/group.
Fig. 6.

Illustration of metabolite changes in adipose tissue of obese mice. Metabolic changes in adipose tissue that occur with HFD are shown in the context of known metabolic pathways. In gray are those metabolites that increased, and in black (and underlined) are those metabolites that were diminished in obese mice. 12-HETE, 12-hydroxyeicosatetraenoic acid; CoA, coenzyme A; CMP, cytidine 5′-monophosphate; DHAP, dihydroxyacetone phosphate; FAs, fatty acids; F6P, fructose-6-phosphate; F-1,6-P2, fructose-1,6-bisphosphate; G3P, glycerol 3-phosphate; G6P, glucose 6-phosphate; GA3P, glyceraldehyde-3-phosphate; GPC, glyerophosphorylcholine; GPE, glycerophosphoethanolamine; IMP, inosine 5′-monophosphate; UMP, uridine monophosphate (5′ or 3′). *Isobaric entity; metabolite identity is unclear from the data and could be either F-1,6-P2, glucose-1,6-diphosphate, or myo-inositol 1,4- or 1,3-diphosphate. #Organic osmolytes that changed in abundance.
Effects of obesity on oxygen consumption and mitochondrial remodeling in WAT.
The changes in energy metabolism found in our metabolomic analyses suggested that HFD might have altered adipose tissue bioenergetics. Importantly, these changes occurred in the absence of inflammatory cell infiltration (see Fig. 3), which could otherwise confound adipocyte-specific changes in metabolism. To determine how obesity affects mitochondrial function, WAT explants from mice fed a LFD or HFD were subjected to extracellular flux analysis. As shown in Fig. 7, A and B, the apparent basal mitochondrial OCR of adipose tissue derived from mice fed a LFD was approximately twofold higher when compared with adipose explants derived from HFD-fed mice (P < 0.05); however, after correction for protein content, there was no difference in mitochondrial oxygen consumption. Nevertheless, explants derived from mice fed a HFD responded more strongly to FCCP (Fig. 7C). Although citrate synthase activity was lower by >50% in WAT derived from these mice (Fig. 7D), relative abundance of mtDNA, as assessed by qPCR of mtDNA normalized to nDNA, was not affected with 6 wk of HFD (Fig. 7E).
Fig. 7.

Obesity-related energetic changes in white adipose tissue. Metabolic analysis of adipose tissue from mice fed a LFD or HFD for 6 wk: A: extracellular flux analysis. After 3 basal oxygen consumption rate (OCR) measurements, FCCP (10 μM) was injected, followed by injection of antimycin A (AA; 25 μM) and rotenone (Rot; 5 μM). Gray circles indicate values from HFD-fed mice that were corrected on the basis of differences in protein content compared with LFD-fed mice. For clarity, error bars were omitted from the HFD-corrected group. Dotted lines indicate ±2 SE from corrected values. B: detailed changes in basal, maximal (Max.), nonmitochondrial (N.M.), basal mitochondrial (Mitobasal), and maximum mitochondrial (Mitomax) rates of oxygen consumption. C: FCCP response: the FCCP response in each explant was calculated using the equation (OCRMAX/OCRBASAL) × 100; n = 10 mice/group. D: citrate synthase activity; n = 3–6 mice/group. E: relative mtDNA content; n = 6/group. *P < 0.05 vs. LFD group.
The observed changes in metabolite profile, increased mitochondrial responsiveness to FCCP, and diminished citrate synthase activity suggested that HFD might affect the expression of genes associated with metabolic activity. Indeed, the expression of Cox7a1, a subunit of cytochrome oxidase, was more than twofold higher in adipose tissue from mice fed a HFD, whereas the expression of Pgc1a, Sirt3, and Pdk4 was diminished (Fig. 8A), indicating mitochondrial remodeling despite preserved basal mitochondrial activity.
Fig. 8.

Obesity-related changes in mitochondrial protein abundance in white adipose tissue. Analysis of gene expression and protein abundance in adipose tissue from mice fed a LFD or HFD for 6 or 12 wk. A: expression of metabolic genes after 6 wk of diet. B: representative Western blots of mitochondrial matrix proteins and respiratory chain subunits. C: quantification of aldehyde dehydrogenase (ALDH2). D: quantification of sirtuin 3 (Sirt3). E: quantification of respiratory subunit abundance. All blots were normalized to ATP5A, which showed no change in abundance in any group; n = 4/group. *P < 0.05 vs. 6-wk LFD; #P < 0.05 vs. 12 wk LFD. NDUFB8, NADH dehydrogenase-1β subcomplex subunit 8; SDHB, succinate dehydrogenase complex subunit B; UQCRC2, ubiquinol-cytochrome c reductase core protein 2; MTCO1, cytochrome c oxidase subunit 1; COX4I1, cytochrome c oxidase subunit 4 isoform 1; ATP5A, ATP synthase H+ transporting mitochondrial F1 complex α-subunit.
To further understand how WAT mitochondria change with obesity, we assessed the relative abundance of several mitochondrial complex proteins as well as mitochondrial matrix proteins. Although no changes in mitochondrial protein abundance were observed at 6 wk of HFD, the protein levels of NDUFB8, SDHB, and COX4I1 subunits of complexes I, II, and IV, respectively, were diminished significantly by 12 wk of HFD (Fig. 8, B–E). The levels of the matrix proteins ALDH2 and Sirt3 showed similar trends, with ALDH2 being significantly lower after 12 wk of HFD.
Assessment of adipose tissue ultrastructure.
To determine subcellular changes that occur in adipose tissue of nutrient-stressed mice, we examined adipocyte ultrastructure using electron microscopy. As shown in Fig. 9A, adipose tissue from mice fed a LFD showed mitochondria with three distinct morphologies: a round morphology of small size that was located near the nucleus (Fig. 9A, images i and ii), a typical elongated shape up to ∼0.7 μm in length located in small protrusions along the adipocyte cell membrane (Fig. 9A, image iii), and long mitochondria (3 μm and above), which were located in juxtaposition to the fat locule (Fig. 9A, image iv). In adipocytes derived from HFD-fed mice, autophagosomes, defined by a double-membrane and comprising cytoplasmic constituents, were found next to mitochondria (Fig. 9B, image i), and large vacuoles of electron-dense material were found adjacent to autophagosomes (Fig. 9B, images ii and iii). In addition, many mitochondria in adipose tissue from HFD-fed mice appeared to be undergoing fission (Fig. 9B, images iv and v).
Fig. 9.

Ultrastructure of white adipose tissue from lean and obese mice. Representative transmission electron micrographs of epididymal adipose tissues derived from mice fed a LFD or HFD for 6 wk. A: ultrastructure of mitochondria in adipose tissues from LF-fed mice. Image i: micrograph of adipocytes in areas close to the nucleus; image ii: a higher magnification of image i; image iii: a cytosolic compartment containing a mitochondrion found protruding into the fat locule; image iv: an elongated mitochondrion in juxtaposition to the fat locule. B: ultrastructure of adipose tissue derived from HFD-fed mice. Image i: an elongated mitochondrion next to an autophagosome; image ii: an autophagosome in close proximity to a vacuole containing electron-dense material; image iii: a higher magnification of image ii; image iv: protrusion of cytosolic compartment containing an atypical mitochondrion; image v: mitochondrion that appears to be undergoing fission. *Autophagosomes. Small arrows indicate collagen. M, mitochondria; N, nucleus; LD, vacuole lipid droplet.
Effects of HFD on autophagy.
Changes in mitochondrial proteins, along with the ultrastructural alterations found to occur in the adipose tissue, suggest that HFD may promote mitochondrial remodeling and activate mitophagy in WAT. To examine this possibility, we measured markers of mitophagy and autophagy in adipose tissues from mice fed a LFD or HFD for 6 wk. Levels of the E3 ubiquitin ligase Parkin, which has been shown to accumulate in mitochondria destined for degradation (24), were 2.3-fold higher with 6 wk of HFD and nearly twofold higher after 12 wk of HFD (Fig. 10, A and B). Furthermore, the kinase Pink1, critical for identifying mitochondria destined for autophagy (24), was also nearly 40% higher in the HFD groups. In combination with the presence of autophagosomes and mitochondrial alterations observed by EM (Fig. 9), this observation suggests that metabolic remodeling of adipocytes in the expanding adipose organ may be related to autophagy. To address this possibility, we measured changes in protein indicators of autophagy. As shown in Fig. 10, C–H, levels of p62 and LC3-I were diminished significantly, and the LC3-II/LC3-I ratio was more than twofold higher in mice fed a HFD for 6 wk compared with those placed on LFD. However, there was no significant difference in total protein abundance of protein-ubiquitin and LC3-II.
Fig. 10.

Evidence for activation of mitophagy in white adipose tissue of obese mice. Immunoblot analysis of markers of mitophagy and autophagy. A: Western blots of Parkin and Pink 1 in adipose tissues from mice fed a LFD or HFD for 6 (left) or 12 wk (right). B: quantification of Parkin and Pink1 abundance from E; n = 4/group. C: representative Western blots of ubiquitinated proteins, p62, and LC3 in mice fed a LFD or HFD for 6 wk. D–H: quantification of protein-ubiquitin abundance, p62, LC3-I, LC3-II abundance, and LC3-II/LC3-I ratio; n = 10/group. *P < 0.05 vs. LFD.
DISCUSSION
The results of this study demonstrate coordinated restructuring of adipose tissue metabolism during adipocyte hypertrophy. Using metabolomics analysis, we identified several HFD-induced changes in key metabolites involved in energy, carbohydrate, nucleotide, lipid, and amino acid metabolism. Importantly, these changes were relatively independent of inflammatory cell infiltration, and the data were corrected for adipocyte content, both of which help eliminate confounding features related to tissue and adipocyte composition. Our analysis showed an early decrease in citrate synthase activity that coincided with reduced expression of Pgc1a, although mitochondrial abundance was not affected. Ultrastructural and immunological evidence suggested that whitening of the adipose tissue, evident after 12 wk of HFD, may also be related to autophagy, which appears to occur early after adipocytes attain near-maximum size. Taken together, these results reveal that conditions of nutrient excess promote progressive metabolic remodeling, decreased capacity for mitochondrial biogenesis, and potential activation of the autophagic program in WAT.
The major goal of this study was to understand how HFD affects adipose tissue biology and metabolism. This is important because key metabolic changes that occur during adipocyte hypertrophy could be targets for antiobesity or insulin-sensitizing therapies. We found that 6 wk of HFD feeding led to a profound increase in fat mass, which was accompanied by a decrease in systemic V̇o2, V̇co2, and RER and insulin resistance. Although adipose tissue inflammation was evident by the 10th to 12th wk of HFD (41, 66), we found no significant increase in inflammatory cell infiltration with 6 wk of HFD (e.g., see Fig. 3 and Refs. 30 and 41). This lack of extensive tissue infiltration by energetic inflammatory cells allowed us to examine HFD-induced changes specific to the adipose tissue.
Moreover, in our metabolic measurements, we accounted for changes in the composition of the adipocytes themselves. Because white adipocytes expand by storing triglycerides in the central lipid droplet, an equivalent tissue mass of WAT from a HFD-fed mouse comprises fewer cells (and more triglyceride) than adipose tissue from a LFD-fed mouse. Thus, normalization of metabolite levels by wet weight alone could lead to erroneous conclusions. An example of this is shown in Figs. 4 and 6, where uncorrected values indicate that most metabolites are decreased in WAT from HFD-fed mice (Fig. 4C, inset) and that mitochondrial OCR is diminished in adipocytes from the obese mice (Fig. 7, A and B). Correcting for protein content in samples from HFD-fed mice normalized the distribution of metabolic changes and rectified the leftward skew caused by decreased adipocyte content. Similarly, when values were normalized for protein content (rather than mg wet wt), there was no difference in tissue oxygen consumption between explants from LFD and HFD groups. This indicates that with 6 wk of HFD there was no change in basal mitochondrial oxygen consumption per adipocyte.
Our metabolic profiling indicates that HFD affects several metabolic pathways. In mice placed on HFD for 6 wk, we found significant changes in lipid, amino acid, energy, nucleotide, and carbohydrate metabolism. Higher levels of several fatty acids and lipid metabolites belonging to the linoleic acid metabolism subfamily were observed in WAT from HFD-fed mice. In this tissue there was also a higher abundance of arachidonic acid as well as metabolites involved in arachidonic acid synthesis, including linolenic acid and dihomo-linolenic acid, which indicates a coordinated upregulation of this pathway. WAT from HFD-fed mice also contained higher levels of the eicosanoid 12-hydroxyeicosatetraenoic acid (12-HETE), which is formed from arachidonate by 12/15-lipoxygenase (12/15-LO). Interestingly, 12/15-LO is upregulated in white adipocytes of both HFD-fed C57BL/6J mice (10) and Zucker obese rats (11). Thus, upregulation of 12/15-LO and increased production of its product (12-HETE) are consistent features of hypertropic WAT. This may be particularly significant in mediating adipose tissue dysfunction because two separate studies have shown that genetic deletion of 12/15-LO prevents HFD-induced adipose tissue inflammation and insulin resistance (42, 59).
The elevated levels of glycerophosphoethanolamine in WAT from obese mice could be due to an increase in the breakdown of phosphatidylethanolamine or a decrease in its rate of hydrolysis. Previous studies have shown that glycerophosphoethanolamine is hydrolyzed by enzymes such as glycerophosphodiester phosphodiesterase to form glycerol 3-phosphate (G3P) (84), which is required for triglyceride synthesis and thus would likely be in high demand in expanding adipocytes (46). Glycerophosphorylcholine, which was elevated in WAT from HFD-fed mice, could also be utilized to form G3P. However, a major function of glycerophosphorylcholine in the cell is to act as an organic osmolyte (19), suggesting that there might be an increase in osmotic stress in hypertrophic adipocytes. This view is supported by our observation that the levels of several major osmolytes such as taurine, hypotaurine, and creatine were higher in WAT from obese mice. Changes in osmolality could trigger a form of cell death called pyroptosis (4), which has been linked with inflammasome activity (57). Interestingly, the NLRP3 inflammasome is activated by cholesterol crystals, and, consistent with previous observations in hypertrophied adipocytes (20, 85), our analyses also showed an elevation of cholesterol in adipose tissue of obese mice. Collectively, these observations suggest an early increase in key metabolic instigators that could contribute to osmotic stress and inflammation in adipose tissue.
The HFD-induced increase in stearoyl sphingomyelin could further prime adipocytes for inflammation and metabolic dysfunction. Sphingomyelin (d18:1/18:0), which in humans is the only membrane phospholipid not derived from glycerol, is a type of sphingolipid found in cell membranes that consists of oleic acid attached to the C1 position and stearic acid attached to the C2 position. Genetic deletion of enzymes involved in sphingomyelin synthesis has been shown to protect against diet-induced obesity and insulin resistance (35, 38), and the breakdown of sphingomyelin could yield significant amounts of ceramide, which inhibits insulin signaling (12). Hence, the elevated levels of sphingomyelin observed in our study could prime adipocytes to release significant amounts of ceramide, which has been shown to be increased by 300% in both the plasma and the adipose tissue by HFD (61). Although ceramide levels were not measured in our analyses, we did observe a decrease in Pgc1a expression and a decrease in citrate synthase activity, both of which can be regulated by ceramide (56, 71). However, further studies are required to determine whether changes in mitochondrial biogenetic capacity and citrate synthase activity are related to high levels of stearoyl sphingomyelin.
Our metabolomic data suggest significant changes in intermediary metabolism related to energy expenditure and fat storage. We found that many carnitine derivatives were increased in WAT from HFD-fed mice. These include acetylcarnitine, proprionylcarnitine, and deoxycarnitine, although 3-dehydrocarnitine, an intermediate in carnitine degradation, was diminished. These changes suggest the presence of a “flooded” pool of adipocyte metabolites, which could be used either for energy provision or, more likely, as a temporary repository for substrates for fatty acid synthesis. In other tissues such as the heart, acetylcarnitine functions as a buffer of the acetyl-CoA pool, although it could be used as an energetic substrate as well (58). Similarly, the propionylcarnitine could buffer the propionyl CoA pool (63). Thus, it appears that, in expanding adipocytes, short-chain acylcarnitine pools could help buffer excess carbon until it is assimilated into fatty acids for triglyceride storage.
The observation that tissue levels of BCAAs such as leucine and valine were decreased, whereas propionylcarnitine and succinate levels were increased with HFD, suggests an increase in BCAA catabolism. Catabolism of BCAAs results in the formation of both acetyl-CoA and succinyl-CoA, and the latter can be converted to succinate, which in our analysis was elevated in WAT from HFD-fed mice. Thus, it is likely that in hypertrophic adipocytes there is increased utilization of BCAAs. In contrast to the decrease in BCAAs, we found that levels of Gln and Glu were increased with HFD. This increase may be particularly important given the multifaceted uses of glutamine, e.g., in anaplerotic metabolism, as a nitrogen donor in anabolic processes as well as a precursor for glutathione synthesis (69, 80). In addition, Gln and Asp are required for synthesis of several nucleotides, such as IMP, UMP, and CMP, all of which were increased in these studies. Interestingly, adipose tissue is a major site of glutamine synthesis, and its uptake can be regulated by changes in osmolality as well as by insulin (49).
Although there is a clear relationship between amino acids, insulin resistance, and obesity in animal models and humans (1, 13, 39), the mechanistic basis of this link is less clear. BCAA metabolism in adipose tissue modulates plasma BCAA levels (23), and interestingly, increasing circulating BCAAs in mice by preventing BCAA catabolism prevents diet-induced obesity and insulin resistance in mice (62), whereas feeding BCAAs to HFD-fed rats increases insulin resistance (39). Although further studies are required to test the full significance of such metabolic changes in BCAAs and other amino acid pathways, it is possible that changes in amino acid catabolism and downstream metabolites have novel roles in adipose tissue biology such as the regulating of lipid storage and release. This possibility is supported by a recent report showing that succinate, which can be derived from BCAA breakdown, inhibits lipolysis by activating Gpr91 (a succinate receptor) (48).
Metabolites related to glucose metabolism were also markedly affected by HFD. We found a nearly 50% decrease in the abundance of 1,5-AG in WAT samples from HFD-fed mice. Plasma 1,5-AG is a validated marker of short-term glycemic control and has been suggested to be in equilibrium in tissue and plasma pools (67, 83). Hence, although 6 wk of HFD does not appear to significantly affect fasting blood glucose levels (54), the decreased abundance of 1,5-AG is consistent with systemic insulin resistance and poor glycemic control (30). With respect to glycolysis, glucose 6-phosphate was decreased, and adipose tissue glucose was increased, suggesting that hexokinase might be a major rate-limiting step in the provision of glucose carbons required for triglyceride synthesis. Further evidence of changes in glucose metabolism is shown by diminished pdk4 expression, which regulates pyruvate dehydrogenase activity. This decrease in pdk4 could potentially promote glyceroneogenic formation of G3P (8) via cataplerosis (45).
Interestingly, although we observed no change in basal mitochondrial OCR when corrected for protein content, WAT from HFD-fed mice showed an increased response to FCCP. We speculate that once metabolic restraint due to Δp is lifted (by addition of uncoupler), the increased substrate available for oxidative phosphorylation, e.g., succinate, malate, glutamate, or short-chain acylcarnitines, results in increased rates of oxygen consumption. Also, in data sets corrected for protein content, there was a trend toward an increased rate of nonmitochondrial oxygen consumption, which might suggest an increase in adipocyte superoxide production due to cytosolic oxidase activity (9, 24). This possibility is consistent with previous studies showing an increase in oxidative stress in adipose tissue of obese mice, which is caused predominantly by an increase in NADPH oxidase and downregulation of antioxidant enzymes (15, 18, 36, 37, 43).
Further support for coordinated remodeling of mitochondrial and cytosolic metabolism is provided by the observation that expression of Cox7a1 was increased in the adipose tissue of obese mice. A similar increase in Cox7a1 gene has been shown in the WAT of fattening cattle (3), although the implications of this increase are currently unclear. Cox7a1 is considered to be a heart- and muscle-specific subunit of cytochrome oxidase, but it is also present in brown adipose tissue; these tissues are highly metabolic organs capable of utilizing large amounts of substrate. Thus, it is possible that the increase in Cox7a1 may be an adaptive response to dissipate excessive energy in the adipocyte. The decrease in citrate synthase activity, which provides additional evidence for remodeling of mitochondrial metabolism during adipocyte hypertrophy, is similar to that observed in db/db mice, where citrate synthase activity is diminished remarkably with no change in mtDNA/nDNA (52). Why citrate synthase decreases in expanding adipocytes is currently unclear; however, it could be an adaptation to prevent excessive influx of carbon into the Krebs cycle, which would be consistent with the view that during obesity there is an overall shift in metabolism from energy provision to lipid storage.
The notion that HFD remodels mitochondrial metabolism in adipocytes is further supported by diminished mitochondrial biogenetic capacity and later decreases in electron transport chain protein abundance. With 6 wk of HFD, mitochondrial abundance per se was not affected; however, we did find a significant decrease in Pgc1a expression, which was followed by loss of mitochondrial protein abundance after 12 wk of HFD. This relatively late decrease in mitochondrial protein abundance may also be due to early changes in mitochondrial metabolism that promote removal of mitochondria by autophagy. Autophagy has been shown previously to be increased in adipocytes from obese humans and mice (28, 33, 44). Consistent with those studies, our biochemical results show an increase in the LC3-II/LC3-I ratio and a decrease in p62, the combination of which would appear to be consistent with an increase in autophagic flux (5, 31). Ultrastructural data showing autophagosomes and vacuolated structures containing electron-dense material further suggest changes in autophagy consistent with its activation. However, a limitation of this study is that autophagic flux was not measured directly, making it difficult to ascribe with certainty directional changes to the autophagy pathway. Nevertheless, an activation of autophagy in adipose tissue would be expected to decrease the abundance of both LC3-I and p62 (32, 64, 76, 86), which is consistent with our results. Hence, our cumulative data are supported by evidence in the literature and suggest that autophagy is activated by HFD. Although we cannot clearly distinguish whether mitochondria are removed by autophagy, the increase in key proteins involved in mitophagy, Parkin and Pink1 (24, 74), would be consistent with a significant role of mitophagy in mediating mitochondrial remodeling and turnover in the WAT of obese mice.
In summary, in this study we identified key metabolic changes that occur during WAT expansion. These coordinated changes occur before the infiltration of inflammatory cells and include loss of mitochondrial biogenetic capacity, dysregulation of lipid, amino acid, glucose, nucleotide, and amino acid metabolism, changes in mitochondrial gene expression and protein abundance, and evidence of alterations in autophagy and possibly mitophagy. Our results are consistent with the view that constant nutrient excess shifts adipocyte metabolic activity from energy provision to lipid storage. We speculate that the decrease in mitochondrial biogenetic capacity and later removal of mitochondria may also be a response to limit lipid oxidation and oxidative stress and to promote fat storage. Further studies are required to assess the significance of each of the metabolic alterations and to test whether therapeutically targeting these pathways is a gainful strategy for preventing obesity and its unfortunate metabolic consequences.
GRANTS
This work was supported in part by grants from the National Institutes of Health (GM-103492, HL-55477, HL-59378, and HL-106173).
DISCLOSURES
The authors declare no competing or relevant financial interests.
AUTHOR CONTRIBUTIONS
T.D.C., C.R.H., B.E.S., and B.G.H. conception and design of research; T.D.C., C.R.H., B.E.S., A.A.G., N.Z., Y.T., J.H., and B.G.H. performed experiments; T.D.C., C.R.H., B.E.S., A.A.G., J.S., N.Z., Y.T., J.H., S.N.R., and B.G.H. analyzed data; T.D.C., C.R.H., B.E.S., J.S., J.H., S.N.R., M.S., and B.G.H. interpreted results of experiments; T.D.C., C.R.H., B.E.S., A.A.G., and B.G.H. prepared figures; T.D.C. and B.G.H. drafted manuscript; T.D.C., C.R.H., B.E.S., A.A.G., J.S., N.Z., Y.T., J.H., S.N.R., M.S., A.B., and B.G.H. approved final version of manuscript; M.S., A.B., and B.G.H. edited and revised manuscript.
REFERENCES
- 1.Adams SH. Emerging perspectives on essential amino acid metabolism in obesity and the insulin-resistant state. Adv Nutr 2: 445–456, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ahima RS. Digging deeper into obesity. J Clin Invest 121: 2076–2079, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Asano H, Yamada T, Hashimoto O, Umemoto T, Sato R, Ohwatari S, Kanamori Y, Terachi T, Funaba M, Matsui T. Diet-induced changes in Ucp1 expression in bovine adipose tissues. Gen Comp Endocrinol 184: 87–92, 2013 [DOI] [PubMed] [Google Scholar]
- 4.Bergsbaken T, Fink SL, Cookson BT. Pyroptosis: host cell death and inflammation. Nat Rev Microbiol 7: 99–109, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bjørkøy G, Lamark T, Pankiv S, Øvervatn A, Brech A, Johansen T. Monitoring autophagic degradation of p62/SQSTM1. Methods Enzymol 452: 181–197, 2009 [DOI] [PubMed] [Google Scholar]
- 6.Bogacka I, Xie H, Bray GA, Smith SR. Pioglitazone induces mitochondrial biogenesis in human subcutaneous adipose tissue in vivo. Diabetes 54: 1392–1399, 2005 [DOI] [PubMed] [Google Scholar]
- 7.Briefel RR, Johnson CL. Secular trends in dietary intake in the United States. Annu Rev Nutr 24: 401–431, 2004 [DOI] [PubMed] [Google Scholar]
- 8.Cadoudal T, Distel E, Durant S, Fouque F, Blouin JM, Collinet M, Bortoli S, Forest C, Benelli C. Pyruvate dehydrogenase kinase 4: regulation by thiazolidinediones and implication in glyceroneogenesis in adipose tissue. Diabetes 57: 2272–2279, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chacko BK, Kramer PA, Ravi S, Johnson MS, Hardy RW, Ballinger SW, Darley-Usmar VM. Methods for defining distinct bioenergetic profiles in platelets, lymphocytes, monocytes, and neutrophils, and the oxidative burst from human blood. Lab Invest 93: 690–700, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chakrabarti SK, Cole BK, Wen Y, Keller SR, Nadler JL. 12/15-lipoxygenase products induce inflammation and impair insulin signaling in 3T3-L1 adipocytes. Obesity (Silver Spring) 17: 1657–1663, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chakrabarti SK, Wen Y, Dobrian AD, Cole BK, Ma Q, Pei H, Williams MD, Bevard MH, Vandenhoff GE, Keller SR, Gu J, Nadler JL. Evidence for activation of inflammatory lipoxygenase pathways in visceral adipose tissue of obese Zucker rats. Am J Physiol Endocrinol Metab 300: E175–E187, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chavez JA, Summers SA. A ceramide-centric view of insulin resistance. Cell Metab 15: 585–594, 2012 [DOI] [PubMed] [Google Scholar]
- 13.Cheng S, Rhee EP, Larson MG, Lewis GD, McCabe EL, Shen D, Palma MJ, Roberts LD, Dejam A, Souza AL, Deik AA, Magnusson M, Fox CS, O'Donnell CJ, Vasan RS, Melander O, Clish CB, Gerszten RE, Wang TJ. Metabolite profiling identifies pathways associated with metabolic risk in humans. Circulation 125: 2222–2231, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Choo HJ, Kim JH, Kwon OB, Lee CS, Mun JY, Han SS, Yoon YS, Yoon G, Choi KM, Ko YG. Mitochondria are impaired in the adipocytes of type 2 diabetic mice. Diabetologia 49: 784–791, 2006 [DOI] [PubMed] [Google Scholar]
- 15.Curtis JM, Grimsrud PA, Wright WS, Xu X, Foncea RE, Graham DW, Brestoff JR, Wiczer BM, Ilkayeva O, Cianflone K, Muoio DE, Arriaga EA, Bernlohr DA. Downregulation of adipose glutathione S-transferase A4 leads to increased protein carbonylation, oxidative stress, and mitochondrial dysfunction. Diabetes 59: 1132–1142, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cypess AM, Lehman S, Williams G, Tal I, Rodman D, Goldfine AB, Kuo FC, Palmer EL, Tseng YH, Doria A, Kolodny GM, Kahn CR. Identification and importance of brown adipose tissue in adult humans. N Engl J Med 360: 1509–1517, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Flegal KM, Carroll MD, Ogden CL, Curtin LR. Prevalence and trends in obesity among US adults, 1999–2008. JAMA 303: 235–241, 2010 [DOI] [PubMed] [Google Scholar]
- 18.Furukawa S, Fujita T, Shimabukuro M, Iwaki M, Yamada Y, Nakajima Y, Nakayama O, Makishima M, Matsuda M, Shimomura I. Increased oxidative stress in obesity and its impact on metabolic syndrome. J Clin Invest 114: 1752–1761, 2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gallazzini M, Burg MB. What's new about osmotic regulation of glycerophosphocholine. Physiology 24: 245–249, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Giordano A, Murano I, Mondini E, Perugini J, Smorlesi A, Severi I, Barazzoni R, Scherer PE, Cinti S. Obese adipocytes show ultrastructural features of stressed cells and die of pyroptosis. J Lipid Res 54: 2423–2436, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Haslam DW, James WP. Obesity. Lancet 366: 1197–1209, 2005 [DOI] [PubMed] [Google Scholar]
- 22.Hellmann J, Tang Y, Kosuri M, Bhatnagar A, Spite M. Resolvin D1 decreases adipose tissue macrophage accumulation and improves insulin sensitivity in obese-diabetic mice. FASEB J 25: 2399–2407, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Herman MA, She P, Peroni OD, Lynch CJ, Kahn BB. Adipose tissue branched chain amino acid (BCAA) metabolism modulates circulating BCAA levels. J Biol Chem 285: 11348–11356, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hill BG, Benavides GA, Lancaster JR, Jr, Ballinger S, Dell'Italia L, Jianhua Z, Darley-Usmar VM. Integration of cellular bioenergetics with mitochondrial quality control and autophagy. Biol Chem 393: 1485–1512, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Horrillo R, González-Périz A, Martínez-Clemente M, López-Parra M, Ferré N, Titos E, Morán-Salvador E, Deulofeu R, Arroyo V, Clària J. 5-lipoxygenase activating protein signals adipose tissue inflammation and lipid dysfunction in experimental obesity. J Immunol 184: 3978–3987, 2010 [DOI] [PubMed] [Google Scholar]
- 26.Hotamisligil GS, Arner P, Caro JF, Atkinson RL, Spiegelman BM. Increased adipose tissue expression of tumor necrosis factor-alpha in human obesity and insulin resistance. J Clin Invest 95: 2409–2415, 1995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hu FB, Li TY, Colditz GA, Willett WC, Manson JE. Television watching and other sedentary behaviors in relation to risk of obesity and type 2 diabetes mellitus in women. JAMA 289: 1785–1791, 2003 [DOI] [PubMed] [Google Scholar]
- 28.Jansen HJ, van Essen P, Koenen T, Joosten LA, Netea MG, Tack CJ, Stienstra R. Autophagy activity is up-regulated in adipose tissue of obese individuals and modulates proinflammatory cytokine expression. Endocrinology 153: 5866–5874, 2012 [DOI] [PubMed] [Google Scholar]
- 29.Kant AK, Graubard BI. Eating out in America, 1987–2000: trends and nutritional correlates. Prev Med 38: 243–249, 2004 [DOI] [PubMed] [Google Scholar]
- 30.Kim F, Pham M, Maloney E, Rizzo NO, Morton GJ, Wisse BE, Kirk EA, Chait A, Schwartz MW. Vascular inflammation, insulin resistance, and reduced nitric oxide production precede the onset of peripheral insulin resistance. Arterioscler Thromb Vasc Biol 28: 1982–1988, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Klionsky DJ, Abeliovich H, Agostinis P, Agrawal DK, Aliev G, Askew DS, Baba M, Baehrecke EH, Bahr BA, Ballabio A, Bamber BA, Bassham DC, Bergamini E, Bi X, Biard-Piechaczyk M, Blum JS, Bredesen DE, Brodsky JL, Brumell JH, Brunk UT, Bursch W, Camougrand N, Cebollero E, Cecconi F, Chen Y, Chin LS, Choi A, Chu CT, Chung J, Clarke PG, Clark RS, Clarke SG, Clave C, Cleveland JL, Codogno P, Colombo MI, Coto-Montes A, Cregg JM, Cuervo AM, Debnath J, Demarchi F, Dennis PB, Dennis PA, Deretic V, Devenish RJ, Di Sano F, Dice JF, Difiglia M, Dinesh-Kumar S, Distelhorst CW, Djavaheri-Mergny M, Dorsey FC, Droge W, Dron M, Dunn WA, Jr, Duszenko M, Eissa NT, Elazar Z, Esclatine A, Eskelinen EL, Fesus L, Finley KD, Fuentes JM, Fueyo J, Fujisaki K, Galliot B, Gao FB, Gewirtz DA, Gibson SB, Gohla A, Goldberg AL, Gonzalez R, Gonzalez-Estevez C, Gorski S, Gottlieb RA, Haussinger D, He YW, Heidenreich K, Hill JA, Hoyer-Hansen M, Hu X, Huang WP, Iwasaki A, Jaattela M, Jackson WT, Jiang X, Jin S, Johansen T, Jung JU, Kadowaki M, Kang C, Kelekar A, Kessel DH, Kiel JA, Kim HP, Kimchi A, Kinsella TJ, Kiselyov K, Kitamoto K, Knecht E, Komatsu M, Kominami E, Kondo S, Kovacs AL, Kroemer G, Kuan CY, Kumar R, Kundu M, Landry J, Laporte M, Le W, Lei HY, Lenardo MJ, Levine B, Lieberman A, Lim KL, Lin FC, Liou W, Liu LF, Lopez-Berestein G, Lopez-Otin C, Lu B, Macleod KF, Malorni W, Martinet W, Matsuoka K, Mautner J, Meijer AJ, Melendez A, Michels P, Miotto G, Mistiaen WP, Mizushima N, Mograbi B, Monastyrska I, Moore MN, Moreira PI, Moriyasu Y, Motyl T, Munz C, Murphy LO, Naqvi NI, Neufeld TP, Nishino I, Nixon RA, Noda T, Nurnberg B, Ogawa M, Oleinick NL, Olsen LJ, Ozpolat B, Paglin S, Palmer GE, Papassideri I, Parkes M, Perlmutter DH, Perry G, Piacentini M, Pinkas-Kramarski R, Prescott M, Proikas-Cezanne T, Raben N, Rami A, Reggiori F, Rohrer B, Rubinsztein DC, Ryan KM, Sadoshima J, Sakagami H, Sakai Y, Sandri M, Sasakawa C, Sass M, Schneider C, Seglen PO, Seleverstov O, Settleman J, Shacka JJ, Shapiro IM, Sibirny A, Silva-Zacarin EC, Simon HU, Simone C, Simonsen A, Smith MA, Spanel-Borowski K, Srinivas V, Steeves M, Stenmark H, Stromhaug PE, Subauste CS, Sugimoto S, Sulzer D, Suzuki T, Swanson MS, Tabas I, Takeshita F, Talbot NJ, Talloczy Z, Tanaka K, Tanaka K, Tanida I, Taylor GS, Taylor JP, Terman A, Tettamanti G, Thompson CB, Thumm M, Tolkovsky AM, Tooze SA, Truant R, Tumanovska LV, Uchiyama Y, Ueno T, Uzcategui NL, van der Klei I, Vaquero EC, Vellai T, Vogel MW, Wang HG, Webster P, Wiley JW, Xi Z, Xiao G, Yahalom J, Yang JM, Yap G, Yin XM, Yoshimori T, Yu L, Yue Z, Yuzaki M, Zabirnyk O, Zheng X, Zhu X, Deter RL. Guidelines for the use and interpretation of assays for monitoring autophagy in higher eukaryotes. Autophagy 4: 151–175, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Komatsu M, Wang QJ, Holstein GR, Friedrich VL, Jr, Iwata J, Kominami E, Chait BT, Tanaka K, Yue Z. Essential role for autophagy protein Atg7 in the maintenance of axonal homeostasis and the prevention of axonal degeneration. Proc Natl Acad Sci USA 104: 14489–14494, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kovsan J, Bluher M, Tarnovscki T, Kloting N, Kirshtein B, Madar L, Shai I, Golan R, Harman-Boehm I, Schon MR, Greenberg AS, Elazar Z, Bashan N, Rudich A. Altered autophagy in human adipose tissues in obesity. J Clin Endocrinol Metab 96: E268–E277, 2011 [DOI] [PubMed] [Google Scholar]
- 34.Leibel RL, Rosenbaum M, Hirsch J. Changes in energy expenditure resulting from altered body weight. N Engl J Med 332: 621–628, 1995 [DOI] [PubMed] [Google Scholar]
- 35.Li Z, Zhang H, Liu J, Liang CP, Li Y, Teitelman G, Beyer T, Bui HH, Peake DA, Zhang Y, Sanders PE, Kuo MS, Park TS, Cao G, Jiang XC. Reducing plasma membrane sphingomyelin increases insulin sensitivity. Mol Cell Biol 31: 4205–4218, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Long EK, Olson DM, Bernlohr DA. High-fat diet induces changes in adipose tissue trans-4-oxo-2-nonenal and trans-4-hydroxy-2-nonenal levels in a depot-specific manner. Free Radic Biol Med 63: 390–398, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Matsuzawa-Nagata N, Takamura T, Ando H, Nakamura S, Kurita S, Misu H, Ota T, Yokoyama M, Honda M, Miyamoto K, Kaneko S. Increased oxidative stress precedes the onset of high-fat diet-induced insulin resistance and obesity. Metabolism 57: 1071–1077, 2008 [DOI] [PubMed] [Google Scholar]
- 38.Mitsutake S, Zama K, Yokota H, Yoshida T, Tanaka M, Mitsui M, Ikawa M, Okabe M, Tanaka Y, Yamashita T, Takemoto H, Okazaki T, Watanabe K, Igarashi Y. Dynamic modification of sphingomyelin in lipid microdomains controls development of obesity, fatty liver, and type 2 diabetes. J Biol Chem 286: 28544–28555, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Newgard CB, An J, Bain JR, Muehlbauer MJ, Stevens RD, Lien LF, Haqq AM, Shah SH, Arlotto M, Slentz CA, Rochon J, Gallup D, Ilkayeva O, Wenner BR, Yancy WS, Jr, Eisenson H, Musante G, Surwit RS, Millington DS, Butler MD, Svetkey LP. A branched-chain amino acid-related metabolic signature that differentiates obese and lean humans and contributes to insulin resistance. Cell Metab 9: 311–326, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Nielsen SJ, Popkin BM. Patterns and trends in food portion sizes, 1977–1998. JAMA 289: 450–453, 2003 [DOI] [PubMed] [Google Scholar]
- 41.Nishimura S, Manabe I, Nagasaki M, Eto K, Yamashita H, Ohsugi M, Otsu M, Hara K, Ueki K, Sugiura S, Yoshimura K, Kadowaki T, Nagai R. CD8+ effector T cells contribute to macrophage recruitment and adipose tissue inflammation in obesity. Nat Med 15: 914–920, 2009 [DOI] [PubMed] [Google Scholar]
- 42.Nunemaker CS, Chen M, Pei H, Kimble SD, Keller SR, Carter JD, Yang Z, Smith KM, Wu R, Bevard MH, Garmey JC, Nadler JL. 12-Lipoxygenase-knockout mice are resistant to inflammatory effects of obesity induced by Western diet. Am J Physiol Endocrinol Metab 295: E1065–E1075, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Okada S, Kozuka C, Masuzaki H, Yasue S, Ishii-Yonemoto T, Tanaka T, Yamamoto Y, Noguchi M, Kusakabe T, Tomita T, Fujikura J, Ebihara K, Hosoda K, Sakaue H, Kobori H, Ham M, Lee YS, Kim JB, Saito Y, Nakao K. Adipose tissue-specific dysregulation of angiotensinogen by oxidative stress in obesity. Metabolism 59: 1241–1251, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ost A, Svensson K, Ruishalme I, Brännmark C, Franck N, Krook H, Sandström P, Kjolhede P, Strålfors P. Attenuated mTOR signaling and enhanced autophagy in adipocytes from obese patients with type 2 diabetes. Mol Med 16: 235–246, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Owen OE, Kalhan SC, Hanson RW. The key role of anaplerosis and cataplerosis for citric acid cycle function. J Biol Chem 277: 30409–30412, 2002 [DOI] [PubMed] [Google Scholar]
- 46.Prentki M, Madiraju SR. Glycerolipid metabolism and signaling in health and disease. Endocr Rev 29: 647–676, 2008 [DOI] [PubMed] [Google Scholar]
- 47.Redman LM, Heilbronn LK, Martin CK, de Jonge L, Williamson DA, Delany JP, Ravussin E. Metabolic and behavioral compensations in response to caloric restriction: implications for the maintenance of weight loss. PLoS One 4: e4377, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Regard JB, Sato IT, Coughlin SR. Anatomical profiling of G protein-coupled receptor expression. Cell 135: 561–571, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ritchie JW, Baird FE, Christie GR, Stewart A, Low SY, Hundal HS, Taylor PM. Mechanisms of glutamine transport in rat adipocytes and acute regulation by cell swelling. Cell Physiol Biochem 11: 259–270, 2001 [DOI] [PubMed] [Google Scholar]
- 50.Robinson TN. Reducing children's television viewing to prevent obesity: a randomized controlled trial. JAMA 282: 1561–1567, 1999 [DOI] [PubMed] [Google Scholar]
- 51.Roger VL, Go AS, Lloyd-Jones DM, Benjamin EJ, Berry JD, Borden WB, Bravata DM, Dai S, Ford ES, Fox CS, Fullerton HJ, Gillespie C, Hailpern SM, Heit JA, Howard VJ, Kissela BM, Kittner SJ, Lackland DT, Lichtman JH, Lisabeth LD, Makuc DM, Marcus GM, Marelli A, Matchar DB, Moy CS, Mozaffarian D, Mussolino ME, Nichol G, Paynter NP, Soliman EZ, Sorlie PD, Sotoodehnia N, Turan TN, Virani SS, Wong ND, Woo D, Turner MB; American Heart Association Statistics Committee and Stroke Statistics Subcommittee. Heart disease and stroke statistics—2012 update: a report from the American Heart Association. Circulation 125: e2–e220, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Rong JX, Qiu Y, Hansen MK, Zhu L, Zhang V, Xie M, Okamoto Y, Mattie MD, Higashiyama H, Asano S, Strum JC, Ryan TE. Adipose mitochondrial biogenesis is suppressed in db/db and high-fat diet-fed mice and improved by rosiglitazone. Diabetes 56: 1751–1760, 2007 [DOI] [PubMed] [Google Scholar]
- 53.Rothwell NJ, Stock MJ. Luxuskonsumption, diet-induced thermogenesis and brown fat: the case in favour. Clin Sci (Lond) 64: 19–23, 1983 [DOI] [PubMed] [Google Scholar]
- 54.Sansbury BE, Cummins TD, Tang Y, Hellmann J, Holden CR, Harbeson MA, Chen Y, Patel RP, Spite M, Bhatnagar A, Hill BG. Overexpression of endothelial nitric oxide synthase prevents diet-induced obesity and regulates adipocyte phenotype. Circ Res 111: 1176–1189, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Sansbury BE, De Martino AM, Xie Z, Brooks AC, Brainard RE, Watson LJ, Defilippis AP, Cummins TD, Harbeson MA, Brittian KR, Prabhu SD, Bhatnagar A, Jones SP, Hill BG. Metabolomic Analysis of Pressure-overloaded and Infarcted Mouse Hearts. Circ Heart Fail. In press [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Schmitz-Peiffer C. Targeting ceramide synthesis to reverse insulin resistance. Diabetes 59: 2351–2353, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Schroder K, Tschopp J. The inflammasomes. Cell 140: 821–832, 2010 [DOI] [PubMed] [Google Scholar]
- 58.Schroeder MA, Atherton HJ, Dodd MS, Lee P, Cochlin LE, Radda GK, Clarke K, Tyler DJ. The cycling of acetyl-coenzyme A through acetylcarnitine buffers cardiac substrate supply: a hyperpolarized 13C magnetic resonance study. Circ Cardiovasc Imaging 5: 201–209, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Sears DD, Miles PD, Chapman J, Ofrecio JM, Almazan F, Thapar D, Miller YI. 12/15-lipoxygenase is required for the early onset of high fat diet-induced adipose tissue inflammation and insulin resistance in mice. PLoS One 4: e7250, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Sewter CP, Digby JE, Blows F, Prins J, O'Rahilly S. Regulation of tumour necrosis factor-alpha release from human adipose tissue in vitro. J Endocrinol 163: 33–38, 1999 [DOI] [PubMed] [Google Scholar]
- 61.Shah C, Yang G, Lee I, Bielawski J, Hannun YA, Samad F. Protection from high fat diet-induced increase in ceramide in mice lacking plasminogen activator inhibitor 1. J Biol Chem 283: 13538–13548, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.She P, Reid TM, Bronson SK, Vary TC, Hajnal A, Lynch CJ, Hutson SM. Disruption of BCATm in mice leads to increased energy expenditure associated with the activation of a futile protein turnover cycle. Cell Metab 6: 181–194, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Siliprandi N, Di Lisa F, Menabo R. Propionyl-l-carnitine: biochemical significance and possible role in cardiac metabolism. Cardiovasc Drugs Ther 5, Suppl 1: 11–15, 1991 [DOI] [PubMed] [Google Scholar]
- 64.Singh R, Xiang Y, Wang Y, Baikati K, Cuervo AM, Luu YK, Tang Y, Pessin JE, Schwartz GJ, Czaja MJ. Autophagy regulates adipose mass and differentiation in mice. J Clin Invest 119: 3329–3339, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Smorlesi A, Frontini A, Giordano A, Cinti S. The adipose organ: white-brown adipocyte plasticity and metabolic inflammation. Obes Rev 13, Suppl 2: 83–96, 2012 [DOI] [PubMed] [Google Scholar]
- 66.Spite M, Hellmann J, Tang Y, Mathis SP, Kosuri M, Bhatnagar A, Jala VR, Haribabu B. Deficiency of the leukotriene B4 receptor, BLT-1, protects against systemic insulin resistance in diet-induced obesity. J Immunol 187: 1942–1949, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Stickle D, Turk J. A kinetic mass balance model for 1,5-anhydroglucitol: applications to monitoring of glycemic control. Am J Physiol Endocrinol Metab 273: E821–E830, 1997 [DOI] [PubMed] [Google Scholar]
- 68.Sutherland LN, Capozzi LC, Turchinsky NJ, Bell RC, Wright DC. Time course of high-fat diet-induced reductions in adipose tissue mitochondrial proteins: potential mechanisms and the relationship to glucose intolerance. Am J Physiol Endocrinol Metab 295: E1076–E1083, 2008 [DOI] [PubMed] [Google Scholar]
- 69.Tapiero H, Mathe G, Couvreur P, Tew KD., 2nd Glutamine and glutamate. Biomed Pharmacother 56: 446–457, 2002 [DOI] [PubMed] [Google Scholar]
- 70.Tseng YH, Cypess AM, Kahn CR. Cellular bioenergetics as a target for obesity therapy. Nat Rev Drug Discov 9: 465–482, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Ussher JR, Koves TR, Cadete VJ, Zhang L, Jaswal JS, Swyrd SJ, Lopaschuk DG, Proctor SD, Keung W, Muoio DM, Lopaschuk GD. Inhibition of de novo ceramide synthesis reverses diet-induced insulin resistance and enhances whole-body oxygen consumption. Diabetes 59: 2453–2464, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.van Marken Lichtenbelt WD, Vanhommerig JW, Smulders NM, Drossaerts JM, Kemerink GJ, Bouvy ND, Schrauwen P, Teule GJ. Cold-activated brown adipose tissue in healthy men. N Engl J Med 360: 1500–1508, 2009 [DOI] [PubMed] [Google Scholar]
- 73.Villena JA, Hock MB, Chang WY, Barcas JE, Giguere V, Kralli A. Orphan nuclear receptor estrogen-related receptor alpha is essential for adaptive thermogenesis. Proc Natl Acad Sci USA 104: 1418–1423, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Vincow ES, Merrihew G, Thomas RE, Shulman NJ, Beyer RP, MacCoss MJ, Pallanck LJ. The PINK1-Parkin pathway promotes both mitophagy and selective respiratory chain turnover in vivo. Proc Natl Acad Sci USA 110: 6400–6405, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Virtanen KA, Lidell ME, Orava J, Heglind M, Westergren R, Niemi T, Taittonen M, Laine J, Savisto NJ, Enerback S, Nuutila P. Functional brown adipose tissue in healthy adults. N Engl J Med 360: 1518–1525, 2009 [DOI] [PubMed] [Google Scholar]
- 76.Wang QJ, Ding Y, Kohtz DS, Mizushima N, Cristea IM, Rout MP, Chait BT, Zhong Y, Heintz N, Yue Z. Induction of autophagy in axonal dystrophy and degeneration. J Neurosci 26: 8057–8068, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Wang YC, Bleich SN, Gortmaker SL. Increasing caloric contribution from sugar-sweetened beverages and 100% fruit juices among US children and adolescents, 1988–2004. Pediatrics 121: e1604–e1614, 2008 [DOI] [PubMed] [Google Scholar]
- 78.Wells JC. Thrift: a guide to thrifty genes, thrifty phenotypes and thrifty norms. Int J Obes (Lond) 33: 1331–1338, 2009 [DOI] [PubMed] [Google Scholar]
- 79.Wilson-Fritch L, Nicoloro S, Chouinard M, Lazar MA, Chui PC, Leszyk J, Straubhaar J, Czech MP, Corvera S. Mitochondrial remodeling in adipose tissue associated with obesity and treatment with rosiglitazone. J Clin Invest 114: 1281–1289, 2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Wu G. Amino acids: metabolism, functions, and nutrition. Amino Acids 37: 1–17, 2009 [DOI] [PubMed] [Google Scholar]
- 81.Xia J, Wishart DS. MetPA: a web-based metabolomics tool for pathway analysis and visualization. Bioinformatics 26: 2342–2344, 2010 [DOI] [PubMed] [Google Scholar]
- 82.Xia J, Wishart DS. Web-based inference of biological patterns, functions and pathways from metabolomic data using MetaboAnalyst. Nat Protoc 6: 743–760, 2011 [DOI] [PubMed] [Google Scholar]
- 83.Yamanouchi T, Ogata N, Tagaya T, Kawasaki T, Sekino N, Funato H, Akaoka L, Miyashita H. Clinical usefulness of serum 1,5-anhydroglucitol in monitoring glycaemic control. Lancet 347: 1514–1518, 1996 [DOI] [PubMed] [Google Scholar]
- 84.Yanaka N. Mammalian glycerophosphodiester phosphodiesterases. Biosci Biotechnol Biochem 71: 1811–1818, 2007 [DOI] [PubMed] [Google Scholar]
- 85.Yu BL, Zhao SP, Hu JR. Cholesterol imbalance in adipocytes: a possible mechanism of adipocytes dysfunction in obesity. Obes Rev 11: 560–567, 2010 [DOI] [PubMed] [Google Scholar]
- 86.Zhang Y, Goldman S, Baerga R, Zhao Y, Komatsu M, Jin S. Adipose-specific deletion of autophagy-related gene 7 (atg7) in mice reveals a role in adipogenesis. Proc Natl Acad Sci USA 106: 19860–19865, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]