

Abstract
Since individual cells from freshly isolated white adipose tissue (WAT) exhibit variable levels of fat accumulation, we attempted to determine which factor(s) cause this variation. We used primary WAT cells from adult mice and the mouse 3T3-L1 cell-line of preadipocytes for these studies. Cells were labeled with BODIPY (boron-dipyrromethene) lipid probe, a marker for fat accumulation in live cells, and sorted on a fluorescence-activated cell sorter into two populations exhibiting low or high BODIPY fluorescence intensity. After more than 12 doublings as dedifferentiated cells in growth medium, the sorted populations were exposed to adipogenic medium for 7 days and analyzed for BODIPY accumulation and mRNA expression of adipogenic markers. WAT-derived cells initially sorted to have low or high BODIPY fluorescence intensity maintained a similar low or high lipid phenotype after redifferentiation. Cell surface TSH receptor expression, which is known to increase when preadipocytes are differentiated, correlated with BODIPY staining in all states. mRNA levels of Pparγ, Srebp1c, aP2, and Pref1, key regulators of adipogenesis, and leptin, Glut4, Fasn, and Tshr, markers of adipocyte differentiation, correlated with the levels of fat accumulation. Overexpression of Pparγ in 3T3-L1 cells, as expected, caused cells from low- and high-BODIPY populations to accumulate more fat. More importantly, prior to differentiation, the endogenous Pparγ promoter exhibited higher levels of acetylated histone H3, an activatory modification, in high-BODIPY- compared with low-BODIPY-derived populations. We conclude that fat accumulation is a heritable trait in WAT and that epigenetic modification on the Pparγ promoter contributes to this heritability.
Keywords: white adipose tissue, differentiation, fluorescence-activated cell sorter, epigenetics, Pparγ, fat
mature adipocytes can revert to a preadipocyte phenotype and gain proliferative ability (4, 19, 22). Preadipocytes from different depots were shown to be inherently distinct in their capacities for replication, differentiation, and susceptibility to apoptosis (2, 37). A subpopulation of stem/stromal cells derived from adipose tissue was shown to proliferate and differentiate into multiple lineages, including adipocytes, osteoblasts, chondrocytes, myocytes, neuronal cells, endothelial cells, and hepatocytes (27). Other studies show that mature adipocytes that were adherent to a top/floating surface (“ceiling culture”) both in vitro and in vivo (29, 34, 35, 39) were able to dedifferentiate and redifferentiate into adipocytes, indicating that these cells possess characteristics of adipocyte progenitors.
Peroxisome proliferator-activated receptor-γ (Pparγ), a nuclear receptor, is a key regulator of preadipocyte differentiation toward adipocytes (adipogenesis) and is both necessary and sufficient for adipogenesis (8, 24, 25). Upon binding to its endogenous ligands (free fatty acids and eicosanoids), Pparγ can act as a key transcription factor to regulate development of adipose tissue (7, 33). Histone acetylation is generally correlated with transcriptional activation (3). Previous studies suggest that histone H3 acetylation (26) on the Pparγ promoter region positively regulates Pparγ expression during adipogenesis. Trimethylation on Lys4 of histone H3 (H3K4me3) on the Pparγ promoter has been associated with increased Pparγ expression also (1, 9, 20).
In this paper, we demonstrate that fat cells derived from white adipose tissue (WAT) can dedifferentiate and redifferentiate. Moreover, the cells that undergo redifferentiation inherit a similar phenotype to the parental cell from which they were derived. The extent of fat accumulation is correlated with histone acetylation of the Pparγ promoter that is heritable and maintained even in dedifferentiated adipocytes.
MATERIALS AND METHODS
All animal work was conducted according to the National Institutes of Health guidelines.
Cell culture and primary white adipocyte isolation.
3T3-L1 preadipocytes were maintained at no higher than 70% confluence in growth medium (GM; DMEM containing 10% FCS, 25 mM glucose, 100 U/ml penicillin, 100 mg/ml streptomycin, and 2 mM glutamine) at 37°C in a 6% CO2 incubator (13).
To isolate primary white adipocytes, inguinal WAT from 6- to 12-wk-old female C57BL/6 mice was excised, minced, and digested with 1 mg/ml collagenase V (Sigma Aldrich) in Hanks' balanced salt solution (Corning-Cellgro, Manassas, VA) supplemented with 100 U/ml penicillin and 100 mg/ml streptomycin at 37°C for 30 min. After digestion, 20% FCS was added for 5 min, cells were centrifuged at 150 g for 5 min, and floating cells were collected using a pipette. Cells were labeled with 10 μM boron-dipyrromethene (BODIPY) 493/505 (Molecular Probes D3922) containing HBSS for 30 min at 37°C and sorted based on fluorescence intensity. Sorted populations were then cultured in GM at 37°C with 6% CO2.
Adipogenesis induction.
Cells were plated at about 30% confluence in GM 4 days before induction of adipogenesis. After 4 days, cells were fully confluent and were treated with adipogenic medium [AM; GM supplemented with 1 nM T3, 0.5 mM 3-isobutyl-1-methylxanthine, 2 μg/ml dexamethasone, and 0.125 mM indomethacin]. After an additional 2 days, the medium was changed back to GM and replenished at 2-day intervals. Six to eight days after initiation of differentiation, adipocytes were collected for either chromatin immunoprecipitation (ChIP), fluorescence-activated cell sorting (FACS) analysis, stained with Oil red O, or subjected to gene expression analysis by quantitative (q)RT-PCR (see below). For dedifferentiation of adipocytes, cells were kept at no higher than 70% confluence (by regularly splitting the cells) in GM. During dedifferentiation, the cells reverted to a fibroblast-like morphology.
qRT-PCR.
qRT-PCR was performed on DNA and cDNAs. All reactions were conducted in 96-well plates in 25-μl volume. Each reaction contained cDNA from 100 ng of total RNA or DNA precipitated from 100 mg of chromatin extracts for ChIP experiments, and appropriate amounts of Universal PCR Master Mix (Applied Biosystems) and Primers/Probe mix. All primers and probes are from Applied Biosystems Assay On-Demand. Cycle threshold (CT) values >40 were considered undetectable and calculated as a CT of 41.
Western blot.
Cells were lysed in RIPA buffer (50 mM Tris, pH 8.0, 150 mM NaCl, 1.0% Nonidet P-40, 0.5% deoxycholate, 0.1% SDS, 0.2 mM NaVO4, 10 mM NaF, 0.4 mM EDTA, 10% glycerol) with protease inhibitors (Roche). Lysates were sonicated for 20 s on ice and centrifuged at 10,000 g for 5 min. Lysates were then placed in a boiling water bath for 5 min with Laemmli loading buffer followed by electrophoresis on 10% SDS polyacrylamide gels. Western analyses were performed with Pparγ and GAPDH (Abcam) antibodies at the recommended antibody dilutions. The blots were scanned using the LiCor laser-based image detection method.
Oil red O staining.
3T3-L1 preadipocytes and adipocytes were stained with Oil red O. Confluent cells were first washed twice with PBS and incubated in 200 μl of Oil red O solution (0.33% wt/vol in 60% isopropanol) for 20 min at room temperature. Images of Oil red O-stained adipocytes were acquired using a Zeiss Axiovert 25, ×20 objective and Olympus E620 camera.
To quantify staining in fat droplets, Oil red O was extracted with 100% isopropanol. Cells were incubated with isopropanol for 10 min at room temperature. OD of two aliquots was measured at 510 nm, 0.5-s reading.
Pparγ overxpression.
Overexpression of Pparγ was accomplished using a retrovirus. 3T3-L1 cells were infected at indicated multiplicities of infection (MOIs). Retrovirus was kindly provided by Kai Ge, NIH, NIDDK (10).
ChIP.
ChIP assays were performed with 100 mg of cell chromatin extracts from 20 × 106 3T3-L1 cells. DNA was obtained with the Active Motif (Carlsbad, CA) chromatin shearing kit. Chromatin was precipitated by incubation with antibodies to acetylated histone H3 (06-599), acetylated H4 (06-866), histone H3K4me3 (07-473) at the recommended dilutions (all from Millipore) or a 1:10,000 dilution of rabbit IgG (Abcam) followed by separation with protein G magnetic beads (Active Motif, Carlsbad, CA). Binding was analyzed by real-time PCR with the following sets of PCR primers and 6FAM-labeled probes: Pparγ F: CTCGGCTCGGCTCCTC, R: GGCTGCCGCTCTGAGT, 6FAM: CCGGCCGCGGACCG; GAPDH F: ACCTTAGTCTGTGGTGATCTGATAGG, R: ACAAAACAGGCCTCAACAGATACAA, 6FAM: ATGCATGGGACAATTT.
Cell labeling with BODIPY and TSHR antibody.
Fully confluent cells were labeled with BODIPY 493/503 lipid probe solution (10 μM in HBSS; Molecular Probes/Life Technologies, Grand Island, NY) for 30 min in 37°C to measure fat accumulation in live cells. Cells were then washed with PBS and collected with 0.5 mM EDTA in PBS, 10% FCS was then added, and cells were spun down at 150 g for 5 min and labeled with 1:100 2C11 antibody against thyrotropin receptor (TSHR) (AbD Serotec, Raleigh, NC) labeled with Alexa fluor 647 (labeling kit; Molecular probes-Life Technologies, Grand Island, NY) in HBSS with 10% FCS for 1 h on ice.
FACS.
Flow cytometry was performed using a BD FACS Aria II cell sorter (Becton-Dickinson) with the following settings: a 70-μm nozzle with a sheath pressure of 65 psi was used, and populations were sorted for purity. Debris and clustered cells were excluded from gated populations.
Statistical analyses.
Statistical analyses were performed by two-tailed paired t-test or one-way analysis of variance (ANOVA). Significance was for P < 0.05.
RESULTS
WAT cells were isolated from the inguinal pads of adult mice by collagenase treatment and stained with BODIPY neutral lipid probe and Alexa fluor 647-labeled antibody to Tshr; Tshr was used as a cell surface marker of adipogenesis, since its expression increases during adipogenesis (18, 31).
We sorted the cells into two populations according to their BODIPY content, either high or low (Fig. 1A). BODIPY content of the cells had a direct correlation with the size of the cell, as can be determined by the side and forward scatter of the cells. Cells with low BODIPY content had a very low side and forward scatter profile compared with cells with high BODIPY. The average cell diameter was 24.7 ± 5.0 μm for the low BODIPY and 36.1 ± 7.2 μm for the high BODIPY cells (P < 0.001). The sorted populations were then cultured in GM for 6 wk, undergoing approximately one doubling every 5 days. After 6 wk in culture, cells were either induced to undergo adipogenesis or continued to be cultured in GM. On day 8 of adipogenesis, cells were stained again with both BODIPY and TSHR antibody and analyzed by FACS. The redifferentiated cells derived from the original low BODIPY fraction, expressed relatively low BODIPY fluorescence and were smaller than the redifferentiated cells derived from the high-BODIPY population (Fig. 1, B and C). Both cell populations that remained in GM showed similar BODIPY content and exhibited lower levels of BODIPY than the groups cultured in AM (Fig. 1C). Tshr levels correlated positively with the BODIPY content of the cells (Fig. 1, A and B).
Fig. 1.

Differentiated white adipose tissue (WAT) cells retain the fat accumulation capability of the cells from which they were derived. Primary WAT cells were isolated from mice, dispersed, and labeled for fat accumulation using the BODIPY (boron-dipyrromethene) lipid probe as well as TSH receptor (TSHR) as a marker for adipogenesis and sorted into low- or high-BODIPY populations. A: representative profiles of cells from the initial sort. B and C: cells were allowed to dedifferentiate/proliferate in culture in growth medium (GM) for 6 wk and then redifferentiate in adipogenic medium (AM). After 7 wk in culture, both undifferentiated cells in GM and differentiated adipocytes in AM were labeled with BODIPY and TSHR antibody and analyzed by FACS. Representative profiles from one mouse of BODIPY and TSHR antibody fluorescence from FACS analysis of the indicated populations (B). Mean values of BODIPY fluorescence of cells obtained from 3 mice (C). Values are means ± SD of 3 separate experiments. Levels were higher in adipocyte-differentiated cells from high- vs. low-BODIPY-derived cells (*P < 0.05).
mRNA levels for Pparγ, leptin (Lep), and Tshr were measured in these cell populations. In freshly isolated WAT cells, the high-BODIPY population expressed higher levels of Pparγ, Lep, and Tshr relative to the low-BODIPY group (Fig. 2). The higher level of Tshr mRNA expression is consistent with our FACS analysis (Fig. 1B). In contrast, the cells cultured in GM for 7 wk resembled preadipocytes and had lower levels of these mRNAs, findings consistent with a dedifferentiated phenotype. There was no significant difference in expression levels of the mRNAs when high- and low-BODIPY populations were compared during the dedifferentiated, proliferative phase. In cells recultured in AM, mRNA expression increased in both groups; however, cells from the high-BODIPY population showed higher expression than cells from the low-BODIPY population (Fig. 2). In addition, we measured other markers of adipocyte differentiation: insulin receptor (InsR) levels were unchanged in all conditions tested; Glut4 and fatty acid synthase (Fasn) mRNA levels were higher in cells derived from the high-BODIPY population in AM and not significantly different in cells in GM (preadipocytes); sterol-regulatory element-binding protein-1c (SREBP1c), a transcription factor that plays a central role in several aspects of adipocyte development including the induction of PPARγ, the generation of an endogenous PPARγ ligand (15, 16), and the expression of several genes critical for lipid biosynthesis (6), adipocyte protein-2 (aP2, commonly known as fatty acid-binding protein-4) levels were lower in the low-BODIPY population than in the high-BODIPY in redifferentiated adipocytes; and preadipocyte factor (Pref1, commonly known as Dlk1), a preadipocyte secreted factor that inhibits adipogenesis (36), levels were higher in preadipocytes than in adipocytes and in the low-BODIPY population compared with the high-BODIPY population (Fig. 2).
Fig. 2.

Redifferentiated WAT cells with High BODIPY staining express higher levels of Pparγ, Leptin (Lep), and Tshr mRNAs than cells with Low BODIPY. These mRNA expression patterns are heritable over numerous passages. Left 2 bars: mRNA levels of Pparγ, Lep, Tshr, InsR, Glut4, Fasn, Srebp1c, aP2, and Pref1 in primary WAT from the original Low and High BODIPY populations (day of sorting); values are means ± SD of 3 separate experiments from WAT from 3 mice. All 3 mRNA levels were higher in lysates of cells from high- vs. low-BODIPY populations (*P < 0.05). Right 4 bars: mRNA levels in low- and high-BODIPY cells cultured in GM for 7 wk or cultured for 6 wk in GM and then redifferentiated with the adipogenic protocol (AM) described in materials and methods. Values are means ± SD of 3–6 cell populations from tissues extracted from 3 mice (*P < 0.05).
To gain insight into the molecular details of heritability and show that these findings were not unique to primary WAT cells, we studied 3T3-L1 cells, a mouse cell line of preadipocytes (20, 23). A similar heritability pattern was observed with 3T3-L1 cells as with the primary adipocytes. Cells maintained in GM for 12 doublings (doubling times of 27 h for both cell populations) dedifferentiated and accumulated little fat in both populations (Fig. 3, A and B). When induced to differentiate, cells derived from the high-BODIPY population maintained their capacity to accumulate more fat after undergoing adipogenesis than cells from the low-BODIPY population for at least 12 doublings (∼6 wk from the initial sort). Even when the adipogenesis protocol was extended to 18 days (instead of 8 days), the cells derived from the low-BODIPY population were substantially smaller and lower in fat content compared with the high-BODIPY cells (data not shown). Moreover, 3T3-L1 mRNA expression of Pparγ, Lep, Tshr, InsR, Glut4, Fasn, Srebp1c, aP2, and Pref1 were similar in all populations (Fig. 4) to the results obtained with primary mouse adipocytes (Fig. 2); that is, the levels in adipocyte-differentiated cells from the high-BODIPY population were higher than in cells from the low-BODIPY population for Pparγ, Lep, TshR, Glut4, Fasn, Srebp1c, and ap2 and lower for Pref1.
Fig. 3.

Level of fat accumulation is heritable in 3T3-L1 cells. Unsorted and sorted 3T3-L1 cells were grown in GM and then redifferentiated in AM. A: representative profiles of BODIPY fluorescence from FACS analysis of adipocyte-differentiated 3T3-L1 cells (unsorted, top) and cells that had been sorted into low- and high-BODIPY populations, cultured for 12 passages in GM and then redifferentiated. B: mean values of BODIPY fluorescence (AU) of sorted 3T3-L1 cells incubated in GM or redifferentiated in AM. Values are means ± SD of 3 experiments. Levels were higher in lysates of adipocyte-differentiated cells from high- vs. low-BODIPY populations (*P < 0.05).
Fig. 4.

Differentiated 3T3-L1 cells with high-BODIPY content express higher levels of Pparγ, Lep, and Tshr mRNAs than cells with Low BODIPY. These mRNA expression patterns are heritable over numerous passages. A: left 2 bars: mRNA levels of Pparγ, Lep, Tshr, InsR, Glut4, Fasn, Srebp1c, aP2, and Pref1 mRNAs in 3T3-L1 cells after initial differentiation to adipocytes and sorting into low- and high-BODIPY populations (day of sorting). Values are means ± SD in 1 of 3 experiments. Right 4 bars: mRNA levels of Pparγ, Lep, and Tshr mRNAs in low- and high-BODIPY 3T3-L1 cells cultured in GM for 7 wk or cultured for 6 wk in GM and then redifferentiated with AM. Values are means ± SD of triplicates from 1 of 3 experiments (*P < 0.05). B: representative Western blot for Pparγ and GAPDH expression.
Expression levels of Pparγ protein were monitored by Western blot. The low-BODIPY population expressed lower levels of Pparγ protein than the high-BODIPY population, both in the differentiated and the dedifferentiated states, and dedifferentiated cells had lower Pparγ protein levels compared with the differentiated condition (Fig. 4B).
3T3-L1 cells were stained with Oil red O for triglycerides to confirm the results obtained by BODIPY staining. Cells cultured in GM for 12 wk exhibited very low Oil red O staining, confirming the dedifferentiated state, whereas cells cultured in AM for the last week were higher in Oil red O staining. Cells derived from the initial high-BODIPY population exhibited higher Oil red O staining relative to the low-BODIPY-derived population (Fig. 5).
Fig. 5.

Adipocyte-differentiated 3T3-L1 cells from the high-BODIPY population exhibit higher Oil red O staining than cells from the low-BODIPY population. Oil red O staining of 3T3-L1 cells derived from low- and high-BODIPY populations were grown for ≥12 passages in GM or in GM for 6 wk and then in AM for 1 wk. A: images of cells stained with Oil red O. B: Oil red O quantification according to the protocol in materials and methods of cells obtained from 3 separate sorts. Values are means ± SD. The levels were higher in extracts of cells from high- vs. low-BODIPY content populations after adipogenic differentiation (*P < 0.05).
To directly show that increasing Pparγ expression in these cells would increase fat accumulation, 3T3-L1 cells were infected with increasing MOIs of Pparγ-expressing retrovirus and then differentiated in AM (Fig. 6A). Oil red O staining in cells not infected with retrovirus was 3.2-fold higher in cells from the high-BODIPY population than in cells from the low-BODIPY population (Fig. 6, B and C, MOI = 0). Pparγ overexpression resulted in increased Oil red O staining in cells from both BODIPY populations. Pparγ at an MOI of 100 resulted in an increase of 10.4 ± 0.06% in Oil red O staining in cells derived from the low-BODIPY population and a similar percentage increase of 13.3 ± 0.19% in cells from the high-BODIPY population. Similar percentage increases were obtained when BODIPY staining was quantified. Pparγ overexpression caused BODIPY staining to increase 12.1 ± 2.6 and 13.5 ± 4.7% in low- and high-BODIPY populations, respectively.
Fig. 6.
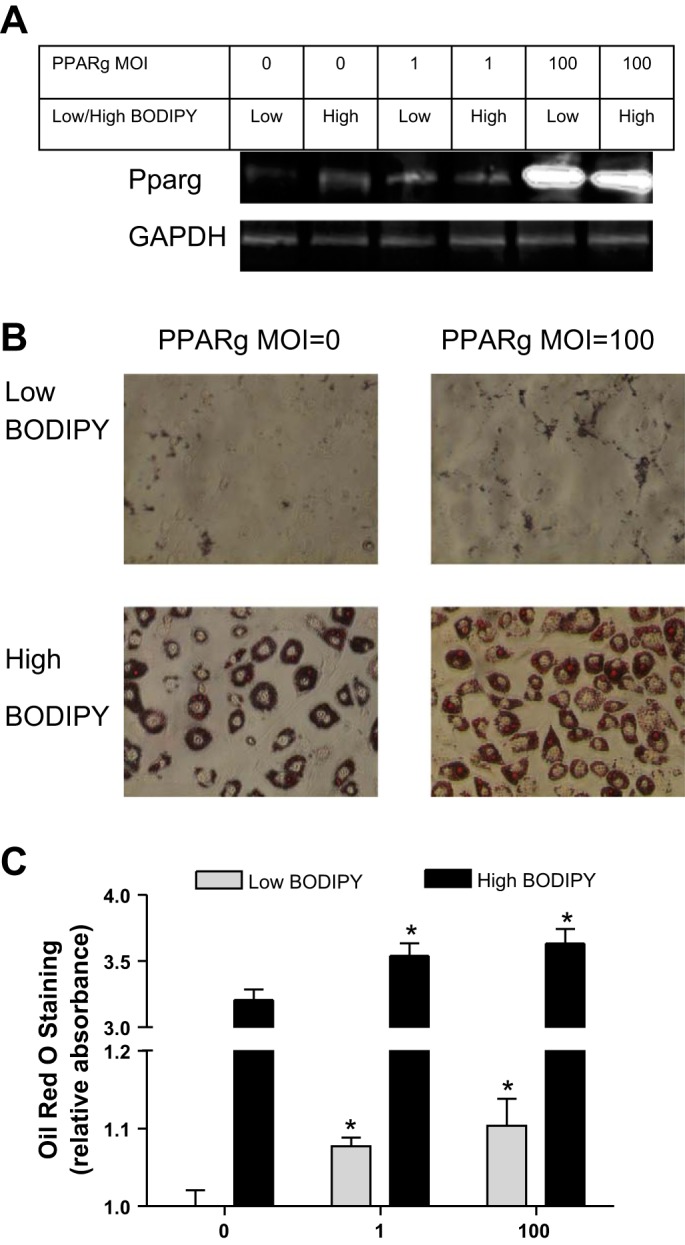
Overexpression of Pparγ results in increased fat content in both low- and high-BODIPY-derived 3T3-L1 cell populations. A: Western blot for Pparγ and GAPDH expression in 3T3L-1 cells infected with the indicated multiplicities of infection (MOI) of Pparγ retrovirus. B: images of cells stained with Oil red O. C: Oil red O quantification according to the protocol in materials and methods. Values are means ± SD (vs. MOI = 0 *P < 0.05).
We hypothesized that epigenetic modification of Pparγ, a master regulator of white and brown adipogenesis (8, 25, 33), might account for the heritability of the adipocyte phenotype upon induction of adipogenesis. H3 acetylation of the Pparγ promoter has been demonstrated previously to account, at least partially, for differentiation of adipocytes. We used ChIP analysis of the Pparγ promoter in chromatin from 3T3-L1 cells 12 doublings after the initial sort to measure three histone modifications that are associated with transcriptional activation: H3 acetylation, H4 acetylation, and H3K4 methylation (Fig. 7). The levels of H4 acetylation and H3K4 methylation in undifferentiated cells in GM were not different in the high- and low-BODIPY cells, whereas the level of H3 acetylation was higher in undifferentiated high-BODIPY cells compared with low-BODIPY cells. All three histone modifications increased after differentiation to adipocytes in cells from both the low- and high-BODIPY populations, and all were higher in cells from the high-BODIPY population than in the low-BODIPY population. We could not perform ChIP experiments in primary isolated or sorted adipocytes because this analysis requires 107 cells. These data show a direct correlation between the levels of activatory histone modifications on the Pparγ promoter and the cell's ability to accumulate fat and express differentiated markers upon induction of adipogenesis. Of note, the level of H3 acetylation was higher in undifferentiated cells from the high-BODIPY population than that in undifferentiated cells from the low-BODIPY population and may therefore be the critical heritable histone modification.
Fig. 7.
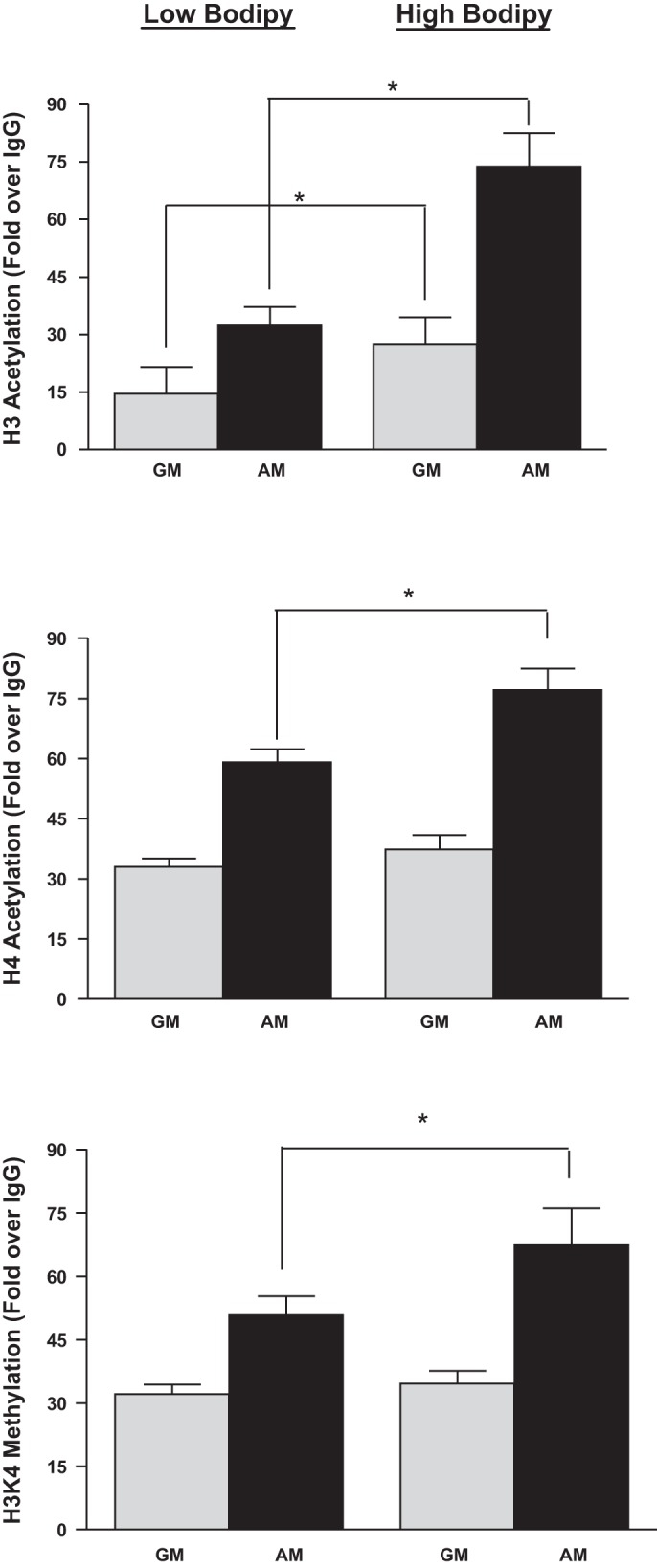
high-BODIPY cells exhibit higher histone H3 acetylation levels than low-BODIPY cells after adipocyte differentiation. Chromatin immunoprecipitation (ChIP) of indicated 3T3-L1 cell populations with antibodies targeted at acetylated histone H3, acetylated H4, and methylated H3K4 on the Pparγ gene promoter. Undifferentiated cells were cultured in GM and differentiated cells in AM. These modifications on the GAPDH promoter, by contrast, remained unchanged in all cell population conditions (data not shown).Values are means ± SD of 3 independent experiments. Levels of H3 and H4 acetylation and H3K4 methylation were higher in both high- and low-BODIPY populations in AM vs. GM (P < 0.02 for H3 acetylation; P < 0.01 for H4 acetylation; P < 0.03 for H3K4 trimethylation). H3 and H4 acetylation were higher in high- vs. low-BODIPY populations in AM (P < 0.02) but not in H3K4 trimethylation (P > 0.05). Importantly, levels of H4 acetylation and H3K4 methylation in cells in GM were not different in high- and low-BODIPY populations (P > 0.05) but were higher in H3 acetylation in chromatin from cells from high- vs. low-BODIPY content populations in GM (*P < 0.01).
DISCUSSION
This study demonstrates that subpopulations reside within WAT and 3T3-L1 cells that vary in their capability to accumulate fat and that these differences are heritable. We have shown that the extent of the cell's ability to accumulate fat correlated positively with expression levels of Pparγ, a master regulator of adipogenesis, and with other markers of differentiated adipocytes, including Lep, Tshr, InsR, Glut4, Fasn, Srebp1c, aP2, and Pref1; exogenous expression of Pparγ in 3T3-L1 cells increased fat accumulation; and the levels of histone H3 acetylation of the Pparγ promoter in preadipocytes was a predictor of the extent of fat accumulation upon induction of adipogenesis. We thus suggest that epigenetic modification of the Pparγ promoter is, in part, the mediator of the heritability of adipocyte differentiation.
Primary adipose tissue contains multiple cell types, including mature adipocytes, preadipocytes, stromal vascular and stem-stromal cells, etc. (21, 27). By initiating our cultures using cells that were floating because of their fat content, we enriched the starting cell populations with adipocytes. Nevertheless, we cannot exclude the presence of other cell types that have different fat-accumulating capabilities in these cultures. However, our finding that 3T3-L1 cells, which were derived from a preadipocyte clone (12), behaved similarly to the cells in the primary WAT-derived cultures confirms our conclusions that within the preadipocyte population there are cells that have inherited different abilities to accumulate fat. It was previously thought that differentiation of cells, including adipocytes, was a terminal, nonreversible state (5, 17, 23). Our experimental models show that both primary adipocytes and preadipocytes from the 3T3-L1 cell line can dedifferentiate, proliferate, and thereafter undergo redifferentiation in accord with other recent studies (4, 11, 19, 22).
Pparγ, Lep, and Tshr are markers of mature adipocytes (14, 18, 28, 30–32, 38), and the levels of these mRNAs decreased during proliferation of primary WATs in culture (dedifferentiation). Upon redifferentiation to an adipocyte phenotype, high-BODIPY-derived cells consistently expressed higher levels of these adipocyte markers than the low-BODIPY-derived population even after many doublings. In cells proliferating in GM, mRNA expression was lower with no significant difference between the high- or low-BODIPY-derived populations. Similar changes in the levels of Pparγ, Lep, and Tshr mRNAs were found in 3T3-L1 cells. These results indicate that white adipocytes can dedifferentiate and upon redifferentiation inherit the ability to upregulate adipocyte-associated genes in a manner similar to the heritability of fat accumulation. The extent to which a cell can accumulate fat is heritable. Both the low- and high-BODIPY populations were able to undergo differentiation and exhibit upregulation of both fat and adipogenic markers. Yet, although both cell populations are differentiated, they are different in their abilities to accumulate fat, which correlates with mRNA levels of adipogenic markers. Indeed, the difference in the extent of fat accumulation was maintained when the cells were maintained in AM for 18 days. It remains possible that the smaller, low-BODIPY cells differentiate more slowly and are at an earlier stage than the larger, high-BODIPY cells. Nevertheless, this difference is maintained generation after generation and is an inherited phenotype.
Overexpression of Pparγ, a key transcription factor that controls adipogenesis (8, 32, 38), resulted in increases of the cell's ability to accumulate fat, consistent with the idea that epigenetic regulation of Pparγ could account for the inherited capability of the cell to accumulate fat. We therefore measured three epigenetic modifications of the Pparγ promoter, histone H3 and H4 acetylation and H3K4 trimethylation, that are correlated with transcription activation. Although all three epigenetic modifications tested were increased at the Pparγ promoter upon induction of adipogenesis and higher levels were found in the high-BODIPY compared with the low-BODIPY population, only histone H3 acetylation was different between the two cell populations in undifferentiated preadipocytes. Although other epigenetic mechanisms and regulation of other genes (such as Pref1, which we showed to be differentially expressed in preadipocytes) could also be involved, we suggest that histone H3 acetylation on the Pparγ promoter as a possible mechanism to explain the inheritance of fat accumulation capability and the higher expression of adipocyte markers. Further studies should address the mechanism(s) that initiates the H3 acetylation of the Pparγ promoter in WAT.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
AUTHOR CONTRIBUTIONS
Author contributions: L.S.K., E.G.-R., and M.C.G. conception and design of research; L.S.K. performed experiments; L.S.K. and M.C.G. analyzed data; L.S.K., E.G.-R., and M.C.G. interpreted results of experiments; L.S.K. and M.C.G. prepared figures; L.S.K. and M.C.G. drafted manuscript; L.S.K., E.G.-R., and M.C.G. edited and revised manuscript; L.S.K., E.G.-R., and M.C.G. approved final version of manuscript.
REFERENCES
- 1.Cho YW, Hong S, Jin Q, Wang L, Lee JE, Gavrilova O, Ge K. Histone methylation regulator PTIP is required for PPARgamma and C/EBPalpha expression and adipogenesis. Cell Metab 10: 27–39, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Cinti S. Transdifferentiation properties of adipocytes in the adipose organ. Am J Physiol Endocrinol Metab 297: E977–E986, 2009 [DOI] [PubMed] [Google Scholar]
- 3.Clayton AL, Hazzalin CA, Mahadevan LC. Enhanced histone acetylation and transcription: a dynamic perspective. Mol Cell 23: 289–296, 2006 [DOI] [PubMed] [Google Scholar]
- 4.Dodson MV, Fernyhougha ME, Viercka JL, GJH Adipocytes may not be a terminally differentiated cell type: implications for animal production. Anim Sci 80: 239–240, 2005 [Google Scholar]
- 5.Dodson MV, Mir PS, Hausman GJ, Guan LL, Du M, Jiang Z, Fernyhough ME, Bergen WG. Obesity metabolic syndrome, adipocytes. J Lipids 2011: 721686, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Eberle D, Hegarty B, Bossard P, Ferre P, Foufelle F. SREBP transcription factors: master regulators of lipid homeostasis. Biochimie 86: 839–848, 2004 [DOI] [PubMed] [Google Scholar]
- 7.Farmer SR. Regulation of PPARgamma activity during adipogenesis. Int J Obes (Lond) 29, Suppl 1: S13–S16, 2005 [DOI] [PubMed] [Google Scholar]
- 8.Farmer SR. Transcriptional control of adipocyte formation. Cell Metab 4: 263–273, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ge K. Epigenetic regulation of adipogenesis by histone methylation. Biochim Biophys Acta 1819: 727–732, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ge K, Cho YW, Guo H, Hong TB, Guermah M, Ito M, Yu H, Kalkum M, Roeder RG. Alternative mechanisms by which mediator subunit MED1/TRAP220 regulates peroxisome proliferator-activated receptor gamma-stimulated adipogenesis and target gene expression. Mol Cell Biol 28: 1081–1091, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Granneman JG, Li P, Zhu Z, Lu Y. Metabolic and cellular plasticity in white adipose tissue. I. Effects of β3-adrenergic receptor activation. Am J Physiol Endocrinol Metab 289: E608–E616, 2005 [DOI] [PubMed] [Google Scholar]
- 12.Green H, Kehinde O. An established preadipose cell line and its differentiation in culture. II. Factors affecting the adipose conversion. Cell 5: 19–27, 1975 [DOI] [PubMed] [Google Scholar]
- 13.Green H, Meuth M. An established pre-adipose cell line and its differentiation in culture. Cell 3: 127–133, 1974 [DOI] [PubMed] [Google Scholar]
- 14.Hwang CS, Loftus TM, Mandrup S, Lane MD. Adipocyte differentiation and leptin expression. Annu Rev Cell Dev Biol 13: 231–259, 1997 [DOI] [PubMed] [Google Scholar]
- 15.Kim JB, Spiegelman BM. ADD1/SREBP1 promotes adipocyte differentiation and gene expression linked to fatty acid metabolism. Genes Dev 10: 1096–1107, 1996 [DOI] [PubMed] [Google Scholar]
- 16.Kim JB, Wright HM, Wright M, Spiegelman BM. ADD1/SREBP1 activates PPARgamma through the production of endogenous ligand. Proc Natl Acad Sci USA 95: 4333–4337, 1998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kokta TA, Dodson MV, Gertler A, Hill RA. Intercellular signaling between adipose tissue and muscle tissue. Domest Anim Endocrinol 27: 303–331, 2004 [DOI] [PubMed] [Google Scholar]
- 18.Lu S, Guan Q, Liu Y, Wang H, Xu W, Li X, Fu Y, Gao L, Zhao J, Wang X. Role of extrathyroidal TSHR expression in adipocyte differentiation and its association with obesity. Lipids Health Dis 11: 17, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Matsumoto T, Kano K, Kondo D, Fukuda N, Iribe Y, Tanaka N, Matsubara Y, Sakuma T, Satomi A, Otaki M, Ryu J, Mugishima H. Mature adipocyte-derived dedifferentiated fat cells exhibit multilineage potential. J Cell Physiol 215: 210–222, 2008 [DOI] [PubMed] [Google Scholar]
- 20.Mikkelsen TS, Xu Z, Zhang X, Wang L, Gimble JM, Lander ES, Rosen ED. Comparative epigenomic analysis of murine and human adipogenesis. Cell 143: 156–169, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mitchell JB, McIntosh K, Zvonic S, Garrett S, Floyd ZE, Kloster A, Di Halvorsen Y, Storms RW, Goh B, Kilroy G, Wu X, Gimble JM. Immunophenotype of human adipose-derived cells: temporal changes in stromal-associated and stem cell-associated markers. Stem Cells 24: 376–385, 2006 [DOI] [PubMed] [Google Scholar]
- 22.Nobusue H, Endo T, Kano K. Establishment of a preadipocyte cell line derived from mature adipocytes of GFP transgenic mice and formation of adipose tissue. Cell Tissue Res 332: 435–446, 2008 [DOI] [PubMed] [Google Scholar]
- 23.Park BO, Ahrends R, Teruel MN. Consecutive positive feedback loops create a bistable switch that controls preadipocyte-to-adipocyte conversion. Cell Rep 2: 976–990, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rosen ED, MacDougald OA. Adipocyte differentiation from the inside out. Nat Rev Mol Cell Biol 7: 885–896, 2006 [DOI] [PubMed] [Google Scholar]
- 25.Rosen ED, Spiegelman BM. PPARgamma: a nuclear regulator of metabolism, differentiation, and cell growth. J Biol Chem 276: 37731–37734, 2001 [DOI] [PubMed] [Google Scholar]
- 26.Salma N, Xiao H, Mueller E, Imbalzano AN. Temporal recruitment of transcription factors and SWI/SNF chromatin-remodeling enzymes during adipogenic induction of the peroxisome proliferator-activated receptor gamma nuclear hormone receptor. Mol Cell Biol 24: 4651–4663, 2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Schaffler A, Buchler C. Concise review: adipose tissue-derived stromal cells—basic and clinical implications for novel cell-based therapies. Stem Cells 25: 818–827, 2007 [DOI] [PubMed] [Google Scholar]
- 28.Semple RK, Chatterjee VK, O'Rahilly S. PPAR gamma and human metabolic disease. J Clin Invest 116: 581–589, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Shigematsu M, Watanabe H, Sugihara H. Proliferation and differentiation of unilocular fat cells in the bone marrow. Cell Struct Funct 24: 89–100, 1999 [DOI] [PubMed] [Google Scholar]
- 30.Shimura H, Miyazaki A, Haraguchi K, Endo T, Onaya T. Analysis of differentiation-induced expression mechanisms of thyrotropin receptor gene in adipocytes. Mol Endocrinol 12: 1473–1486, 1998 [DOI] [PubMed] [Google Scholar]
- 31.Sorisky A, Bell A, Gagnon A. TSH receptor in adipose cells. Horm Metab Res 32: 468–474, 2000 [DOI] [PubMed] [Google Scholar]
- 32.Soukas A, Cohen P, Socci ND, Friedman JM. Leptin-specific patterns of gene expression in white adipose tissue. Genes Dev 14: 963–980, 2000 [PMC free article] [PubMed] [Google Scholar]
- 33.Spiegelman BM, Hu E, Kim JB, Brun R. PPAR gamma and the control of adipogenesis. Biochimie 79: 111–112, 1997 [DOI] [PubMed] [Google Scholar]
- 34.Sugihara H, Yonemitsu N, Miyabara S, Toda S. Proliferation of unilocular fat cells in the primary culture. J Lipid Res 28: 1038–1045, 1987 [PubMed] [Google Scholar]
- 35.Sugihara H, Yonemitsu N, Miyabara S, Yun K. Primary cultures of unilocular fat cells: characteristics of growth in vitro and changes in differentiation properties. Differentiation 31: 42–49, 1986 [DOI] [PubMed] [Google Scholar]
- 36.Sul HS. Minireview: Pref-1: role in adipogenesis and mesenchymal cell fate. Mol Endocrinol 23: 1717–1725, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tchkonia T, Giorgadze N, Pirtskhalava T, Thomou T, DePonte M, Koo A, Forse RA, Chinnappan D, Martin-Ruiz C, von Zglinicki T, Kirkland JL. Fat depot-specific characteristics are retained in strains derived from single human preadipocytes. Diabetes 55: 2571–2578, 2006 [DOI] [PubMed] [Google Scholar]
- 38.Tontonoz P, Spiegelman BM. Fat and beyond: the diverse biology of PPARgamma. Annu Rev Biochem 77: 289–312, 2008 [DOI] [PubMed] [Google Scholar]
- 39.Yagi K, Kondo D, Okazaki Y, Kano K. A novel preadipocyte cell line established from mouse adult mature adipocytes. Biochem Biophys Res Commun 321: 967–974, 2004 [DOI] [PubMed] [Google Scholar]