

Abstract
Increased leptin levels have been suggested to contribute to cardiac hypertrophy and attenuate cardiac lipid accumulation in obesity, although it has been difficult to separate leptin's direct effects from those caused by changes in body weight and adiposity. To determine whether leptin attenuates cardiac lipid accumulation in obesity or directly causes left ventricular hypertrophy (LVH), we generated a novel mouse model in which the long form of the leptin receptor (LepR) was “rescued” only in cardiomyocytes of obese db/db mice. Reexpression of cardiomyocyte leptin receptors in db/db mice did not cause LVH but reduced cardiac triglycerides and improved cardiac function. Compared with lean wild-type (WT) or db/db-cardiac LepR rescue mice, db/db mice exhibited significantly lower E/A ratio, a measurement of early to late diastolic filling, which averaged 1.5 ± 0.07 in db/db vs. 1.9 ± 0.08 and 1.8 ± 0.11 in WT and db/db-cardiac LepR rescue mice, respectively. No differences in systolic function were observed. Although db/db and db/db-cardiac LepR rescue mice exhibited similar increases in plasma triglycerides, insulin, glucose, and body weight, cardiac triglycerides were significantly higher in db/db compared with WT and db/db cardiac LepR rescue mice, averaging 13.4 ± 4.2 vs. 3.8 ± 1.6 vs. 3.8 ± 0.7 mg/g, respectively. These results demonstrate that despite significant obesity and increases in plasma glucose and triglycerides, db/db cardiac LepR rescue mice are protected against myocardial lipid accumulation. However, we found no evidence that leptin directly causes LVH.
Keywords: lipotoxicity, obesity, heart disease, transgenic mice, lipids, diabetes
obesity is associated with metabolic derangements such as diabetes and dyslipidemia that contribute to the development of cardiovascular dysfunction and heart failure. Furthermore, obesity is a significant risk factor for the development of cardiac dysfunction independent of hypertension and diabetes (13). One mechanism by which obesity may lead to cardiac dysfunction is lipid accumulation in the heart. This ectopic lipid deposition in the heart (myocardial steatosis) appears to have adverse effects on myocardial structure and function and has been referred to as “cardiac lipotoxicity” (14, 44).
Myocardial triglyceride accumulation, lipotoxic cardiac injury, and subsequent myocardial dysfunction have been observed in murine models of obesity, including db/db and ob/ob mice that have nonfunctional leptin signaling pathways (4, 10, 11, 45). Significant intramyocardial lipid accumulation has also been found in obese patients with nonischemic heart failure (40).
Increased levels of leptin, a hormone derived from adipocytes, have been suggested to protect against myocardial triglyceride accumulation in obesity. Leptin not only regulates food consumption and metabolic rate through central nervous system (CNS) actions but also stimulates β-oxidation of fatty acids in peripheral tissues through its CNS actions as well as direct leptin receptor (LepR)-mediated effects on peripheral tissues (1). This increase in β-oxidation of fatty acids shunts them away from storage, thus reducing tissue lipid accumulation (43). Several studies have demonstrated that leptin administration reverses steatosis in the liver and pancreas (47). Leptin has also been reported to lower cardiac triglyceride levels and preserve systolic function in a mouse model of severe lipotoxic cardiomyopathy (28). This evidence suggests that leptin may play an important role in protecting the heart against lipotoxic cardiomyopathy. However, in these studies leptin administration also had important CNS actions that reduced body weight and overall adiposity, which could reduce cardiac lipids independent of direct actions of leptin on the heart (32). In addition, leptin administration could also elicit other CNS actions that increase tissue fatty acid oxidation. To our knowledge, there have been no studies that assessed the importance of leptin's direct effects on the heart in attenuating obesity-induced cardiac dysfunction and lipid accumulation.
To determine directly the role of leptin in preventing cardiac lipid accumulation in obesity, we generated a murine model in which the LepR was expressed specifically in cardiomyocytes under the control of the α-myosin heavy chain promoter. This transgene was then bred onto the background of the LepR-resistant db/db mouse to create mice lacking functional LepRs everywhere except in the heart. An important goal of this study was to characterize the cardiovascular and metabolic phenotypes of these db/db cardiac LepR “rescue” mice. We hypothesized that restoration of leptin signaling in the hearts of obese db/db mice would attenuate myocardial steatosis and protect against myocardial injury and cardiac dysfunction despite severe obesity, dyslipidemia, and other features of the metabolic syndrome. Additionally, we assessed whether leptin exerts a prohypertrophic response in the myocardium, as this has been difficult to determine in previously studied rodent models due to confounding factors such as changes in body weight and blood pressure associated with obesity or chronic leptin administration.
METHODS
Mice.
All animal protocols were approved by the Institutional Animal Care and Use Committee at the University of Mississippi Medical Center. Mice were maintained on a standard laboratory diet that contained 17% calories from fat, 29% calories from protein, 54% calories from carbohydrates, and 0.4% sodium chloride (Teklad 22/5 rodent diet no. 8640; Harlan Laboratories, Indianapolis, IN). Experiments were performed on male mice every 6 wk beginning at age 12 wk until 30 wk of age.
Cardiac-specific LepR transgenic mice (Myh6-LepR) were generated by placing the long form of the rat LepR behind the cardiac-specific myosin heavy chain 6 (Myh6) promoter. The rat LepR (3.53 Kb) was excised from the plasmid pCX6-LepR by digestion with the restriction enzyme HindIII and ligated to HindIII digested plasmid Myh6-MerCreMer to create the transgenic construct (Myh6-LepR). The plasmid Myh6-LepR was then digested with the restriction enzyme NotI and then purified and injected into fertilized mouse embryos using established pronuclear injection techniques. Myh6-LepR mice were derived on a mixed B6SJL genetic background and bred back to C57BL/6J mice for more than 10 generations. To create cardiac-specific LepR rescue mice, heterozygous Myh6-LepR mice were crossed with heterozygous mice containing the LepRdb mutation, which were also on the C57BL/6J genetic background (Jackson Laboratories, Bar Harbor, ME). To generate db/db mice with and without the Myh6-LepR transgene, heterozygous lepRdb mice were mated with heterozygous transgenic Myh6-LepR, heterozygous lepRdb mice. The offspring were then genotyped for the Myh6-LepR transgene using the primers 5′-GGTGTAGGAAAGTCAGGACTTCA-3′ and 5′-GATAGGCCAGGTTAAGTGCAG-3′ as well as the lepRdb mutation using a restriction enzyme digestion (RsaI) of PCR reaction. db/db Mice that inherited the Myh6-LepR transgene are referred to as “rescue” mice. Experiments were also performed on age-matched db/db mice on the C57BL/6J background purchased from Jackson Laboratories.
Blood glucose measurements.
Blood glucose levels were obtained from mice that were fasted for 8 h with access to water. Blood was obtained from lightly anesthetized mice via retroorbital bleeding. Blood glucose concentration was determined immediately from a 5-μl blood sample with the Accuchek Advantage glucometer (Roche, Madison, WI).
Plasma insulin, leptin, and triglyceride measurements.
Plasma insulin levels were measured by ELISA with the Ultra Sensitive Mouse Insulin ELISA Kit (Crystal Chem, Downers Grove, IL). Plasma leptin levels were also measured by ELISA with the mouse Quantikine leptin immunoassay (R & D Systems, Minneapolis, MN). Plasma triglycerides were measured using a triglyceride reagent (GPO Trinder; Sigma-Aldrich, St. Louis, MO) and read at an optical density of 500 nm, with standards ranging from 0 to 200 mg/dl. Triglyceride concentration of each sample was determined using linear regression analysis and reported as milligrams per deciliter. All samples were measured in triplicate for each of the measurements.
Cardiac triglyceride measurement.
All triglyceride measurements were made after the mice were euthanized at 30 wk of age. Triglycerides were measured from 100 mg of heart tissue homogenized in 200 μl of homogenization buffer (18 mM Tris, pH 7.5, 300 mM mannitol, and 50 mM EGTA). After homogenization, 4 ml of 2:1 chloroform-methanol mixture was added to the samples, and the samples were incubated for 1 h at room temperature and mixed every 15 min. After this time, 800 μl of water was added to the samples, and the samples were mixed thoroughly and centrifuged for 5 min at 4,700 rpm at 4°C. The organic phase was then collected and dried under vacuum. After this time, samples were resuspended in 60 μl of T-butanol, followed by 40 μl of 2:1 Triton X-114-methanol mixture and sampled mixed for 10 s. To perform the triglyceride measurements, 10 μl of this mixture was added to 1 ml of triglyceride reagent (GPO Trinder) and incubated for 5 min at 37°C. Optical densities of the samples along with standards ranging from 0 to 200 mg/dl were measured at 500 nM. Triglyceride concentration of each sample was determined using linear regression analysis and reported as milligrams per deciliter. All samples and standards were measured in triplicate.
Echocardiography.
Echocardiographic assessment of cardiac function was conducted using a Vevo 770 High Resolution In Vivo Imaging System (Visualsonics, Toronto, ON, Canada), as described previously (16, 17). Measurements were made with a 710B RMV scanhead with a center frequency of 25 MHz. Echocardiography was performed on each mouse beginning at 12 wk of age and subsequently at 6-wk intervals until 30 wk of age. All echocardiographic data were obtained within a 15-min session and were made according to the American Society of Echocardiography Guidelines using the leading edge technique (27). Mice were anesthetized with 1.5% isoflurane gas, and body temperature was maintained on a prewarmed pad. Heart rate was monitored via an EKG transducer pad throughout each echocardiography session. Two-dimensional B-Mode parasternal long-axis views were obtained first to visualize the aortic and mitral valves, and then the transducer was rotated clockwise 90° to obtain the parasternal short-axis view. At least three M-Mode images were obtained at the midpapillary muscle level from this view. Ejection fraction (EF%) and fractional shortening (FS%) were analyzed and calculated using the Visualsonics Advanced Cardiovascular Measurements Package, using the mean of 12–15 cardiac cycles derived from three separate M-Mode images during each session. Left ventricular (LV) wall dimensions were determined from the M-Mode images and included end-diastolic dimension, end-systolic dimension, diastolic left ventricular wall dimensions, and systolic left ventricular wall dimensions. An apical four-chamber view was then obtained for Doppler analysis of the mitral valve inflow. Mitral E wave (early ventricular filling wave) and A wave (late diastolic filling from atrial contraction) velocities and isovolumetric relaxation (IVRT) and E wave deceleration times (DT) were obtained by Doppler analysis from at least five representative waveforms.
Acute leptin treatment.
Mice were treated with an intraperitoneal injection of leptin (100 μg in a 0.1-ml volume). After 1 h, mice were euthanized and hearts collected and stored at −80°C until use for assessment of phosphorylation of STAT3 (p-STAT3) protein.
Western blots.
Western blots were performed on lysates prepared from whole hearts collected at the end of the experimental protocol (30 wk). Samples of 50 μg of protein were boiled in Laemmli sample buffer (Bio-Rad, Hercules, CA) for 5 min and electrophoresed on 10% SDS-polyacrylamide gels and blotted onto a nitrocellulose membrane. Membranes were blocked with Odyssey blocking buffer (LI-COR, Lincoln, NE) for 2 h at room temperature and then incubated with primary antibodies overnight at 4°C. Membranes were incubated with either Alexa (fluor) 680 (Molecular Probes) or IRDye 800 (Rockland, Gilbertsville, PA) secondary antibodies for 1 h at room temperature. Membranes were visualized using an Odyssey infrared imager (Li-COR), which allows for simultaneous detection of two proteins. Densitometry analysis was performed using Odyssey software (LI-COR).
Antibodies for Western blots were as follows: p-STAT3-Tyr705 (no. 9145, 1:1,000 dilution; Cell Signaling Technology, Danvers, MA), STAT3 (no. 9139, 1:1,000; Cell Signaling Technology), phospho-ERK1/2-Thr202/Tyr204 (no. 4370, 1:1,000; Cell Signaling Technology), ERK1/2 antibodies (no. 4695, 1:1,000; Cell Signaling Technology), p-Akt-Ser473 (no. 9271, 1:1,000; Cell Signaling Technology), p-Akt-Thr308 (no. 9275 1:1,000; Cell Signaling Technology), total Akt (no. 4685, 1:1,000; Cell Signaling Technology), mouse anti-glucose transporter 4 (GLUT4; ab645, 1:2,500; Abcam, Cambridge, MA), glyceraldehyde-3-phosphate dehydrogenase (GAPDH; ab8245, 1:10,000; Abcam), sheep anti-Cu/Zn SOD (K90077C, 1:1,000; Amsbio, Lake Forest, CA), mouse anti-heme oxygenase-1 (HO-1; ALX-210–116, 1:2,000; Enzo Life Sciences, Plymouth Meeting, PA), rabbit anti-NF-κB (sc-109, 1:1,000; Santa Cruz Biotechnology, Dallas, TX), and β-tubulin (T4026, 1:1,000; Sigma-Aldrich, St. Louis, MO). Protein levels were normalized to β-tubulin and expressed as a ratio.
PCR.
RNA was isolated from whole hearts using a commercially available kit according to the manufacturer's guidelines (Tri-Reagent; Molecular Research Center, Cincinnati, OH). Following DNase treatment, RNA (1 μl) was used in a reverse transcription reaction using an oligo-dT primer with AMV reverse transcriptase in a 20-μl final volume. Reverse transcriptase PCR was performed on 1 μl of the reverse transcription reaction in a 25-μl volume using standard PCR reaction conditions. The PCR reactions for leptin receptor and GAPDH were cycled as follows: 94°C for 30 s, 60°C for 1 min, and 72°C for 1 min for a total of 35 cycles. Primers used to detect the leptin receptor were as follows: LepR+, 5′-AGTGATATTTGGTCCTCTTCT-3′; LepR−, 5′-GAGTGGTCAAAAGAAGTGAG-3′. The primers amplified a 400-bp fragment. The primers used to detect GAPDH were as follows: GAPDH+, 5′-AAGAAGGTGGTGAAGCAGGCAT-3′; GAPDH−, 5′GATGGTATTCAAGAGAGTAGGGA. The primers amplified a 405-bp fragment. Real-time PCR was performed on 1 μl of reverse transcription reaction prepared as above using a SYBR Green-containing PCR mix (Bio-Rad, Hercules, CA). All samples were normalized to levels of endogenous 18s rRNA. Data are presented as the difference in threshold cycles between the leptin receptor and 18s within each group compared with the difference in threshold cycle of the control mice. Real-time PCR was performed three separate times with RNA from each of the four groups. The primers for the leptin receptor were identical to those listed above. The primers for the 18s were as follows: 18s+, 5′-TAAGTCCCTTTGTACACA; 18s−, 5′-GATCCGAGGGCCTCACTAAAC-3′.
Statistics.
All results are presented as means ± SE. One-way analysis of variance (ANOVA) was performed on heart weight/tibia length ratios. One-way ANOVA was performed on all echocardiographic measurements with post hoc testing using Tukey's Studentized Range Test. Unpaired Student's t-test was used for fasting glucose levels, plasma triglyceride levels, myocardial triglyceride and ATP levels, and protein expression. All statistics were performed using GraphPad Prism 5 (La Jolla, CA). Results were considered statistically significant at a level of P < 0.05.
RESULTS
Cardiomyocyte-specific expression of the rat LepR was achieved in transgenic mice using the α-myosin heavy chain promoter (Fig. 1A). Analysis of LepR expression by reverse-transcriptase PCR in Myh6-LepR mice indicated higher levels of the LepR mRNA compared with nontransgenic mice (Fig. 1B). The Myh6-LepR transgenic mice were then bred onto the db/db background to create mice that lack functional LepRs throughout the body, with the exception of the heart (db/db cardiac LepR rescue mice). LepR expression in the heart was determined in Myh6-LepR transgenic mice, db/db mice, and db/db LepR rescue and compared with control and wild-type (WT) mice by real-time PCR. Levels of LepR were increased significantly in transgenic and db/db LepR cardiac rescue mice and decreased in db/db compared with WT control mice (Fig. 1B). The functionality of the cardiac LepR levels was determined in WT and Myh6-LepR mice by examining the levels of phosphorylated STAT3 in the heart following acute treatment with leptin. Acute leptin treatment resulted in greater levels of p-STAT3 in both male and female Myh6-LepR mice compared with WT mice (Fig. 1C).
Fig. 1.

A: schematic of the myosin heavy chain (MHC)/rat leptin receptor (LepR) transgene. RT-PCR of RNA from the heart of control and transgenic MHC-LepR mice. B: representative real-time PCR for cardiac leptin receptor and 18s rRNA from control, MHC-LepR, db/db, and db/db-cardiac LepR rescue mice. Arrows indicate threshold cycles (CT) for 18s and LepR. Expression level is presented as the difference in CT between LepR and 18s compared with control mice. C: effect of acute leptin treatment on cardiac phosphorylated (p)-STAT3 and STAT3 levels. Mice were injected with 100 μg of leptin, and hearts were harvested 1 h later. *P < 0.05 compared with db/db; n = 3.
Rescue of cardiac LepR protects against myocardial triglyceride accumulation despite obesity, hyperglycemia, and high plasma triglyceride levels.
At 30 wk of age, body weight, plasma insulin, plasma triglyceride, and plasma leptin levels were measured in lean WT control, db/db, and db/db-cardiac LepR rescue mice. db/db and db/db-cardiac LepR rescue mice were significantly obese compared with control mice (body weights: 62.2 ± 4.4, 58.3 ± 7.7, and 29.8 ± 2.3 g, respectively; Fig. 2A). Blood insulin levels in the db/db and the db/db-cardiac lepR rescue mice were similar and significantly higher than in controls (Fig. 2B; P < 0.05). Fasting plasma triglyceride levels were also similar between db/db and db/db-cardiac LepR rescue mice and were significantly elevated (P < 0.05) compared with control mice (Fig. 2C). Plasma leptin levels were significantly higher (P < 0.05) in both db/db and db/db-cardiac LepR rescue mice compared with controls (Fig. 2D). Fasting blood glucose levels measured every 6 wk starting from week 12 were significantly higher in db/db and rescue mice compared with lean mice (Fig. 3). There was no significant difference in fasting blood glucose levels observed between db/db and rescue mice at any time point measured (Fig. 3). db/db Mice exhibited significantly higher myocardial triglyceride levels compared with control mice (13.5 ± 4.5 vs. 3.9 ± 1.6, P < 0.05); however, myocardial triglyceride levels in db/db-cardiac LepR rescue mice were completely normalized and were not significantly different than in lean controls [3.8 ± 0.7, P = not significant (NS)] (Fig. 4).
Fig. 2.

Metabolic profile of lean (n = 10), db/db (n = 9), and db/db-cardiac LepR rescue mice (n = 8). A: body weight. B: plasma insulin. C: plasma triglyceride. D: plasma leptin. E: heart weight to tibia length. *P < 0.05 compared with lean mice.
Fig. 3.
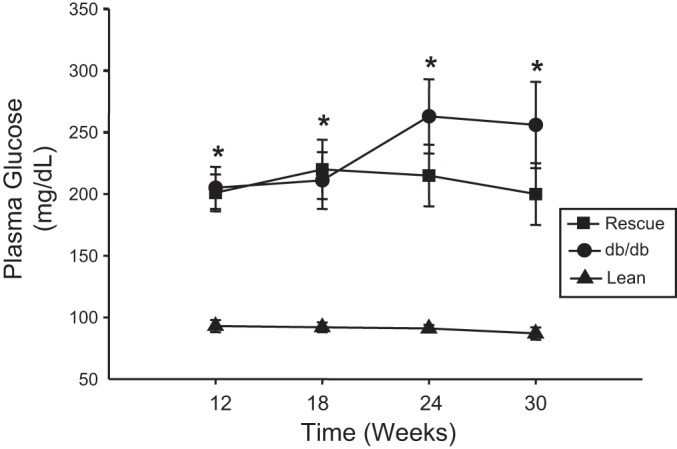
Time course of plasma glucose measurements in lean (n = 10), db/db (n = 9), and db/db-cardiac LepR rescue mice (n = 8). Plasma samples were measured in fasted mice every 6 wk starting at 12 wk of age. *P < 0.05 vs. lean mice.
Fig. 4.
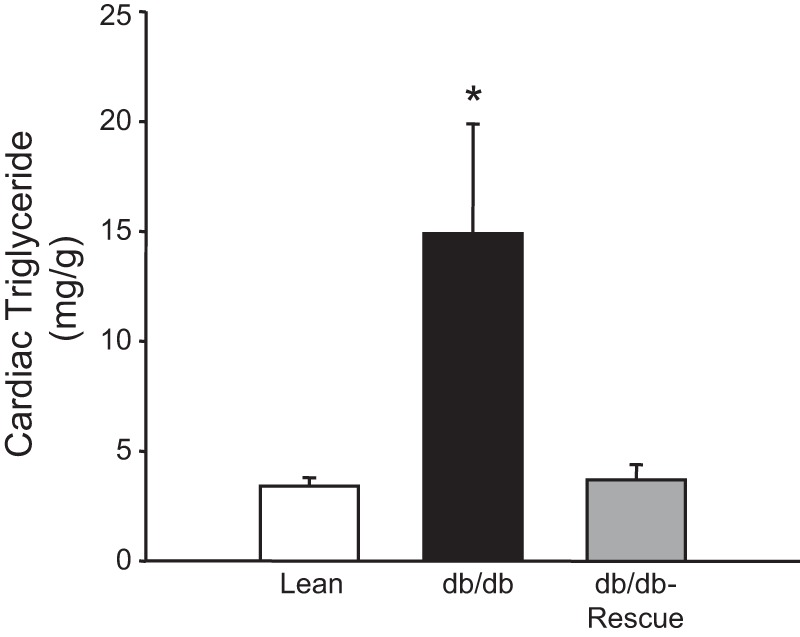
Cardiac triglyceride levels in lean (n = 10), db/db (n = 9), and db/db-cardiac LepR rescue mice (n = 8). db/db Mice exhibited a significant increase in myocardial triglyceride levels, which were completely normalized in db/db-cardiac LepR mice. *P < 0.05 vs. lean and db/db-cardiac LepR rescue mice.
Effect of cardiomyocyte overexpression of leptin on cardiac hypertrophy.
Heart weight/tibial length (HW/TL) ratios were measured after euthanization at age 30 wk in each mouse (Fig. 2E). db/db Mice and db/db-cardiac LepR rescue mice had higher HW/TL ratios compared with lean controls (125.8 ± 4.4 and 115.8 ± 3.6 mg/cm compared with 94.8 ± 3.6 mg/cm respectively, P < 0.05; Fig. 2E), although this effect was attenuated by nearly 10% in db/db-cardiac LepR rescue mice compared with db/db mice (P = NS). Additionally, diastolic posterior wall thickness (PWTd) was reduced in db/db-cardiac LepR rescue mice compared with db/db mice, although it was not statistically significant.
Left ventricular systolic function is normal in 30-wk-old obese db/db mice.
Echocardiographic parameters of left ventricular (LV) systolic function, including EF, FS, cardiac output, and stroke volume, were obtained every 6 wk from 12 wk of age until 30 wk of age. There were no significant differences in any measurements of LV systolic performance among the three groups (Table 1). The mean EF of db/db-cardiac LepR rescue mice was 63.1 ± 2.1 vs. 65.7 ± 1.7% in db/db mice and 62.5 ± 1.5% in lean controls (P = NS). Cardiac dimensions were also similar among the three groups. There were no significant differences in chamber dimensions.
Table 1.
Echocardiographic parameters in lean, db/db, and db/db rescue mice at 30 wk of age
| Parameter | Lean (n = 10) | db/db (n = 9) | db/db-Cardiac Rescue (n = 8) | P Value (Lean vs. Others) |
|---|---|---|---|---|
| Ejection fraction, % | 62.5 ± 1.5 | 65.7 ± 1.7 | 63.1 ± 2.1 | 0.397 |
| Fractional shortening, % | 33 ± 1 | 36 ± 1.3 | 34.6 ± 1.7 | 0.344 |
| Cardiac output, μl/min | 22.6 ± 1.3 | 27.7 ± 1.5 | 25.8 ± 1.6 | 0.092 |
| Stroke volume, μl | 48.6 ± 2.4 | 57.2 ± 2.8 | 56.9 ± 2.7 | 0.078 |
| LVAWd, mm | 0.87 ± 0.03 | 0.94 ± 0.04 | 0.98 ± 0.06 | 0.431 |
| LVPWd, mm | 0.79 ± 0.04 | 0.88 ± 0.03 | 0.85 ± 0.02 | 0.218 |
| LVIDd, mm | 4.13 ± 0.11 | 4.33 ± 0.1 | 4.22 ± 0.14 | 0.527 |
| LVIDs, mm | 2.98 ± 0.1 | 2.94 ± 0.11 | 2.96 ± 0.12 | 0.975 |
| IVRT, ms | 18.1 ± 0.9 | 17.5 ± 0.4 | 17.5 ± 0.6 | 0.899 |
| DT, ms | 17.5 ± 1.6 | 23.8 ± 2 | 22.3 ± 2.4 | 0.134 |
Values are means ± SE. LVAWd, left ventricular anterior wall diastole; LVPWd, left ventricular posterior wall diastole; LVIDd, left ventricular internal dimension diastole; LVIDs, left ventricular internal dimension systole; IVRT, isovolumic relaxation time; DT, deceleration time.
Rescue of cardiac LepR prevents diastolic dysfunction in obese db/db mice.
Doppler analysis of the mitral valve inflow pattern was obtained to evaluate LV diastolic function. The mitral early-to-late (E/A) diastolic filling ratios were measured every 6 wk starting at 12 wk of age. E/A ratios were significantly lower in the db/db mice compared with lean control mice at 12, 24, and 30 wk, suggesting impaired relaxation (Fig. 5). Diastolic function was normalized in db/db rescue mice compared with db/db mice at weeks 24 and 30 (Fig. 5). There were no significant differences in IVRT or DT among the three groups (Table 1).
Fig. 5.
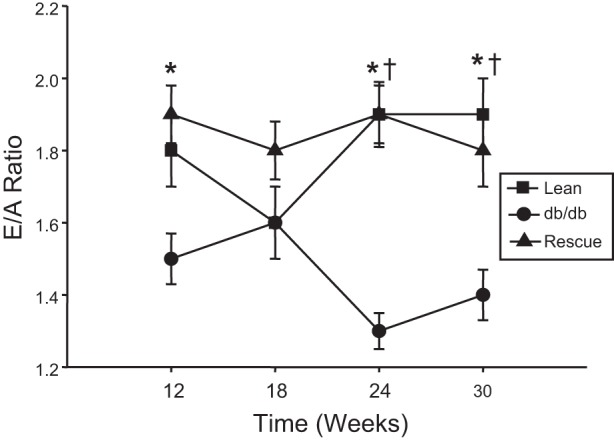
Time course of early-to-late (E/A) diastolic filling ratio in lean (n = 10), db/db (n = 9), and db/db-cardiac LepR rescue mice (n = 8). *P < 0.05 lean mice compared with db/db; †P < 0.05 db/db mice compared with rescue mice.
Rescue of LepR normalizes cardiac p-STAT3 and p-ERK level and increases GAPDH, insulin signaling, and GLUT4 levels in db/db rescue mice.
To determine the effect of rescue of the LepR on cardiac cell signaling, we performed Western blot analysis on proteins of the STAT and ERK pathways. db/db Mice exhibited a significant decrease in the levels of p-STAT3, which was normalized by rescue of the cardiac LepR (Fig. 6, A, B, and D). Cardiac total STAT3 protein was increased in db/db-cardiac LepR rescue mice compared with control and db/db mice (Fig. 6, A and C). db/db Mice exhibited significant decreases in p-ERK1/2, which was normalized in db/db-cardiac LepR rescue mice (Fig. 6, A, E, and G); however, there were no significant differences in total cardiac ERK1/2 in the three groups of mice (Fig. 6, A and F). Levels of GAPDH were increased in db/db-cardiac LepR rescue mice compared with control and db/db mice (Fig. 6, A and H).
Fig. 6.

A: representative Western blots in lean, db/db, and db/db-cardiac LepR rescue mice. B: levels of p-STAT3-Tyr. C: levels of STAT3. D: levels of p-STAT3 normalized to total STAT3. E: levels of p-ERK1/2. F: levels of ERK. G: levels of p-ERK1/2 normalized to total ERK. H: levels of GAPDH. *P < 0.05 compared with db/db mice; †P < 0.05 compared with lean and db/db mice; n = 3. ‡P < 0.05 compared with db/db and rescue mice. AU, arbitrary units.
The effect of rescue of the cardiac LepR on insulin signaling and glucose uptake was also examined by Western blot analysis. Rescue of the cardiac LepR was associated with increases in the levels of p-Akt. Both p-Akt-Ser473 and -Thr308 levels were significantly elevated in the hearts of the rescue mice compared with both db/db and control mice (Fig. 7, B and C). Next, the effect of rescue of the cardiac LepR on glucose uptake was examined by measuring the levels of GLUT4 from cardiac protein lysates. Rescue mice exhibited a significant increase in cardiac GLUT4 levels compared with both db/db and control mice (Fig. 7D).
Fig. 7.

A: representative Western blots in lean, db/db, and db/db-cardiac LepR rescue mice. B: levels of p-Akt-Ser473. C: levels of p-Akt-Thr308. D: levels of glucose transporter 4 (GLUT4). *P < 0.05 compared with lean and db/db mice, †P < 0.05 compared with lean mice; n = 3.
Rescue of LepR does not alter cardiac inflammation or oxidative stress markers in db/db mice.
To determine the effect of rescue of the LepR on cardiac inflammation and oxidative stress, we performed Western blot analysis on proteins associated with cardiac inflammation and oxidative stress. Levels of nuclear factor κ-light-chain enhancer of activated B cells (NF-κB) were measured as an index of cardiac inflammation in control, db/db, and rescue mice. No significant differences in NF-κB levels were detected between any of the groups (Fig. 8B). To determine whether alterations in myocardial fatty acid oxidation resulted in increased oxidative stress, we measured the levels of two markers of oxidative stress, Cu/Zn superoxide dismutase (SOD) and HO-1. No differences in Cu/Zn (Fig. 8C) or HO-1 (Fig. 8D) levels were found between any of the groups.
Fig. 8.

A: representative Western blots in lean, db/db, and db/db-cardiac LepR rescue mice. B: levels of NF-κB. C and D: levels of Cu/Zn superoxide dismutase (SOD; C) and heme oxygenase 1 (HO-1; D); n = 3.
DISCUSSION
Our results demonstrate that transgenic “rescue” of the cardiac LepR in obese db/db mice prevents myocardial lipid accumulation and subsequent cardiac diastolic dysfunction. These results support the hypothesis that intact leptin signaling in the heart may attenuate myocardial steatosis in obesity independent of central leptin signaling and changes in body weight or adiposity. Our results also provide no evidence that leptin has a prohypertrophic effect, as suggested previously (35).
db/db Mice in our study exhibited many of the metabolic derangements observed in human “metabolic syndrome,” including morbid obesity, severe hyperglycemia, high circulating plasma triglycerides, and hyperleptinemia. However, the hearts of db/db mice in which the LepR was selectively rescued in cardiomyocytes were protected from triglyceride accumulation. Although db/db mice did not develop systolic dysfunction in the time frame in which they were studied, our findings are in agreement with previous studies indicating that obese db/db mice exhibit cardiac diastolic dysfunction compared with lean controls (5, 12, 39, 53). However, the db/db-cardiac LepR rescue mice with intact cardiac leptin signaling were protected from myocardial triglyceride accumulation and associated left ventricular diastolic dysfunction. In fact, db/db-cardiac LepR rescue mice actually had slightly higher (albeit not statistically significant) E/A ratios compared with lean controls. This finding suggests that restoration of leptin receptors in the hearts of db/db mice normalized cardiac function as well as myocardial lipids despite the continued presence of severe obesity.
Diastolic dysfunction in the db/db mouse has been associated previously with increased diastolic sarcoplasmic reticulum calcium leak and decreased sarcoplasmic reticulum activity (5, 42). Thus, it is possible that rescue of the cardiac LepR could result in improvement of intracellular calcium handling due to activation of the calcium ATPase, sarcoendoplasmic reticulum Ca2-ATPase, or alterations of contractile proteins such as phospholamban. The relationship between restoration of leptin signaling and intracellular calcium handling and its effects on contractile proteins needs to be examined further, as this could represent a novel pathway by which restoration of leptin signaling improves diastolic function in db/db cardiac LepR rescue mice.
Enhancement of inflammation and increases in oxidative stress are also potential pathways for altered diastolic function in db/db mice. Several studies have linked increases in inflammation to development of cardiac dysfunction in obesity (51, 52). The role of leptin in contributing to cardiac inflammation in obesity is controversial, with studies from leptin-deficient rodents suggesting that leptin may be proinflammatory (29). However, studies from obese human patients did not find any effect of recombinant leptin treatment on levels of inflammatory markers (23). In the present study, we found no significant differences in cardiac NF-κB protein, an index of cardiac inflammation, in any of the groups. Likewise, we examined two important proteins that are markers for increases in oxidative stress: SOD and HO-1. We failed to detect any differences in the cardiac levels of these two proteins in any of the groups. These findings would argue against alterations in inflammation or oxidative stress as potential mechanisms for the effect of rescuing cardiac LepR to improve diastolic function or myocardial lipid accumulation in the present study.
Previous studies have also reported that lipid accumulation in the heart is associated with cardiac dysfunction (34, 37, 54). High myocardial triglyceride levels are associated with impaired biventricular strain in diabetic humans (33). Furthermore, caloric restriction in obese diabetic patients has decreased myocardial triglyceride content and improved myocardial diastolic function (18, 19, 46). Short-term high-fat diet in mice has resulted in increased myocardial lipid accumulation and reductions in left ventricular strain (20). Our results demonstrate that restoration of cardiac leptin signaling in obese db/db mice prevents myocardial lipid accumulation and improves diastolic function. These results are consistent with the hypothesis that the cardiac LepR signaling plays an important role in attenuating myocardial lipid accumulation independent of CNS and actions of leptin, changes in body weight, or blood levels of triglycerides and glucose, which has been studied extensively by Unger (43).
The mechanisms by which myocardial lipid accumulation induces cardiac dysfunction are poorly understood. One mechanism that has received increased attention is an imbalance in delivery and oxidation of fatty acids to the heart resulting in “lipotoxic” injury (43). The normal heart utilizes a combination of predominantly fatty acids and glucose for ATP generation and contractile work. In obesity, or states of an overabundance of fatty acids, there may be increased fatty acid oxidation if the heart is programmed to preferentially utilize these energy sources (7, 30, 50). As fatty acid delivery to the heart exceeds oxidative capabilities, lipid accumulation or myocardial steatosis develops. Myocardial steatosis may lead to the accumulation of toxic intermediates, such as ceramide or diacylglycerol, that contribute to oxidative stress, mitochondrial dysfunction, tissue fibrosis, and apoptosis (8, 34, 48). Alternatively, myocardial steatosis or triglyceride accumulation could serve as a storage reservoir for, and therefore protect against, the adverse effects of fatty acid metabolites such as ceramides and diacylglycerols on the heart (7). In the current study, we did not evaluate these lipid intermediates in the heart, nor did we examine whether the hearts of the rescue mice exhibited significant increases in enzymes associated with fatty acid oxidation or transport of fatty acids in the heart. This is a limitation of the present study and will require further investigation.
Leptin signaling in the heart occurs through three main pathways, including Jak2/IRS/PI3K, Shp2/MAP kinase, and STAT3 (15). Disruption of these leptin signaling pathways causes dilated cardiomyopathies (21, 24, 26). Previous studies from our laboratory and others have also demonstrated the protective role of leptin against the development of heart failure, and this appears to be mediated at least in part by cardiac metabolic effects (17, 32). In the present study, we demonstrated significant increases in p-STAT3 and STAT3 in the hearts of db/db-cardiac LepR rescue mice compared with db/db mice and lean controls, indicative of increased leptin signaling in the hearts of these mice. We also demonstrated that STAT3 signaling in response to acute leptin administration is restored in the hearts of the db/db-cardiac LepR rescue mice, whereas there was no effect in the hearts of db/db mice. However, it is unclear whether STAT3 signaling itself or perhaps one of the other signaling pathways is responsible for the attenuated myocardial lipid accumulation in our db/db-cardiac rescue mice. Previous studies support the possibility that intact STAT3 signaling may be important in mediating leptin's effects on cardiac fatty acid metabolism. For example, in isolated perfused rat hearts, leptin stimulated fatty acid oxidation via a STAT3/nitric oxide-dependent mechanism (41). Additionally, targeted deletion of leptin receptors in the heart resulted in loss of cardiac STAT3 signaling and worsened heart failure in a mouse model of myocardial infarction (31).
We also found evidence for enhanced signaling through the insulin pathway in the db/db-cardiac LepR rescue mice. These mice exhibited significant elevations in the levels of p-Akt compared with both db/db and lean controls. The increased Akt activity along with increased levels of GLUT4 transporters in the hearts of the db/db cardiac LepR rescue mice suggest that restoration of cardiac leptin signaling may also improve glucose uptake and utilization in these hearts. However, more direct measurements of glucose metabolism are needed to establish this mechanism.
Although db/db mice did not exhibit systolic dysfunction, the left ventricular diastolic functional derangements observed may be similar to those found in obese patients with heart failure and preserved ejection fraction (49). Obesity-induced cardiomyopathy is an increasingly recognized cause of nonischemic cardiac dysfunction thought to be related to impaired myocardial metabolism (2, 22). The db/db mouse displays a cardiac phenotype similar to this cardiomyopathy that may predispose patients to adverse cardiac outcomes. Obesity is also strongly associated with risk factors for coronary artery disease and heart failure such as hypertension, diabetes, and dyslipidemia (9, 25). Therefore, our transgenic mouse model provides important insights into mechanisms by which obesity-mediated adverse cardiac outcomes may be prevented. However, further studies evaluating leptin signaling in the myocardium, particularly in relation to myocardial metabolism, are warranted.
Although our results suggest a protective role of leptin against obesity-mediated cardiac dysfunction, other studies have associated high leptin levels with cardiovascular diseases such as coronary atherosclerosis and congestive heart failure (6, 38). It is possible that high or low leptin levels may have detrimental effects on the heart (36). Although our observations imply a direct anti-steatotic effect of leptin on cardiomyocytes, it is conceivable that restoration of leptin signaling in the heart could affect neurohumoral or metabolic regulatory systems that may alter cardiac function and lipid content (1).
The present study also provides insight into the potential role of leptin in cardiac hypertrophy. There have been discrepant results in previous studies related to leptin's effect on LVH. Studies in isolated rat cardiomyocytes and human pediatric ventricular myocytes suggest a prohypertrophic effect of leptin (35). This is in contrast with studies in ob/ob mice, which have demonstrated that adiminstration of leptin was associated with a decrease in LVH independent of changes in body weight (3). We observed a nearly 10% reduction in HW/TL ratio in db/db cardiac LepR rescue mice compared with db/db mice in addition to reduced diastolic posterior wall thickness (PWTd); however, this change was not statistically significant. The observation that db/db LepR rescue mice, despite intact cardiac LepR signaling and high circulating leptin levels, exhibited reduced wall thicknesses and lower normalized heart weights compared with db/db mice of similar weights provides no evidence that leptin directly causes cardiomyocyte hypertrophy. Our observations suggest that cardiac hypertrophy associated with obesity is unlikely to be related directly to increased levels of leptin; instead, other factors associated with obesity, such as hypertension and increased preload due to increased venous return, appear to be more important. It should be noted, however, that we did not directly examine cardiomyocyte diameters in the present study.
In summary, we developed and characterized a novel transgenic model in which cardiac LepRs were specifically rescued in obese lepR-resistant db/db mice. These cardiac LepR rescue mice are protected from myocardial triglyceride accumulation and left ventricular diastolic dysfunction associated with obesity. Despite systemic metabolic derangements, these mice have normal cardiac diastolic function compared with lean WT mice and improved cardiac function compared with obese db/db mice with an identical metabolic profile of morbid obesity, hyperglycemia, hypertriglyceridemia, and hyperleptinemia. Furthermore, our results suggest that leptin does not directly cause cardiac hypertrophy.
Perspectives and significance.
Obesity causes an increase in fatty acid delivery and oxidation in the heart, which can have deleterious effects on cardiac function. Adipokines such as leptin appear to regulate cardiac energy metabolism and may have effects on cardiac structure and function. The role of the cardiac leptin signaling pathway in preventing lipid accumulation, lipotoxicity, and its effects on cardiac function is not fully understood. In the present study, we characterized a novel mouse model in which cardiac leptin receptors (LepR) were “rescued” specifically in the hearts of obese db/db mice with defective LepR. Despite severe obesity and major metabolic abnormalities, these db/db cardiac LepR rescue mice were protected against cardiac triglyceride accumulation and diastolic dysfunction observed in db/db mice. These results directly demonstrate that cardiac LepR signaling plays an important role in protecting against excessive cardiac lipid accumulation and possibly myocardial diastolic dysfunction in obesity independent of body weight or circulating lipid levels. Additionally, although leptin has been suggested to have both pro- and antihypertrophic effects on the heart, our results indicate that expression of cardiac leptin receptors in obese db/db mice with high levels of leptin does not cause cardiac hypertrophy. Further investigations should evaluate the mechanisms by which leptin protects the heart against lipid accumulation in obesity.
GRANTS
The research reported in this publication was supported by grants from the National Heart, Lung, and Blood Institute (PO1-HL-051971) and the National Institute of General Medical Sciences (P20-GM-104357) as well as a seed grant from the American Medical Association (M. E. Hall). Transgenic mice were made through the Transgenic Core, Department of Physiology and Biophysics, University of Mississippi Medical Center.
DISCLOSURES
The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
M.E.H., J.E.H., and D.E.S. conception and design of research; M.E.H., M.W.M., and D.E.S. performed experiments; M.E.H., M.W.M., and D.E.S. analyzed data; M.E.H., M.W.M., J.E.H., and D.E.S. interpreted results of experiments; M.E.H., M.W.M., and D.E.S. prepared figures; M.E.H. and D.E.S. drafted manuscript; M.E.H., M.W.M., J.E.H., and D.E.S. edited and revised manuscript; M.E.H., M.W.M., J.E.H., and D.E.S. approved final version of manuscript.
ACKNOWLEDGMENTS
We thank Dr. Takashi Murakami, Department of Molecular Functional Analysis, Kanazawa University Graduate School of Medical Science, Kanazawa, Japan, for the gift of the rat leptin receptor cDNA.
REFERENCES
- 1.Abel ED, Sweeney G. Modulation of the cardiovascular system by leptin. Biochimie 94: 2097–2103, 2012 [DOI] [PubMed] [Google Scholar]
- 2.Banerjee S, Peterson LR. Myocardial metabolism and cardiac performance in obesity and insulin resistance. Curr Cardiol Rep 9: 143–149, 2007 [DOI] [PubMed] [Google Scholar]
- 3.Barouch LA, Berkowitz DE, Harrison RW, O'Donnell CP, Hare JM. Disruption of leptin signaling contributes to cardiac hypertrophy independently of body weight in mice. Circulation 108: 754–759, 2003 [DOI] [PubMed] [Google Scholar]
- 4.Belke DD, Severson DL. Diabetes in mice with monogenic obesity: the db/db mouse and its use in the study of cardiac consequences. Methods Mol Biol 933: 47–57, 2012 [DOI] [PubMed] [Google Scholar]
- 5.Belke DD, Swanson EA, Dillmann WH. Decreased sarcoplasmic reticulum activity and contractility in diabetic db/db mouse heart. Diabetes 53: 3201–3208, 2004 [DOI] [PubMed] [Google Scholar]
- 6.Bobbert P, Jenke A, Bobbert T, Kuhl U, Rauch U, Lassner D, Scheibenbogen C, Poller W, Schultheiss HP, Skurk C. High leptin and resistin expression in chronic heart failure: adverse outcome in patients with dilated and inflammatory cardiomyopathy. Eur J Heart Fail 14: 1265–1275, 2012 [DOI] [PubMed] [Google Scholar]
- 7.Brindley DN, Kok BP, Kienesberger PC, Lehner R, Dyck JR. Shedding light on the enigma of myocardial lipotoxicity: the involvement of known and putative regulators of fatty acid storage and mobilization. Am J Physiol Endocrinol Metab 298: E897–E908, 2010 [DOI] [PubMed] [Google Scholar]
- 8.Buchanan J, Mazumder PK, Hu P, Chakrabarti G, Roberts MW, Yun UJ, Cooksey RC, Litwin SE, Abel ED. Reduced cardiac efficiency and altered substrate metabolism precedes the onset of hyperglycemia and contractile dysfunction in two mouse models of insulin resistance and obesity. Endocrinology 146: 5341–5349, 2005 [DOI] [PubMed] [Google Scholar]
- 9.Burke GL, Bertoni AG, Shea S, Tracy R, Watson KE, Blumenthal RS, Chung H, Carnethon MR. The impact of obesity on cardiovascular disease risk factors and subclinical vascular disease: the Multi-Ethnic Study of Atherosclerosis. Arch Intern Med 168: 928–935, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Carley AN, Severson DL. Fatty acid metabolism is enhanced in type 2 diabetic hearts. Biochim Biophys Acta 1734: 112–126, 2005 [DOI] [PubMed] [Google Scholar]
- 11.Christoffersen C, Bollano E, Lindegaard ML, Bartels ED, Goetze JP, Andersen CB, Nielsen LB. Cardiac lipid accumulation associated with diastolic dysfunction in obese mice. Endocrinology 144: 3483–3490, 2003 [DOI] [PubMed] [Google Scholar]
- 12.Demarco VG, Ford DA, Henriksen EJ, Aroor AR, Johnson MS, Habibi J, Ma L, Yang M, Albert CJ, Lally JW, Ford CA, Prasannarong M, Hayden MR, Whaley-Connell AT, Sowers JR. Obesity-related alterations in cardiac lipid profile and nondipping blood pressure pattern during transition to diastolic dysfunction in male db/db mice. Endocrinology 154: 159–171, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Field AE, Coakley EH, Must A, Spadano JL, Laird N, Dietz WH, Rimm E, Colditz GA. Impact of overweight on the risk of developing common chronic diseases during a 10-year period. Arch Intern Med 161: 1581–1586, 2001 [DOI] [PubMed] [Google Scholar]
- 14.Goldberg IJ, Trent CM, Schulze PC. Lipid metabolism and toxicity in the heart. Cell Metab 15: 805–812, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hall JE, da Silva AA, do Carmo JM, Dubinion J, Hamza S, Munusamy S, Smith G, Stec DE. Obesity-induced hypertension: role of sympathetic nervous system, leptin, and melanocortins. J Biol Chem 285: 17271–17276, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hall ME, Smith G, Hall JE, Stec DE. Systolic dysfunction in cardiac-specific ligand-inducible MerCreMer transgenic mice. Am J Physiol Heart Circ Physiol 301: H253–H260, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hall ME, Smith G, Hall JE, Stec DE. Cardiomyocyte-specific deletion of leptin receptors causes lethal heart failure in Cre-recombinase-mediated cardiotoxicity. Am J Physiol Regul Integr Comp Physiol 303: R1241–R1250, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hammer S, Snel M, Lamb HJ, Jazet IM, van der Meer RW, Pijl H, Meinders EA, Romijn JA, de Roos A, Smit JW. Prolonged caloric restriction in obese patients with type 2 diabetes mellitus decreases myocardial triglyceride content and improves myocardial function. J Am Coll Cardiol 52: 1006–1012, 2008 [DOI] [PubMed] [Google Scholar]
- 19.Hammer S, van der Meer RW, Lamb HJ, Schär M, de Roos A, Smit JW, Romijn JA. Progressive caloric restriction induces dose-dependent changes in myocardial triglyceride content and diastolic function in healthy men. J Clin Endocrinol Metab 93: 497–503, 2008 [DOI] [PubMed] [Google Scholar]
- 20.Hankiewicz JH, Banke NH, Farjah M, Lewandowski ED. Early impairment of transmural principal strains in the left ventricular wall after short-term, high-fat feeding of mice predisposed to cardiac steatosis. Circ Cardiovasc Imaging 3: 710–717, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hilfiker-Kleiner D, Hilfiker A, Fuchs M, Kaminski K, Schaefer A, Schieffer B, Hillmer A, Schmiedl A, Ding Z, Podewski E, Podewski E, Poli V, Schneider MD, Schulz R, Park JK, Wollert KC, Drexler H. Signal transducer and activator of transcription 3 is required for myocardial capillary growth, control of interstitial matrix deposition, and heart protection from ischemic injury. Circ Res 95: 187–195, 2004 [DOI] [PubMed] [Google Scholar]
- 22.Horwich TB, Fonarow GC. Glucose, obesity, metabolic syndrome, and diabetes relevance to incidence of heart failure. J Am Coll Cardiol 55: 283–293, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hukshorn CJ, Lindeman JH, Toet KH, Saris WH, Eilers PH, Westerterp-Plantenga MS, Kooistra T. Leptin and the proinflammatory state associated with human obesity. J Clin Endocrinol Metab 89: 1773–1778, 2004 [DOI] [PubMed] [Google Scholar]
- 24.Jacoby JJ, Kalinowski A, Liu MG, Zhang SS, Gao Q, Chai GX, Ji L, Iwamoto Y, Li E, Schneider M, Russell KS, Fu XY. Cardiomyocyte-restricted knockout of STAT3 results in higher sensitivity to inflammation, cardiac fibrosis, and heart failure with advanced age. Proc Natl Acad Sci USA 100: 12929–12934, 2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kenchaiah S, Evans JC, Levy D, Wilson PW, Benjamin EJ, Larson MG, Kannel WB, Vasan RS. Obesity and the risk of heart failure. N Engl J Med 347: 305–313, 2002 [DOI] [PubMed] [Google Scholar]
- 26.Kontaridis MI, Yang W, Bence KK, Cullen D, Wang B, Bodyak N, Ke Q, Hinek A, Kang PM, Liao R, Neel BG. Deletion of Ptpn11 (Shp2) in cardiomyocytes causes dilated cardiomyopathy via effects on the extracellular signal-regulated kinase/mitogen-activated protein kinase and RhoA signaling pathways. Circulation 117: 1423–1435, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lang RM, Bierig M, Devereux RB, Flachskampf FA, Foster E, Pellikka PA, Picard MH, Roman MJ, Seward J, Shanewise JS, Solomon SD, Spencer KT, Sutton MS, Stewart WJ. Recommendations for chamber quantification: a report from the American Society of Echocardiography's Guidelines and Standards Committee and the Chamber Quantification Writing Group, developed in conjunction with the European Association of Echocardiography, a branch of the European Society of Cardiology. J Am Soc Echocardiogr 18: 1440–1463, 2005 [DOI] [PubMed] [Google Scholar]
- 28.Lee Y, Naseem RH, Duplomb L, Park BH, Garry DJ, Richardson JA, Schaffer JE, Unger RH. Hyperleptinemia prevents lipotoxic cardiomyopathy in acyl CoA synthase transgenic mice. Proc Natl Acad Sci USA 101: 13624–13629, 2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Loffreda S, Yang SQ, Lin HZ, Karp CL, Brengman ML, Wang DJ, Klein AS, Bulkley GB, Bao C, Noble PW, Lane MD, Diehl AM. Leptin regulates proinflammatory immune responses. FASEB J 12: 57–65, 1998 [PubMed] [Google Scholar]
- 30.Lopaschuk GD, Folmes CD, Stanley WC. Cardiac energy metabolism in obesity. Circ Res 101: 335–347, 2007 [DOI] [PubMed] [Google Scholar]
- 31.McGaffin KR, Witham WG, Yester KA, Romano LC, O'Doherty RM, McTiernan CF, O'Donnell CP. Cardiac-specific leptin receptor deletion exacerbates ischaemic heart failure in mice. Cardiovasc Res 89: 60–71, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Minokoshi Y, Kim YB, Peroni OD, Fryer LG, Müller C, Carling D, Kahn BB. Leptin stimulates fatty-acid oxidation by activating AMP-activated protein kinase. Nature 415: 339–343, 2002 [DOI] [PubMed] [Google Scholar]
- 33.Ng AC, Delgado V, Bertini M, van der Meer RW, Rijzewijk LJ, Hooi Ewe S, Siebelink HM, Smit JW, Diamant M, Romijn JA, de Roos A, Leung DY, Lamb HJ, Bax JJ. Myocardial steatosis and biventricular strain and strain rate imaging in patients with type 2 diabetes mellitus. Circulation 122: 2538–2544, 2010 [DOI] [PubMed] [Google Scholar]
- 34.Park TS, Hu Y, Noh HL, Drosatos K, Okajima K, Buchanan J, Tuinei J, Homma S, Jiang XC, Abel ED, Goldberg IJ. Ceramide is a cardiotoxin in lipotoxic cardiomyopathy. J Lipid Res 49: 2101–2112, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Rajapurohitam V, Gan XT, Kirshenbaum LA, Karmazyn M. The obesity-associated peptide leptin induces hypertrophy in neonatal rat ventricular myocytes. Circ Res 93: 277–279, 2003 [DOI] [PubMed] [Google Scholar]
- 36.Ren J. Leptin and hyperleptinemia - from friend to foe for cardiovascular function. J Endocrinol 181: 1–10, 2004 [DOI] [PubMed] [Google Scholar]
- 37.Rijzewijk LJ, van der Meer RW, Smit JW, Diamant M, Bax JJ, Hammer S, Romijn JA, de Roos A, Lamb HJ. Myocardial steatosis is an independent predictor of diastolic dysfunction in type 2 diabetes mellitus. J Am Coll Cardiol 52: 1793–1799, 2008 [DOI] [PubMed] [Google Scholar]
- 38.Sattar N, Wannamethee G, Sarwar N, Chernova J, Lawlor DA, Kelly A, Wallace AM, Danesh J, Whincup PH. Leptin and coronary heart disease: prospective study and systematic review. J Am Coll Cardiol 53: 167–175, 2009 [DOI] [PubMed] [Google Scholar]
- 39.Semeniuk LM, Kryski AJ, Severson DL. Echocardiographic assessment of cardiac function in diabetic db/db and transgenic db/db-hGLUT4 mice. Am J Physiol Heart Circ Physiol 283: H976–H982, 2002 [DOI] [PubMed] [Google Scholar]
- 40.Sharma S, Adrogue JV, Golfman L, Uray I, Lemm J, Youker K, Noon GP, Frazier OH, Taegtmeyer H. Intramyocardial lipid accumulation in the failing human heart resembles the lipotoxic rat heart. FASEB J 18: 1692–1700, 2004 [DOI] [PubMed] [Google Scholar]
- 41.Sharma V, Mustafa S, Patel N, Wambolt R, Allard MF, McNeill JH. Stimulation of cardiac fatty acid oxidation by leptin is mediated by a nitric oxide-p38 MAPK-dependent mechanism. Eur J Pharmacol 617: 113–117, 2009 [DOI] [PubMed] [Google Scholar]
- 42.Stolen TO, Hoydal MA, Kemi OJ, Catalucci D, Ceci M, Aasum E, Larsen T, Rolim N, Condorelli G, Smith GL, Wisloff U. Interval training normalizes cardiomyocyte function, diastolic Ca2+ control, and SR Ca2+ release synchronicity in a mouse model of diabetic cardiomyopathy. Circ Res 105: 527–536, 2009 [DOI] [PubMed] [Google Scholar]
- 43.Unger RH. Hyperleptinemia: protecting the heart from lipid overload. Hypertension 45: 1031–1034, 2005 [DOI] [PubMed] [Google Scholar]
- 44.Unger RH, Orci L. Lipotoxic diseases of nonadipose tissues in obesity. Int J Obes Relat Metab Disord 24, Suppl 4: S28–S32, 2000 [DOI] [PubMed] [Google Scholar]
- 45.Van den Bergh A, Vanderper A, Vangheluwe P, Desjardins F, Nevelsteen I, Verreth W, Wuytack F, Holvoet P, Flameng W, Balligand JL, Herijgers P. Dyslipidaemia in type II diabetic mice does not aggravate contractile impairment but increases ventricular stiffness. Cardiovasc Res 77: 371–379, 2008 [DOI] [PubMed] [Google Scholar]
- 46.van der Meer RW, Hammer S, Smit JW, Frölich M, Bax JJ, Diamant M, Rijzewijk LJ, de Roos A, Romijn JA, Lamb HJ. Short-term caloric restriction induces accumulation of myocardial triglycerides and decreases left ventricular diastolic function in healthy subjects. Diabetes 56: 2849–2853, 2007 [DOI] [PubMed] [Google Scholar]
- 47.Wang MY, Koyama K, Shimabukuro M, Mangelsdorf D, Newgard CB, Unger RH. Overexpression of leptin receptors in pancreatic islets of Zucker diabetic fatty rats restores GLUT-2, glucokinase, and glucose-stimulated insulin secretion. Proc Natl Acad Sci USA 95: 11921–11926, 1998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wende AR, Abel ED. Lipotoxicity in the heart. Biochim Biophys Acta 1801: 311–319, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wong C, Marwick TH. Obesity cardiomyopathy: pathogenesis and pathophysiology. Nat Clin Pract Cardiovasc Med 4: 436–443, 2007 [DOI] [PubMed] [Google Scholar]
- 50.Yang R, Barouch LA. Leptin signaling and obesity: cardiovascular consequences. Circ Res 101: 545–559, 2007 [DOI] [PubMed] [Google Scholar]
- 51.Yudkin JS, Juhan-Vague I, Hawe E, Humphries SE, di Minno G, Margaglione M, Tremoli E, Kooistra T, Morange PE, Lundman P, Mohamed-Ali V, Hamsten A. Low-grade inflammation may play a role in the etiology of the metabolic syndrome in patients with coronary heart disease: the HIFMECH study. Metabolism 53: 852–857, 2004 [DOI] [PubMed] [Google Scholar]
- 52.Yudkin JS, Kumari M, Humphries SE, Mohamed-Ali V. Inflammation, obesity, stress and coronary heart disease: is interleukin-6 the link? Atherosclerosis 148: 209–214, 2000 [DOI] [PubMed] [Google Scholar]
- 53.Yue P, Arai T, Terashima M, Sheikh AY, Cao F, Charo D, Hoyt G, Robbins RC, Ashley EA, Wu J, Yang PC, Tsao PS. Magnetic resonance imaging of progressive cardiomyopathic changes in the db/db mouse. Am J Physiol Heart Circ Physiol 292: H2106–H2118, 2007 [DOI] [PubMed] [Google Scholar]
- 54.Zhou YT, Grayburn P, Karim A, Shimabukuro M, Higa M, Baetens D, Orci L, Unger RH. Lipotoxic heart disease in obese rats: implications for human obesity. Proc Natl Acad Sci USA 97: 1784–1789, 2000 [DOI] [PMC free article] [PubMed] [Google Scholar]