

Abstract
Muscle-specific RING finger-1 (MuRF-1), a ubiquitin ligase and key regulator of proteasome-dependent protein degradation, is highly expressed during skeletal muscle atrophy. The transcription factor forkhead box O3 (FoxO3) induces MuRF-1 expression, but the direct role of other major atrophy-related transcription factors, such as SMAD3, is largely unknown. The goal of this study was to determine whether SMAD3 individually regulates, or with FoxO3 coordinately regulates, MuRF-1 expression. In cultured myotubes or human embryonic kidney cells, MuRF-1 mRNA content and promoter activity were increased by FoxO3 but not by SMAD3 overexpression. However, FoxO3 and SMAD3 coexpression synergistically increased MuRF-1 mRNA and promoter activity. Mutation of the SMAD-binding element (SBE) in the proximal MuRF-1 promoter or overexpression of a SMAD3 DNA-binding mutant attenuated FoxO3-dependent MuRF-1 promoter activation, showing that SMAD binding to DNA is required for optimal activation of FoxO3-induced transcription of MuRF-1. Using chromatin immunoprecipitation, SMAD3 DNA binding increased FoxO3 abundance and SBE mutation reduced FoxO3 abundance on the MuRF-1 promoter. Furthermore, SMAD3 overexpression dose-dependently increased FoxO3 protein content, and coexpression of FoxO3 and SMAD3 synergistically increased FoxO-dependent gene transcription [assessed with a FoxO response element (FRE)-driven reporter]. Collectively, these results show that SMAD3 regulates transcription of MuRF-1 by increasing FoxO3 binding at a conserved FRE-SBE motif within the proximal promoter region, and by increasing FoxO3 protein content and transcriptional activity. These data are the first to indicate that two major transcription factors regulating protein degradation, FoxO3 and SMAD3, converge to coordinately and directly regulate transcription of MuRF-1.
Keywords: protein degradation, protein-DNA interaction, transcription factors, ubiquitin ligase, cell culture
severe loss of muscle mass, or muscle atrophy, is characterized by an increased protein degradation rate, especially via the ubiquitin proteasome system (UPS) (14). Numerous forms of muscle atrophy (e.g., cancer cachexia, denervation, and fasting) activate a common transcriptional program to induce expression of genes involved in protein degradation, including the ubiquitin ligases Atrogin-1 (MAFbx) and muscle-specific RING finger-1 (MuRF-1) (16, 22, 25). Atrogin-1 promotes degradation of eukaryotic translation initiation factor 3F (eIF3-F) (8) and proteins of the intermediate filament such as desmin and vimentin (19). During muscle atrophy, MuRF-1 targets contractile proteins of the thick filament for degradation (7), thereby contributing to both decreased skeletal muscle mass and presumably decreased capacity to generate force. Furthermore, these ubiquitin ligases are essential for rapid muscle atrophy (4). Unfortunately, the cellular mechanisms regulating transcription of Atrogin-1 and MuRF-1 are incompletely understood.
Atrogin-1 and MuRF-1 gene expression are both regulated at least in part by the transcription factor forkhead box type O (FoxO) (24, 28, 34). FoxO1 and FoxO3 are sufficient to induce Atrogin-1 (26) and MuRF-1 gene expression (28, 34), protein degradation (26, 36), and ultimately, muscle atrophy. FoxO binds to the FoxO response element [FRE; (A/G)TAAA(T/C)A] within the proximal promoter region to induce transcription of Atrogin-1 and MuRF-1 (26, 34). Interestingly, the synthetic glucocorticoid dexamethasone synergistically increases FoxO-induced MuRF-1 promoter activity (34), suggesting that FoxO may require DNA-binding partners to fully activate the transcription of MuRF-1.
One potential FoxO DNA-binding partner is SMAD3, a transcription factor activated by the transforming growth factor-β (TGF-β) pathway. A recent meta-analysis of human and rodent microarray studies implicated the TGF-β pathway as a core pathway and SMAD3 as a major signaling node of muscle atrophy (6). Gene expression of Atrogin-1 and MuRF-1 is increased following administration of the TGF-β family member myostatin in vitro (17, 18). However, overexpression of SMAD3 (11) or activation of the TGF-β pathway by constitutively active activin receptor-like kinase 5 (ALK5) (27) induces muscle atrophy and Atrogin-1 gene expression, but is insufficient to induce MuRF-1 gene transcription in vivo. Therefore, the direct role of SMAD3 controlling the transcriptional regulation of ubiquitin ligases is unclear. It has been suggested that SMAD3 may play a role in regulating FoxO activity by inhibiting Akt-mediated phosphorylation (and cytosolic sequestration) of FoxO (2, 21, 33). FoxO and SMAD transcription factors have previously been shown to physically associate and synergistically regulate expression of the cyclin-dependent kinase inhibitor p21Cip1 (29). Furthermore, of 115 genes regulated by TGF-β in keratinocytes, 11 require FoxO as a DNA-binding partner (10). Therefore, it is possible that SMAD3 may modulate FoxO-induced transcription of muscle-specific ubiquitin ligases.
Both SMAD and FoxO transcription factors are major regulators of the transcriptional program that induces muscle atrophy (12). The effects of these transcription factors on atrophy have largely been studied independently; the effects of the interaction between these transcription factors have not been well characterized. The purpose of the present study was to determine whether SMAD3 individually regulates or with FoxO coordinately regulates MuRF-1 gene expression. Because it has been previously reported that SMAD3 and FoxO coordinately regulate several genes (1, 10, 29), we hypothesized that SMAD3 functions to augment FoxO-dependent gene transcription.
MATERIALS AND METHODS
Cell culture.
Primary human skeletal muscle myoblasts (HSkM) were generated as previously described (3) by isolating satellite cells from muscle biopsies of the vastus lateralis from one African American and 13 Caucasian female subjects (age 29.1 ± 0.5 yr, height 1.67 ± 0.01 m, weight 94.0 ± 9.4 kg, body mass index 33.6 ± 0.7 kg/m2). Written informed consent was obtained from all subjects, and the study was approved by the East Carolina University and Medical Center Institutional Review Board. Myoblasts were cultured in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal bovine serum (no. 16140-071; Life Technologies), recombinant human epidermal growth factor, dexamethasone, gentamycin, BSA, and fetuin (SkGM Singlequots; Lonza). Upon reaching confluence, growth medium was switched to low-serum medium (2% horse serum) supplemented with 3% BSA and 0.1% fetuin to induce differentiation into myotubes. Experiments were performed on day 6 of differentiation. Four pools, each with three or four different subjects, were tested on different occasions to verify the reproducibility of results. Therefore, results and statistics are from four independent observations.
Human embryonic kidney cells (HEK293a) were cultured in DMEM supplemented with 10% fetal bovine serum and 100 IU/ml penicillin-streptomycin. For all experiments, cells were seeded into six-well plates at a density of 2.5 × 105 cells per well and allowed to adhere to the plate overnight.
Mouse skeletal muscle myoblasts (C2C12; American Type Culture Collection, Manassas, VA) were cultured in DMEM supplemented with 10% fetal bovine serum and 100 IU/ml penicillin-streptomycin and maintained at subconfluent density prior to differentiation. For all experiments, myoblasts were seeded into six-well plates at a density of 2.5 × 105 cells per well and allowed to adhere overnight. Growth medium was switched to low-serum medium (2% horse serum) to induce differentiation into myotubes. When necessary, myotubes (day 4 of differentiation) were infected with adenovirus-expressing human wild-type SMAD3 (wtSMAD3) (Vector Biosystems) for 24 h, then coinfected with adenovirus-encoding human wild-type FoxO3 (wtFoxO3) (36) for an additional 24 h. As a control, C2C12 myotubes were infected with adenoviruses encoding green fluorescent protein (GFP). All cell cultures were maintained at 37°C and 5% CO2, and media were changed at least every 48 h.
Protein degradation rate.
Protein degradation rate was determined by release of a radiolabeled amino acid as previously described (5). Myotubes were radiolabeled with l-[3,5-3H]tyrosine (5 μCi/ml, PerkinElmer Life Sciences) for 24 h. Cells were thoroughly washed and radioactivity was chased for 2 h using differentiation media supplemented with 2 mM nonradioactive l-tyrosine to allow degradation of short-lived proteins and limit reincorporation of the radiolabel. Myotubes then were incubated in differentiation media (DMEM + 2% horse serum), media lacking serum (DMEM only), or media lacking both serum and amino acids (Hank's balanced salt solution). Four serial media samples were collected over the course of 5 h. At the end of the experiments, culture medium was removed completely and myotubes were solubilized in 0.2 N NaOH. Radioactivity of cells and trichloroacetic acid (TCA)-precipitated media samples were measured using scintillation counting. Total radioactivity was calculated as the sum of the residual radioactivity in the myotubes plus radioactivity of all time points. Protein degradation rate was calculated as a percentage of total [3H]tyrosine incorporation from the regression line of serial samples.
Real-time PCR.
After myotubes were washed twice with PBS, RNA was collected using TRIzol (Ambion) and then stored at −80°C until further processing. RNA integrity was determined by agarose gel electrophoresis, and RNA concentration and purity (Ab260/280) were assessed spectrophometrically using a NanoDrop UV-Vis spectrophometer (ND-1000; ThermoScientific). RNA was reversed transcribed to cDNA (Quanta Biosciences) at a final concentration of 0.10 μg/μl. Large ribosomal protein (RPLPO) was used as an endogenous control gene, and fold expression was calculated using the ΔΔCt method. None of the treatment conditions significantly altered expression of the normalizing gene (RPLPO). Custom RT-PCR primers (Table 1) were selected using National Center for Biotechnology Information (NCBI) BLAST algorithms (http://blast.ncbi.nlm.nih.gov/Blast.cgi). Specificity of these primers was confirmed using PCR followed by agarose gel electrophoresis of a single band at the predicted amplicon size. All RT-PCR experiments were run with a standard curve of eight orders of magnitude, and amplification efficiency was near 100%.
Table 1.
Primers used for RT-PCR
| Gene | Sequence (5′–3′) |
|---|---|
| Mouse | |
| FoxO1 | Fwd TGCTGTGAAGGGACAGATTG |
| Rev GAGTGGATGGTGAAGAGCGT | |
| FoxO3 | Fwd CTTCATTCTGAACGCGCA |
| Rev CTTCAAGGATAAGGGCGACA | |
| Atrogin-1 | Fwd TCAGGGATGTGAGCTGTGAC |
| Rev GACTGGACTTCTCGACTGCC | |
| MuRF-1 | Fwd TGGCACTTGAGAGGAAGGTAGCCC |
| Rev TGCAGCGGATCACGCAGGAG | |
| RPLPO | Fwd CCGATCTGCAGACACACACT |
| Rev ACCCTGAAGTGCTCGACATC | |
| Human | |
| Atrogin-1 | Fwd TCAGGGATGTGAGCTGTGAC |
| Rev GGGGGAAGCTTTCAACAGAC | |
| MuRF-1 | Fwd CTTCGTGCTCCTTGCACAT |
| Rev ATCGTCACGGAGTGTACGG | |
| RPLPO | Fwd AGGCGTCCTCGTGGAAGTGACA |
| Rev TGCTGCATCTGCTTGGAGCCC |
FoxO, forkhead box O; MuRF-1, muscle-specific RING finger-1; RPLPO, large ribosomal protein; Fwd, forward; Rev, reverse.
Plasmid vectors.
Plasmids encoding wild-type human FoxO1 (32), FoxO3 (23), SMAD3 (15), and dominant-negative SMAD3 (dnSMAD3) (35) were purchased from Addgene (catalog no. 13507, 10710, 11742, and 12626, respectively). The FRE reporter (FRE-luc) has been described previously (26). The 5-kb pGL3 wtMuRF-1 reporter (mouse) has been used to examine MuRF-1 promoter activity both in vivo (5, 27) and in vitro (34). We confirmed the entire promoter region of this plasmid by DNA sequencing. To determine sequence homology between the mouse and human MuRF-1 promoter region, we aligned these sequences using NCBI algorithms (http://blast.ncbi.nlm.nih.gov). Site-directed mutagenesis (Stratagene) of two putative SMAD-binding elements (SBE, AGAC) within the proximal promoter region (5 and 11 bp upstream of the transcriptional start site) was performed to generate a 5-kb mutant MuRF-1 reporter (SBE-mut). These AGAC sequences were mutated to AGgg and AtcC as described by Seoane et al. (29). Both the 5-kb wild-type and 5-kb SBE-mut reporters were truncated to ∼2 kb (digested with NheI and NdeI) and 0.25 kb (digested with NheI and PstI) followed by blunt-end ligation (New England Biolabs, Ipswich, MA). The promoter regions of these mutated and truncated reporters were sequenced and aligned against the wild-type reporter to confirm effective mutagenesis.
Plasmid transfection.
HEK cells were plated into six-well plates at a density of 2.5 × 105 cells/well, allowed to adhere to the plate overnight, and transfected with 2.0 μg (including 0.25 μg reporter DNA) of total plasmid DNA per well using FuGene 6 per the manufacturer's recommendations (Promega, Madison, WI). An enhanced GFP (eGFP) plasmid was used as a transfection control and to balance total DNA between conditions. Interwell transfection efficiency was confirmed by equal GFP expression between wells. Experiments were performed 48 h after transfection.
Luciferase reporter assay.
Cells were washed twice with PBS and collected in 1× passive lysis buffer per the manufacturer's recommendation (Promega). Samples were spun at 20,000 g for 5 min and 20 μl of the supernatant pipetted into a white 96-well plate with 100 μl of substrate. Luminescence was measured using a microplate reader (Spectramax M4). Total protein content was measured by bicinchoninic acid assay (ThermoScientific) and did not differ between treatment conditions. Luciferase values were normalized to total protein and expressed as fold relative to eGFP-only transfected cells.
Chromatin immunoprecipitation.
HEK cells were plated in 10-cm dishes (n = 6 per group) at a density of 2.0 × 106 cells per dish, allowed to adhere overnight, and then transfected with plasmid DNA encoding 1) either wild-type or SBE-mut 0.25 kb MuRF-1 reporter and 2) eGFP, 3) wtFoxO3, 4) SMAD3, or 5) wtFoxO3 + wtSMAD3. Forty-eight hours after transfection, proteins were cross-linked with chromatin using 1% formaldehyde and chromatin was collected. Chromatin was sheared by sonication to approximately 1-kb fragments, and antibodies against FoxO3 (no. 2497; Cell Signaling), SMAD3 (no. 9523; Cell Signaling), or IgG (negative control) were used to immunoprecipitate chromatin. After cross-linking was reversed, quantitative real-time PCR was performed for the MuRF-1 reporter using primers flanking the FRE-SBE motif: (5′ to 3′) GCGATTGCTCATCCCTGCAT (forward) and ATGCCAAGCTTAGCGTTTCC (reverse). Using primers specific for the reporter accomplished our threefold goal: 1) confirming mutagenesis eliminated SMAD3 binding within the MuRF-1 promoter region; 2) bypassing epigenetic modifications such as CpG methylation that may prevent MuRF-1 expression in HEK cells; and 3) eliminating effects of secondary and tertiary DNA structure on DNA binding. Data are expressed relative to the amount of DNA in a nonprecipitated (i.e., nonantibody treated) aliquot of the same sample (Input). All chromatin immunoprecipitation assays (starting from cell culture/transfections) were repeated at least twice with the same pattern of response observed each time.
Western blotting.
Cells were washed twice with PBS and protein was collected using freshly made radioimmunoprecipitation assay (RIPA) buffer containing 1% NP-40, 0.5% sodium deoxycholate, 0.1% SDS, and protease inhibitor cocktail (Roche Complete). Samples were vortexed for 1 min, rotated end-over-end for 30 min, then centrifuged at 12,000 g for 10 min, and the pellet was discarded. Proteins were separated by SDS-PAGE, transferred to a polyvinylidene difluoride membrane, and incubated with primary antibodies against FoxO3 (no. 2497; Cell Signaling, 1:1,000), SMAD3 (no. 9523; Cell Signaling, 1:5,000), β-tubulin (no. 2128; Cell Signaling, 1:5,000), or FLAG (no. F1804; Sigma-Aldrich, 1:3,000) overnight. Secondary antibodies were conjugated to horseradish peroxidase and detected using an ECL substrate (Millipore). Band imaging and analysis were performed using a Bio-Rad Chemi Doc XRS and Image Lab software.
Statistics.
Data are presented as means ± SE. Statistical significance (P < 0.05) was assessed using ANOVA followed by Tukey's honestly significant difference post hoc test when appropriate. All analyses were performed using SPSS software (version 19.0).
RESULTS
Starvation increases protein degradation rate and MuRF-1 gene expression in muscle cells.
To demonstrate the response of ubiquitin ligases to muscle atrophy, we measured protein degradation rate and gene expression of Atrogin-1 and MuRF-1 in primary HSkM myotubes and mouse myotubes (C2C12) in response to atrophic stimuli. Serum starvation or combined serum and amino acid starvation for 24 h increased the rate of protein degradation in both human and mouse cells (Fig. 1, A and B). Likewise, these conditions increased gene expression of both Atrogin-1 (Fig. 1, C and D) and MuRF-1 (Fig. 1, E and F). Thus increased gene expression of Atrogin-1 and MuRF-1 is a robust response to atrophy stimulation in skeletal muscle cells. This finding supports previous research (4, 16) and suggests that increased transcription of these muscle-specific ubiquitin ligases is a major component of increased protein degradation and muscle atrophy in skeletal muscle.
Fig. 1.

Atrophy stimulation increases the rate of protein degradation and increases the expression of Atrogin-1 and MuRF-1 in cultured myotubes. Primary human skeletal muscle (HSkM) (A, C, and E) or mouse skeletal myoblast (C2C12) (B, D, and F) cells were cultured in media lacking serum or lacking both serum and amino acids (AA). For HSkM experiments, four independent pools, each containing myotubes from three or four women, were tested on separate occasions. A and B: protein degradation rate was determined by [3H]tyrosine release over the course of 5 h. C and D: Atrogin-1 gene expression. E and F: MuRF-1 gene expression. *Significantly different from basal (P < 0.05). †Significantly different from no serum (P < 0.05). n = 4 per experiment.
SMAD3 and FoxO3 synergistically increase Atrogin-1 and MuRF-1 expression.
Since both FoxO3 and SMAD3 are major transcriptional regulators of muscle protein degradation, we hypothesized that these transcription factors act synergistically to increase transcription of Atrogin-1 and MuRF-1. To test this hypothesis, we coinfected C2C12 myotubes with ad-SMAD3 and ad-FoxO3 and then measured Atrogin-1 and MuRF-1 mRNA levels. C2C12 myotubes were used owing to the fact that we observed more consistent infection efficiency than HSkM myotubes. As shown in Fig. 2, FoxO3 but not SMAD3 significantly increased gene expression of Atrogin-1 (Fig. 2A) and MuRF-1 (Fig. 2B). Coexpression of FoxO3 and SMAD3 increased Atrogin-1 and MuRF-1 gene expression to a greater extent than FoxO3 alone. None of the treatment conditions significantly altered expression of the normalizing gene, RPLPO. These data indicate that FoxO3 and SMAD3 synergistically increase gene expression of Atrogin-1 and MuRF-1.
Fig. 2.

FoxO and SMAD3 synergistically increase Atrogin-1 and MuRF-1 mRNA levels. C2C12 myotubes were infected with adenoviruses encoding SMAD3 and/or FoxO3, RNA was collected 24 h later, and RT-PCR was performed. A: Atrogin-1 gene expression. B: MuRF-1 gene expression. GFP, green fluorescent protein. *Significantly different from basal (P < 0.05). †Significantly different from FoxO only (P < 0.05). n = 4 per experiment.
Examining the promoter regions of Atrogin-1 and MuRF-1 in the mouse genome revealed that putative SBEs occur within close proximity (9–31 bp) of consensus FREs within the proximal promoter regions of both genes (FRE-SBE motif). The spacing between these binding sites is similar to that found for other genes regulated by both SMAD and FoxO transcription factors (1, 10). In the mouse Atrogin-1 promoter region, we found three such FRE-SBE motifs within 1.5 kb upstream of the transcriptional start site (TSS). However, we did not find any putative FRE-SBE motifs within the human Atrogin-1 promoter region. Likewise, in the mouse MuRF-1 promoter region, we identified three FRE-SBE motifs within 1.5 kb of the TSS, located 1.3 kb, 0.2 kb, and 2 bp upstream of the TSS. We identified two FRE-SBE motifs in the human MuRF-1 promoter region (0.2 kb and 2 bp upstream of the TSS). Alignment of the promoter regions of the mouse and human MuRF-1 promoter regions (TRIM62) revealed that distal regions are highly divergent and no FRE-SBE is found in the human promoter region near 1.3 kb upstream of the TSS. However, the proximal 0.25-kb sequence is exceptionally conserved (83% homology) (Fig. 3A), including a 38-bp sequence containing the FRE-SBE motif at 2 bp upstream from the TSS that is identical between mice and humans. Due to the high degree of sequence homology within the proximal promoter region and the presence of a conserved FRE-SBE motif, we focused on the mouse MuRF-1 promoter region to determine the mechanistic action mediating the synergistic effects of SMAD3 and FoxO3.
Fig. 3.

The proximal 0.25 kb of the MuRF-1 promoter is highly conserved and accounts for over one-third of basal transcriptional activity. A: the 0.25-kb reporter sequence is highly conserved between human and mouse genomes. The FoxO response element (FRE; black box) and SMAD-binding elements (SBE; gray boxes) of the FRE-SBE motif appear in bold. B: human embryonic kidney cells (HEK293a) were transfected with plasmid DNA encoding a 5-kb MuRF-1 promoter reporter, SMAD3, FoxO1, and/or FoxO3. Protein was collected 48 h later and luciferase activity was measured. *Significantly different from basal (P < 0.05). †Significantly different from respective FoxO only (P < 0.05). C: schematic of mouse MuRF-1 promoter reporters generated for the current study. D: basal luciferase activity of the reporters transfected in HEK293a cells. *Significantly different from 5-kb wild-type (wt). n = 4 per experiment.
To determine whether the synergistic effect of SMAD3 and FoxO is mediated by increased MuRF-1 promoter activity, we cotransfected HEK cells with a 5-kb wtMuRF-1 reporter, FoxO1, FoxO3, and/or SMAD3 plasmid DNA [either alone or in combination (Fig. 3B)]. Use of HEK cells eliminates any additional muscle-specific factors that may affect MuRF-1 gene transcription and thus provides a specific model to study the effects of these transcription factors in the regulation of muscle-specific genes. Overexpression of FoxO3, but unexpectedly not FoxO1, significantly increased MuRF-1 promoter activity. As previously reported (11, 27), overexpression of SMAD3 had no effect on MuRF-1 promoter activity. Coexpression of FoxO1 + SMAD3 or FoxO3 + SMAD3 increased MuRF-1 gene transcription to a greater extent than FoxO1 or FoxO3 alone, indicating a synergistic effect of these transcription factors on MuRF-1 promoter activity.
Mutation of the proximal SMAD-binding site does not affect MuRF-1 basal transcriptional activity.
To determine the contribution of the SMAD binding motif to MuRF-1 promoter activity, we created a series of luciferase reporters through site-directed mutagenesis of the putative SMAD binding region (SBE-mut, AGACCAAGAC → AGggCAAtcC) and restriction digestion (5, 2, and 0.25 kb) of the 5-kb MuRF-1 luciferase reporter (Fig. 3C). These reporters were cotransfected into HEK cells and promoter activity was measured (Fig. 3D). The wild-type 2-kb reporter retained 100% of basal activity, whereas the wild-type 0.25-kb reporter retained 37% of basal promoter activity. Therefore, the highly conserved proximal 0.25-kb promoter region, representing only 5% of the length of the 5-kb region, accounts for a disproportionately greater amount of basal MuRF-1 promoter activity. Importantly, the SBE-mut reporters (5, 2, and 0.25 kb) retained basal promoter activity similar to their respective wild-type reporters.
SMAD3 augments FoxO3-induced MuRF-1 promoter activity in a DNA-binding-dependent manner.
To determine the mechanism of the synergistic effect of FoxO3 and SMAD3 on MuRF-1 promoter activity, we cotransfected HEK cells with the 0.25-kb MuRF-1 reporter (wild-type or SBE-mut), FoxO3 and/or SMAD3. As expected, overexpression of FoxO3 significantly increased MuRF-1 promoter activity in the wild-type reporter (Fig. 4A). However, this activity was significantly attenuated in the SBE-mut reporter. Similar results were found with overexpression of FoxO1 (data not shown). Unlike the 5-kb reporter, overexpression of SMAD3 alone significantly increased promoter activity in both the wild-type and SBE-mut 0.25-kb reporters. Furthermore, coexpression of FoxO3 and SMAD3 did not significantly increase MuRF-1 gene transcription compared with FoxO3 alone in either the wild-type or SBE-mut reporters, which may be due to elimination of upstream FREs from the MuRF-1 promoter region. To confirm these results, we overexpressed dnSMAD3 (lacking the DNA binding region) alone or in conjunction with FoxO3. Despite dnSMAD3 and wtSMAD3 proteins being expressed to a similar degree (Fig. 4B), overexpression of dnSMAD3 did not significantly increase MuRF-1 promoter activity, and it significantly attenuated FoxO3-induced MuRF-1 promoter activity. On the basis of these results, we hypothesized that SMAD3 binding to DNA augments and is required for optimal FoxO binding to the MuRF-1 promoter regions at FRE-SBE motifs.
Fig. 4.
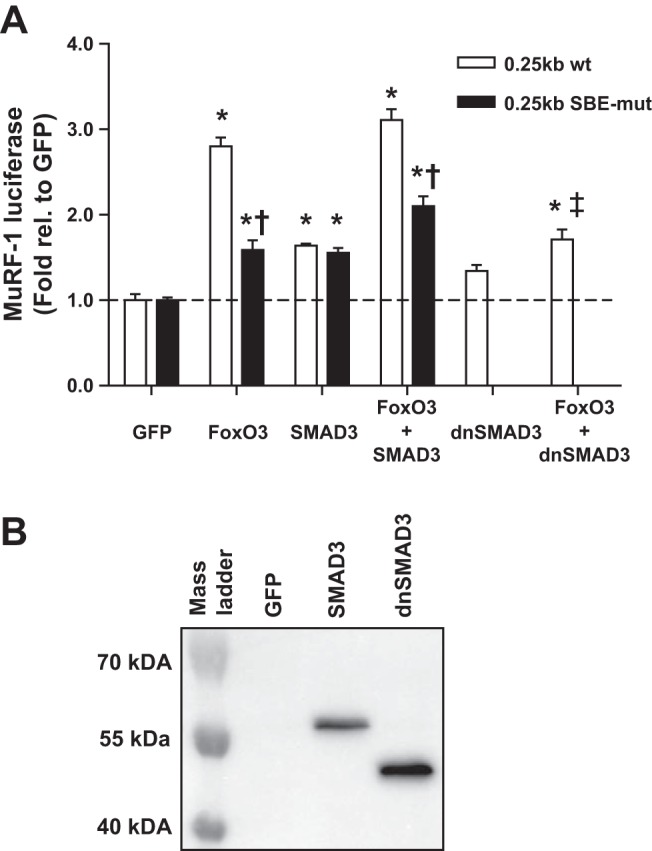
SMAD3 enhances FoxO3-induced MuRF-1 promoter activity in a DNA binding-dependent manner. HEK293a cells were transfected with plasmid DNA encoding a 0.25-kb (wild-type or SBE-mut) MuRF-1 reporter, FoxO3, wtSMAD3, and/or dominant-negative (dn)SMAD3. A: luciferase activity was measured 48 h after transfection. Data were normalized to the respective GFP control. No differences were noted in basal luciferase activity between the wild-type and SBE-mut reporters. B: representative Western blots for FLAG-tagged SMAD3 and dnSMAD3 transfected cells using anti-FLAG primary antibody. *Significantly different from basal (P < 0.05). †Significantly different from wild-type (P < 0.05). ‡Significantly different from FoxO3 alone. n = 4 per experiment.
To determine how SMAD3 affects FoxO3-DNA interactions, we performed chromatin immunoprecipitation of the 0.25-kb MuRF-1 reporter. As expected, SMAD3 overexpression increased SMAD3 binding within the wild-type (Fig. 5A) but not the SBE-mut (Fig. 5B) reporter. Interestingly, coexpression of FoxO3 and SMAD3 dramatically increased SMAD3 binding to DNA in both the wild-type and SBE-mut reporters (Fig. 5, A and B), suggesting that SMAD3 directly interacts with FoxO3 as well as SBEs within the FRE-SBE motif of the proximal promoter region. Overexpression of FoxO3 and, to a lesser extent, SMAD3, independently increased FoxO3 binding within the wild-type reporter (Fig. 5C). In the SBE-mut reporter, FoxO3 overexpression, but not SMAD3 overexpression, increased FoxO3-DNA binding (Fig. 5D). Mutation of the SBE greatly attenuated FoxO3 abundance on the proximal MuRF-1 promoter region (Fig. 5, C and D, note difference in y-axes scale). Coexpression of FoxO3 and SMAD3 increased FoxO3 binding to DNA to a greater extent than overexpression of FoxO3 alone in both the wild-type and SBE-mut reporters (Fig. 5, C and D).
Fig. 5.

SMAD3 augments FoxO3 binding within the proximal promoter region of MuRF-1. HEK293a cells were transfected with plasmid DNA encoding the 0.25-kb MuRF-1 reporter (wild-type or SBE-mut), SMAD3 and/or FoxO3, and chromatin immunoprecipitation was performed. A and B: SMAD3 binding to the wild-type and SBE-mut reporters. C and D: FoxO3 binding to the wild-type and SBE-mut reporters. E and F: IgG (control antibody) binding to the wild-type and SBE-mut reporters. Note the differential y-axes scale bars between A, C, E and B, D, F. Results from this experiment were confirmed on two independent occasions.
Overexpression of SMAD3 increases FoxO3 expression and transcriptional activity.
As indicated above, overexpression of dnSMAD3, which lacks the DNA binding region, was insufficient to induce MuRF-1 gene transcription (Fig. 4A), suggesting that SMAD3-induced MuRF-1 gene transcription is dependent on DNA binding. However, SMAD3 overexpression was sufficient to induce MuRF-1 promoter activity in the SBE-mut 0.25-kb MuRF-1 reporter (Fig. 4A) despite the fact that SMAD binding to the FRE-SBE motif was completely abolished in the SBE-mut reporter (Fig. 5B). Together these data suggest that SMAD3-induced MuRF-1 promoter activity may be dependent on DNA binding at sites outside of the proximal promoter region of MuRF-1. However, SMAD transcription factors have weak affinity for DNA without a DNA-binding cofactor (30). Because FoxO3 is an established SMAD3 partner and FoxO3 is a major regulator of MuRF-1 gene transcription, we hypothesized that SMAD3-induced MuRF-1 gene expression is mediated by increased FoxO expression. To test this hypothesis, we performed immunoblots for FoxO3 in HEK cells transfected with SMAD3 and found increased FoxO3 protein content (images not shown). Furthermore, FoxO3 protein content increased in a dose-dependent manner in C2C12 myotubes infected with increasing doses of ad-wtSMAD3 (Fig. 6A). However, SMAD3 overexpression was insufficient to induce FoxO1 or FoxO3 mRNA (Fig. 6B).
Fig. 6.
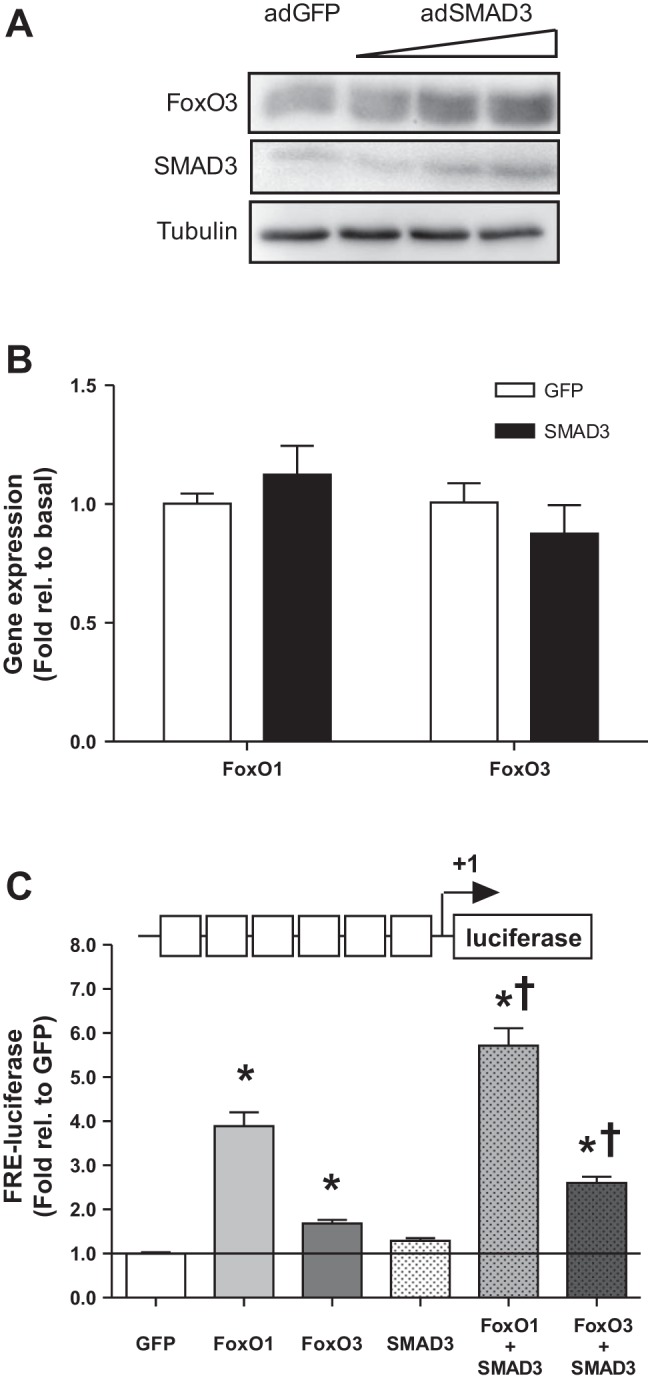
SMAD3 overexpression increases protein amount and transcriptional activity of FoxO3. A: C2C12 myotubes were infected with increasing dosages of adenovirus encoding SMAD3 (adSMAD3) for 24 h, protein was collected, and Western blotting was performed for total FoxO3 protein. B: FoxO3 mRNA was determined by RT-PCR in adSMAD3-infected C2C12 myotubes. C: HEK293a cells were transfected with plasmid DNA encoding an FRE luciferase reporter, SMAD3, FoxO1, and/or FoxO3. Protein was collected following 48 h of transfection and luciferase activity was measured. *Significantly different from basal (P < 0.05). †Significantly different from FoxO alone (P < 0.05). n = 4 per experiment.
To determine whether SMAD3-induced increases in FoxO protein content affect global FoxO-dependent gene transcription, we cotransfected HEK293a cells with plasmid DNA encoding an FRE reporter and wtFoxO1, wtFoxO3, or wtSMAD3, either alone or in combination. As shown in Fig. 6C, both FoxO1 and FoxO3 independently increased FRE-luciferase activity. Interestingly, although wtSMAD3 alone was insufficient to induce FRE-luciferase activity (1.29-fold; P = 0.14), coexpression of FoxO1 + wtSMAD3 or FoxO3 + SMAD3 increased FRE-luciferase activity to a greater extent than FoxO1 or FoxO3 alone.
DISCUSSION
The major finding of the present study is that SMAD3 synergistically augments FoxO-induced expression of muscle-specific ubiquitin ligases Atrogin-1 and MuRF-1 by increasing FoxO binding to DNA at FRE-SBE motifs within the promoter region of these genes. Although a few studies have found that these transcription factors coordinately regulate gene expression (1, 10, 29), to our knowledge, this is the first study to demonstrate a synergistic effect of these transcription factors on transcription of E3 ubiquitin ligases. Our data indicate that two major signaling pathways (IGF-1/Akt/FoxO and TGF-β/myostatin/SMAD) regulating skeletal muscle mass converge to synergistically upregulate genes involved in ubiquitin proteasome system (UPS)-mediated protein degradation.
Both FoxO and SMAD transcription factors are known to play a role in modulating protein degradation (17, 26, 27, 36). FoxO transcription factors upregulate transcription of both Atrogin-1 (5, 26) and MuRF-1 (5, 34) by binding the FRE within the promoter regions of these genes, but until now, the role of SMAD3 has been less clear. Goodman et al. (11) showed that SMAD3 was sufficient to induce expression of Atrogin-1 and induce muscle atrophy, but was insufficient to induce MuRF-1 gene transcription. Our data confirm that SMAD3 overexpression is insufficient to increase 5-kb MuRF-1 promoter activity or MuRF-1 mRNA content, but demonstrate that SMAD3 directly augments FoxO-induced gene expression of Atrogin-1 and MuRF-1. Genetic knockdown of FoxO (24, 28, 31) or myostatin, an upstream activator of SMAD3 (9, 13), prevents muscle atrophy in numerous chronic conditions. Because both Atrogin-1 and MuRF-1 are required for rapid muscle atrophy (4), it is possible that disrupting the synergistic effects of FoxO and SMAD3 may preserve muscle mass during cachectic conditions by preventing upregulation of these ubiquitin ligases.
Furthermore, we have demonstrated that SMAD3 binding to the FRE-SBE motif is essential for optimal FoxO-induced MuRF-1 promoter activity. Overexpression of a dnSMAD3 or a mutation of the proximal SBE elements of the MuRF-1 reporter significantly attenuated FoxO-induced MuRF-1 promoter activity (see Fig. 4A). Additionally, FoxO3-DNA binding was dramatically reduced in the SBE-mut reporter (see Fig. 5, C and D). Interestingly, SMAD3-DNA binding increased with coexpression of FoxO3 and SMAD3, even in the SBE-mut reporter (see Fig. 5, A and B), indicating that SMAD3 directly interacts with FoxO3. These data suggest that FoxO and SMAD3 colocalize at FRE-SBE motifs thus increasing the stability of both transcription factors at these sites and augmenting each other's transcriptional activity. This presents a model in which SMAD3 directly interacts with both FoxO3 and DNA at the FRE-SBE motif to stabilize FoxO3 DNA interaction, thereby augmenting FoxO3-induced gene transcription. It has previously been shown that FoxO and SMAD transcription factors physically associate with each other (29) and that FREs and SBEs lie within close proximity within promoter regions of genes regulated by both FoxO and SMAD3 (10). Therefore, it is possible that disrupting SMAD3-DNA interactions may attenuate FoxO-dependent gene transcription by decreasing FoxO3 affinity for FREs.
In contrast to the present findings, Lokireddy et al. (17) demonstrated that myostatin administration increased UPS-mediated protein degradation in a SMAD3-dependent manner, but SMAD3 knockdown had no effect on myostatin-induced MuRF-1 expression. Because myostatin can activate both SMAD2 and SMAD3 (33), it is possible that SMAD2 and SMAD3 have redundant roles in regulating MuRF-1 gene expression and that knockdown of both of these transcription factors may be necessary to decrease myostatin-induced MuRF-1 expression. This is supported by the fact that the DNA recognition sequence is conserved between SMAD isoforms (20).
In our analysis of the Atrogin-1 and MuRF-1 gene sequences, we identified FRE-SBE motifs in the proximal promoter region of both genes. In the mouse genome, three such motifs are found within the proximal 1.5 kb of both genes. In the human genome, we did not find any putative FRE-SBE motifs within the proximal 2-kb Atrogin-1 promoter region. Conversely, we found two such sequences (0.2 kb and 2 bp upstream of the TSS) within the proximal MuRF-1 promoter region. It is important to note that the present study used primarily the mouse MuRF-1 promoter region. Since the proximal 0.25 kb is 83% conserved and the proximal FRE-SBE motifs are 100% conserved between mouse and human, we expect the mechanistic synergism between FoxO and SMAD3 transcription factors is retained in humans. However, because of divergent distal regions of the promoters, we cannot discount additional regulatory mechanisms that differ between species.
In addition to Atrogin-1 and MuRF-1, FoxO3 is known to bind to the promoter region and induce expression of LC3b, Gabarapl1, and Atg12l, genes involved in degrading proteins via the autophagic/lysosomal pathway (36). Interestingly, LC3b, Gabarapl1, and Atg12l all have at least one putative SBE within 100 bp of these verified FRE regions. This raises the possibility that the synergistic effect of FoxO and SMAD3 on gene expression may be a general mechanism that regulates both major protein degradative systems of skeletal muscle atrophy, the UPS and the autophagic/lysosomal pathway. Future studies should aim to determine which specific genes are coordinately regulated by FoxO and SMAD3 in this manner.
In addition to augmenting FoxO-induced MuRF-1 gene transcription, we demonstrated that SMAD3 increases FoxO protein content and global FoxO-induced transcriptional activity. Protein content of FoxO1 and FoxO3 has been shown to increase following myostatin administration in HSkM myotubes (18) and SMAD3 inhibition may prevent this effect. In the present study, the whole cell amount of FoxO3 protein increased with increasing dosages of SMAD3 (Fig. 6A). However, the precise mechanism of this effect is not known. It has previously been suggested that SMAD3 directly induces gene transcription of FoxO1 (18). However, in the present study, we found no increase in FoxO1 or FoxO3 mRNA content after SMAD3 overexpression, indicating that SMAD3 increases FoxO protein content downstream of gene transcription or mRNA stability. The activity of FoxO transcription factors is also determined by intracellular localization (nuclear vs. cytosolic) by phosphorylation by Akt (32). Several studies have suggested that SMAD3 decreases Akt-mediated phosphorylation (inhibition) of FoxO (2, 21, 33), which would be expected to increase global FoxO-dependent transcriptional activity. However, in the present study, increased total FoxO protein presumably did not lead to increased functional FoxO in the nucleus because the activity of the FoxO DNA binding reporter was not increased with SMAD3 overexpression (Fig. 6C). Therefore, the increase in FoxO appears to be restricted to the cytosol. The role in differing levels of Akt activity on the synergistic effects of FoxO3 and SMAD3 remain to be tested.
In summary, our data indicate that SMAD3 synergistically increases, and is essential for, FoxO-induced transcription of ubiquitin ligases through a dualistic mechanism. SMAD3 increases FoxO protein content and directly interacts with DNA at the FRE-SBE motif within the proximal promoter region and stabilizes FoxO binding to enhance its transcriptional activity. These data indicate that two major transcription factors regulating protein degradation converge at the proximal promoter region to induce gene transcription of MuRF-1.
GRANTS
This work was supported by start-up funds from East Carolina University (to J. J. Brault), National Institutes of Health Grant R00 AR-05629 (National Institute of Arthritis and Musculoskeletal and Skin Diseases), start-up funds from East Carolina University (to C. A. Witczak), National Institutes of Health/National Institute of Diabetes and Digestive and Kidney Disease Grant R01 DK-056112 (to J. A. Houmard), and a doctoral grant from the American College of Sport Medicine Foundation (to L. M. Bollinger).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
AUTHOR CONTRIBUTIONS
Author contributions: L.M.B., J.A.H., and J.J.B. conception and design of research; L.M.B. and J.J.B. performed experiments; L.M.B. and J.J.B. analyzed data; L.M.B., C.A.W., J.A.H., and J.J.B. interpreted results of experiments; L.M.B. and J.J.B. prepared figures; L.M.B. drafted manuscript; L.M.B., C.A.W., J.A.H., and J.J.B. edited and revised manuscript; L.M.B., C.A.W., J.A.H., and J.J.B. approved final version of manuscript.
ACKNOWLEDGMENTS
We thank Sanghee Park for excellent technical contributions.
Present address of L. M. Bollinger: Department Kinesiology and Health Promotion, University of Kentucky, Lexington, KY 40506.
REFERENCES
- 1.Allen DL, Unterman TG. Regulation of myostatin expression and myoblast differentiation by FoxO and SMAD transcription factors. Am J Physiol Cell Physiol 292: C188–C199, 2007 [DOI] [PubMed] [Google Scholar]
- 2.Amirouche A, Durieux AC, Banzet S, Koulmann N, Bonnefoy R, Mouret C, Bigard X, Peinnequin A, Freyssenet D. Down-regulation of Akt/mammalian target of rapamycin signaling pathway in response to myostatin overexpression in skeletal muscle. Endocrinology 150: 286–294, 2009 [DOI] [PubMed] [Google Scholar]
- 3.Berggren JR, Tanner CJ, Houmard JA. Primary cell cultures in the study of human muscle metabolism. Exerc Sport Sci Rev 35: 56–61, 2007 [DOI] [PubMed] [Google Scholar]
- 4.Bodine SC, Latres E, Baumhueter S, Lai VK, Nunez L, Clarke BA, Poueymirou WT, Panaro FJ, Na E, Dharmarajan K, Pan ZQ, Valenzuela DM, DeChiara TM, Stitt TN, Yancopoulos GD, Glass DJ. Identification of ubiquitin ligases required for skeletal muscle atrophy. Science 294: 1704–1708, 2001 [DOI] [PubMed] [Google Scholar]
- 5.Brault JJ, Jespersen JG, Goldberg AL. Peroxisome proliferator-activated receptor gamma coactivator 1alpha or 1beta overexpression inhibits muscle protein degradation, induction of ubiquitin ligases, and disuse atrophy. J Biol Chem 285: 19460–19471, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Calura E, Cagnin S, Raffaello A, Laveder P, Lanfranchi G, Romualdi C. Meta-analysis of expression signatures of muscle atrophy: gene interaction networks in early and late stages. BMC Genomics 9: 630, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cohen S, Brault JJ, Gygi SP, Glass DJ, Valenzuela DM, Gartner C, Latres E, Goldberg AL. During muscle atrophy, thick, but not thin, filament components are degraded by MuRF1-dependent ubiquitylation. J Cell Biol 185: 1083–1095, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Csibi A, Leibovitch MP, Cornille K, Tintignac LA, Leibovitch SA. MAFbx/Atrogin-1 controls the activity of the initiation factor eIF3-f in skeletal muscle atrophy by targeting multiple C-terminal lysines. J Biol Chem 284: 4413–4421, 2009 [DOI] [PubMed] [Google Scholar]
- 9.Gilson H, Schakman O, Combaret L, Lause P, Grobet L, Attaix D, Ketelslegers JM, Thissen JP. Myostatin gene deletion prevents glucocorticoid-induced muscle atrophy. Endocrinology 148: 452–460, 2007 [DOI] [PubMed] [Google Scholar]
- 10.Gomis RR, Alarcón C, He W, Wang Q, Seoane J, Lash A, Massagué J. A FoxO-Smad synexpression group in human keratinocytes. Proc Natl Acad Sci USA 103: 12747–12752, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Goodman CA, McNally RM, Hoffmann FM, Hornberger TA. Smad3 induces Atrogin-1, inhibits mTOR and protein synthesis, and promotes muscle atrophy in vivo. Mol Endocrinol 27: 1946–1957, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Han HQ, Zhou X, Mitch WE, Goldberg AL. Myostatin/activin pathway antagonism: molecular basis and therapeutic potential. Int J Biochem Cell Biol 45: 2333–2347, 2013 [DOI] [PubMed] [Google Scholar]
- 13.Heineke J, Auger-Messier M, Xu J, Sargent M, York A, Welle S, Molkentin JD. Genetic deletion of myostatin from the heart prevents skeletal muscle atrophy in heart failure. Circulation 121: 419–425, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jagoe RT, Goldberg AL. What do we really know about the ubiquitin-proteasome pathway in muscle atrophy? Curr Opin Clin Nutr Metab Care 4: 183–190, 2001 [DOI] [PubMed] [Google Scholar]
- 15.Labbé E, Silvestri C, Hoodless PA, Wrana JL, Attisano L. Smad2 and Smad3 positively and negatively regulate TGF beta-dependent transcription through the forkhead DNA-binding protein FAST2. Mol Cell 2: 109–120, 1998 [DOI] [PubMed] [Google Scholar]
- 16.Lecker SH, Jagoe RT, Gilbert A, Gomes M, Baracos V, Bailey J, Price SR, Mitch WE, Goldberg AL. Multiple types of skeletal muscle atrophy involve a common program of changes in gene expression. FASEB J 18: 39–51, 2004 [DOI] [PubMed] [Google Scholar]
- 17.Lokireddy S, McFarlane C, Ge X, Zhang H, Sze SK, Sharma M, Kambadur R. Myostatin induces degradation of sarcomeric proteins through a Smad3 signaling mechanism during skeletal muscle wasting. Mol Endocrinol 25: 1936–1949, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 18.Lokireddy S, Mouly V, Butler-Browne G, Gluckman PD, Sharma M, Kambadur R, McFarlane C. Myostatin promotes the wasting of human myoblast cultures through promoting ubiquitin-proteasome pathway-mediated loss of sarcomeric proteins. Am J Physiol Cell Physiol 301: C1316–C1324, 2011 [DOI] [PubMed] [Google Scholar]
- 19.Lokireddy S, Wijesoma IW, Sze SK, McFarlane C, Kambadur R, Sharma M. Identification of atrogin-1-targeted proteins during the myostatin-induced skeletal muscle wasting. Am J Physiol Cell Physiol 303: C512–C529, 2012 [DOI] [PubMed] [Google Scholar]
- 20.Massagué J, Seoane J, Wotton D. Smad transcription factors. Genes Dev 19: 2783–2810, 2005 [DOI] [PubMed] [Google Scholar]
- 21.Morissette MR, Cook SA, Buranasombati C, Rosenberg MA, Rosenzweig A. Myostatin inhibits IGF-I-induced myotube hypertrophy through Akt. Am J Physiol Cell Physiol 297: C1124–C1132, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Raffaello A, Laveder P, Romualdi C, Bean C, Toniolo L, Germinario E, Megighian A, Danieli-Betto D, Reggiani C, Lanfranchi G. Denervation in murine fast-twitch muscle: short-term physiological changes and temporal expression profiling. Physiol Genomics 25: 60–74, 2006 [DOI] [PubMed] [Google Scholar]
- 23.Ramaswamy S, Nakamura N, Sansal I, Bergeron L, Sellers WR. A novel mechanism of gene regulation and tumor suppression by the transcription factor FKHR. Cancer Cell 2: 81–91, 2002 [DOI] [PubMed] [Google Scholar]
- 24.Reed SA, Sandesara PB, Senf SM, Judge AR. Inhibition of FoxO transcriptional activity prevents muscle fiber atrophy during cachexia and induces hypertrophy. FASEB J 26: 987–1000, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sacheck JM, Hyatt JP, Raffaello A, Jagoe RT, Roy RR, Edgerton VR, Lecker SH, Goldberg AL. Rapid disuse and denervation atrophy involve transcriptional changes similar to those of muscle wasting during systemic diseases. FASEB J 21: 140–155, 2006 [DOI] [PubMed] [Google Scholar]
- 26.Sandri M, Sandri C, Gilbert A, Skurk C, Calabria E, Picard A, Walsh K, Schiaffino S, Lecker SH, Goldberg AL. Foxo transcription factors induce the atrophy-related ubiquitin ligase atrogin-1 and cause skeletal muscle atrophy. Cell 117: 399–412, 2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sartori R, Milan G, Patron M, Mammucari C, Blaauw B, Abraham R, Sandri M. Smad2 and 3 transcription factors control muscle mass in adulthood. Am J Physiol Cell Physiol 296: C1248–C1257, 2009 [DOI] [PubMed] [Google Scholar]
- 28.Senf SM, Dodd SL, Judge AR. FOXO signaling is required for disuse muscle atrophy and is directly regulated by Hsp70. Am J Physiol Cell Physiol 298: C38–C45, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Seoane J, Le HV, Shen L, Anderson SA, Massagué J. Integration of Smad and forkhead pathways in the control of neuroepithelial and glioblastoma cell proliferation. Cell 117: 211–223, 2004 [DOI] [PubMed] [Google Scholar]
- 30.Shi Y, Wang YF, Jayaraman L, Yang H, Massagué J, Pavletich NP. Crystal structure of a Smad MH1 domain bound to DNA: insights on DNA binding in TGF-beta signaling. Cell 94: 585–594, 1998 [DOI] [PubMed] [Google Scholar]
- 31.Stitt TN, Drujan D, Clarke BA, Panaro F, Timofeyva Y, Kline WO, Gonzalez M, Yancopoulos GD, Glass DJ. The IGF-1/PI3K/Akt pathway prevents expression of muscle atrophy-induced ubiquitin ligases by inhibiting FOXO transcription factors. Mol Cell 14: 395–403, 2004 [DOI] [PubMed] [Google Scholar]
- 32.Tang ED, Nuñez G, Barr FG, Guan KL. Negative regulation of the forkhead transcription factor FKHR by Akt. J Biol Chem 274: 16741–16746, 1999 [DOI] [PubMed] [Google Scholar]
- 33.Trendelenburg AU, Meyer A, Rohner D, Boyle J, Hatakeyama S, Glass DJ. Myostatin reduces Akt/TORC1/p70S6K signaling, inhibiting myoblast differentiation and myotube size. Am J Physiol Cell Physiol 296: C1258–C1270, 2009 [DOI] [PubMed] [Google Scholar]
- 34.Waddell DS, Baehr LM, den Brandt van J, Johnsen SA, Reichardt HM, Furlow JD, Bodine SC. The glucocorticoid receptor and FOXO1 synergistically activate the skeletal muscle atrophy-associated MuRF1 gene. Am J Physiol Endocrinol Metab 295: E785–E797, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhang Y, Feng X, We R, Derynck R. Receptor-associated Mad homologues synergize as effectors of the TGF-beta response. Nature 383: 168–172, 1996 [DOI] [PubMed] [Google Scholar]
- 36.Zhao J, Brault JJ, Schild A, Cao P, Sandri M, Schiaffino S, Lecker SH, Goldberg AL. FoxO3 coordinately activates protein degradation by the autophagic/lysosomal and proteasomal pathways in atrophying muscle cells. Cell Metab 6: 472–483, 2007 [DOI] [PubMed] [Google Scholar]