

Abstract
At birth, asphyxial stressors such as hypoxia and hypercapnia are important physiological stimuli for adrenal catecholamine release that is critical for the proper transition to extrauterine life. We recently showed that chronic opioids blunt chemosensitivity of neonatal rat adrenomedullary chromaffin cells (AMCs) to hypoxia and hypercapnia. This blunting was attributable to increased ATP-sensitive K+ (KATP) channel and decreased carbonic anhydrase (CA) I and II expression, respectively, and involved μ- and δ-opioid receptor signaling pathways. To address underlying molecular mechanisms, we first exposed an O2- and CO2-sensitive, immortalized rat chromaffin cell line (MAH cells) to combined μ {[d-Arg2,Ly4]dermorphin-(1–4)-amide}- and δ ([d-Pen2,5,P-Cl-Phe4]enkephalin)-opioid agonists (2 μM) for ∼7 days. Western blot and quantitative real-time PCR analysis revealed that chronic opioids increased KATP channel subunit Kir6.2 and decreased CAII expression; both effects were blocked by naloxone and were absent in hypoxia-inducible factor (HIF)-2α-deficient MAH cells. Chronic opioids also stimulated HIF-2α accumulation along a time course similar to Kir6.2. Chromatin immunoprecipitation assays on opioid-treated cells revealed the binding of HIF-2α to a hypoxia response element in the promoter region of the Kir6.2 gene. The opioid-induced regulation of Kir6.2 and CAII was dependent on protein kinase A, but not protein kinase C or calmodulin kinase, activity. Interestingly, a similar pattern of HIF-2α, Kir6.2, and CAII regulation (including downregulation of CAI) was replicated in chromaffin tissue obtained from rat pups born to dams exposed to morphine throughout gestation. Collectively, these data reveal novel mechanisms by which chronic opioids blunt asphyxial chemosensitivity in AMCs, thereby contributing to abnormal arousal responses in the offspring of opiate-addicted mothers.
Keywords: opioid, hypoxia-inducible factor-2α, adenosine 5′-triphosphate-sensitive potassium channels, carbonic anhydrase
catecholamine (CAT) secretion from adrenomedullary chromaffin cells (AMCs) in response to asphyxial stressors at birth is a crucial physiological response during the transition of the fetus to extrauterine life (18, 33). In particular, CAT secretion in the neonate aids in metabolic regulation, cardiac function, and in the transformation of the lungs into an air-breathing epithelium (18). This nonneurogenic response is triggered by the direct action of stressors such as low O2 (hypoxia) and high CO2/H+ (acid hypercapnia) on AMCs, leading to membrane depolarization, voltage-gated Ca2+ entry, and CAT secretion (23, 25, 34). These direct chemosensing mechanisms are suppressed postnatally, in parallel with the development of splanchnic innervation (after the first postnatal week in the rat), and return following denervation of adult AMCs (19, 23, 25, 31, 32, 34). Thus, it is plausible that neurochemicals released from the splanchnic nerve during innervation activate signaling cascades that lead to the suppression of hypoxia and hypercapnia chemosensitivity. In recent tests of this hypothesis, we considered the potential involvement of nicotinic cholinergic and opioid receptor signaling pathways (29), given that acetylcholine (ACh) and opiate peptides are among the presynaptic neurochemicals released from the splanchnic nerve (12, 13, 17). Interestingly, we found that exposure of neonatal rat AMCs to chronic nicotine suppressed only hypoxia chemosensitivity (6), whereas exposure to μ- and/or δ-opioid agonists led to the suppression of both hypoxia and hypercapnia sensitivity (30). In both instances, the suppression of hypoxia sensitivity was attributable to the increased expression of functional ATP-sensitive K+ (KATP) channels, which induce membrane hyperpolarization and decreased excitability during hypoxia (5, 30). On the other hand, the suppression of hypercapnia sensitivity in opioid-treated AMCs was associated with decreased expression of the CO2-hydrating enzymes carbonic anhydrase (CA) I and II (30).
In the present study, we were interested in the intracellular signaling mechanisms leading to the blunting of O2 and CO2 chemosensitivity in opioid-treated chromaffin cells, and therefore focused on factors regulating KATP channel and CA expression. As a first step, we considered the potential role of the transcription factor hypoxia-inducible factor (HIF)-2α, because: 1) HIF-2α stabilization in nicotine-treated AMCs mediated the transcriptional upregulation of KATP channel subunit Kir6.2 (28); and 2) the HIF pathway has previously been implicated in the signaling cascade activated in neuroblastoma cells during chronic opioid exposure (8). To facilitate these studies, we used a fetal-derived, immortalized rat chromaffin cell line, i.e., MAH cells, that is sensitive to both hypoxia and hypercapnia (7, 9) and express Kir6.2 and CAII mRNA and protein (5, 27, 28). Moreover, the availability of a stable HIF-2α-deficient (>90% knockdown) MAH cell line (shMAH) allowed us to investigate the role of HIF-2α in the opioid signaling cascade. Because protein kinases [e.g., protein kinase C (PKC) and calmodulin kinase (CaMK)] were implicated in the upregulation of Kir6.2 in nicotine-treated chromaffin cells (5), we also investigated their potential involvement in opioid receptor signaling in MAH cells using pharmacological inhibitors. Finally, to validate the predictions of this in vitro model, we used a physiologically relevant in vivo model where pregnant dams received daily injections of morphine just before and throughout gestation. The expression patterns of HIF-2α, Kir6.2, and CAI and -II were then compared in chromaffin tissues from the adrenal glands of neonates born to morphine- vs. saline-exposed dams. This in vivo model was of additional interest because the use of opiates during pregnancy whether for drug abuse (e.g., heroin) or replacement therapy (e.g., methadone and naltrexone) has been linked to a number of negative pregnancy outcomes associated with increased fetal and neonatal mortality, as well as the incidence of sudden infant death syndrome (SIDS) (4).
MATERIALS AND METHODS
Cell Culture
v-myc immortalized chromaffin cells.
The v-myc immortalized rat chromaffin cell line (MAH) was grown in L-15/CO2 medium containing 0.6% glucose, 1% penicillin/streptomycin, 10% fetal bovine serum, and 5 μM dexamethasone, as previously described (9). A stable HIF-2α-deficient MAH cell line (shMAH), generated using interference RNAi techniques (2), was used in some experiments and grown under similar conditions. All cultures were incubated in a humidified atmosphere of 95% air-5% CO2 at 37°C for varying periods up to ∼7 days in vitro. Cells were fed every 1–2 days and routinely passaged every 3–4 days when cell density reached ∼70% confluency. When passaging cells, medium was removed, and cells were detached using 0.25% trypsin-EDTA. Suspended cells were pelleted by centrifugation, and the pellet was resuspended in prewarmed medium. Cells were then plated on 35-mm culture dishes coated with poly-d-lysine and laminin.
Adrenal Gland Tissues
All animal experiments were approved by the Animal Research Ethics Board at McMaster University, in accordance with the guidelines of the Canadian Council for Animal Care. Nulliparous 200- to 250-g female Wistar rats (Harlan, Indianapolis, IN) were maintained under controlled lighting (12:12 light-dark) and temperature (22°C) with ad libitum access to food and water. Dams were randomly assigned (n = 10/group) to receive saline (vehicle) or morphine sulfate (Medisca Pharmaceutique, St. Laurent PQ) via subcutaneous injection. Dams were given 5 mg·kg−1·day−1 morphine for 3 days and then 10 mg·kg−1·day−1 for 4 days until mating. Control dams received the same volume of saline daily. Seven days after the initiation of treatment, dams were mated 1:1 with unexposed males. Morphine and saline administration continued throughout pregnancy until tissue collection soon after birth [i.e., postnatal day 0 (PND0)]. For each dam, litter size, litter weight, sex ratio (no. of male offspring/no. of female offspring), birth weight, and live birth index [(no. of live offspring/no. of offspring delivered) × 100] were calculated, and the number of stillbirths was recorded (Table 1). Both adrenal glands were removed from neonates (PND0) as previously described (28); most of the surrounding adrenal cortex (AC) was trimmed and isolated from the central adrenal medulla (AM) for separate molecular analysis of the two tissues.
Table 1.
Effects of chronic morphine exposure on pregnancy outcomes
| Pregnancy Outcomes | Saline (n =10) | Morphine (n =17) | P Value |
|---|---|---|---|
| Litter size, n | 13.0 ± 0.7 | 8.5 ± 0.9 | 0.004* |
| Litter wt, g | 79.4 ± 5.0 | 48.5 ± 4.9 | <0.001* |
| Birth wt, g | 6.3 ± 0.1 | 6.0 ± 0.1 | 0.123 |
| Proportion of SGA pups, % | 13.1 | 39.2 | <0.0001* |
| Live birth index, % | 100 | 84.6 ± 6.0 | 0.025* |
| Proportion of dams delivering stillborns, % | 0 | 53 | 0.0088* |
| Sex ratio (M/F) | 1.1 ± 0.1 | 2.1 ± 0.4 | 0.114 |
Values are means ± SE; n, no. of animals. SGA, small for gestational age; F, female; M, male.
P < 0.05.
Immunofluorescence
MAH cells were grown on modified Nunc 35-mm dishes with central wells to which glass cover slips were attached as previously described (6). Immunofluorescence procedures were performed as outlined in our previous study (30). Briefly, medium was removed, and cells were washed with prewarmed PBS, pH 7.2, and fixed with ice-cold 4% paraformaldehyde in PBS for 1 h at 4οC. Cells were then washed with PBS and incubated with 100 μl of primary antibodies (rabbit polyclonal anti-μ-opioid receptor; rabbit polyclonal anti-δ-opioid receptor; Alomone) diluted in 1% BSA/PBS overnight. For preadsorption control, primary antibodies were incubated in the presence of 3× excess antigen overnight at 4οC. Following incubation with primary antibodies, samples were washed the next day with PBS and incubated with FITC-conjugated secondary antibody (1:50; Jackson ImmunoResearch Laboratories, West Grove, PA) in the dark for 1 h at room temperature. Samples were then washed with PBS and covered with Vectashield to prevent photobleaching. Fluorescence visualization and images were obtained using a Zeiss inverted microscope (IM 35).
Quantitative Real-Time PCR
Quantitative real-time PCR (QPCR) analysis was performed using the Stratagene (Mx3000p) detection system and ABsolute QPCR SYBR Green Mix. Primers were designed using Gene Fisher, and specificity was confirmed using BLAST. Thermal cycling conditions included Platinum Taq DNA polymerase activation at 95°C for 2 min, 40 cycles of denaturing at 95°C for 3 s, and annealing and extension at 60°C for 30 s, followed by routine melting curve analysis. Samples with no template were used as a negative control. Data were compared quantitatively using the arithmetric equation 2−ΔΔCT (20); mRNA levels were normalized to Lamin A/C expression and expressed as transcript fold change relative to mRNA from untreated control MAH cells. Each experiment was repeated three to four times. Primers used were as follows: Kir6.2 subunit, forward: 5′-ACA AGA ACA TCC GAG AGC A-3′, reverse: 5′-CTG CAC GAT CAG AAT AAG GA-3′ (accession no. NM_008428); CAII, forward: 5′-CCG ACA GTC CCC TGT GGA-3′, reverse: 5′-GCG GAG TGG TCA GAG AGC CA-3′ (accession no. NM_010602); vascular endothelial growth factor (VEGF), forward: 5′-AATGATGAAGCCCTGGAGTG-3′, reverse: 5′-AATGCTTTCTCCGCTCTGAA-3′ (14); and lamin, forward: 5′-GCAGTACAAGAAGGAGCTA-3′, reverse: 5′-CAGCAATTCCTGGTACTCA-3′ (accession no. NM_008084).
Chromatin Immunoprecipitation Assay
Chromatin immunoprecipitation (ChIP) assay was performed using a standard protocol provided by a ChIP assay kit (Millipore) as previously described (2, 28). Briefly, MAH cells were plated on 100-mm dishes (Corning) at a confluency of ∼0.75 × 106 cells/dish and treated with opioid agonists for 7 days in culture. Following chronic treatments, the cultures were treated with 1% formaldehyde for 10 min at 37°C to cross-link histones to DNA. Medium was removed, and cells were washed with ice-cold PBS (pH 7.4) containing protease inhibitors, scraped, and pelleted at 2,000 rpm for 4 min at 4°C. The pellet was then resuspended in 200 μl SDS lysis buffer. Cross-linked DNA was sheared and pelleted at 13,000 rpm for 10 min at 4°C. The lysate supernatant was diluted 10× using the ChIP dilution buffer, and a small portion (1%) was kept for DNA quantification and used as an input control. For immunoprecipitation, lysates were precleared by the addition of protein A agarose/salmon sperm DNA (50% slurry) and maintained under constant agitation for 30 min at 4°C. Samples were incubated with rabbit polyclonal antibody against HIF-2α (Novus Biologicals) overnight at 4°C with constant rotation. For negative controls, the antibody was omitted from the samples. Following immunoprecipitation, agarose beads were pelleted and washed, and the DNA was eluted and reverse cross-linked by adding 5 M NaCl and heating at 65°C for 4 h. DNA was recovered by phenol/chloroform extraction and ethanol precipitation. PCR analysis was used to detect HIF-2α binding using primers specific for a putative hypoxia-responsive element (HRE) present in KCNJ11 (Kir6.2 subunit) sequence (forward: 5′-CGG ACT CTC AGA GCA GTG TA-3′; and reverse 5′-GCA GAC TCT GAC AGT GCC TTT-3). PCR products were sequenced, and the sequencing results were further matched to Kir6.2 gene by a BLAST search (http://blast.ncbi.nlm.nih.gov/Blast.cgi).
Western Immunoblot Analysis
MAH cell cultures and adrenal gland tissues were lysed so as to obtain cytoplasmic and nuclear fractions as previously described (3). Protein samples were boiled at 95–100°C for 5 min. Total protein concentration was determined using Bradford assay (1:5 dilution reagent and 1 mg/ml BSA). Samples were loaded and resolved on 10% SDS-PAGE and transferred onto PVDC membranes. Membranes were then washed and incubated with either primary rabbit polyclonal antibody against Kir6.2 (catalog no. APC-020; 1:1,000 dilution; Alomone Labs, Jerusalem, Israel), rabbit polyclonal anti-human CAI antibody (catalog no. Ab112522; 1:1,000 dilution; Abcam, Cambridge, MA), sheep polyclonal anti-human CAII antibody (catalog no. AHP 206; 1:1,000 dilution; AbD Serotec, Kidlington, UK), HIF-1α mouse monoclonal antibody (catalog no. NB 100–105; 1:1,000 dilution; Novus Biologicals, Littleton, CO), or HIF-2α rabbit polyclonal antibody (catalog no. NB 100–122; 1:1,000 dilution; Novus Biologicals), primary rabbit monoclonal β-actin antibody as loading control for cytoplasmic extracts (1:10,000 dilution; Millipore, Billerica, MA), or primary rabbit polyclonal TATA-binding protein antibody as a loading control for nuclear extracts (1:25,000 dilution; Santa Cruz) at 4°C overnight. Membranes were then washed in PBS and incubated in a goat anti-rabbit horseradish peroxidase-linked secondary antibody (1:10,000 dilution; Jackson Labs, Bar Harbor, ME) for 1 h at room temperature. Immunoreactions were visualized using ECL and exposed to XAR film.
Detection of Dopamine and Norepinephrine Release by ELISA
To determine dopamine (DA) and norepinephrine (NE) basal release, medium was removed from the cultures and replaced with 500 μl of extracellular (normoxic) solution containing (in mM): 110 NaCl, 5 KCl, 2 CaCl2, 1 MgCl2, 10 glucose, 12 sucrose, and 24 NaHCO3, bubbled with 5% CO2 at pH 7.4, for 15 min. For hypoxic stimulation, the extracellular solution was replaced with an equal volume of the same solution previously bubbled with N2 gas, and cells were then placed in a hypoxic incubator (2% O2; 5% CO2) for 15 min at 37°C. For isohydric hypercapnia exposure (10% CO2; pH 7.4), the extracellular bicarbonate concentration was increased to 48 mM (equimolar substitution with NaCl) so as to maintain the pH at 7.4, and cells were placed in an incubator set at 10% CO2 for 15 min at 37°C. Following hypoxic and hypercapnic stimulation, the extracellular medium was collected for determination of DA and NE release. In addition, DA and NE stores were determined after removal of the “release” medium as follows. MAH cells were gently washed with 1× PBS and lysed in 0.1 N HCl. Cells were then sonicated at 50% power with two sets of 10-s pulses. Release and store samples were then analyzed for DA and NE content using ELISA (Rocky Mountain Diagnostics) as outlined in the manufacturer's protocol.
Drugs
All drugs were purchased from Sigma-Aldrich (St. Louis, MO), except morphine sulfate, which was purchased from Medisca Pharmaceutique. In cell culture studies, fresh drugs were added to the growth medium every 2 days. For chronic opioid treatments, a combination of the μ-opioid agonist [d-Arg2,Ly4]dermorphin-(1–4)-amide (DALDA) and the δ-opioid agonist [d-Pen2,5,P-Cl-Phe4]encephalin (DPDPE) was used at a concentration of 2 μM each. In some experiments, naloxone hydrochloride dehydrate (2 μM) was used as a general opioid antagonist and added at the same time as the agonists. Protein kinase inhibitors used in this study include: 2 μM of H-89 [protein kinase A (PKA) inhibitor], 2 μM of GF-109203X (PKC inhibitor), and 3 μM of KN-62 (CaMK inhibitor).
Data Analysis
Statistical analyses were performed using GraphPad Prism (version 4.0; GraphPad). Molecular data were normalized to loading control, and results were compared using one-way ANOVA followed by Tukey's post hoc multiple-comparison test and expressed as means ± SE. For pregnancy outcomes, statistical analyses were performed using Student's t-test (SigmaStat, version 2.03; SPSS, Chicago, IL) by comparing the morphine-injected group with the control saline-injected group. Fisher's exact test (α = 0.05) was used when categorical variables were compared. Littermates were used as an experimental unit, and values are presented as means ± SE. Asterisk indicates P < 0.05.
RESULTS
In the in vitro experiments reported below, all opioid exposures were performed on immortalized chromaffin (MAH) cell cultures that were incubated with combined μ- and δ-opioid agonists, i.e., DALDA and DPDPE, respectively, at a concentration of 2 μM each. The duration of the exposure period varied from 24 h to 7 days as indicated in the text. Similar to primary neonatal rat AMCs (30), MAH cells express both μ- and δ-opioid receptors as exemplified in the immunocytochemical experiments shown in Fig. 1 (n = 3). In addition, microarray data from our laboratory suggest that MAH cells express μ- and δ-opioid, but not κ-opioid, receptors (data not shown). Thus, MAH cells represent a simple surrogate model to study signaling pathways activated by μ- and δ-opioid receptors in chromaffin cells.
Fig. 1.
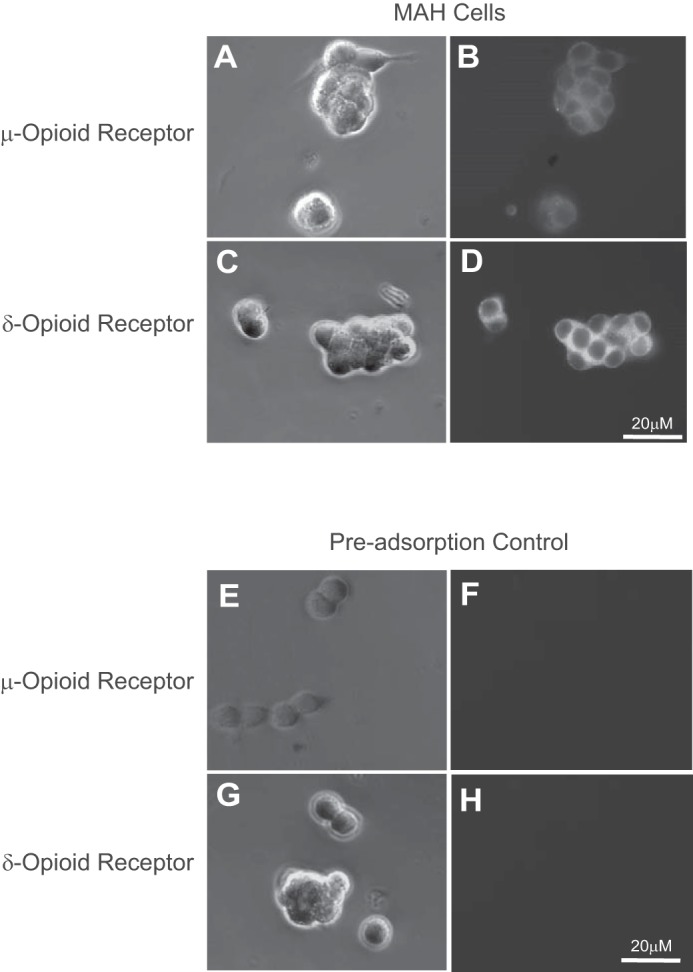
Immunofluorescence staining of μ- and δ-opioid receptors in MAH cell cultures. Corresponding phase-contrast (top left) and fluorescence (top right) images of MAH cells showing positive immunostaining for μ-opioid (A and B) and δ-opioid (C and D) receptors. In control experiments for μ-opioid (E and F) and δ-opioid (G and H) receptor, the primary antibody was preincubated with excess antigen before application to the cells, followed by FITC-conjugated secondary antibody.
Effects of Chronic Opioid Exposure on KATP Channel Expression in MAH Cells
In our recent study, chronic exposure of primary neonatal rat AMCs to combined μ (DALDA)- and δ (DPDPE)-opioid agonists (2 μM) for ∼7 days in vitro caused an increased expression of the KATP channel subunit, Kir6.2, as determined by Western blot analysis (30). Using both QPCR and Western blotting, we first investigated whether upregulation of Kir6.2 subunit also occurs in MAH cells exposed for ∼7 days to medium containing combined opioids. As illustrated in Fig. 2A, the transcript level of Kir6.2 subunit was significantly increased (>2-fold) in opioid-treated MAH cells compared with untreated controls. Moreover, this enhanced Kir6.2 expression was mediated via opioid receptor signaling pathways because it was prevented in MAH cells exposed to combined opioids plus the general opioid receptor blocker naloxone (2 μM; Fig. 2A). Western blot analysis also confirmed the upregulation of Kir6.2 expression at the protein level in opioid-treated MAH cells and its prevention during coincubation with naloxone (Fig. 2B). These data are consistent with those we previously reported for primary neonatal AMCs (30) and suggest that the MAH cell line is a suitable model for probing opioid-mediated signaling pathways in rat chromaffin cells.
Fig. 2.
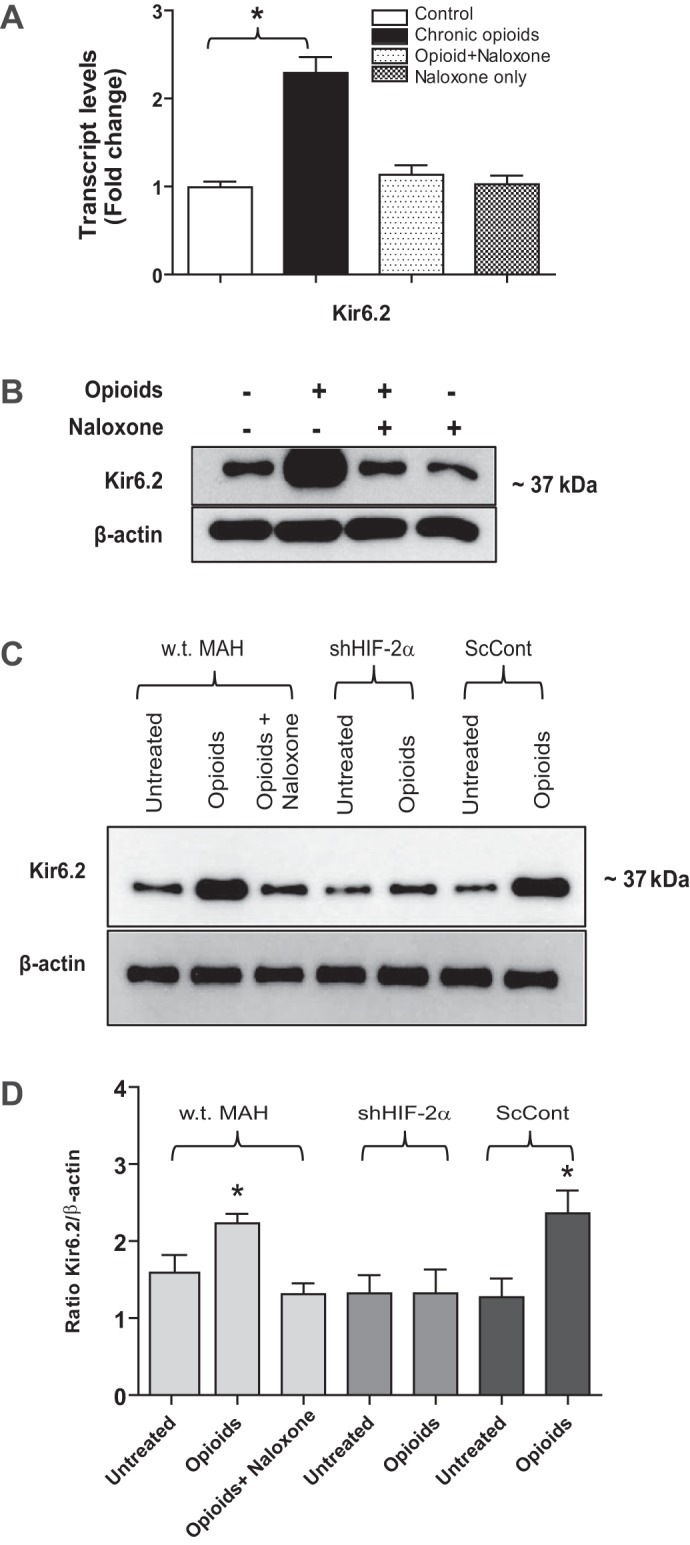
Quantitative real-time PCR (QPCR) and Western blot analyses of ATP-sensitive K+ (KATP) channel subunit Kir6.2 expression in MAH cells. A: QPCR analysis of Kir6.2 mRNA in control MAH cells, MAH cells cultured with combined μ- and δ- opioid agonists (2 μM) ± naloxone (2 μM), and MAH cells cultured with naloxone for 7 days. Note significant upregulation of Kir6.2 transcript in opioid-treated cells (*P < 0.05; n = 4 for each group); one-way ANOVA was used for multiple comparisons within groups. B: Western blot analysis showing increased expression of Kir6.2 protein in opioid-treated MAH cells and its prevention in the presence of naloxone; β-actin was used as an internal control (n = 3). Western blot analyses of Kir6.2 subunit expression in wild-type (wt), hypoxia inducible factor-2α (HIF-2α)-deficient (shHIF-2α), and scrambled control (ScCont) MAH cells cultured with combined opioid agonists (2 μM) or in the presence of naloxone (2 μM) for 7 days (C). Note absence of Kir6.2 subunit upregulation in shHIF-2α MAH cells. Densitometric analysis of changes in Kir6.2 subunit expression normalized β-actin (D); n = 3 for each group, *P < 0.05.
HIF-2α Stabilization is Required for Opioid-Induced Upregulation of KATP Channel Subunit (Kir6.2) Expression in MAH Cells
We previously demonstrated that exposure of both perinatal rat AMCs and immortalized MAH cells to chronic nicotine causes stabilization of the transcription factor HIF-2α that in turn led to the transcriptional upregulation of Kir6.2 subunit and KATP channel expression (5, 28). In other cell types, e.g., neuroblastoma cells, stimulation of μ- and δ-opioid receptors can lead to HIF activation under nonhypoxic conditions (8). It was therefore of interest to investigate whether or not the opioid-induced upregulation of Kir6.2 subunit expression in MAH cells was HIF-2α dependent. To test this, we used Western blots to compare Kir6.2 expression in a HIF-2α-deficient (>90% knockdown) MAH cell line (shHIF-2α MAH; see Ref. 2) following exposure to chronic opioids. As shown in Fig. 2, C and D, the opioid-induced upregulation of Kir6.2 subunit relative to β-actin was present in nontransfected control (wt MAH) and transfected scrambled control (ScCont MAH) cells, but was absent in HIF-2α-deficient (shHIF-2α MAH) cells. These results demonstrate that HIF-2α is required for the opioid-mediated upregulation of Kir6.2 subunit in MAH cells.
We next investigated whether chronic opioid exposure could stabilize HIF-2α in nuclear extracts of wt MAH cells. As illustrated in Fig. 3A, there was a robust accumulation of HIF-2α in MAH cells exposed to chronic opioids for ∼7 days in vitro, and this effect was abolished during coincubation with the opioid receptor blocker naloxone (2 μM).
Fig. 3.
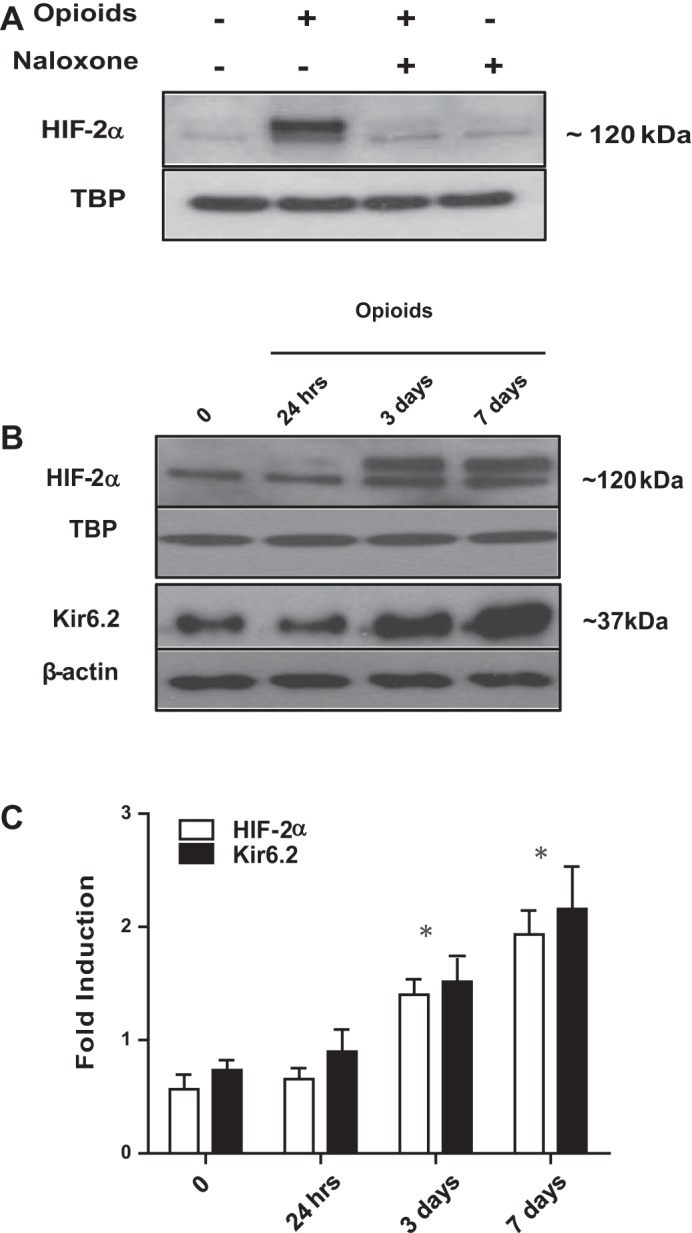
Effects of chronic opioid exposure on Kir6.2 and HIF-2α expression in MAH cells. A: Western blot analysis of HIF-2α accumulation in MAH cells cultured with combined μ- and δ- opioid agonists (2 μM) ± naloxone (2 μM), or with naloxone only (2 μM), for 7 days. B: time-dependent HIF-2α and Kir6.2 protein expression in MAH cells exposed to combined opioids (2 μM) for 24 h, 3 days, and 7 days in culture. β-Actin was used as loading control for cytoplasmic extracts in the case of Kir6.2 and TATA-binding protein (TBP) for nuclear extracts in the case of HIF-2α. Results show progressive increase in Kir6.2 expression that parallels HIF-2α accumulation following chronic opioids; for both Kir6.2 and HIF-2α, the increase is significant at 3 and 7 days (B and C). C: densitometric analysis demonstrating the fold induction in Kir6.2 subunit expression and HIF-2α accumulation normalized to loading control. Data are expressed as means ± SE for three independent experiments for each group, *P < 0.05.
Time-Dependent Effects of Chronic Opioids on HIF-2α Accumulation and Kir6.2 Subunit Expression in MAH Cells
In a previous study, we found that chronic nicotine exposure caused a parallel, progressive, and time-dependent accumulation of HIF-2α and Kir6.2 subunit expression in MAH cells (28). To test whether a similar pattern occurs during opioid exposure, we monitored HIF-2α and Kir6.2 subunit expression in opioid-treated MAH cells at 0 h, 24 h, 3 days, and 7 days in nuclear and cytoplasmic extracts. As illustrated in Fig. 3, B and C, Western blot analysis revealed a slow progressive increase in HIF-2α accumulation that occurred in parallel with the increase in Kir6.2 subunit over the 7-day treatment period. Both increases were significant at exposure periods of 3 and 7 days but not at 24 h (Fig. 3C); by contrast, exposure of MAH cells to chronic hypoxia (2% O2) normally results in a robust HIF-2α accumulation at 24 h (data not shown) (3, 28). This expression pattern of HIF-2α and Kir6.2 subunit is reminiscent of that seen in MAH cells during exposure to chronic nicotine over a similar time period (28).
HIF-2α Binds to the Promoter Region of Kir6.2 Gene in Opioid-Treated Cells
The promoter region of the Kir6.2 gene in both rats and mice contains a putative HRE, where the HIF core site (GCGTG) spans nucleotides −1087 to −1083 and the HIF ancillary site (CACAG) spans nucleotides −1065 to −1061 (Fig. 4A, top). Using a ChIP assay, we previously demonstrated that HIF-2α bound to the promoter region of Kir6.2 gene, following its stabilization in MAH cells exposed to chronic nicotine (28). To test whether opioid receptor signaling promotes binding of HIF-2α to the promoter region of Kir6.2 gene, we performed ChIP assays on control and HIF-2α-deficient MAH cells exposed to chronic opioids for ∼7 days. Indeed, we found that, under these conditions, HIF-2α bound to the Kir6.2 promoter region in wt and ScCont MAH cells but not in shHIF-2α MAH cells, as illustrated in Fig. 4A, bottom. These data complement those demonstrating that HIF-2α deficiency prevents upregulation of Kir6.2 in opioid-treated MAH cells (Fig. 2, C and D) and imply that the mechanism of HIF-2α action most likely involves increased expression of Kir6.2 at the transcriptional level.
Fig. 4.
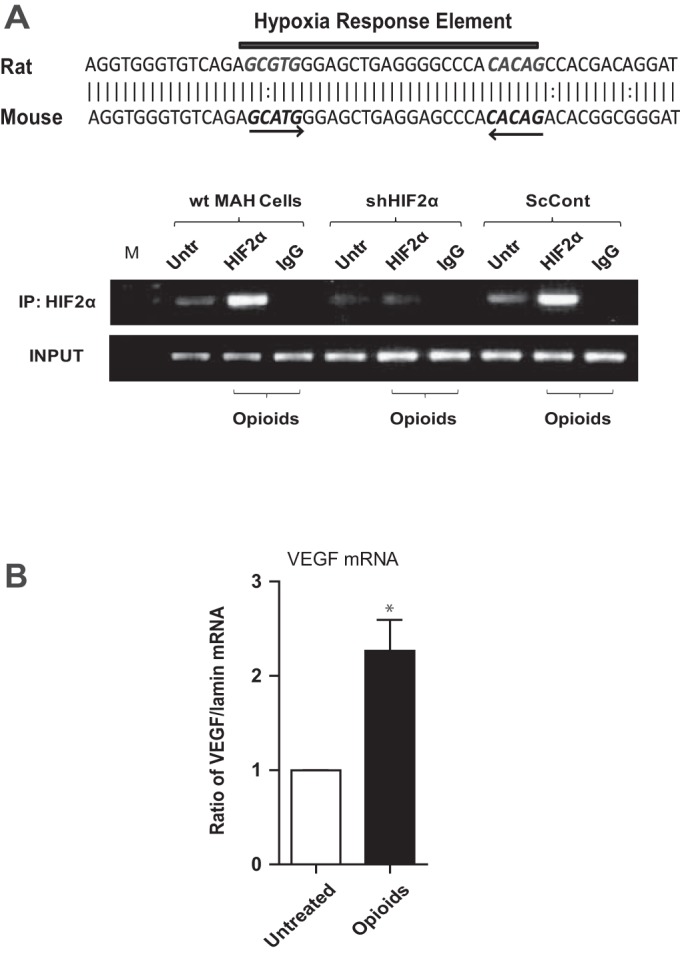
Chromatin immunoprecipitation (ChIP) assay demonstrating binding of HIF-2α to the promoter region of Kir6.2 gene in opioid-treated MAH cells. Note hypoxia response element (HRE) within the promoter region of rat and mouse Kir6.2 gene (A, top); the HIF core site (GCGTG) spans nucleotides −1087 to −1083 and HIF ancillary site (CACAG) spans nucleotides −1065 to −1061. Lysates from untreated control (Untr) and opioid-treated wt, shHIF2α, and ScCont MAH cells were subjected to ChIP assay using a HIF-2α polyclonal antibody (A, bottom). PCR analysis was performed using a primer pair designed to span the putative HRE upstream of the promoter region. Technical controls include a ChIP performed using nonspecific IgG monoclonal antibody (IgG) and a starting material control (Input). B: vascular endothelial growth factor (VEGF) mRNA expression as determined by QPCR analysis in control (untreated) and opioid-treated MAH cells (n = 3). Data are expressed as means ± SE for three independent experiments for each group, *P < 0.05.
To confirm transcriptional activity of HIF-2α in opioid-treated MAH cells, we measured mRNA expression levels of a common HIF-2α target gene, VEGF, using QPCR analysis. Data indicate that VEGF mRNA is significantly increased in opioid-treated MAH cells (Fig. 4B).
Chronic Opioid Exposure Downregulates the Expression of CAII in MAH Cells: Role of HIF-2α
Sensitivity of neonatal rat AMCs to high CO2 (hypercapnia) is dependent on the activity of the CO2-hydrating enzymes CAI and -II (23). We previously showed that exposure of primary neonatal rat AMC to chronic opioids blunts hypercapnia sensitivity, in association with the downregulation of CAI and CAII (30). Although MAH cells express only CAII, they show CO2 sensitivity similar to primary neonatal AMC (7), thereby providing a model for investigating opioid-mediated blunting of CO2 sensing. Indeed, exposure of MAH cells to chronic opioids for ∼7 days resulted in a significant reduction in CAII transcript as determined by QPCR; furthermore, this reduction was prevented during continuous coincubation with the opioid receptor blocker naloxone (2 μM; Fig. 5A). Western blot analysis revealed that chronic opioids also caused a naloxone-sensitive downregulation of CAII at the protein level, as illustrated in Fig. 5B.
Fig. 5.
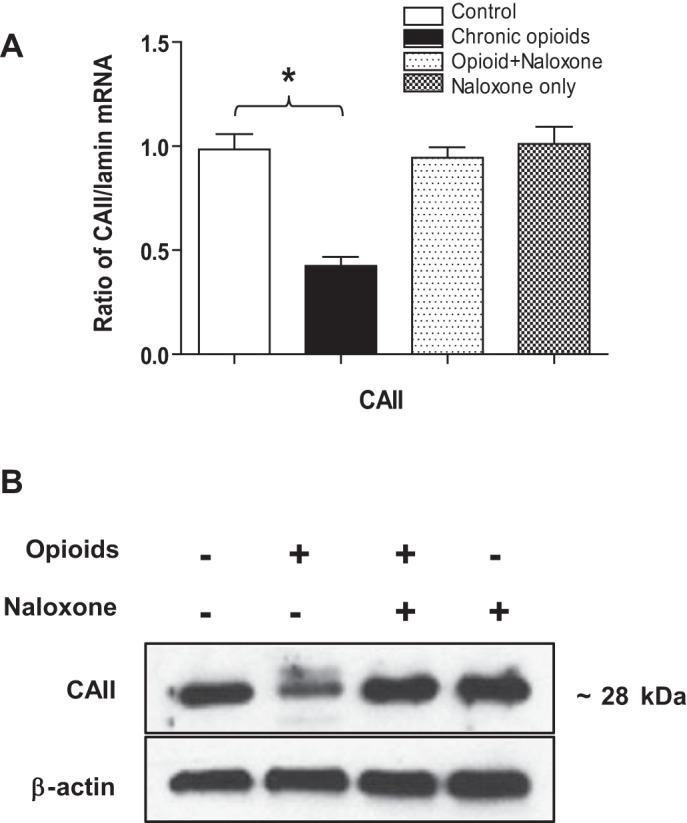
Effects of chronic opioid exposure on carbonic anhydrase II (CAII) expression in MAH cells. A: mRNA expression analysis of CAII in control MAH cells, in MAH cells cultured with combined μ- and δ-opioid agonists (2 μM) ± naloxone (2 μM), and in MAH cells cultured with naloxone (2 μM) only for 7 days. *P < 0.05, n = 3 for each group. One-way ANOVA was used for multiple comparisons within groups. B: Western blot analysis showing expression of CAII protein in MAH cells grown under similar conditions. β-Actin was used as an internal control. Note coincubation with naloxone prevented the chronic opioid-induced decrease in CAII expression in A and B.
Because chronic opioids stabilize HIF-2α in MAH cells (Fig. 3), we wondered whether or not the opioid-mediated downregulation of CAII expression was HIF-2α dependent. To test this, we used Western blot analysis to compare CAII expression in shHIF-2α MAH cells following exposure to chronic opioids for ∼7 days. As shown in Fig. 6, A and B, the opioid-mediated downregulation of CAII relative to β-actin was present in wt MAH and transfected ScCont MAH cells but was absent in shHIF-2α MAH cells. These results demonstrate that HIF-2α is required for the opioid-mediated downregulation of CAII in MAH cells.
Fig. 6.
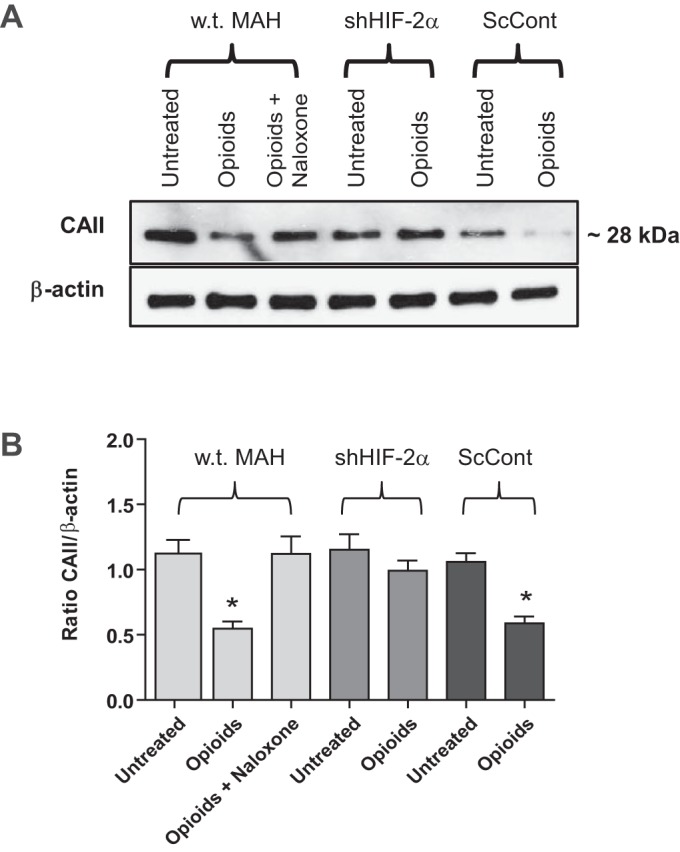
Effects of HIF-2α-deficiency on CAII expression in opioid-treated MAH cells. A, top: Western blots of CAII protein expression in control MAH cells, shHIF2α MAH cells, and ScCont MAH cells, cultured with or without combined opioid agonists (2 μM) or in the presence of naloxone (2 μM) for 7 days. B: densitometric analysis of CAII protein expression normalized to β-actin under the various conditions indicated. Note the opioid-mediated downregulation of CAII is absent in HIF-2α-deficient MAH cells. Data are expressed as means ± SE for three independent experiments for each group, *P < 0.05.
Central Role of PKA in Mediating the Downstream Effects of Opioid Receptor Signaling in MAH Cells
Various protein kinases are known to be activated during opioid receptor signaling (10). We therefore used pharmacological inhibitors to probe for the potential involvement of three common kinases, i.e., PKC, CaMK, and PKA, in mediating the opposing effects of chronic opioid exposure on Kir6.2 and CAII expression in MAH cells. As illustrated in Fig. 7A, Western blot analysis revealed that inhibition of PKA by H-89 (2 μM) prevented the opioid-induced upregulation of Kir6.2 evident in MAH cells following chronic opioid exposure. By contrast, inhibition of PKC and CaMK by GF-109203X (2 μM) and KN-62 (3 μM), respectively, had no effect on the upregulation of Kir6.2 protein in comparable experiments (Fig. 7A). Likewise PKA, but not PKC or CaMK, inhibition prevented the opioid-induced downregulation of CAII evident in MAH cells following chronic opioid exposure (Fig. 7B). These data suggest a central role of PKA in the opioid receptor signaling pathway, leading to the regulation of key proteins involved in the blunting of O2 and CO2 chemosensitivity in chromaffin cells.
Fig. 7.

Effects of protein kinase inhibitors on expression of Kir6.2, CAII, and HIF-2α in MAH cells following chronic opioid exposure. A: Western blot analysis showing the effects of protein kinase inhibition on Kir6.2 expression in opioid-treated MAH cells. Inhibitors of protein kinase A (PKA) (H-89) (2 μM), but not inhibitors of protein kinase C (GF-109203X) (2 μM) or calmodulin (CaM) kinase (KN-62) (3 μM), prevented the upregulation of Kir6.2 in opioid-treated cells. Densitometric analysis of Kir6.2 subunit expression normalized to β-actin is shown in histogram (right). A similar protocol was used to study the effects of protein kinase inhibitors on CAII expression relative to β-actin (B), and HIF-2α accumulation relative to TBP (C), in opioid-treated MAH cells. Note the PKA inhibitor (H-89) prevented the opioid-induced downregulation of CAII expression, but not HIF-2α accumulation, in MAH cells. Data are expressed as means ± SE for three independent experiments for each group, *P < 0.05.
Given that both PKA activity (Fig. 7, A and B) and HIF-2α accumulation (Figs. 2 and 6) are necessary for the regulation of Kir6.2 and CAII in opioid-treated MAH cells, the question arises whether HIF-2α accumulation is dependent on PKA activity. To address this, we investigated whether the opioid-induced HIF-2α accumulation could occur in the presence of PKA inhibition. As illustrated in Fig. 7C, inhibition of PKA by H-89 did not prevent the opioid-induced HIF-2α accumulation in MAH cells. Furthermore, inhibition of PKC by KN-62 and CaMK by GF-109203X was similarly ineffective (Fig. 7C). Taken together, these data suggest that the regulation of Kir6.2 and CAII in opioid-treated MAH cells requires the separate actions of PKA and HIF-2α.
Chronic Opioids Blunt Hypoxia and Hypercapnia Chemosensing in MAH Cells as Monitored by DA and NE Release
In regards to CAT biosynthesis, MAH cells synthesize predominantly DA and NE because they lack expression of the epinephrine-producing enzyme phenylethanolamine N-methyltransferase (2, 7, 36). To test whether or not chronic opioids blunt hypoxia- and hypercapnia-evoked secretion in MAH cells, we monitored DA and NE release using ELISAs in control vs. opioid-treated cells plated at similar densities. In these experiments, exposure of control (untreated) MAH cells to hypoxia (2% O2; Fig. 8A) and isohydric hypercapnia (10% CO2, pH 7.4; Fig. 8B) for ∼15 min significantly stimulated DA and NE release (≥2× basal). By contrast, when the same stimuli were applied to MAH cells treated with opioids for ∼7 days, there was no significant increase in DA and NE levels above basal (Fig. 8, A and B). HIF-2α-deficient MAH cells were not investigated because they synthesize negligible amounts of DA and NE, attributable to the markedly reduced expression of DOPA decarboxylase and dopamine-β-hydroxylase (2). These data suggest that chronic opioids blunt hypoxia and hypercapnia chemosensitivity in MAH cells as determined by a functional assay based on CAT release.
Fig. 8.
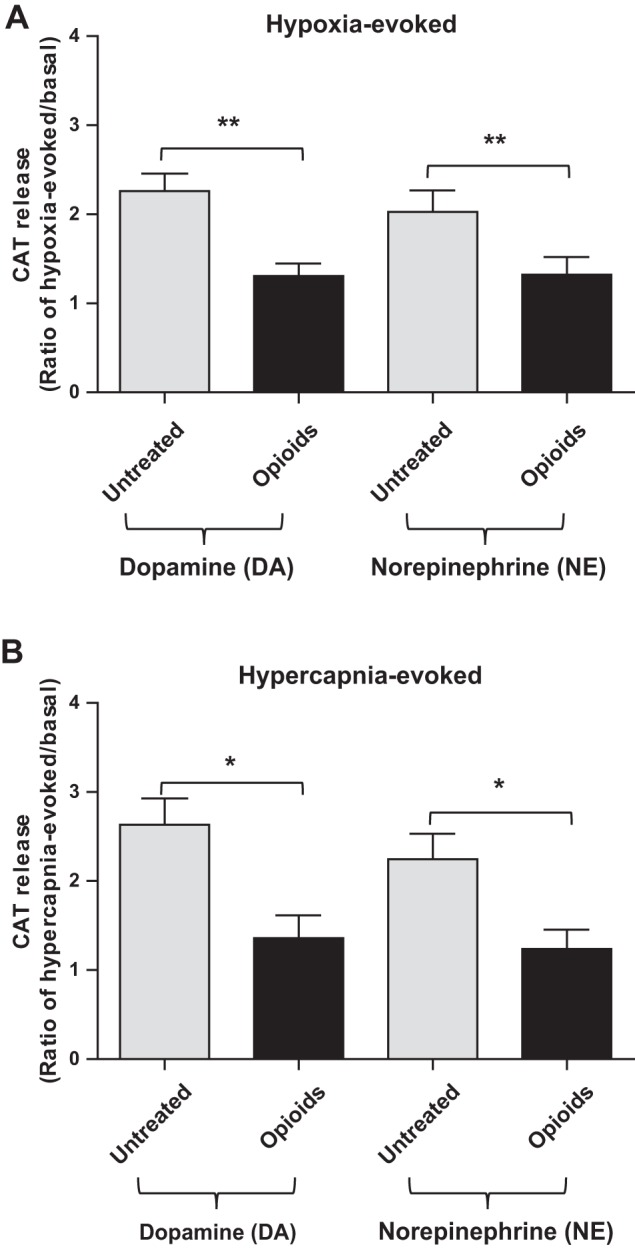
Effects of chronic opioids on hypoxia- and hypercapnia-evoked dopamine (DA) and norepinephrine (NE) release from MAH cells. ELISA was used to measure DA and NE release, and data were expressed as a ratio of evoked relative to basal release. Chemostimulation with hypoxia (2% O2; 15 min) (A) and isohydric hypercapnia (10% CO2; pH 7.4) (B) caused a significant increase in DA and NE release in control (untreated) MAH cells. Following exposure of MAH cells to chronic opioids for ∼1 wk in vitro, the effect of hypoxia and hypercapnia on DA and NE release was significantly reduced. Data are represented as means ± SE, where n = 5 for A and 4 for B (*P < 0.05 and **P < 0.01).
Effects of Chronic Maternal Morphine Exposure on Kir6.2, CAI, CAII, and HIF-2α Expression in the Adrenal Glands of Affected Neonates
To validate the main conclusions from our in vitro model in a physiologically relevant in vivo system, we administered morphine or saline (control) to pregnant Wistar rats throughout gestation. We then compared the expression pattern of Kir6.2, CAI and -II, HIF-1α, and HIF-2α in separate “enriched” tissue fractions of the AM and AC, obtained from the adrenal glands of the offspring at birth. Before tissue collection, it was evident that morphine administration had an adverse effect on the well-being and viability of the offspring, as revealed by measures of several parameters indicated in Table 1. As illustrated in Fig. 9, A–C, Western blot analysis revealed there was a significant increase in expression of Kir6.2 and HIF-2α, and a decrease in expression of CAI and CAII, in the medullary-rich regions of glands taken from morphine-exposed pups relative to saline-exposed controls; by contrast, expression of HIF-1α was unchanged. Interestingly, this change in expression pattern for Kir6.2, HIF-2α, and CAII was similar to that seen in opioid-treated MAH cells in vitro. On the other hand, the changes in expression pattern seen in the medullary region were not replicated in cortical-rich regions of glands. Notably, CAI and CAII expression was robust in the medullary-rich regions of control (saline-exposed) glands, but was almost absent from cortical-rich regions, confirming there was minimal cross-contamination during separation of cortex from medulla. These data suggest that, in this in vivo model, the morphine-induced changes in expression pattern were confined to the medullary chromaffin cells, which are the predominant cell type expressing opioid receptors in the adrenal gland (30).
Fig. 9.
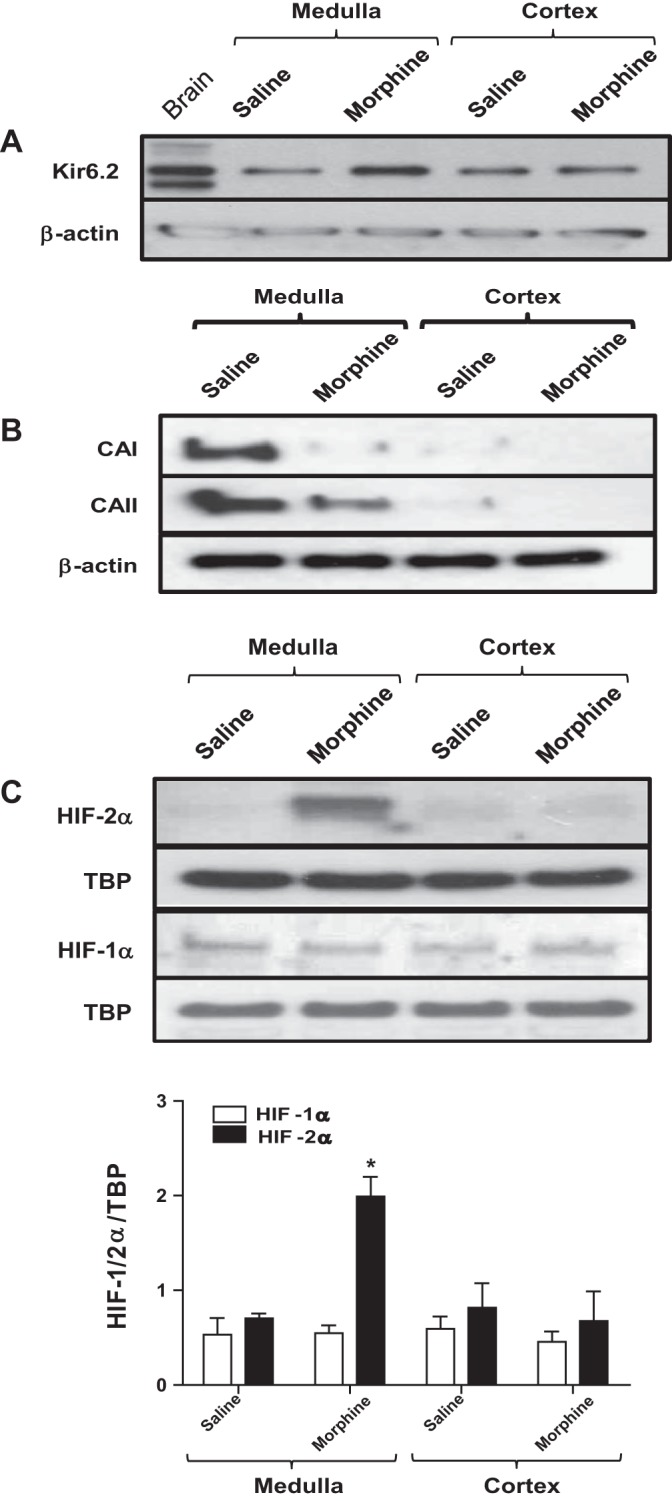
Effects of maternal morphine injections on the expression of KATP channel subunit (Kir6.2), carbonic anhydrase I (CAI) and CAII enzymes, and HIFs in adrenal gland tissues of affected neonates. Western blot analyses of KATP channel subunit Kir6.2 (A), CAI and -II (B), and HIF-1α and -2α (C) expression in adrenal medulla (AM) and adrenal cortex (AC) of saline- and morphine-exposed rat pups. Note the increased Kir6.2 subunit and reduced CAI and CAII expression (relative to β-actin) in AM, but not AC, of morphine-exposed pups. Also, note the selective increase in HIF-2α, but not HIF-1α, accumulation (relative to TBP) in AM of morphine-exposed pups as shown in C. Data are expressed as means ± SE for three independent experiments for each group, *P < 0.05.
DISCUSSION
In a recent study, we reported that chronic exposure of neonatal rat AMCs to μ- and δ-opioid receptor agonists in vitro blunts hypoxia and hypercapnia chemosensitivity via upregulation of KATP channels and downregulation of CAI and CAII, respectively (30). The goals of the present study were to obtain a more complete understanding of the underlying molecular mechanisms and signaling pathways using an in vitro model, and to validate key aspects of these molecular mechanisms in a relevant in vivo model. Using an immortalized, fetal-derived rat chromaffin (MAH) cell line, we found that chronic opioid exposure led to increased expression of Kir6.2 subunit of the KATP channel, and decreased expression of CAII, the only one of the CA isoforms expressed in MAH cells. Both effects were mediated via PKA, and were dependent on the transcription factor HIF-2α, because they were abrogated in HIF-2α-deficient MAH cells. Strong support for this schema was obtained from an in vivo model where adrenal tissue from pups born to morphine-exposed dams was probed for Kir6.2, CAI, CAII, and HIF-2α expression. Interestingly, compared with saline-treated controls, adrenal tissue from morphine-exposed pups showed increased expression of Kir6.2 and HIF-2α and decreased expression of both CAI and CAII.
Role of HIF-2α in Opioid-Induced Regulation of Kir6.2 and CAII Expression
One key factor involved in gene regulation in MAH cells following exposure to chronic μ- and δ-opioids was the transcription factor HIF-2α. Whereas the opioid-induced regulation of Kir6.2 and CAII was observed in wt and transfected scrambled control MAH cells, it was absent in HIF-2α-deficient MAH cells. Moreover, following opioid exposure, there was a gradual increase in HIF-2α accumulation that occurred along a time course similar to the increase in Kir6.2 expression in MAH cells. The importance of HIF-2α in this signaling pathway was further demonstrated using a ChIP assay, which revealed the direct binding of HIF-2α to the promoter region of the Kir6.2 gene in opioid-treated MAH cells. These data suggest a molecular explanation for the increased expression of Kir6.2 subunit and increased KATP current density previously reported in primary neonatal rat AMCs exposed to chronic opioids in vitro (28). In the latter study, the increased expression of KATP channels accounted largely for the opioid-mediated blunting of hypoxia sensitivity, which could be restored by simply adding the KATP channel blocker glibenclamide to the extracellular solution. Thus, the ability of chronic opioid exposure to blunt hypoxia sensing in chromaffin cells appears to be dependent on the induction of HIF-2α, leading to the transcriptional upregulation of Kir6.2 and increased expression of functional KATP channels. The opening of these KATP channels in response to a fall in ATP concentration during hypoxia favors membrane hyperpolarization, and therefore a decreased sensitivity to the hypoxic stimulus.
Evidence that a nonhypoxic stimulus such as opioid exposure could lead to HIF induction has been obtained in a previous study on human SH-SY5Y neuroblastoma cells (8). These authors reported that chronic exposure of SH-SY5Y cells to μ- and δ-opioid receptor agonists led to the induction of HIF-1α protein and an increase in HIF-1 target gene expression. Whereas these data demonstrated a link between HIF and the biological effects of chronic opioid exposure, there are notable differences when compared with the present study. In particular, HIF-2α protein (and not HIF-1α) was the major HIF involved in the regulation of Kir6.2 and CAII in MAH cells, and, moreover, its induction was not apparent at 24 h exposure. By contrast, in SH-SY5Y cells, the induction of HIF-1α protein was apparent as early as 8 h and peaked at 24 h exposure (8). Although the fate of HIF-1α in MAH cells is unknown in the present study, it is noteworthy that, in the adrenal tissue of morphine-exposed pups, we found that HIF-2α was elevated (relative to saline-exposed controls), whereas HIF-1α remained unchanged. Thus, it still remains plausible that, at earlier time points after opioid exposure, other target genes, specific to HIF-1, may be regulated in chromaffin cells; however, this idea remains to be tested. Alternatively, the opioid-induced regulation of HIFs may occur via the same opioid receptors but in a cell-type-specific manner, depending on the intracellular signaling pathways. In this regard, chronic exposure of different cells to another nonhypoxic stimulus may lead to the differential regulation of HIF-1α and HIF-2α. For example, activation of nicotinic ACh receptor signaling pathways by exposure to chronic nicotine led to HIF-1α accumulation in human small cell lung cancers (42). On the other hand, a similar exposure led to HIF-2α accumulation in MAH cells (28), although there was increased expression of the general HIF target gene, VEGF, in both studies.
A particularly novel aspect of the present study was the demonstration that the opioid-induced downregulation of CAII seen in control MAH cells was absent in HIF-2α-deficient MAH cells. Functional CA activity is critical for the ability of neonatal AMCs to sense elevated CO2 (hypercapnia), and CAII is one of only two CAs expressed in these cells [Munoz-Cabello et al. (23)]. To our knowledge, this is the first demonstration that CAII expression can be regulated by HIF, and particularly by HIF-2α. In this regard, HIF-1α has been reported to act as a transcriptional repressor of a number of genes such as α-fetoprotein and LIFR under hypoxia, by direct binding to a reverse HRE located on the antisense strand (15, 22, 24). This raises the attractive possibility that the CAII gene may act as a novel transcriptional target of HIF-2α, however, this requires validation. Interestingly, a recent study identified a large number of genes that are repressed by HIF-1α in arterial endothelial cells under hypoxia in which no specific pattern of HRE was identified (21). In addition, it is well known that the tumor-associated CAs, i.e., CAIX and CAXII, represent a class of HIF-1-dependent genes that is strongly induced in tumor cell lines under hypoxia, and a HRE has also been identified on the antisense strand of CAIX gene (38). Therefore, further studies are required to clarify the mechanisms underlying the HIF-2α-dependent downregulation of the CAII gene as observed in the present study.
Role of PKA in Opioid-Induced Regulation of Kir6.2 Subunit and CAII Expression
Using inhibitors of three common protein kinases, we found that PKA (but not PKC or CaMK) activation was necessary for the opioid-induced regulation of Kir6.2 subunit and CAII expression in MAH cells. Because HIF-2α induction was also necessary for this regulation, the question arose whether the requirement for PKA activation occurred upstream or downstream of HIF-2α induction. Our results showed that PKA inhibition did not significantly affect HIF-2α accumulation in opioid-treated MAH cells, suggesting that PKA activation was required downstream of (or in parallel with) HIF-2α accumulation. It remains to be determined whether PKA activation during opioid-induced signaling is involved in the transcriptional or posttranslational modification of Kir6.2 subunit and CAII expression. In a recent report, activation of μ- and δ-opioid receptors in SH-SY5Y neuroblastoma cells led to HIF-1α accumulation via the phosphatidylinositol 3-kinase/protein kinase B (Akt)/mammalian target of rapamycin (mTOR) signaling pathway (8). It is tempting to speculate that at least part of the opioid receptor signaling in MAH cells is mediated via activation of this pathway, given the evidence that HIF-2α expression may also be regulated via a distinct mTOR complex and Akt isoform, as shown in renal carcinoma cells (35). However, further experiments are required to test this idea.
Effects of Chronic Morphine In Utero on Expression of Kir6.2, CAII, and HIF-2α in AM vs. AC of Affected Neonates
It was of interest to test the predictions of the in vitro opioid-treated MAH cell model in a physiologically relevant in vivo system. To this end, both medullary and cortical tissues from the adrenal glands of rat pups exposed to chronic morphine in utero were examined for the expression pattern of Kir6.2, CAI and -II, HIF-2α, and HIF-1α. The rat adrenal gland expresses all three types of opioid receptors, and these are restricted to the chromaffin cells of the medulla (30, 37). Interestingly, even though Kir6.2 is expressed in both AC and AM (5, 39), there was a selective upregulation of Kir6.2 protein in the medullary region of morphine-exposed pups compared with saline-exposed controls. This is the expected result if an intact opioid receptor signaling pathway is required for Kir6.2 upregulation and argues against a nonspecific action of morphine following injections in vivo. Moreover, even though the MAH cell model allowed us to test only for the regulation of CAII following opioid exposure, morphine-exposed pups showed downregulation of both CAI and CAII expression. Not surprisingly, this downregulation was again confined to the medullary tissue, the only region of the gland where the two CAs are known to be expressed (23).
Similar to Kir6.2 subunit expression, we detected HIF-2α protein in both medullary and cortical tissues of the adrenal gland. However, chronic morphine exposure led to a selective accumulation of HIF-2α in the medullary tissue only. This result not only validated our in vitro MAH cell model but, once again, emphasized the requirement for an intact opioid receptor signaling pathway for HIF-2α induction, given that the cortical cells do not express μ- and δ-opioid receptors (30). Additionally, the effects of chronic morphine on HIF accumulation were restricted to HIF-2α, for though HIF-1α was expressed in the medulla, its levels were not significantly affected. Generally, the importance of HIF-2α function is thought to be tissue specific, and HIF-2α appears to play a major role in cells of the sympathoadrenal lineage (1–3). Although the two transcription factors are paralogs, they seem to play distinct target-specific roles even when expressed in the same tissue. In fact, in studies using partially deficient HIF-1α and HIF-2α mice, a mutual antagonism between HIF-1α and HIF-2α was found to regulate intracellular redox status and hypoxia sensitivity of carotid body glomus cells and AMCs (40). Using the MAH cell model, we demonstrated that chronic opioids led to HIF-2α accumulation and that HIF-2α deficiency prevented the opioid-induced upregulation of Kir6.2 and downregulation of CAII. These observations suggest that the increased HIF-2α accumulation seen in morphine-exposed neonatal medullary tissue likely led to the transcriptional upregulation of Kir6.2 and downregulation of CAII (and probably CAI as well) in the tissue.
Clinical Significance
While our studies contributed to a general understanding of the role of opioid receptor signaling in the developmental regulation of hypoxia and hypercapnia sensitivity in chromaffin cells, they also have clinical significance. Prenatal exposure of pregnant mothers to opiates (e.g., heroin and related illicit drugs) has been linked to higher rates of stillbirths and SIDS that are often characterized by impaired arousal during asphyxial challenges (4, 16). Indeed, in the present study, we noted that chronic morphine exposure in utero was associated with decreased litter size, and an increase in the percentage of dams delivering stillborns (Table 1), as reported in previous studies (41). In addition, infants born small for gestational age have been proposed to have higher risk of becoming victims to SIDS (11). In this regard, morphine exposure in utero also resulted in a significantly higher proportion of offspring that were growth restricted (i.e., body wt <2 SDs below the average weight of control offspring; Table 1). A recent study reported that use of opiates by pregnant mothers has climbed and that the number of neonates born addicted to painkiller medication (e.g., opioid pain relievers) has tripled in the past decades due to the rise in use and abuse of these drugs (26). Because adrenal CAT release in response to perinatal asphyxia is important for proper arousal, the adverse effects of chronic opioid exposure on the chemosensing properties of chromaffin cells may well contribute to the increased morbidity of drug-exposed neonates. If so, further studies on these chromaffin cells should aid in the development of therapeutic strategies that could benefit the offspring of women who use opiates during pregnancy, whether for pain management or drug abuse.
GRANTS
This work was supported by a Discovery grant from the Natural Sciences and Engineering Research Council of Canada and an operating grant from the Canadian Institutes of Health Research (MOP-119501) to C. A. Nurse and a Discovery grant from the Natural Sciences and Engineering Research Council of Canada to A. C. Holloway.
DISCLOSURES
The authors declare no conflict of interest, financial or otherwise.
AUTHOR CONTRIBUTIONS
Author contributions: S.S., A.C.H., and C.A.N. conception and design of research; S.S. and A.C.H. performed experiments; S.S. analyzed data; S.S., A.C.H., and C.A.N. interpreted results of experiments; S.S. prepared figures; S.S. and C.A.N. drafted manuscript; S.S., A.C.H., and C.A.N. edited and revised manuscript; S.S., A.C.H., and C.A.N. approved final version of manuscript.
ACKNOWLEDGMENTS
We thank Stephen Brown for creating the HIF-2α-deficient MAH cell line, Cathy Vollmer for expert technical assistance, Chris Wood for use of the ELISA assay reader, and the McMaster Central Animal Facility for providing assistance with the animal work.
REFERENCES
- 1.Bishop T, Gallagher D, Pascual A, Lygate CA, de Bono JP, Nicholls LG, Ortega-Saenz P, Oster H, Wijeyekoon B, Sutherland AI, Grosfeld A, Aragones J, Schneider M, van Geyte K, Teixeira D, Diez-Juan A, Lopez-Barneo J, Channon KM, Maxwell PH, Pugh CW, Davies AM, Carmeliet P, Ratcliffe PJ. Abnormal sympathoadrenal development and systemic hypotension in PHD3−/− mice. Mol Cell Biol 28: 3386–3400, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Brown ST, Kelly KF, Daniel JM, Nurse CA. Hypoxia inducible factor (HIF)-2 alpha is required for the development of the catecholaminergic phenotype of sympathoadrenal cells. J Neurochem 110: 622–630, 2009 [DOI] [PubMed] [Google Scholar]
- 3.Brown ST, Nurse CA. Induction of HIF-2alpha is dependent on mitochondrial O2 consumption in an O2-sensitive adrenomedullary chromaffin cell line. Am J Physiol Cell Physiol 294: C1305–C1312, 2008 [DOI] [PubMed] [Google Scholar]
- 4.Burns L, Conroy E, Mattick RP. Infant mortality among women on a methadone program during pregnancy. Drug Alcohol Rev 29: 551–556, 2010 [DOI] [PubMed] [Google Scholar]
- 5.Buttigieg J, Brown S, Holloway AC, Nurse CA. Chronic nicotine blunts hypoxic sensitivity in perinatal rat adrenal chromaffin cells via upregulation of KATP channels: role of alpha7 nicotinic acetylcholine receptor and hypoxia-inducible factor-2alpha. J Neurosci 29: 7137–7147, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Buttigieg J, Brown S, Zhang M, Lowe M, Holloway AC, Nurse CA. Chronic nicotine in utero selectively suppresses hypoxic sensitivity in neonatal rat adrenal chromaffin cells. FASEB J 22: 1317–1326, 2008 [DOI] [PubMed] [Google Scholar]
- 7.Buttigieg J, Brown ST, Lowe M, Zhang M, Nurse CA. Functional mitochondria are required for O2 but not CO2 sensing in immortalized adrenomedullary chromaffin cells. Am J Physiol Cell Physiol 294: C945–C956, 2008 [DOI] [PubMed] [Google Scholar]
- 8.Daijo H, Kai S, Tanaka T, Wakamatsu T, Kishimoto S, Suzuki K, Harada H, Takabuchi S, Adachi T, Fukuda K, Hirota K. Fentanyl activates hypoxia-inducible factor 1 in neuronal SH-SY5Y cells and mice under non-hypoxic conditions in a mu-opioid receptor-dependent manner. Eur J Pharmacol 667: 144–152, 2011 [DOI] [PubMed] [Google Scholar]
- 9.Fearon IM, Thompson RJ, Samjoo I, Vollmer C, Doering LC, Nurse CA. O2-sensitive K+ channels in immortalised rat chromaffin-cell-derived MAH cells. J Physiol 545: 807–818, 2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Feng B, Li Z, Wang JB. Protein kinase C-mediated phosphorylation of the mu-opioid receptor and its effects on receptor signaling. Mol Pharmacol 79: 768–775, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Getahun D, Amre D, Rhoads GG, Demissie K. Maternal and obstetric risk factors for sudden infant death syndrome in the United States. Obstet Gynecol 103: 646–652, 2004 [DOI] [PubMed] [Google Scholar]
- 12.Holgert H, Aman K, Cozzari C, Hartman BK, Brimijoin S, Emson P, Goldstein M, Hokfelt T. The cholinergic innervation of the adrenal gland and its relation to enkephalin and nitric oxide synthase. NeuroReport 6: 2576–2580, 1995 [DOI] [PubMed] [Google Scholar]
- 13.Holgert H, Dagerlind A, Hokfelt T. Immunohistochemical characterization of the peptidergic innervation of the rat adrenal gland. Horm Metab Res 30: 315–322, 1998 [DOI] [PubMed] [Google Scholar]
- 14.Huang YF, Yang CH, Huang CC, Tai MH, Hsu KS. Pharmacological and genetic accumulation of hypoxia-inducible factor-1alpha enhances excitatory synaptic transmission in hippocampal neurons through the production of vascular endothelial growth factor. J Neurosci 30: 6080–6093, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jeong CH, Lee HJ, Cha JH, Kim JH, Kim KR, Yoon DK, Kim KW. Hypoxia-inducible factor-1 alpha inhibits self-renewal of mouse embryonic stem cells in Vitro via negative regulation of the leukemia inhibitory factor-STAT3 pathway. J Biol Chem 282: 13672–13679, 2007 [DOI] [PubMed] [Google Scholar]
- 16.Kato I, Franco P, Groswasser J, Scaillet S, Kelmanson I, Togari H, Kahn A. Incomplete arousal processes in infants who were victims of sudden death. Am J Respir Crit Care Med 168: 1298–1303, 2003 [DOI] [PubMed] [Google Scholar]
- 17.Kobayashi S, Miyabayashi T, Uchida T, Yanaihara N. Met-enkephalin-Arg6-Gly7-Leu8 in large-cored vesicles of splanchnic nerve terminals innervating guinea pig adrenal chromaffin cells. Neurosci Lett 53: 247–252, 1985 [DOI] [PubMed] [Google Scholar]
- 18.Lagercrantz H, Slotkin TA. The “stress” of being born. Sci Am 254: 100–107, 1986 [DOI] [PubMed] [Google Scholar]
- 19.Levitsky KL, Lopez-Barneo J. Developmental change of T-type Ca2+ channel expression and its role in rat chromaffin cell responsiveness to acute hypoxia. J Physiol 587: 1917–1929, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 25: 402–408, 2001 [DOI] [PubMed] [Google Scholar]
- 21.Manalo DJ, Rowan A, Lavoie T, Natarajan L, Kelly BD, Ye SQ, Garcia JG, Semenza GL. Transcriptional regulation of vascular endothelial cell responses to hypoxia by HIF-1. Blood 105: 659–669, 2005 [DOI] [PubMed] [Google Scholar]
- 22.Mazure NM, Chauvet C, Bois-Joyeux B, Bernard MA, Nacer-Cherif H, Danan JL. Repression of alpha-fetoprotein gene expression under hypoxic conditions in human hepatoma cells: characterization of a negative hypoxia response element that mediates opposite effects of hypoxia inducible factor-1 and c-Myc. Cancer Res 62: 1158–1165, 2002 [PubMed] [Google Scholar]
- 23.Munoz-Cabello AM, Toledo-Aral JJ, Lopez-Barneo J, Echevarria M. Rat adrenal chromaffin cells are neonatal CO2 sensors. J Neurosci 25: 6631–6640, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Narravula S, Colgan SP. Hypoxia-inducible factor 1-mediated inhibition of peroxisome proliferator-activated receptor alpha expression during hypoxia. J Immunol 166: 7543–7548, 2001 [DOI] [PubMed] [Google Scholar]
- 25.Nurse CA, Buttigieg J, Brown S, Holloway AC. Regulation of oxygen sensitivity in adrenal chromaffin cells. Ann NY Acad Sci 1177: 132–139, 2009 [DOI] [PubMed] [Google Scholar]
- 26.Patrick SW, Schumacher RE, Benneyworth BD, Krans EE, McAllister JM, Davis MM. Neonatal abstinence syndrome and associated health care expenditures: United States, 2000–2009. J Am Med Assoc 307: 1934–1940, 2012 [DOI] [PubMed] [Google Scholar]
- 27.Piskuric NA, Brown ST, Zhang M, Nurse CA. Glucosensing in an immortalized adrenomedullary chromaffin cell line: role of ATP-sensitive K+ channels. Neurosci Lett 445: 94–98, 2008 [DOI] [PubMed] [Google Scholar]
- 28.Salman S, Brown ST, Nurse CA. Chronic nicotine induces hypoxia inducible factor-2α in perinatal rat adrenal chromaffin cells: Role in transcriptional upregulation of KATP channel subunit Kir6.2. Am J Physiol Cell Physiol 302: C1531–C1538, 2012 [DOI] [PubMed] [Google Scholar]
- 29.Salman S, Buttigieg J, Nurse CA. Ontogeny of O2 and CO2//H+ chemosensitivity in adrenal chromaffin cells: role of innervation. J Exp Biol 217: 673–681, 2014 [DOI] [PubMed] [Google Scholar]
- 30.Salman S, Buttigieg J, Zhang M, Nurse CA. Chronic exposure of neonatal rat adrenomedullary chromaffin cells to opioids in vitro blunts both hypoxia and hypercapnia chemosensitivity. J Physiol 591: 515–529, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Seidler FJ, Slotkin TA. Adrenomedullary function in the neonatal rat: responses to acute hypoxia. J Physiol 358: 1–16, 1985 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Seidler FJ, Slotkin TA. Ontogeny of adrenomedullary responses to hypoxia and hypoglycemia: role of splanchnic innervation. Brain Res Bull 16: 11–14, 1986 [DOI] [PubMed] [Google Scholar]
- 33.Slotkin TA, Seidler FJ. Adrenomedullary catecholamine release in the fetus and newborn: secretory mechanisms and their role in stress and survival. J Dev Physiol 10: 1–16, 1988 [PubMed] [Google Scholar]
- 34.Thompson RJ, Jackson A, Nurse CA. Developmental loss of hypoxic chemosensitivity in rat adrenomedullary chromaffin cells. J Physiol 498: 503–510, 1997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Toschi A, Lee E, Gadir N, Ohh M, Foster DA. Differential dependence of hypoxia-inducible factors 1 alpha and 2 alpha on mTORC1 and mTORC2. J Biol Chem 283: 34495–34499, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Vandenbergh DJ, Mori N, Anderson DJ. Co-expression of multiple neurotransmitter enzyme genes in normal and immortalized sympathoadrenal progenitor cells. Dev Biol 148: 10–22, 1991 [DOI] [PubMed] [Google Scholar]
- 37.Wittert G, Hope P, Pyle D. Tissue distribution of opioid receptor gene expression in the rat. Biochem Biophys Res Commun 218: 877–881, 1996 [DOI] [PubMed] [Google Scholar]
- 38.Wykoff CC, Beasley NJ, Watson PH, Turner KJ, Pastorek J, Sibtain A, Wilson GD, Turley H, Talks KL, Maxwell PH, Pugh CW, Ratcliffe PJ, Harris AL. Hypoxia-inducible expression of tumor-associated carbonic anhydrases. Cancer Res 60: 7075–7083, 2000 [PubMed] [Google Scholar]
- 39.Xu L, Enyeart JJ. Properties of ATP-dependent K+ channels in adrenocortical cells. Am J Physiol Cell Physiol 280: C199–C215, 2001 [DOI] [PubMed] [Google Scholar]
- 40.Yuan G, Peng YJ, Reddy VD, Makarenko VV, Nanduri J, Khan SA, Garcia JA, Kumar GK, Semenza GL, Prabhakar NR. Mutual antagonism between hypoxia-inducible factors 1alpha and 2alpha regulates oxygen sensing and cardio-respiratory homeostasis. Proc Natl Acad Sci USA 110: E1788–E1796, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 41.Zagon IS, McLaughlin PJ. Effects of chronic morphine administration on pregnant rats and their offspring. Pharmacology 15: 302–310, 1977 [DOI] [PubMed] [Google Scholar]
- 42.Zhang Q, Tang X, Zhang ZF, Velikina R, Shi S, Le AD. Nicotine induces hypoxia-inducible factor-1alpha expression in human lung cancer cells via nicotinic acetylcholine receptor-mediated signaling pathways. Clin Cancer Res 13: 4686–4694, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]