

Abstract
Several serum markers are used to assess hepatocyte damage, but they have limitations related to etiology specificity and prognostication. Identification of novel hepatocyte-specific biomarkers could provide important prognostic information and better pathogenesis classification. We tested the hypothesis that hepatocyte-selective biomarkers are released after subjecting isolated mouse hepatocytes to Fas-ligand-mediated apoptosis. Proteomic analysis of hepatocyte culture medium identified the mitochondrial matrix protein carbamoyl phosphate synthetase-1 (CPS1) among the most readily detected proteins that are released by apoptotic hepatocytes. CPS1 was also detected in mouse serum upon acute challenge with Fas-ligand or acetaminophen and in hepatocytes upon hypoosmotic stress, independent of hepatocyte caspase activation. Furthermore, CPS1 was observed in sera of mice chronically fed the hepatotoxin 3,5-diethoxycarbonyl-1,4-dihydrocollidine. Mouse CPS1 detectability was similar in serum and plasma, and its half-life was 126 ± 9 min. Immune staining showed that CPS1 localized to mouse hepatocytes but not ductal cells. Analysis of a few serum samples from patients with acute liver failure (ALF) due to acetaminophen, Wilson disease, or ischemia showed readily detectable CPS1 that was not observed in several patients with chronic viral hepatitis or in control donors. Notably, CPS1 rapidly decreased to undetectable levels in sera of patients with acetaminophen-related ALF who ultimately recovered, while alanine aminotransferase levels remained elevated. Therefore, CPS1 becomes readily detectable upon hepatocyte apoptotic and necrotic death in culture or in vivo. Its abundance and short serum half-life, compared with alanine aminotransferase, suggest that it may be a useful prognostic biomarker in human and mouse liver injury.
Keywords: hepatocytes, apoptosis, necrosis, acetaminophen, alanine aminotransferase, HMGB1
a multitude of etiologies can cause or predispose to liver injury including toxins (e.g., drugs, alcohol), hepatotropic viruses, and genetic factors. Such injury may be acute or chronic and involves apoptotic or necrotic cell death. Furthermore, some patients with acute or chronic liver injury may develop severe or progressive disease culminating in liver failure or even death unless rescued by liver transplantation (10). There are few biomarkers available that are used in clinical settings to accurately measure the extent of hepatocyte damage. For example, alanine aminotransferase (ALT), which is routinely used as a marker of hepatocellular damage, is expressed in two isoforms (ALT1 and ALT2) and exhibits differential tissue distribution (9). Specifically, ALT 1 is expressed in skeletal muscle, kidney, and heart in addition to liver, whereas ALT2 is not expressed in liver but is found in skeletal muscles, heart, and pancreas (9). However, the standard ALT assay used in clinical settings does not discriminate between the different ALT isoforms or ALT from different tissues (5). In addition, the caspase-cleaved keratin 18-exposed neoepitope (M30) and another keratin 18 protein backbone epitope (M65) have been investigated as serum markers of acute (19) and chronic liver disease (7) and appear to be useful biomarkers. However, they are not specific to liver disease (4, 16, 22) because of the broad distribution of the abundant cytoskeletal keratin 18 in all simple-type epithelia (15). Importantly, it is difficult to predict which patients who present with acute liver failure (ALF) are likely to survive without the need for transplantation. The use of the Model for End-Stage Liver Disease and Kings College Criteria remain imperfect (1, 12) although a proposed index that includes M30 and clinical markers may offer some advantages (19). Therefore, the identification of novel hepatocyte-specific biomarkers will greatly benefit the diagnosis and prognosis of various acute and chronic liver diseases.
Carbamoyl phosphate synthetase-1 (CPS1) is the most abundant protein in liver mitochondria, and it accounts for nearly 20% of the matrix protein mass but it is expressed in much lower levels in intestine, kidney, and fibroblasts (6, 11, 18). In the mitochondrial matrix, CPS1 catalyzes the conversion of ammonia and bicarbonate into carbamoyl phosphate, which is the first and rate-limiting step in the urea cycle (11). CPS1 deficiency has been linked to urea cycle disorders (11) but little is known regarding the potential utility of CPS1 as a liver marker. For example, it was identified as the antigen for hepatocyte paraffin-1 (Hep Par-1) antibody, which is used in some surgical pathology settings to confirm the hepatocellular origin of neoplasms (2). In addition, CPS1 was found in serum or plasma of sepsis animal models and plasma of human septic patients, suggesting that it might serve as a serum marker for detecting mitochondrial injury of the liver and/or small intestine under septic conditions (3, 21, 24). In addition, CPS1 was detected as one of several markers in acetaminophen-intoxicated mouse plasma but not in human patients who have relatively limited hepatotoxicity (6). The mechanism of mitochondrial CPS1 depletion in liver is unknown but it has been suggested that it might be related to removal of damaged mitochondria by autophagy (3, 24). However, whether CPS1 release is a result of hepatocyte cell death due to apoptosis, necrosis, or other form of liver injury is currently unknown. In addition, it is unknown whether CPS1 is released and detected in human conditions beyond sepsis.
In the present study, we used an unbiased approach to identify proteins that are released into culture medium upon exposure of primary mouse hepatocyte cultures to Fas ligand (FasL). CPS1 was identified as a major protein released into the medium at a level similar to that seen with albumin. We then pursued this finding by examining the extent of CPS1 release in mouse and human liver injury. Overall, our findings point to CPS1 as a robust biomarker in acute liver injury (ALI) that is rapidly cleared from serum and may serve as prognostic biomarker in ALI.
MATERIALS AND METHODS
Isolation and treatment of primary mouse hepatocytes.
Hepatocytes were isolated from mice for primary culture as previously described (20) with the following modifications. The liver was perfused with 3–5 ml of perfusion medium through the portal vein with a flow rate of 3 ml/min, followed by perfusion with 15–20 ml of digestion medium containing 150 units/ml of collagenase-II (Worthington) at the same flow rate. After the first wash, the cell pellet was suspended in 6 ml of ice-cold wash medium and separated on ice-cold Percoll gradient (Sigma, 15% in PBS pH 7.4, 500 rpm for 10 min) to remove dead cells. The cell pellet was washed with ice-cold wash medium and suspended in culture medium (Williams' medium E supplemented with 10% heat inactivated FBS and 1% penicillin-streptomycin, 37°C) and plated [0.20 × 106 cells/ml on collagen-I-coated 6-well plates (BD BioCoat)] for subsequent analysis of the released proteins and the biochemical experiments. For immunostaining, cells were plated on four-well collagen-I coated chamber slides (BD BioCoat). After 1 h (37°C, 5% CO2) to allow attachment, the cell culture medium was replaced (to remove any debris or unattached cells) with fresh medium and allowed to equilibrate for an additional 12 h (37°C, 5% CO2). The cultured hepatocytes were then treated with FasL (0.5 μg/ml, BD Pharmingen), acetaminophen (APAP) (1 μM, Sigma) or hypoosmotic medium (200 mosmol/l) in the absence of fetal bovine serum. Notably, the 200 mosmol/l hypoosmotic treatment results in cell leakiness (based on uptake of dye) without any evidence of cell death (25).
Toxin administration and blood analysis in mice.
For the in vivo experiments, FVB nontransgenic mice were fasted overnight followed by intraperitoneal administration of FasL (0.15 μg/g), APAP (700 mg/kg), or vehicle (saline or DMSO, respectively) or fed with 3,5-diethoxycarbonyl-1,4-dihydrocollidine (without fasting). After 4 h (FasL injected) or 8 h (APAP injected) or at the indicated time after 3,5-diethoxycarbonyl-1,4-dihydrocollidine (DDC) feeding, mice were euthanized by CO2 inhalation and livers were isolated and divided into pieces that were stored in liquid nitrogen (for biochemical analysis) or fixed in 10% formalin (for hematoxylin and eosin staining). A total of six mice per treatment (FasL or APAP), and two additional control mice, were used to detect CPS1 release in mouse serum. Three other separate mice were used for the serum and plasma comparisons. To determine the half life of CPS1, five mice were used with serial (12, 24, 28, 32, and 36 h) tail vein blood draws from each mouse (after determination of the likely time range in a pilot experiment). For DDC feeding, three mice per time point were used. Blood samples were collected from the mice by intracardiac puncture and stored overnight (4°C) before enzyme or biochemical analysis. Serum ALT levels were determined with a VetScan VS2 instrument (ABAXIS) using the comprehensive diagnostic profile cartridge or were measured by using the Unit for Laboratory Animal Medicine (ULAM) core facility at the University of Michigan. All mice received humane care, and animal use was performed in accordance with a protocol that was approved by the Committee for the Use and Care of Animals at the University of Michigan.
Preparation of liver and hepatocyte lysates and biochemical analysis.
Total hepatocyte and liver tissue lysates were prepared by homogenizing the cells or tissues, respectively, with 2× Tris-glycine SDS sample buffer. Sera were also mixed with 2× SDS-containing sample buffer before analysis. Proteins were separated by SDS-polyacrylamide gel electrophoresis (SDS-PAGE), then stained with Coomassie blue or transferred to polyvinylidene fluoride membranes followed by blotting with antibodies to CPS1 (Abcam), active caspases 3 and 7 (Cell Signaling), high mobility group box 1 protein (HMGB1) (Abcam), actin (Lab Vision), keratin 18 (8), and keratin 19 (23).
Immunofluorescence staining and confocal microscopy imaging.
Immunostaining of hepatocytes or liver tissue sections were carried out as described (8). In brief, hepatocytes or liver sections were fixed with methanol (10 min, −20°C) and air dried (1 h). Nonspecific binding was blocked by incubation in blocking buffer [phosphate-buffered saline (PBS) with 2.5% wt/vol bovine serum albumin and 2% goat serum] for 10 min. For hepatocyte staining, anti-CPS1 antibody was incubated in blocking buffer for 1 h at 22°C. For triple immunostaining of liver sections, CPS1 antibody was coincubated overnight (4°C) with antibodies to keratin 18 or keratin 19. After three 5-min washes in PBS, slides were incubated with Alexa Fluor-conjugated secondary antibody (30 min). The slides were washed three times (5 min/wash) in PBS, air dried, and mounted in ProLong Gold containing DAPI (Invitrogen) for nuclear staining. Cells were imaged by use of a laser-scanning confocal microscope (FluoView 500; Olympus) with a ×60 oil immersion (1.4 NA) objective.
Analysis of the medium from cultured hepatocytes.
After hepatocyte treatments, the culture plates were centrifuged (1,200 rpm, 5 min) followed by collection of the culture medium without disturbing the cells. The collected medium was repelleted (1,200 rpm, 5 min) and the supernatant was concentrated by using Centricon YM-10 filters (Millipore; typically, 2 ml was concentrated to 0.1 ml) then mixed with 2× SDS-PAGE sample buffer for subsequent immunoblotting.
Human serum samples.
Serum samples were obtained from patients hospitalized at the University of Michigan Medical center with the diagnosis of ALI and enrolled in the Acute Liver Failure Study Group Registry. Blood samples were collected daily during hospitalization from patients with ALI due to APAP (4 patients), ischemia-related injury (1 patient), and Wilson disease (1 patient). Serum was also analyzed from patients with chronic hepatitis B (6 patients) and C (6 patients) who attend the Liver Clinic at the University of Michigan. In addition, available randomly selected unused serum was obtained from the Hospital Chemistry Laboratory from individuals who had normal serum ALT levels and was classified as Control Serum. No donor identifiers were collected and the underlying reason for blood collection from these individuals is not known. Serum collection and analysis were carried out under protocols approved by the University of Michigan Human Subjects Committee.
Mass spectrometry.
Coomassie-stained gel bands were excised, destained, and digested with trypsin overnight. The peptides were extracted from the gel bands, ziptipped (C18 cleanup), spotted on the MALDI target, then analyzed by MALDI TOF/TOF. Peptides were identified by searching the UniProt mouse database with the Mascot V2.2 search tool.
Statistical analysis.
Statistical analyses were performed by ANOVA (for cell count experiments) and column statistics (to determine the average half-life) with GraphPad Prism 6 statistical software. For the time-dependent CPS1 and ALT turnover analysis, a linear mixed model was performed using IBM SPSS Statistics software to test the significance of the change-over-time slopes of the CPS1 vs. ALT groups accounting for within-subject correlation. The comparison was done by using log transformed outcome.
Ethics approvals.
The approval to conduct the human and animal studies was obtained from the University of Michigan committees for human and animal use, respectively.
RESULTS
CPS1 is a major protein released to culture medium during apoptotic hepatocyte injury.
To identify potential new biomarkers of liver cell death and injury, primary hepatocytes were isolated from mice and treated with FasL to induce apoptosis. After 6 h, the cell culture medium was collected, concentrated, separated by SDS-PAGE, and stained with Coomassie blue dye. Release of many protein bands was observed in the culture medium collected from FasL-treated compared with untreated hepatocytes (Fig. 1A). We focused on the most intense bands (labeled 1–5, Fig. 1A), which were subjected to mass spectrometry analysis with the identity of the bands included in Fig. 1A. We selected CPS1 for further detailed study because of its high abundance and selective expression in the liver, unique localization in mitochondria, and, importantly, the fact that it has been not reported as a biomarker for apoptotic cell death (albeit it has been described as a released protein in sepsis models). We then confirmed the mass spectrometry prediction by immunoblotting the concentrated hepatocyte culture medium with an antibody specific to CPS1 (Fig. 1B, left). The presence of CPS1 in the culture supernatants occurred in association with the predicted FasL-mediated apoptosis as evident by caspases 3 and 7 activation and generation of the caspase-cleaved keratin 18 apoptotic fragment (Fig. 1B, right, band highlighted by arrow). These findings indicate that primary hepatocytes release CPS1 during apoptotic mouse hepatocellular injury. Importantly, the extracellular released CPS1 is readily visualized by Coomassie stain and is similarly abundant to the released albumin (Fig. 1A).
Fig. 1.

Primary mouse hepatocytes release carbamoyl phosphate synthetase-1 (CPS1) during Fas ligand (FasL)-induced apoptotic injury. A: Coomassie stain of concentrated culture medium collected from FasL-treated and untreated hepatocytes. FasL-treated hepatocytes showed many proteins that were released into the culture medium, compared with untreated cells. Mass spectrometry identified the 5 most intense protein bands (1–5) as indicated. B, left: CPS1 immunoblot of culture medium confirms the mass spectrometry prediction of band 1. B, right: immunoblot of total cell lysates from FasL-treated and untreated hepatocytes. The antibodies were directed to active caspases 3 and 7 (cas-3 and cas-7, respectively); to actin; and to keratin 18 (K18), recognizing K18 and its apoptotic fragment (arrow). Equal proportions of the concentrated culture medium and an equal amount of the total lysate protein were analyzed. The displayed Coomassie stain and immunoblot findings were from a single mouse hepatocyte isolation experiment, with the findings being essentially identical in 2 additional separate experiments.
Mouse primary hepatocytes and intact liver release CPS1 during different types of cell injury.
We compared CPS1 release in response to FasL with other different types of hepatocyte injury that do not involve apoptosis per se. For this, isolated mouse primary hepatocytes were challenged with hypoosmotic stress (selected as a form of mechanical stress), APAP (selected as a form of necrotic stress), or FasL. Analysis of the concentrated culture medium showed a Coomassie-stained band that is at the predicted molecular size of CPS1 (nearly 160 kDa), which was confirmed by immunoblotting with anti-CPS1 antibody (Fig. 2A). APAP treatment of hepatocytes results in cell death via necrosis, which is confirmed by absence of caspase activation and release of the nuclear protein HMGB1 (Fig. 2A). Notably, CPS1 is released during hypoosmotic conditions without any evidence for necrosis or apoptosis (Fig. 2, A and B), thereby suggesting that the hepatocytes become leaky though the mechanism of release is not known. We assessed the extent of hepatocyte death after each treatment (performed in Fig. 2A) by Trypan blue staining. In the absence of HMGB1 release or caspase activation, hypoosmotic treatment showed no detectable cell death (Fig. 2B), whereas CPS1 release was readily observed (Fig. 2A). In contrast, FasL and APAP treatment showed significant cell death (Fig. 2B) in association with CPS1 release. Immune staining further verified the release of CPS1 from hepatocytes in response to the different challenges shown in Fig. 2C. As shown in Fig. 2C, untreated control hepatocytes showed punctae that are dense and uniformly distributed throughout the cytoplasm (Fig. 2C). All three challenges resulted in loss of cytoplasmic CPS1 staining (Fig. 2C, arrows). Collectively these data suggest that CPS1 release occurs in response to different modes of hepatocyte injury that may (FasL, APAP) or may not (hypoosmosis) result in cell death.
Fig. 2.

Primary hepatocytes release CPS1 in response to different types of liver injury. A: primary mouse hepatocytes were cultured in the presence of hypoosmotic medium (200 mosmol/l), FasL (0.5 μg/ml), and acetaminophen (APAP, 1 mM). The concentrated cell culture medium (25 μl) and total cell lysates were used for immunoblotting with antibodies to the indicated proteins. B: after each treatment, viable cell count was determined by Trypan blue staining after trypsinization. Each bar represents mean counts of 10–15 fields. ***P < 0.001 (compared with control cells). C: mouse hepatocytes were challenged with FasL, APAP, and hypoosmosis as in A. Cells were fixed and immunostained with CPS1 (red) and counterstained with DAPI (blue). Arrows highlight areas with less dense CPS1 staining. Scale bar = 20 μm. The displayed results were from a single mouse hepatocyte isolation with similar results obtained from 2 additional independent experiments.
Next, we examined which epithelial cell in the liver (hepatocytes and/or ductal cells) expresses CPS1. As shown in Fig. 3Ac, CPS1 clearly colocalizes with the simple epithelial marker keratin 18 (14) but some keratin 18 staining cells (green stain) were clearly CPS1 negative. The CPS1-negative cells were shown to be ductal based on the absence of CPS1 colocalization with the ductal cell selective marker, keratin 19 (23; Fig. 3Bc). These results suggest that CPS1 is selectively expressed in hepatocytes in the liver.
Fig. 3.

Hepatocyte-selective CPS1 expression in the liver. A: liver sections from normal mice were fixed in methanol (−20°C, 10 min) then stained with antibodies to CPS1 (red) and keratin 18 (green) followed by DAPI (blue, nuclei). B: duplicate slides to those used in A were stained with antibodies to CPS1 (red) and keratin 19 (green). Note that ductal cells exclude CPS1 staining (merged, C). Scale = 50 μm. All images are representative of similar images obtained from 3 separate livers.
We then tested whether CPS1 can be detected in mouse serum in response to apoptotic and necrotic liver injury. Mice were injected with FasL (to induce apoptosis) or with APAP (to induce necrosis) or were injected with vehicle. The serum and liver histology were then analyzed. CPS1 was readily detected in the serum of the mice when serum ALT levels are significantly elevated (Fig. 4A). Activation of caspases in the total liver lysates confirms the apoptosis in FasL-treated mice but not in the APAP-intoxicated mice (Fig. 4A). The relatively low serum CPS1 levels in APAP-treated mice compared with FasL-treated mice (Fig. 4A) likely reflect the extent of liver injury and the rapid dynamics of CPS1 (see discussion of CPS1 half-life below). Histological analysis of liver sections showed marked liver hemorrhage in mice treated with FasL (Fig. 4B, upper right, arrows) and necrotic cell death in mice treated with APAP (Fig. 4B, lower right, arrows) compared with control livers.
Fig. 4.

CPS1 is released into serum during FasL- and APAP-induced mouse liver injury. Mice were injected with FasL (0.15 μg/g) and APAP (700 mg/kg) and euthanized after 4 h and 8 h, respectively. Liver damage was analyzed serologically, biochemically, and histologically. A: immunoblots of serum CPS1, high mobility group box 1 protein (HMGB1), and caspase activation using total liver lysate (each lane represents an independent serum and liver sample from separate mice). The Coomassie stain of serum IgG heavy chain (just above the 50-kDa marker) and IgG light chain (25 kDa), and an actin blot of total liver lysates are included as loading controls. Serum alanine aminotransferase (ALT) of the individual mice is also indicated (bottom). The experiment was repeated using 3 additional mice and demonstrated similar results (not shown). Con, control. B: hematoxylin and eosin-stained liver sections. FasL treatment (b) shows severe hemorrhage formation (arrows), whereas APAP (d) induces necrotic cell death (arrows) compared with control vehicle treatments (a and c). Scale bar = 40 μm.
A time course analysis shows that CPS1 is readily detected in serum as early as 2 h after mouse apoptotic liver injury (Fig. 5A) with parallel activation of caspases (Fig. 5B), thereby suggesting that CPS1 is released during apoptotic hepatocyte injury. Both serum and plasma are equally effective in detecting CPS1 in the blood after FasL-induced liver injury (Fig. 5C). Collectively, these data clearly indicate that CPS1 can be detected in serum during apoptotic and necrotic liver injury in mice. We then determined the approximate half-life of CPS1 in mouse serum in response to FasL treatment by collecting serial serum samples from five independent animals (Fig. 6, A–D; results from four animals are shown). The mean half-half was estimated to be 126 ± 9 min, which reflects (in the utilized experimental system) combined turnover and any ongoing release.
Fig. 5.

Kinetics of CPS1 release in apoptotic mouse liver injury. Mice were injected with FasL (0.15 μg/g), and euthanized at the indicated time points followed by serum collection. A: time-dependent serum CPS1 release into blood. Serum CPS1 was detected as early as 2 h after FasL injection and the levels increased with time. Parallel to CPS1 levels, serum ALT also increased in a time-dependent manner. Coomassie stain of serum IgG is included to show equal protein loading. Each lane represents serum from an individual animal. B: livers were isolated from the same animals used in A and homogenized in SDS-containing sample buffer to obtain a total liver homogenate. The liver lysates were analyzed by blotting with antibodies to the indicated antigens. The keratin 18 (K18) blot is included as a loading control. Collectively these findings indicate that CPS1 release is commensurate with onset of apoptosis. C: serum and plasma are equally suited to detect CPS1 release into the circulation. Mice were injected with FasL (0.15 μg/g) and blood was collected from the euthanized mice after 4 h. A separate mouse (Con) was injected with saline and used as a control. Blood from each mouse was divided and processed to obtain serum (S) and plasma (P). H, heavy chain; L, light chain. Coomassie stain of serum IgG is included as loading control. The serum ALT of mice is indicated.
Fig. 6.

CPS1 has a short half-life in mouse serum. To determine the half-life of CPS1 in mouse serum, 5 mice were injected with FasL (0.075 μg/g) and blood was collected from the same animal via tail vain at the indicated time points. Sera were used to determine serum ALT or mixed with 2 × SDS sample buffer to measure circulating serum CPS1 by immunoblot analysis. Relative CPS1 pixel intensity (CPS1 OD) at each time point was determined by densitometry scanning. A–D: immunoblots of time-dependent serum CPS1 levels of 4 individual mice are shown (the 5th mouse analysis gave a similar overall rapid dropoff of CPS1, not shown). CPS1 OD and serum ALT values at each time point are indicated. A Coomassie stain of serum is included as loading control. To determine the half-life of serum CPS1, the OD values of the 24 h to 28 h and 28 h to 32 h time points of all 5 mice were used. Assuming an exponential decay, the half-life of CPS1 for each time interval was calculated by using the equation Ending amount = Beginning amount/2n; where n = number of half-lives. The average of 10 half-lives of CPS1 in mouse serum was determined to be 126 ± 9 min (mean ± SE).
CPS1 is present in human sera of patients with acute liver failure.
We evaluated the possibility of detecting CPS1 in human sera from patients suffering from ALF due to APAP, ischemia, or Wilson disease. Serial daily serum samples (from day 1 of hospitalization) were obtained and analyzed for CPS1 by immunoblotting. In the APAP patient, serum CPS1 was readily detectable at day 1 then decreased at day 2 and became completely undetectable from days 3 to 5 (Fig. 7A). Notably, while CPS1 was undetectable in this patient at days 3–5, serum ALT levels remained significantly elevated (Fig. 7A). In the case of the patient with ALF related to ischemia, the serum CPS1 was elevated on day 1 (Fig. 7B) and also became undetectable while serum ALT remained very high. Both patients recovered from their ALF without the need for liver transplantation. In contrast, the patient with Wilson disease developed a rapid rise in serum CPS1 and ALT on day 6 (Fig. 7C) and unfortunately subsequently died from complications related to ALI. These data suggest that serum CPS1 is readily detectable in sera of human patients with ALF, while serum ALT levels are also very high, but CPS1 level drop precipitously with ALT values remaining elevated.
Fig. 7.

CPS1 is detected in sera of human patients with acute liver failure (ALF). The presence of CPS1 in serum of human patients with different forms of ALF was tested by using serum collected from day 1 up to day 6 of hospitalization. A: analysis of CPS1 in serial serum samples collected from a patient with APAP. Sera were separated on duplicate gels by SDS-PAGE, with 1 gel stained by Coomassie blue to visualize the immunoglobulin heavy (H) and light (L) chains. The second gel was transferred to a membrane for subsequent blotting using anti-CPS1 antibody. ALT values from the same serum samples are also included. B and C: serial serum samples from a patient with ischemic hepatitis and with Wilson disease, respectively, were analyzed as in A.
Serum CPS1 turns over rapidly in patients with APAP-related ALF.
We explored further the rapid drop in serum CPS1 that was suggested in the two patients with APAP and ischemia (Fig. 7, A and B) and compared this to changes in serum ALT. For this we analyzed serial serum samples from three additional patients with APAP-related ALF. As shown in Fig. 8, A–C, serum CPS1 was readily detectable on day 1 of hospitalization when serum ALT is significantly elevated. Consistently, CPS1 disappeared rapidly while serum ALT levels remained elevated. Densitometric analysis of CPS1 immunoblots was performed and the percent remaining intensity of CPS1 (relative to day 1) was compared with the percent remaining ALT values by log transformed outcome analysis. As shown in Fig. 8D, the change-over-time slope of the decrease in serum CPS1 level occurs at a significantly faster rate compared with that of ALT (P = 0.019) [all four patients (one shown in Fig. 7 and three shown in Fig. 8) recovered]. This analysis, albeit preliminary, suggests that CPS1 might be a sensitive prognostic marker for recovery in some cases of ALI.
Fig. 8.

CPS1 turns over more rapidly than ALT in patients with APAP-related ALF. A–C: immunoblots of serum CPS1 from patients with APAP-related ALF (sera were collected on days 1 to 7 of hospitalization; days 4–7 were not available for the patient in C). The Coomassie stain of serum indicates equal loading (H, heavy chain; L, light chain). ALT values from the same serum samples are also shown. D: %CPS1 and %ALT values compared with day 1 values are displayed as loge to achieve a linear turnover pattern, and the significance of the change-over-time slopes of the CPS1 vs. ALT groups was determined (P = 0.019). CPS1 relative levels were obtained by densitometry scanning. The serial serum samples from 4 APAP patients (3 shown in panels A–C, and 1 shown in Fig. 7A) were used for quantification.
Serum CPS1 in mouse and human chronic liver diseases.
We tested the possibility of detecting serum CPS1 in chronic liver injury in mice and humans. To test CPS1 release during chronic mouse liver injury, mice were fed 0.1% DDC (a porphyrinogenic hepatotoxin) for 10 days, 15 days, 6 wk, and 3 mo and serum CPS1 levels were analyzed. Serum CPS1 levels were highest at the earlier and more acute period of liver injury (10 days), which correlated in general terms with the ALT levels (Fig. 9A). As expected, control mouse serum showed no detectable CPS1, and there were no detectable levels of the necrosis marker HMGB1 at any of the time points tested (Fig. 9A). Histological assessment of the livers showed early evidence of cell dropoff at days 10 and 15 with subsequent stabilization as injury becomes chronic (Fig. 9B).
Fig. 9.

Serum CPS1 in chronic liver injury. A: the presence of serum CPS1 and HMGB1 was tested in animals exposed to 3,5-diethoxycarbonyl-1,4-dihydrocollidine (DDC) for 10 days (10D), 15 days (15D), 6 wk (6W), and 3 mo (3M) (2 mice per treatment duration). The corresponding serum ALT values are included. Coomassie stain of serum IgG shows equal serum protein loading. Each lane corresponds to serum from separate mice. B: hematoxylin and eosin staining of livers from control and DDC-administered mice. Scale bar = 40 μm. C and D: human sera from patients with chronic hepatitis B virus infection (HBV), chronic hepatitis C virus infection (HCV), or controls were analyzed for the presence of CPS1 and HMGB1. Serum from a patient with APAP-related ALF and from control donors with normal ALT were used as positive and negative controls, respectively. Coomassie stain of serum showed similar protein loading. Each lane represents separate hepatitis B or C patient samples (the controls and APAP samples in C and D are identical).
We also tested the presence of CPS1 in the sera of patients with chronic hepatitis B and C, compared with serum from controls who had normal ALT. In contrast to DDC-induced mouse chronic liver injury, there was no readily detectable CPS1 in any of the serum sample of patients with hepatitis B or C (Fig. 9, C and D). In these patients, serum ALT varied but was elevated in most of the cases (Fig. 9, C and D) although not to the same extent as in the ALF cases shown in Figs. 7 and 8. As expected, sera from the controls also showed no detectable serum CPS1. Consistent with the mouse data, there was no measurable serum HMGB1 in any of the patient serum samples (Fig. 9, C and D). These data suggest that serum CPS1 is unlikely to be a useful marker in the context of chronic human viral hepatitis, although this may be reflected by the extent of liver injury.
DISCUSSION
Our findings from the ex vivo and in vivo mouse liver injury models provide strong evidence that the hepatocyte-selective and most abundant mitochondrial matrix protein CPS1 is a marker for apoptotic and necrotic forms of hepatocyte death and injury. The marked release of CPS1 during hypoosmotic stress (Fig. 2), without evidence of cell death per se, suggests that such release may represent a state of organelle and cell leakiness that does not require cell death. The mechanism of release of CPS1 from the hepatocyte may be multifactorial and likely reflects organelle spillage and cell rupture (e.g., during necrosis) or a nonclassical release that is vesicle (e.g., exosome or microparticle) or nonvesicle related. In addition, prominent release of CPS1 by primary hepatocytes in response to hypoosmosis, apoptosis (FasL) and necrosis (APAP) may be associated with distinct mechanism of release depending on the injury model. This is supported by the observed different combinations of caspase activation and HMGB1 release associated with each hepatocyte injury model (Fig. 2A). During sepsis, it has been suggested that the CPS1 release occurs via autophagy (3, 24); however, the exact mechanism of CPS1 release from hepatocytes during apoptosis, necrosis, and other forms of injury and how CPS1 spills into blood during ALI remain to be investigated. The abundance of CPS1 and relative selective expression in hepatocytes (Ref. 13; Fig. 3) make it an attractive candidate liver injury biomarker. The liver is the dominant human organ for CPS1 expression, with the small intestine having the next highest expression level (13). For example, mRNA Ct values (using qRT-PCR) for CPS1 in human liver were 22.85 whereas those for small intestine were 25.86 (13).
The utility of CPS1 as a potential biomarker of liver injury appears to be limited to acute rather than chronic liver injury in both mice and humans. For example, a sharp rise in serum or plasma CPS1 is observed in ALI in mice due to FasL or APAP administration (Ref. 6; Fig. 4) and has been also described in animals during sepsis (3, 21) or lipopolysaccharide toxicity (24). Similar findings of a dramatic increase in serum CPS1 were noted in human ALF due to APAP, ischemia, and Wilson disease (Fig. 7). In contrast, CPS1 was not detected in patients with chronic viral hepatitis (Fig. 9). Furthermore, serum CPS1 levels decreased in the DDC mouse toxicity model as the injury moved from acute to subacute then chronic (Fig. 9). It is important to highlight that the chronic hepatitis B virus and hepatitis C virus human sera we tested all had ALTs ≤543 U/l (Fig. 9) without the detection of serum CPS1 or HMGB1. It is possible that sera from patients with viral hepatitis (or other chronic liver diseases) with higher ALT levels, or at specific disease activities, may harbor detectable serum CPS1. Therefore, some chronic livers diseases may still be associated with CPS1 release in a context- and ALT level-related manner.
A recent report showed that CPS1 can be detected in mouse serum in response to APAP-induced ALI but not in human serum (6). It is important to note that the human serum samples tested in that study had relatively mild ALT elevation that ranged from 182 to 632 U/l (6). Our findings here clearly demonstrate that to detect CPS1 in human serum, the ALT level need to be markedly elevated, indicating that CPS1 is a biomarker that reflects the severity of liver injury. Based on the APAP-associated human sera in our study, an ALT value ≥2,500 U/l allows detection of serum CPS1 by immunoblotting (derived from the ALT values shown in Fig. 7A and Fig. 8, A–C). However, we caution that any cutoff CPS1 value would need to be taken in the dynamic context of serum ALT and CPS1 turnover and the clinical situation. Analysis of a large cohort of ALI patients will be needed to evaluate the full spectrum of CPS1 potential clinical utility.
Another relevant observation of this study is how rapidly CPS1 was eliminated from serum in patients with ALF (Figs. 7 and 8). It remains to be determined whether this elimination represents degradation by serum proteases or uptake by circulating leukocytes or by endothelial cells. Regardless of the mode of elimination, this finding provides a distinct advantage to CPS1 as a potential early marker of recovery from ALI since all patients who manifested a rapid drop in CPS1 recovered. If validated in a large cohort of patients, then one clear advantage that CPS1 may offer compared with other serum biomarkers such as ALT is its switchlike disappearance whereas ALT levels decrease more gradually (Fig. 10). Such rapid disappearance of CPS1 during ALI patient recovery could be clinically beneficial to predict the subgroup of ALI patients who are likely to survive without the need for liver transplantation and who can have a shortened stay in the intensive care unit. Indeed, the half-life of CPS1 was estimated in rats to be 67 min (based on intravenous injection of purified CPS1; Ref. 17) and estimated in the present study in mice to be 126 min. The estimation of mouse CPS1 half-life (Fig. 6) is likely to be less than 126 min, since our measurement may include potential continuous CPS1 release, but it does reflect a physiological injury context. The half-life of human CPS1 in serum is not known but is likely to be very short based on its rapid disappearance in patients with ALF. Taken together, our findings suggest that CPS1 could prove to be a prognostic biomarker of ALI, but further validation studies are required using a large cohort of patients representing different types of ALI.
Fig. 10.
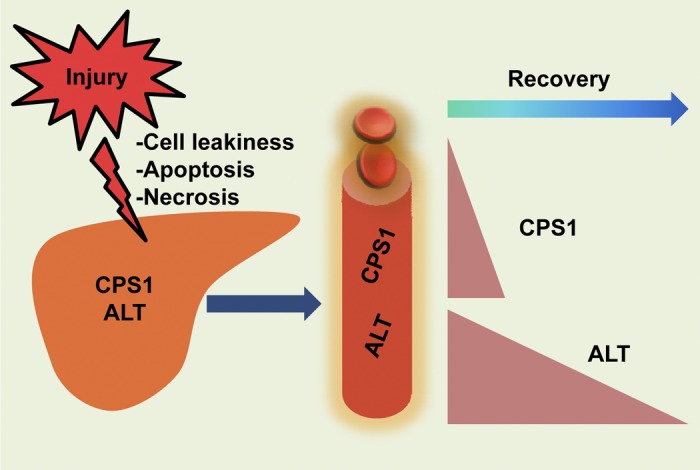
Proposed model for CPS1 and ALT release in the context of acute liver injury. The mitochondrial matrix protein CPS1 and the cytoplasmic protein ALT are released to the blood circulation upon acute liver injury. This occurs in the context of apoptotic (e.g., via FasL) or necrotic (e.g., via APAP) liver injury, or other conditions that cause cell leakiness without cell death (as reflected by the hypoosmotic stress of primary cultured mouse hepatocytes). During recovery from liver injury, serum CPS1 levels drop rapidly while the serum ALT decline is more gradual.
GRANTS
This work was supported by NIH grant DK47918 and the Department of Veterans Affairs (M. B. Omary) and NIH grant DK58369 (R. J. Fontana).
DISCLOSURES
The authors together with the Technology Transfer Office at the University of Michigan have submitted a provisional patent application for the use of CPS1 as a prognostic marker in liver injury.
AUTHOR CONTRIBUTIONS
S.V.W., R.J.F., and M.B.O. conception and design of research; S.V.W. and Y.-J.J. performed experiments; S.V.W. and M.B.O. analyzed data; S.V.W. and M.B.O. interpreted results of the experiments; S.V.W. prepared figures; S.V.W. drafted the manuscript; S.V.W., R.J.F., and M.B.O. edited and revised the manuscript; S.V.W., Y.-J.J., R.J.F., and M.B.O. approved final version of manuscript.
ACKNOWLEDGMENTS
We thank Xi Xia from the Center for Statistical Consultation and Research (CSCAR), University of Michigan, for essential assistance with statistical analysis.
REFERENCES
- 1.Blei AT. Selection for acute liver failure: have we got it right? Liver Transpl 11: 30–34, 2005 [DOI] [PubMed] [Google Scholar]
- 2.Butler SL, Dong H, Cardona D, Jia M, Zheng R, Zhu H, Crawford JM, Liu C. The antigen for Hep Par 1 antibody is the urea cycle enzyme carbamoyl phosphate synthetase 1. Lab Invest 88: 78–88, 2008 [DOI] [PubMed] [Google Scholar]
- 3.Crouser ED, Julian MW, Huff JE, Struck J, Cook CH. Carbamoyl phosphate synthase-1: a marker of mitochondrial damage and depletion in the liver during sepsis. Crit Care Med 34: 2439–2446, 2006 [DOI] [PubMed] [Google Scholar]
- 4.Greystoke A, Dean E, Saunders MP, Cummings J, Hughes A, Ranson M, Dive C, Renehan AG. Multi-level evidence that circulating CK18 is a biomarker of tumor burden in colorectal cancer. Br J Cancer 107: 1518–1524, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Glinghammar B, Rafter I, Lindström AK, Hedberg JJ, Andersson HB, Lindblom P, Berg AL, Cotgreave I. Detection of the mitochondrial and catalytically active alanine aminotransferase in human tissue and plasma. Int J Mol Med 23: 621–631, 2009 [DOI] [PubMed] [Google Scholar]
- 6.Hu Z, Lausted C, Yoo H, Yan X, Brightman A, Chen J, Wang W, Bu X, Hood L. Quantitative liver-specific protein fingerprint in blood: a signature for hepatotoxicity. Theranostics 4: 215–228, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Joka D, Wahl K, Moeller S, Schlue J, Vaske B, Bahr MJ, Manns MP, Schulze-Osthoff K, Bantel H. Prospective biopsy-controlled evaluation of cell death biomarkers for prediction of liver fibrosis and nonalcoholic steatohepatitis. Hepatology 55: 455–464, 2012 [DOI] [PubMed] [Google Scholar]
- 8.Ku NO, Toivola DM, Zhou Q, Tao GZ, Zhong B, Omary MB. Studying simple epithelial keratins in cells and tissues. Methods Cell Biol 78: 489–517, 2004 [DOI] [PubMed] [Google Scholar]
- 9.Lindblom P, Rafter I, Copley C, Andersson U, Hedberg JJ, Berg AL, Samuelsson A, Hellmold H, Cotgreave I, Glinghammar B. Isoforms of alanine aminotransferases in human tissue and serum-differential tissue expression using novel antibodies. Arch Biochem Biophys 466: 66–77, 2007 [DOI] [PubMed] [Google Scholar]
- 10.Malhi H, Guicciardi ME, Gores GJ. Hepatocyte death: a clear and present danger. Physiol Rev 90: 1165–1194, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Martínez AI, Pérez-Arellano I, Pekkala S, Barcelona B, Cervera J. Genetic, structural and biochemical basis of carbamoyl phosphate synthetase 1 deficiency. Mol Genet Metab 101: 311–323, 2010 [DOI] [PubMed] [Google Scholar]
- 12.McPhail MJ, Wendon JA, Bernal W. Meta-analysis of performance of Kings's College Hospital Criteria in prediction of outcome in non-paracetamol-induced acute liver failure. J Hepatol 53: 492–499, 2010 [DOI] [PubMed] [Google Scholar]
- 13.Neill MA, Aschner J, Barr F, Summar ML. Quantitative RT-PCR comparison of the urea and nitric oxide cycle gene transcripts in adult human tissues. Mol Genet Metab 97: 121–127, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Omary MB, Ku NO, Toivola DM. Keratins: guardians of the liver. Hepatology 35: 251–257, 2002 [DOI] [PubMed] [Google Scholar]
- 15.Omary MB, Ku NO, Strnad P, Hanada S. Toward unraveling the complexity of simple epithelial keratins in human disease. J Clin Invest 119: 1794–805, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Oyama K, Fushida S, Kinoshita J, Okamoto K, Makino I, Nakamura K, Hayashi H, Inokuchi M, Nakagawara H, Tajima H, Fujita H, Takamura H, Ninomiya I, Kitagawa H, Fujimura T, Ohta T. Serum cytokeratin 18 as a biomarker for gastric cancer. Clin Exp Med 13: 289–295, 2013 [DOI] [PubMed] [Google Scholar]
- 17.Ozaki M, Terada K, Kanazawa M, Fujiyama S, Tomita K, Mori M. Enzyme-linked immunosorbent assay of carbamoyl phosphate synthetase 1: plasma enzyme in rat experimental hepatitis and its clearance. Enzyme Protein 48: 213–221, 1994 [DOI] [PubMed] [Google Scholar]
- 18.Rapp B, Häberle J, Linnebank M, Wermuth B, Marquardt T, Harms E, Koch HG. Genetic analysis of carbamoylphosphate synthetase I and ornithine transcarbamylase deficiency using fibroblasts. Eur J Pediatr 160: 283–287, 2001 [DOI] [PubMed] [Google Scholar]
- 19.Rutherford A, King LY, Hynan LS, Vedvyas C, Lin W, Lee WM, Chung RT ALF. Study Group. Development of an accurate index for predicting outcomes of patients with acute liver failure. Gastroenterology 143: 1237–1243, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Snider NT, Weerasinghe SV, Singla A, Leonard JM, Hanada S, Andrews PC, Lok AS, Omary MB. Energy determinants GAPDH and NDPK act as genetic modifiers for hepatocyte inclusion formation. J Cell Biol 195: 217–229, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Struck J, Uhlein M, Morgenthaler NG, Fürst W, Höflich C, Bahrami S, Bergmann A, Volk HD, Redl H. Release of the mitochondrial enzyme carbamoyl phosphate synthase 1 under septic condition. Shock 23: 533–538, 2005 [PubMed] [Google Scholar]
- 22.Tas F, Karabulut S, Bilgin E, Sen F, Yildiz I, Tastekin D, Ciftci R, Duranyildiz D. Clinical significance of serum M30 and M65 levels in metastatic pancreatic adenocarcinoma. Tumour Biol 34: 3529–3536, 2013 [DOI] [PubMed] [Google Scholar]
- 23.Toivola DM, Krishnan S, Binder HJ, Singh HK, Omary MB. Keratins modulate colonocyte electrolyte transport via protein mistargeting. J Cell Biol 164: 911–921, 2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Unuma K, Aki T, Matsuda S, Funakoshi T, Yoshida K, Uemura K. Elimination and active extrusion of liver mitochondrial proteins during lipopolysaccharide administration in rat. Hepatol Res 43: 526–534, 2013 [DOI] [PubMed] [Google Scholar]
- 25.Weerasinghe SVW, Ku NO, Altshuler PJ, Kwan R, Omary MB. Mutation of caspase-digestion sites in keratin 18 interferes with filament reorganization, and predisposes to hepatocyte necrosis and loss of membrane integrity. J Cell Sci 127: 1464–1475, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]