

Abstract
Intervening in angiotensin (Ang)-II type 2 receptor (AT2) signaling may have therapeutic potential for bronchopulmonary dysplasia (BPD) by attenuating lung inflammation and preventing arterial hypertension (PAH)-induced right ventricular hypertrophy (RVH). We first investigated the role of AT2 inhibition with PD123319 (0.5 and 2 mg·kg−1·day−1) on the beneficial effect of AT2 agonist LP2–3 (5 μg/kg twice a day) on RVH in newborn rats with hyperoxia-induced BPD. Next we determined the cardiopulmonary effects of PD123319 (0.1 mg·kg−1·day−1) in two models: early treatment during continuous exposure to hyperoxia for 10 days and late treatment starting on day 6 in rat pups exposed postnatally to hyperoxia for 9 days, followed by a 9-day recovery period in room air. Parameters investigated included lung and heart histopathology, fibrin deposition, vascular leakage, and differential mRNA expression. Ten days of coadministration of LP2–3 and PD123319 abolished the beneficial effects of LP2–3 on RVH in experimental BPD. In the early treatment model PD123319 attenuated cardiopulmonary injury by reducing alveolar septal thickness, pulmonary influx of inflammatory cells, including macrophages and neutrophils, medial wall thickness of small arterioles, and extravascular collagen III deposition, and by preventing RVH. In the late treatment model PD123319 diminished PAH and RVH, demonstrating that PAH is reversible in the neonatal period. At high concentrations PD123319 blocks the beneficial effects of the AT2-agonist LP2–3 on RVH. At low concentrations PD123319 attenuates cardiopulmonary injury by reducing pulmonary inflammation and fibrosis and preventing PAH-induced RVH but does not affect alveolar and vascular development in newborn rats with experimental BPD.
Keywords: bronchopulmonary dysplasia, angiotensin II type 2 receptor, lung inflammation, right ventricular hypertrophy, pulmonary hypertension
bronchopulmonary dysplasia (BPD) is a chronic lung disease that is frequently seen in very premature infants with interrupted lung development and lung damage caused by mechanical ventilation and/or reactive oxygen species generated by (prolonged) exposure to supplemental oxygen. The hallmark of BPD is alveolar enlargement due to a permanent alveolar simplification secondary to an arrest in alveolar and vascular development, and a subsequent reduction of the alveolar surface and lung function (5, 17, 24). BPD is complicated by inflammation and oxidative stress in the neonatal period, and progressive disease ultimately results in pulmonary arterial hypertension (PAH), which is characterized by persistent vasoconstriction and structural remodeling of the pulmonary blood vessels with increased proliferation of vascular smooth muscle cells and a fixed reduction in blood vessel lumen. PAH induces right ventricular hypertrophy (RVH) and right heart failure (1, 5). PAH-induced right heart failure in children and adults is associated with a high mortality (1, 2, 37, 40). Newborn rats exposed to hyperoxia and premature infants with severe BPD develop chronic lung inflammation, persistent alveolar simplification, fibrosis, PAH, and RVH (5, 13, 14). The rodent hyperoxia model is valuable in identifying candidate interventions for BPD (4, 13, 14, 38, 47, 49).
Normal regulation of cardiovascular and renal function is affected by the renin angiotensin system and an imbalance between the opposing effector molecules angiotensin II (AngII) and angiotensin-(1–7) [Ang-(1–7)] may contribute to cardiovascular pathogenesis and lung fibrosis (19, 33, 41, 42). AngII is stepwise produced by cleavage of the prohormone angiotensinogen into the NH2-terminal decapeptide AngI by renin, and the conversion of AngI to AngII by angiotensin-converting enzyme (ACE). The ligand of the Mas oncogene receptor (MAS), Ang-(1–7), is generated directly via the conversion of AngII by ACE2 or indirectly via AngI by ACE2 and ACE. AngII exerts its biological effects after binding to angiotensin II type 1 (AT1) or -type 2 receptors (AT2). Stimulation of the ACE-AngII-AT1 axis leads to vasoconstriction, proliferation, and fibrosis in multiple tissues, including the lung. Blocking AT1 or stimulation of the ACE2-Ang-(1–7)-MAS axis counterbalances the detrimental biological actions of AngII by inducing vasodilation and by inhibiting fibrinogenesis, thrombogenesis, hypertension, cardiac hypertrophy, and lung injury (8, 10, 19, 22, 25, 27, 29, 30, 32, 34, 39, 42, 45, 48). The biological response of AT1 activation by AngII may be modulated by receptor dimerization, resulting in aggravation of the response by an AT1-bradykinin type 2 heterodimer and attenuation by a dimer of AT1 and MAS oncogene receptor compared with the AngII-induced response of the AT1 homodimer (23). Alternatively, the detrimental effects of AngII binding to AT1 may be affected by binding to AT2 (36, 43). AT2 signaling can result in either beneficial or adverse effects on proliferation, inflammation, and fibrosis in cardiovascular disease (19, 36, 43), suggesting that knowledge of the role of AT2 signaling in cardiopulmonary disease is still incomplete and controversial. This may be explained by low AT2 expression in adult tissues and the complexity of AT2 signaling that can be dependent on the presence of multiple receptors, including AT1 (6). Potential adverse effects of AT2 signaling in cardiopulmonary disease are demonstrated in mice in which pharmacological blocking of AT2 attenuates bleomycin-induced fibrosis (46). Potential beneficial effects of AT2 signaling in cardiopulmonary disease are demonstrated in AT2-deficient mice that suffer from aggravated acute lung and heart injury compared with wild-type controls (3, 15) and by our recent data that demonstrated the therapeutic potential of an AT2 agonist in newborn rats with experimental BPD (45). The newborn rat is an excellent model to study AT2-dependent signaling in cardiopulmonary disease because of the 100-fold higher level of expression of AT2 in neonatal lung compared with adults (45).
To provide mechanistic data that PD123319 acts on the AT2 receptor by blocking activation of AT2 receptors in neonatal cardiopulmonary disease in vivo we investigated the role of AT2 inhibition with PD123319 (0.5 and 2 mg·kg−1·day−1) on the beneficial effect of AT2 agonist LP2–3 (5 μg/kg twice a day) on RVH in newborn rats with hyperoxia-induced BPD. After finding beneficial effects of treatment with the AT2 antagonist PD123319 (0.1–5 mg·kg−1·day−1) on RVH in a pilot experiment we determined the cardiopulmonary effects of daily treatment with the optimal dose of PD123319 (0.1 mg·kg−1·day−1) in the absence of the AT2 agonist in two animal models of hyperoxia-induced experimental BPD: 1) daily treatment during exposure to 100% oxygen for 10 days and 2) treatment starting on day 6 in rat pups exposed postnatally to hyperoxia for 9 days, followed by a 9-day recovery period in room air (RA).
MATERIALS AND METHODS
Animals
The research protocol was approved by the Institutional Animal Care and Use Committee of the Leiden University Medical Center. Adult Wistar rats (6 mo old; N = 8) were exsanguinated after induction of anesthesia with an intraperitoneal injection of ketamine (50 mg/kg) and xylazine (50 mg/kg). Organs were stored at −80°C until isolation of RNA for real-time RT-PCR.
For each experiment, newborn rat pups from two to three litters were pooled and distributed over the experimental group. For the intervention experiments, newborn rat pups were distributed over two experimental groups (N = 12), i.e., an oxygen and an oxygen-intervention (PD123319 and/or LP2–3) group, and two RA-exposed control groups (N = 6) injected with either saline or PD123319. Pups were fed by foster dams. Foster dams were rotated daily between the oxygen-exposed pups and two groups of RA-exposed pups to avoid oxygen toxicity: 24 h in 100% oxygen and 48 h in RA. Oxygen concentration, body weight, evidence of disease, and mortality were monitored daily.
Early concurrent treatment.
Pups were continuously exposed to 100% oxygen for 10 days from birth onward. Starting on day 2 pups received daily subcutaneous injections of either the optimal concentration of 0.1 mg·kg body wt−1·day−1 (135 nmol·kg−1·day−1) of AT2 antagonist PD123319 (1276, Axon Medchem, Groningen, The Netherlands) in 100 μl of 0.9% saline or 100 μl of 0.9% saline (age-matched control). Lung and heart tissue was collected on day 10.
To find the optimal dosing of PD123319 we performed two pilot experiments: 1) hyperoxia-exposed rat pups were treated with 0.5 or 2 mg·kg−1·day−1 PD123319 and/or the optimal dose of LP2–3, 5 μg/kg twice a day (a gift from Lanthio Pharma, Groningen, The Netherlands; Ref. 45) or saline (N = 8) and 2) hyperoxia-exposed rat pups were treated with 0.1–5 mg·kg−1·day−1 PD123319 or saline (N = 10). Because PD123319 and LP2–3 have a similar half-life in vivo in adult rats of ∼20 min (Ref. 7 and Lanthio Pharma, unpublished results), 2 mg/kg of PD123319 will result in a 480-fold molar excess compared with 5 μg/kg of LP2–3 and is therefore expected to block AT2 in the presence of LP2–3, provided that both AT2 interacting compounds are simultaneously administered. We used the ratio of right and left free ventricular wall thickness (RV/LV) in histological sections of the heart as a readout. This parameter was selected for two reasons: 1) hyperoxia-induced experimental BPD results in persistent PAH-induced RVH (13, 14) and 2) stimulation of AT2 reduces PAH-induced RVH (45). Pups exposed to hyperoxia developed RVH, which could be completely prevented by administration of PD123319 (0.1 mg·kg−1·day−1; Fig. 1B). This concentration of PD123319 was then used in separate experiments for 1) histology (N = 10), 2) lung tissue homogenates (N = 10), 3) bronchoalveolar lavage fluid (BALF, N = 12), and measurement of chemokine-induced neutrophilic chemoattractant-1 (CINC1) in BALF (N = 10). To quantify the degree of RVH, hearts were harvested, followed by removal of the atria. Next, the right ventricular (RV) free wall was dissected, weighed separately from the interventricular septum (IVS) and left ventricle (LV), frozen immediately in liquid nitrogen, and stored at −80°C for RNA isolation. The weight ratio RV/(LV + IVS) was calculated as an indicator of RVH (N = 10). In a separate experiment tissues were collected from 6-mo-old adult rats for RT-PCR (N = 8).
Fig. 1.
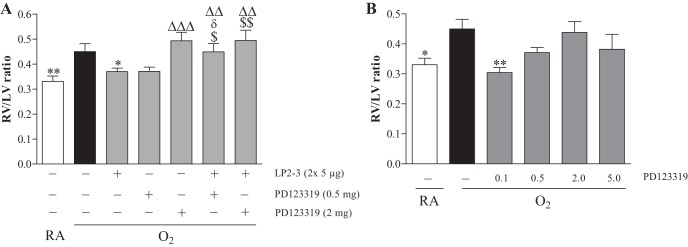
Effect of PD123319 (0.5 and 2 mg·kg−1·day−1) on angiotensin receptor type 2 (AT2) agonist (LP2–3: 5 μg/kg twice a day)-induced prevention of hyperoxia-induced right ventricular hypertrophy (RVH) in newborn rat pups on day 10 after birth by morphometry in paraffin sections stained with hematoxylin and eosin and depicted as right ventricle (RV)/left ventricle (LV) wall thickness ratio (N = 6; A). Pilot experiment to find the optimal dose of PD123319 for treatment of experimental bronchopulmonary dysplasia (BPD) by determining RVH depicted as RV/LV wall thickness ratio on day 10 after birth of rat pups (N = 10; B). Rat pups were exposed to room air (RA) and injected daily with saline (open bars) and of rat pups exposed to hyperoxia (O2) and injected daily with saline (solid bars) or PD123319: 0.1, 0.5, 2, and 5 mg·kg−1·day−1 and/or LP2–3 (shaded bars). Data are expressed as means ± SE. *P < 0.05 and **P < 0.01 vs. own age-matched O2-exposed controls. ΔΔP < 0.05 and ΔΔΔP < 0.001 vs. RA controls. $P < 0.05 and $$P < 0.01 vs. AT2 agonist-treated O2 pups. δP < 0.05 vs. PD123319 (0.5 mg·kg−1·day−1).
Late treatment and recovery.
Lung injury and recovery were investigated by exposing pups to hyperoxia for 9 days, followed by recovery in RA for 9 days. After 6 days of hyperoxia, daily injections with 0.9% saline or 0.1 mg·kg−1·day−1 of PD123319 in 0.9% saline were started and continued throughout the 9-day recovery period in RA. Lung and heart tissue was collected on day 9, after 9 days of hyperoxic lung injury (N = 8), and on day 18, after 9 days of recovery in RA (N = 8).
Histology
Pups were anesthetized with an intraperitoneal injection of ketamine (25 mg/kg body wt; Nimatek, Eurovet Animal Health BV, Bladel, The Netherlands), xylazine (50 mg/kg body wt; Rompun, Bayer, Leverkusen, Germany), and heparin (100 units; Leo Pharma, Breda, The Netherlands, to avoid postmortem fibrin deposition in the lungs) on day 10. After 5 min, pups were exsanguinated by transection of the abdominal blood vessels and the trachea was cannulated (Bioflow 0.6-mm intravenous catheter, Vygon, Veenendaal, The Netherlands) for perfusion fixation of the lungs with buffered formaldehyde (4% paraformaldehyde in PBS, pH 7.4) at 25 cm H2O pressure for 6 min. 15 min after bleeding the heart did not beat anymore when the thorax was opened and lungs and heart were removed. The heart was submerged in cold 0.9% NaCl for cardioplegia, as the heart stops beating and remains in diastole. Hereafter heart and lungs were fixed (additionally) in cold formaldehyde for 24 h at 4°C and embedded in paraffin after dehydration in a graded alcohol series and xylene. Formalin-fixed, paraffin-embedded, 4-μm-thick heart and lung sections were stained with hematoxylin and eosin. Lungs were immunostained additionally with anti-ED-1 (monocytes and macrophages; diluted 1:5), anti-myeloperoxidase (MPO, RB-373-A1, Thermo Fisher Scientific, Fremont, CA; diluted 1:1,500), anti-α smooth muscle actin (ASMA, A2547, Sigma-Aldrich, St. Louis, MO; diluted 1:20,000), anti-von Willebrand factor (vWF, A0082, Dako Cytomation, Glostrup, Denmark; diluted 1:4,000) or anti-COL3A1 (collagen III; no. ab7778; Abcam, Cambridge, UK; diluted 1:3,000) stained with EnVision-HRP (Dako, Glostrup, Denmark), by using the chromogenic substrate NovaRed as recommended by the manufacturer (Vector, Burlingame, CA), and counterstained briefly with hematoxylin by standard methods (14, 45). Extracellular collagen III deposition was quantified on lung sections stained immunohistochemically for collagen III with the NIH ImageJ program by calculating the relative area positive for collagen III at a 400-fold magnification in 10 nonoverlapping fields. Morphometric quantification of alveolar enlargement, vascularization, septal thickness, the pulmonary influx of neutrophils and macrophages, and right ventricular hypertrophy was performed by two independent researchers blinded to the treatment strategy as previously described (20, 45, 49; N = 8).
Fibrin Detection Assay
Quantitative fibrin deposition in lung tissue homogenates was determined by Western blotting, using the monoclonal 59D8 (Oklahoma Medical Research Foundation, Oklahoma City, OK; diluted 1:1,000) and an infrared detection system (Odyssey infrared imaging system, Licor Biosciences) as described previously (44, 45; N = 10).
Bronchoalveolar Lavages, Protein Assay, and Cytokine Measurement
Total protein content in BALF was determined as an indicator of vascular leakage using a standard protein assay (DC Protein Assay, Bio-Rad, Veenendaal, The Netherlands), according to the manufacturer's instructions, using bovine serum albumin (fraction V; Roche Diagnostics, Almere, The Netherlands) as a standard (Ref. 45; N = 12). In BALF CINC1 protein was measured with a commercially available ELISA (catalog no. ADI-900-074, Enzo Life Sciences, Raamsdonksveer, The Netherlands), as recommended by the manufacturer.
Real-time RT-PCR
Total RNA isolation from lung tissue homogenates (RNA-Bee, Tel-Test, Bio-Connect, Huissen, The Netherlands), first-strand cDNA synthesis [SuperScript Choice System (Life Technologies, Breda, The Netherlands)] and real-time quantitative PCR using β-actin as a housekeeping gene reference were performed on a LightCycler 480 (Roche, Almere, The Netherlands) of the Leiden Genome Technology Center (Leiden, The Netherlands) as described previously (Ref. 45; N = 8). Primers are listed in Table 1.
Table 1.
Sequences of oligonucleotides for forward and reverse primers for real-time RT-PCR
| Gene Product | Forward Primer | Reverse Primer |
|---|---|---|
| ACE | 5′-CCCCACCCTCTCGCTACA-3′ | 5′-TGGGCAGATCCCCTGATACT-3′ |
| ACE2 | 5′-ACCCAAAAATGTGTCTGACATCAT-3′ | 5′-GATACGGCCCCGAGACATC-3′ |
| AT1a | 5′-ACAACCCTCCCAGAAAGTGATC-3′ | 5′-TTGTTTTTCTGGGTTGAGTTGGT-3′ |
| AT2 | 5′-CCGTGACCAAGTCTTGAAGATG-3′ | 5′-AGGGAAGCCAGCAAATGATG-3′ |
| CINC1 | 5′-AGAACATCCAGAGTTTGAAGGTGAT-3′ | 5′-GTGGCTATGACTTCGGTTTGG-3′ |
| MAS | 5′-CTGGTCAACCTTTGGGAACCT-3′ | 5′-AAAGGGTTGGCGCTGCTA-3′ |
| MCP1 | 5′-ATGCAGTTAATGCCCCAGTCA-3′ | 5′-TTCTCCAGCCGACTCATTGG-3′ |
| MRGD | 5′-CTGTCTCTCCGTGCTTTTCC-3′ | 5′-GCATGAAGATCCCCAGGATA-3′ |
| PAI-1 | 5′-AGCTGGGCATGACTGACATCT-3′ | 5′-GCTGCTCTTGGTCGGAAAGA-3′ |
| β-Actin | 5′-TTCAACACCCCAGCCATGT-3′ | 5′-AGTGGTACGACCAGAGGCATACA-3′ |
Statistical Analysis
Values are expressed as means ± SE. Differences between groups were analyzed by one-way ANOVA for independent samples, followed by Tukey's multiple comparison test using the GraphPad Prism, version 5, software package (San Diego, CA). Differences at P values < 0.05 were considered statistically significant.
RESULTS
Dose Finding and Effects of PD123319 on AT2-Agonist-Induced Prevention of Right Ventricular Hypertrophy
Development of RVH by exposure of newborn rats to hyperoxia for 10 days, shown by an increase (1.4-fold, Fig. 1A; 1.3-fold, Fig. 1B) in the RV/LV free wall thickness ratio over RA controls, was prevented by twice daily administration of 5 μg/kg of AT2 agonist LP2–3 (Fig. 1A and Ref. 45). This beneficial effect of LP2–3 on RVH was abolished by coadministration of PD123319 (0.5 and 2 mg·kg−1·day−1). Administration of 0.5 and 2 mg·kg−1·day−1 of PD123319 had no significant effect on hyperoxia-induced RVH. Because 0.5 mg·kg−1·day−1 of PD123319 showed a tendency toward lower levels of RVH (P = 0.06; Fig. 1A) we subsequently investigated the beneficial effect of PD123319 on RVH. Administration of 0.1 mg·kg−1·day−1 of PD123319 for 10 days was the most optimal concentration to prevent RVH in hyperoxia-induced BPD, demonstrated by a normalization in relative RV/LV free wall thickness (P < 0.001; Fig. 1B).
Effects of PD123319 on Growth and Survival
Early concurrent treatment.
On day 10, mean body weight of pups was comparable in all RA groups (19 g; Fig. 2A) and all oxygen groups (12–13 g). Administration of PD123319 (0.1 mg·kg−1·day−1) for 10 days had no adverse effect on mean body weight in RA controls and oxygen-exposed pups. Exposure to hyperoxia resulted in a 70% survival on day 10 and was not affected by administration of PD123319 (Fig. 2B). RA-exposed pups showed no morbidity or mortality during the experimental period of 10 days.
Fig. 2.

Growth (A and C) and survival (B and D) on day 10 of prophylactic PD123319 treatment (N = 12; A and B) and after late treatment and recovery (N = 8; C and D) on days 9 and 18 in RA and age-matched O2-exposed pups injected daily with saline PD123319 (0.1 mg·kg−1·day−1). Data are expressed as means ± SE. Open bars, RA-NaCl; hatched bars, RA-PD123319; solid bars, O2-NaCl; shaded bars, O2-PD123319. Data are expressed as means ± SE. *P < 0.05 and ***P < 0.001 vs. age-matched O2-exposed controls. ΔΔΔP < 0.001 vs. own RA-exposed controls. δδδP < 0.001 vs. own treatment controls on day 9.
Late treatment and recovery.
Mean body weight of RA controls was 18.2 g on day 9 (Fig. 2C) and 37.6 g on day 18 and was not influenced by PD123319 treatment. After 9 days of hyperoxia exposure, mean body weight was 11.5 g and this increased to 24.5 g after 9 days of recovery in RA on day 18. Treatment with PD123319 did not affect body weight compared with oxygen-exposed controls on days 9 and 18. On days 9 and 18 all RA controls survived (Fig. 2D), but exposure to hyperoxia resulted in a 70% survival. Treatment with PD123319 for 3 days did not affect survival on day 9, and 80% of the pups that recovered in RA survived until day 18.
Effects of PD123319 on Lung Airway Development and Inflammation
Early concurrent treatment.
In the rat lung development proceeds from the saccular stage at birth toward the alveolar stage in 10 days (Fig. 3A). Administration of PD123319 (0.1 mg·kg−1·day−1) did not have adverse effects on the number of alveolar crests (Fig. 4A), pulmonary vessel density (Figs. 3B and 4B), alveolar septal thickness (Figs. 3B and 4C), arterial medial wall thickness (Figs. 3F and 4D), and influx of macrophages (Figs. 3J and 4E) and neutrophilic granulocytes (Figs. 3N and 4F). Oxygen exposure for 10 days resulted in edema, a heterogeneous distribution of enlarged air spaces with a decreased number of alveolar crests (2.1-fold, P < 0.001; Fig. 4A), surrounded by septa with increased thickness (1.6-fold, P < 0.001; Figs. 3C and 4C), reduced pulmonary vessel density (2.7-fold, P < 0.001; Figs. 3C and 4B), and increased pulmonary arterial medial wall thickness (2.3-fold, P < 0.001; Figs. 3G and 4D). Hyperoxia led to a massive inflammatory reaction, characterized by an overwhelming influx of inflammatory cells, including macrophages (5.4-fold, P < 0.001; Figs. 3K and 4E) and neutrophils (2.8-fold, P < 0.001; Figs. 3O and 4F), compared with RA-exposed controls. Administration of PD123319 reduced alveolar septal thickness by 17% (P < 0.001; Figs. 3D and 4C), arterial medial wall thickness by 25% (P < 0.01; Figs. 3H and 4D), and the influx of macrophages by 29% (P < 0.01; Figs. 3L and 4E) and neutrophils by 54% (P < 0.001; Figs. 3P and 4F) compared with oxygen-exposed controls but had no beneficial effects on hyperoxia-induced inhibition of alveolarization and angiogenesis.
Fig. 3.

Representative lung sections stained for von Willebrand factor (vWF; A–D), α-smooth muscle actin (ASMA; E–H), the monocyte and macrophage marker ED1 (I–L), or myeloperoxidase (MPO) as a marker for neutrophilic granulocytes (M–P) of RA (A, B, E, F, I, J, M, and N) and O2-exposed pups (C, D, G, H, K, L, O, and P) injected daily with saline (A, C, E, G, I, K, M, and O) or PD123319 (PD; 0.1 mg·kg−1·day−1; B, D, F, H, J, L, N, and P) until 10 days of age. a, Alveolus. Arrows in A–D indicate vWF-positive blood vessels.
Fig. 4.

Lung morphometry, including the quantifications of alveolar crests (A), number of pulmonary vessels (B), septal thickness (C), arterial medial wall thickness (D), and influx of macrophages (E) and neutrophilic granulocytes (F) was determined on paraffin sections in RA pups injected daily with saline (open bars) or PD123319 (hatched bars) and O2 pups injected daily with saline (solid bars) or PD123319 (shaded bars): 0.1 mg·kg−1·day−1 until 10 days of age. Values are expressed as means ± SE (N = 10). **P < 0.01 and ***P < 0.001 vs. age-matched O2-exposed controls. ΔΔΔP < 0.001 vs. own RA controls.
Late treatment and recovery.
Treatment of RA-exposed pups with PD123319 had no adverse effect on alveolar (Fig. 6A) and vascular development (Figs. 5B and 6, B and C) on days 9 (Fig. 5B) and 18 (Fig. 5F). Continuous neonatal exposure to hyperoxia for 9 days resulted in enlarged alveoli (Fig. 5C), demonstrated by a 2.3-fold decrease in the number of alveolar crests (P < 0.001; Fig. 6A), disturbed vascular development, demonstrated by a 2.4-fold reduction in blood vessel density (P < 0.001; Figs. 5C and 6B), and a 2.2-fold increase in arterial medial wall thickness (P < 0.001; Figs. 5K and 6C) compared with RA controls. PD123319 treatment during the last 3 days of the injurious 9-day hyperoxic period decreased arterial medial wall thickness by 27% (P < 0.05; Figs. 5L and 6C) but had no beneficial effects on alveolarization (Figs. 5D and 6A) and pulmonary blood vessel density (Figs. 5D and 6B). A recovery period of 9 days in RA after hyperoxia-induced lung injury on day 18 had a beneficial effect on the number of alveolar crests (P < 0.001; Fig. 6A), blood vessel density (P < 0.001; Fig. 6B) and arterial medial wall thickness (P < 0.05; Fig. 6C), but the number of alveoli and blood vessels were still reduced and arterial medial wall thickness was still increased after injury and recovery. Treatment with PD123319 reduced arterial medial wall thickness by 32% (P < 0.05; Figs. 5P and 6C) but did not have a beneficial effect on alveolarization (Fig. 6A) and vascularization (Figs. 5H and 6B), compared with nontreated experimental BPD pups at the end of the recovery period on day 18.
Fig. 6.
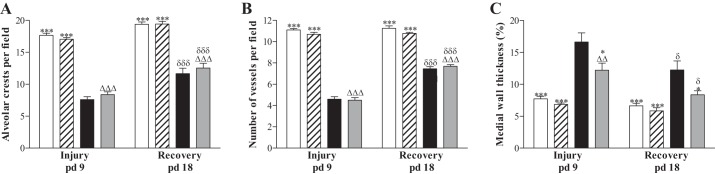
Quantification of alveolar crest (A), number of pulmonary vessels (B), and arterial medial wall thickness (C) determined on paraffin sections after late treatment and recovery on days 9 and 18 in RA-exposed pups injected daily with saline (open bars) or PD123319 (hatched bars) and O2-exposed pups injected daily with saline (solid bars) or PD123319 (shaded bars). Values are expressed as means ± SE (N = 8). *P < 0.05 and ***P < 0.001 vs. age-matched O2-exposed controls. ΔΔP < 0.01 and ΔΔΔP < 0.001 vs. own RA controls. δP < 0.05 and δδδP < 0.001 vs. own treatment controls on day 9.
Fig. 5.

Representative lung sections stained for vWF (A–H) or ASMA (I–P) after late treatment and recovery on days 9 (A–D and I–L) and 18 (E–H and M–P) of RA (A, B, E, F, I, J, M, and N) and O2-exposed pups (C, D, G, H, K, L, O, and P) injected daily with saline (A, C, E, G, I, K, M, and O) or PD123319 (0.1 mg·kg−1·day−1; B, D, F, H, J, L, N, and P) from days 6 to 18. a, Alveolus. Arrows in A–H indicate vWF-positive blood vessels.
Effects of PD123319 on Lung Coagulation, Collagen Deposition, Vascular Leakage, and CINC1 Expression
Early concurrent treatment.
Collagen III was present at high levels in the perivasculature of large and small blood vessels in normal lung in the absence (Fig. 7A) or presence of PD123319 (Fig. 7B). In alveolar septa expression was low or absent. In lungs of pups exposed to hyperoxia for 10 days, collagen III deposition increased 6.3-fold (P < 0.001; Fig. 7E) and was present in the perivasculature of blood vessels and in thick alveolar septa (Fig. 7C). Treatment with PD123319 for 10 days reduced collagen III expression by 52% (P < 0.001; Fig. 7E). Hyperoxia-induced extracellular collagen III expression was only reduced in alveolar septa, but not in the (peri)vascular area (Fig. 7D). Fibrin deposition was at reference levels during normal neonatal pulmonary development on day 10 in the absence or presence of PD123319 (< 15 ng fibrin/mg tissue) and increased 11-fold (P < 0.01) in lungs of pups exposed to 100% oxygen for 10 days (Fig. 7F). PD123319 (0.1 mg·kg−1·day−1) administration did not reduce hyperoxia-induced fibrin deposition. Protein expression of CINC1 was low in BALF of RA controls and increased 6.4-fold (P < 0.05; Fig. 7G) after exposure to hyperoxia for 10 days. Daily administration of 0.1 mg/kg of PD123319 showed a tendency toward lower CINC1 levels. Total protein concentration in BALF was determined to establish the inhibitory effect of PD123319 on pulmonary edema by capillary-alveolar leakage (Fig. 7H). The protein concentration on postnatal day 10 increased 5.3-fold after hyperoxia (P < 0.05) and was not affected by PD123319 administration during normoxia or hyperoxia.
Fig. 7.

Quantification of collagen III (Col3) expression (E) on lung paraffin sections of RA (A and B) and O2-exposed pups (O2; C and D) injected daily with saline (A and C) or PD123319 (PD; B and D) until 10 days of age (N = 8); quantification of extravascular fibrin deposition in lung homogenates (N = 10, F) as a marker for lung injury; CINC1 protein expression in bronchoalveolar lavage fluid (BALF; N = 10, G); and total protein concentration in BALF (N = 12, H) as a marker for vascular leakage on day 10 in RA pups injected daily with saline (open bars) or PD123319 (0.1 mg·kg−1·day−1; hatched bars) and O2 pups injected daily with saline (solid bars) or PD123319 (shaded bars). a, Alveolus; at, arteriole. Values are expressed as means ± SE. *P < 0.05, **P < 0.01, and ***P < 0.001 vs. age-matched O2-exposed controls. ΔP < 0.05 and ΔΔP < 0.01 vs. RA controls.
Effects of PD123319 on mRNA Expression in Lung Tissue
Early concurrent treatment.
Administration of PD123319 (0.1 mg·kg−1·day−1) for 10 days during normal neonatal development in RA did not change mRNA expression (Fig. 8) of the proinflammatory factors monocyte chemoattractant protein (MCP)-1 (Fig. 8A) and CINC1 (Fig. 8B), the anti-fibrinolytic protein plasminogen activator inhibitor 1 (PAI-1; Fig. 8C), angiotensin II type 1a receptor (AT1a; Fig. 8D), AT2 (Fig. 8E), MAS (Fig. 8F), ACE (Fig. 8G), and ACE2 (Fig. 8H). Ten days of oxygen exposure resulted in an increase in mRNA expression of MCP-1 (5.4-fold, P < 0.001), CINC-1 (12.1-fold, P < 0.001), PAI-1 (53-fold P < 0.001), and AT2 (4.1-fold, P < 0.001), whereas mRNA expression was decreased in lungs of oxygen-exposed pups for AT1a (1.8-fold, P < 0.001), MAS (1.6-fold, P < 0.001), and ACE (4.2-fold, P < 0.001) compared with RA controls. Treatment of oxygen-exposed pups with PD123319 for 10 days did not result in changes in mRNA expression compared with oxygen-exposed pups.
Fig. 8.

Relative mRNA expression in lung homogenates after early concurrent treatment (A–H) of monocyte chemoattractant protein 1 (MCP1; A), chemokine-induced neutrophilic chemoattractant-1 (CINC1; B), plasminogen activator inhibitor 1 (PAI-1; C), angiotensin receptor type 1a (AT1a; D), AT2 (E), MAS oncogene receptor (MAS; F), angiotensin converting enzyme (ACE; G), and angiotensin converting enzyme 2 (ACE2; H) on day 10 in RA pups injected daily with saline (open bars) or PD123319 (0.1 mg·kg−1·day−1; hatched bars) and O2-exposed pups injected daily with saline (solid bars) or PD123319 (shaded bars). Relative mRNA expression of Mas-related receptor, type D (MrgD or alamandine receptor) in lung, spleen, thymus, ovary, kidney, brain, heart, liver, and testis (I). Values are expressed as means ± SE (N = 8). *P < 0.05 and ***P < 0.001 vs. age-matched O2-exposed controls. ΔΔP < 0.01 and ΔΔΔP < 0.001 vs. RA controls.
Tissue Distribution of MAS-Related Receptor Type D
Because PD123319 (0.1 mg·kg−1·day−1) may exert its biological effects by inhibiting AT2 and MAS-related receptor type D (MrgD; Ref. 21), we studied the tissue distribution of MrgD and found that MrgD is expressed in testis, but not in the other organs studied, including lung and heart (Fig. 8I and Ref. 35). In addition, MrgD expression was absent in lungs of PD123319-treated rat pups with experimental BPD and controls (data not shown).
Effects of PD123319 on Right Ventricular Hypertrophy
Early concurrent treatment.
Administration of 0.1 mg·kg−1·day−1 of PD123319 for 10 days during normal neonatal development had no adverse effect on the heart (Fig. 9, A–C). Exposure to hyperoxia for 10 days resulted in RVH, affected by an increase (1.3-fold; Fig. 9B) in the ratio RV/LV free wall thickness and an 1.4-fold increase in RV/(LV + IVS) weight ratio (Fig. 9C) compared with RA controls (P < 0.01). Administration of 0.1 mg·kg−1·day−1 of PD123319 prevented RVH, demonstrated by a normalization in relative RV/LV free wall thickness (P < 0.01; 9B) and a tendency toward lower levels of the RV/(LV + IVS) weight ratio after PD123319 treatment compared with oxygen-exposed pups (9C).
Fig. 9.

RVH was determined by morphometry in paraffin sections stained with hematoxylin and eosin (A) and depicted as RV/LV wall thickness ratio (B and D) or by determining the weight ratio RV/(LV+IVS) where IVS is interventricular septum (C) on day 10 of prophylactic PD123319 treatment (A–C) and after late treatment and recovery (D) on days 9 and 18 in RA and age-matched O2-exposed pups injected daily with saline or PD123319 (0.1 mg·kg−1·day−1). White bars, RA-NaCl; hatched bars, RA-PD123319; solid bars, O2-NaCl; shaded bars, O2-PD123319. Data are expressed as means ± SE (N = 8). *P < 0.05, **P < 0.01, and ***P < 0.001 vs. own age-matched O2-exposed controls. ΔP < 0.05 vs. own RA controls.
Late treatment and recovery.
The ratio RV/LV free wall thickness was increased 1.4-fold after 9 days of hyperoxic lung injury compared with RA controls (P < 0.01; Fig. 9D), which was attenuated after 3 days of PD123319 treatment on day 9 (23%; P < 0.05). A recovery period of 9 days did not reduce RVH in the nontreated and PD123319-treated pups (Fig. 9D).
DISCUSSION
PAH and RVH are major causes of mortality and severe morbidity in premature infants with severe BPD (1, 5). The hyperoxia-exposed newborn rat pup is an established in vivo model for severe experimental BPD and RVH (44, 45). Because of the high expression of AT2 in neonatal rat lung (45), neonatal rats with BPD are very suitable to study novel treatment options that target AT2. Treatment of neonatal rats with experimental BPD with the AT2 agonist LP2–3 attenuated lung inflammation and prevented PAH-induced RVH (45). In this study we demonstrate that the beneficial effect of LP2–3 (5 μg·kg−1·day−1) on RVH can be abolished by coadministration with PD123319 at relatively high concentrations (0.5 and 2 mg·kg−1·day−1), which is consistent with the AT2-agonist action of LP2–3 and blocking of AT2 by PD123319. In addition, we show that administration of a relatively low concentration of PD123319 (0.1 mg·kg−1·day−1) to newborn rat pups with BPD reduced cardiopulmonary injury by attenuating arterial medial wall thickness (PAH), alveolar septal thickness, extracellular collagen III expression, and inflammation in the lung and by preventing RVH. PD123319 (0.1 mg·kg−1·day−1) had no beneficial effects on lung alveolarization, vascularization, fibrin deposition, and capillary alveolar leakage and no adverse effects on normal lung and heart development. These findings suggest that PD123319 may be a suitable therapeutic candidate to reduce lung inflammation and fibrosis and prevent PAH-induced RVH in premature infants with severe BPD.
A role for angiotensin II-angiotensin receptor signaling in the pathophysiology of severe experimental BPD, in which alveolar enlargement and pulmonary hypertension play a pivotal role, is suggested by the differential expression of the AT1, AT2, and MAS receptors, the precursor of their ligands angiotensinogen and the converting enzymes ACE and ACE2 in the lung during development and/or in hyperoxia-induced experimental BPD (this study and Ref. 45). In agreement with a role of the renin angiotensin system in experimental BPD we recently demonstrated that treatment of rat pups with the MAS agonist cAng1–7 attenuated hyperoxia-induced BPD. The relatively high expression of AT2 and the adaptive mRNA response in the newborn lung exposed to hyperoxia, result in a relative higher AT2 than AT1 expression (45), and may contribute to the cardiopulmonary effects that we observed after treatment of rat pups with experimental BPD with PD123319 (this study) or the AT2 agonist LP2–3 (45).
Inflammation contributes significantly to lung injury in experimental BPD as demonstrated by 1) the massive influx of macrophages and neutrophils into the lung, 2) the upregulation of chemokines that are involved in the activation and migration of leukocytes in vivo (44, 49) and 3) the beneficial effects of PDE-4 inhibitors in experimental BPD, which are potent anti-inflammatory agents (14, 47). The reduced influx of monocytes, macrophages, and neutrophilic granulocytes in the lung suggests that the beneficial effects of treatment with PD123319 on hyperoxia-induced neonatal lung injury may be explained by reduced inflammation, but this was not confirmed by reduced mRNA expression of proinflammatory cytokines (MCP1 and CINC1), suggesting regulation of gene expression at a posttranscriptional level. A role for the AT2 receptor in the regulation of the inflammatory response in vivo is further supported by the anti-inflammatory and/or antifibrotic effects of AT2 stimulation in neonatal lung disease (45), bleomycin-induced cutaneous injury (31), and renovascular hypertension and inflammation (26), of AT2 inhibition in bleomycin-induced lung fibrosis (46) and the aggravation of acute lung disease in AT2-deficient mice (15).
Treatment of hyperoxia-induced neonatal lung injury with PD123319 results in a significant improvement of severe cardiopulmonary injury, demonstrated by prevention of PAH-induced RVH and reduction of inflammation, but the beneficial effects of PD123319 are probably still too small to result in reduced mortality. The data demonstrate that the development of PAH and RVH can be attenuated by prophylactic PD123319 treatment and by treatment during the last 3 days of an injurious period of 9 days, and that the beneficial effect of PD123319 treatment on PAH is persistent during recovery in RA. However, the beneficial effect on RVH after recovery in RA is absent, which may be explained by an adverse effect of PD123319 on the heart after prolonged treatment in RA.
Although the beneficial effects of MAS stimulation on cardiovascular disease in adult and neonatal animal models in the literature are rather consistent, the role of AT2 continues to be controversial as demonstrated by two observations in our study: 1) beneficial effects of the AT2 antagonist PD123319 on RVH observed in this study were also observed for treatment with AT2 agonist LP2–3 in the same model (45) and 2) these beneficial effects disappear when the PD123319 dose is increased. These unexpected finding are supported by two observations by us and others: Firstly, the beneficial effects of PD123319 administration in rat pups with experimental BPD are in agreement with the beneficial effects observed in PD123319-treated mice with bleomycin-induced fibrosis (46), but in sharp contrast with our previous findings with the AT2 agonist LP2–3 (45) and observations in AT2-deficient mice in which cardiovascular disease is aggravated compared with wild-type controls (3, 15). Secondly, in previous studies we observed that beneficial effects of treatment with MAS, AT2, and APJ receptor-specific agonists cAng1–7, LP2–3, and apelin, respectively, were only found at low concentrations in newborn rats with BPD and disappeared when the dose was increased (13, 45). These discrepancies may be explained by the presence of AT2 on multiple cell types, including endothelial and vascular smooth muscle cells, and may be complicated by its increased expression under pathological conditions compared with healthy controls. We speculate that AT2 activation on the endothelium may lead to vasodilation, whereas AT2 activation on vascular smooth muscle cells may lead to vasoconstriction (43). In the cardiovasculature this behavior is not unique to AT2 but is also observed for other receptors related to the renin angiotensin system, including the apelin receptor APJ, bradykinin B1 and B2 receptors, and muscarinic receptors, and may be complicated by receptor expression on inflammatory cells and cardiomyocytes (9, 12, 16, 18, 28, 43). This makes it difficult to predict the biological effect of an AT2 agonist or antagonist, because it depends not only on the administered dose and diffusion into the tissue, but also on local levels of AngII and its intermediates and on the differential cellular expression of AT1, AT2, and MAS. Alternatively, these discrepancies may be explained by a lack of specificity of PD123319 for the AT2 receptor, as was demonstrated recently by 1) an inhibitory effect of PD123319 on the alamandine receptor MrgD (21) and 2) an increase of AngII-induced abdominal aortic aneurysms by PD123319 in AT2-deficient mice (11), possibly due to MrgD inhibition (21). However, MrgD expression is absent in lung and heart tissue (this study and Ref. 33). It is therefore unlikely that inhibition of MrgD by PD123319 is involved in the beneficial cardiopulmonary effects in rat pups with BPD, but binding of PD123319 to other proteins than AT2 or MrgD cannot be excluded. In addition, PD123319 may act as an AT2 agonist at low concentrations and mimic the beneficial effects of the AT2 agonist LP2–3 in our previous study (45). Also pleiotropic effects of LP2–3 will have to be investigated. Off-target effects of both protective compounds would offer possibilities to discover new therapeutic targets.
AT2-agonist (LP2–3; Ref. 45) and antagonist treatment (PD123319, this study) show similar beneficial effects on septal thickness, pulmonary arterial hypertension and the influx of inflammatory cells (monocytes and macrophages). However, agonist treatment had a more pronounced effect on RVH (Fulton's index) and showed an additional beneficial effect on fibrin deposition in the lung, a marker for lung tissue damage, which was absent after PD123319 treatment. Therefore, we prefer AT2 agonist treatment for (experimental) BPD. The beneficial effects we observed with the AT2 interventions were similar to those we observed after treatment with PDE4 inhibitors, showing beneficial effects on inflammation and pulmonary arterial hypertension, whereas beneficial effects on alveolarization and vascularization were absent (14). The magnitude of response was in favor of the PDE4 inhibitor piclamilast, resulting in prolonged survival as well. However, the major drawback of PDE4 inhibitors that may preclude their use in the clinic are their serious adverse effects, including nausea, headache, and vomiting, and as a result reduced food intake and growth retardation. Although it is difficult to measure vomiting and nausea in rat pups, we did not observe growth retardation after AT2 intervention in experimental BPD, suggesting fewer side effects of LP2–3 or PD123319. Treatment of experimental BPD with sildenafil had similar beneficial effects as well and showed additional beneficial effects on reduced alveolarization and vascularization. However, prolonged sildenafil treatment of children with pulmonary arterial hypertension resulted in increased mortality and let the FDA to give a safety warning against the use (particularly chronic use) of sildenafil in children.
Extrapolation of our experimental findings to the clinic would suggest a beneficial effect of PD123319 on inflammation, fibrosis, PAH, and RVH and the possibility of combating these major causes of mortality or severe morbidity in premature infants with severe BPD.
GRANTS
This study was supported by grants 1R01 HL092158 and 1R01 ES015330 from the National Institutes of Health (F. J. Walther), Dutch Heart Foundation research grant NHS2010B009 (A. J. M. Roks), and a grant from the Stichting Zeldzame Ziekten Fonds (G. T. M. Wagenaar).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
AUTHOR CONTRIBUTIONS
G.T.W., G.F., and F.J.W. conception and design of research; G.T.W., R.M.S., E.H.L., X.C., and M.P.L. performed experiments; G.T.W., R.M.S., E.H.L., X.C., and M.P.L. analyzed data; G.T.W., R.M.S., E.H.L., A.J.M.R., G.F., and F.J.W. interpreted results of experiments; G.T.W. prepared figures; G.T.W., A.J.M.R., and F.J.W. drafted manuscript; G.T.W., A.J.M.R., G.F., and F.J.W. edited and revised manuscript; G.T.W., R.M.S., E.H.L., X.C., M.P.L., A.J.M.R., G.F., and F.J.W. approved final version of manuscript.
ACKNOWLEDGMENTS
The authors gratefully acknowledge I. van Ark and T. A. Leusink-Muis (Department of Pharmacology, Utrecht University, Utrecht, The Netherlands) for expert technical assistance, Dr. E. de Heer (Department of Pathology, Leiden University Medical Center, Leiden, The Netherlands) for providing the ED-1 antibody, and Dr. J. J. Baelde (Department of Pathology, Leiden University Medical Center, Leiden, The Netherlands) for providing the COL3A antibody.
REFERENCES
- 1.Abman SH. Recent advances in the pathogenesis and treatment of persistent pulmonary hypertension of the newborn. Neonatology 91: 283–290, 2007 [DOI] [PubMed] [Google Scholar]
- 2.Abman SH. Role of endothelin receptor antagonists in the treatment of pulmonary arterial hypertension. Annu Rev Med 60: 13–23, 2009 [DOI] [PubMed] [Google Scholar]
- 3.Adachi Y, Saito Y, Kishimoto I, Harada M, Kuwahara K, Takahashi N, Kawakami R, Nakanishi M, Nakagawa Y, Tanimoto K, Saitoh Y, Yasuno S, Usami S, Iwai M, Horiuchi M, Nakao K. Angiotensin II type 2 receptor deficiency exacerbates heart failure and reduces survival after acute myocardial infarction in mice. Circulation 107: 2406–2408, 2003 [DOI] [PubMed] [Google Scholar]
- 4.Alphonse RS, Vadivel A, Coltan L, Eaton F, Barr AJ, Dyck JR, Thébaud B. Activation of Akt protects alveoli from neonatal oxygen-induced lung injury. Am J Respir Cell Mol Biol 44: 146–154, 2011 [DOI] [PubMed] [Google Scholar]
- 5.Baraldi E, Filippone M. Chronic lung disease after premature birth. N Engl J Med 357: 1946–1955, 2007 [DOI] [PubMed] [Google Scholar]
- 6.Batenburg WW, Garrelds IM, Bernasconi CC, Juillerat-Jeanneret L, van Kats JP, Saxena PR, Danser AH. Angiotensin II type 2 receptor-mediated vasodilation in human coronary microarteries. Circulation 109: 2296–2301, 2004 [DOI] [PubMed] [Google Scholar]
- 7.Chai W, Wang W, Dong Z, Cao W, Liu Z. Angiotensin II receptors modulate muscle microvascular and metabolic responses to insulin in vivo. Diabetes 60: 2939–2946, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chou HC, Lang YD, Wang LF, Wu TY, Hsieh YF, Chen CM. Angiotensin II type 1 receptor antagonist attenuates lung fibrosis in hyperoxia-exposed newborn rats. J Pharmacol Exp Ther 340: 169–175, 2012 [DOI] [PubMed] [Google Scholar]
- 9.Chun HJ, Ali ZA, Kojima Y, Kundu RK, Sheikh AY, Agrawal R, Zheng L, Leeper NJ, Pearl NE, Patterson AJ, Anderson JP, Tsao PS, Lenardo MJ, Ashley EA, Quertermous T. Apelin signaling antagonizes AngII effects in mouse models of atherosclerosis. J Clin Invest 118: 3343–3354, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.da Silveira KD, Coelho FM, Vieira AT, Sachs D, Barroso LC, Costa VV, Bretas TL, Bader M, de Sousa LP, da Silva TA, dos Santos RA, Simões e Silva AC, Teixeira MM. Anti-inflammatory effects of the activation of the angiotensin-(1–7) receptor, Mas, in experimental models of arthritis. J Immunol 185: 5569–5576, 2010 [DOI] [PubMed] [Google Scholar]
- 11.Daugherty A, Rateri DL, Howatt DA, Charnigo R, Cassis LA. PD123319 augments angiotensin II-induced abdominal aortic aneurysms through an AT2 receptor-independent mechanism. PLoS One 8: e61849, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Felipe SA, Rodrigues ES, Martin RP, Paiva AC, Pesquero JB, Shimuta SI. Functional expression of kinin B1 and B2 receptors in mouse abdominal aorta. Braz J Med Biol Res 40: 649–655, 2007 [DOI] [PubMed] [Google Scholar]
- 13.de Visser YP, Walther FJ, Laghmani EH, van der Laarse A, Wagenaar GT. Apelin attenuates hyperoxic lung and heart injury in neonatal rats. Am J Respir Crit Care Med 182: 1239–1250, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.de Visser YP, Walther FJ, Laghmani EH, van Wijngaarden D, Nieuwland K, Wagenaar GT. Phosphodiesterase-4 inhibition attenuates pulmonary inflammation in neonatal lung injury. Eur Respir J 31: 633–644, 2008 [DOI] [PubMed] [Google Scholar]
- 15.Imai Y, Kuba K, Rao S, Huan Y, Guo F, Guan B, Yang P, Sarao R, Wada T, Leong-Poi H, Crackower MA, Fukamizu A, Hui CC, Hein L, Uhlig S, Slutsky AS, Jiang C, Penninger JM. Angiotensin-converting enzyme 2 protects from severe acute lung failure. Nature 436: 112–116, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Japp AG, Newby DE. The apelin-APJ system in heart failure: pathophysiologic relevance and therapeutic potential. Biochem Pharmacol 75: 1882–1892, 2008 [DOI] [PubMed] [Google Scholar]
- 17.Jobe AH. The new BPD: an arrest of lung development. Pediatr Res 46: 641–643, 1999 [DOI] [PubMed] [Google Scholar]
- 18.Katugampola SD, Maguire JJ, Matthewson SR, Davenport AP. [125I]-(Pyr1)Apelin-13 is a novel radioligand for localizing the APJ orphan receptor in human and rat tissues with evidence for a vasoconstrictor role in man. Br J Pharmacol 132: 1255–1260, 2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kim S, Iwao H. Molecular and cellular mechanisms of angiotensin II-mediated cardiovascular and renal diseases. Pharmacol Rev 52: 11–34, 2011 [PubMed] [Google Scholar]
- 20.Koppel R, Han RN, Cox D, Tanswell AK, Rabinovitch M. Alpha 1-antitrypsin protects neonatal rats from pulmonary vascular and parenchymal effects of oxygen toxicity. Pediatr Res 36: 763–770, 1994 [DOI] [PubMed] [Google Scholar]
- 21.Lautner RQ, Villela DC, Fraga-Silva RA, Silva N, Verano-Braga T, Costa-Fraga F, Jankowski J, Jankowski V, Sousa F, Alzamora A, Soares E, Barbosa C, Kjeldsen F, Oliveira A, Braga J, Savergnini S, Maia G, Peluso AB, Passos-Silva D, Ferreira A, Alves F, Martins A, Raizada M, Paula R, Motta-Santos D, Klempin F, Pimenta A, Alenina N, Sinisterra R, Bader M, Campagnole-Santos MJ, Santos RA. Discovery and characterization of alamandine: a novel component of the renin-angiotensin system. Circ Res 112: 1104–1111, 2013 [DOI] [PubMed] [Google Scholar]
- 22.Li X, Molina-Molina M, Abdul-Hafez A, Uhal V, Xaubet A, Uhal BD. Angiotensin converting enzyme-2 is protective but downregulated in human and experimental lung fibrosis. Am J Physiol Lung Cell Mol Physiol 295: L178–L185, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lyngsø C, Erikstrup N, Hansen JL. Functional interactions between 7TM receptors in the renin-angiotensin system—dimerization or crosstalk? Mol Cell Endocrinol 302: 203–212, 2009 [DOI] [PubMed] [Google Scholar]
- 24.Madurga A, Mizíková I, Ruiz-Camp J, Morty RE. Recent advances in late lung development and the pathogenesis of bronchopulmonary dysplasia. Am J Physiol Lung Cell Mol Physiol 305: L893–L905, 2013 [DOI] [PubMed] [Google Scholar]
- 25.Marshall RP, Gohlke P, Chambers RC, Howell DC, Bottoms SE, Unger T, McAnulty RJ, Laurent GJ. Angiotensin II and the fibroproliferative response to acute lung injury. Am J Physiol Lung Cell Mol Physiol 286: L156–L164, 2004 [DOI] [PubMed] [Google Scholar]
- 26.Matavelli LC, Huang J, Siragy HM. Angiotensin AT2 receptor stimulation inhibits early renal inflammation in renovascular hypertension. Hypertension 57: 308–313, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Molina-Molina M, Serrano-Mollar A, Bulbena O, Fernandez-Zabalegui L, Closa D, Marin-Arguedas A, Torrego A, Mullol J, Picado C, Xaubet A. Losartan attenuates bleomycin induced lung fibrosis by increasing prostaglandin E2 synthesis. Thorax 61: 604–610, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Obi T, Kabeyama A, Nishio A. Characterization of muscarinic receptor subtype mediating contraction and relaxation in equine coronary artery in vitro. J Vet Pharmacol Ther 17: 226–231, 1994 [DOI] [PubMed] [Google Scholar]
- 29.Podowski M, Calvi C, Metzger S, Misono K, Poonyagariyagorn H, Lopez-Mercado A, Ku T, Lauer T, McGrath-Morrow S, Berger A, Cheadle C, Tuder R, Dietz HC, Mitzner W, Wise R, Neptune E. Angiotensin receptor blockade attenuates cigarette smoke-induced lung injury and rescues lung architecture in mice. J Clin Invest 122: 229–240, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rey-Parra GJ, Vadivel A, Coltan L, Hall A, Eaton F, Schuster M, Loibner H, Penninger JM, Kassiri Z, Oudit GY, Thébaud B. Angiotensin converting enzyme 2 abrogates bleomycin-induced lung injury. J Mol Med 90: 637–647, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rompe F, Artuc M, Hallberg A, Alterman M, Ströder K, Thöne-Reineke C, Reichenbach A, Schacherl J, Dahlöf B, Bader M, Alenina N, Schwaninger M, Zuberbier T, Funke-Kaiser H, Schmidt C, Schunck WH, Unger T, Steckelings UM. Direct angiotensin II type 2 receptor stimulation acts anti-inflammatory through epoxyeicosatrienoic acid and inhibition of nuclear factor kappaB. Hypertension 55: 924–931, 2010 [DOI] [PubMed] [Google Scholar]
- 32.Santos RA, Simoes e Silva AC, Maric C, Silva DM, Machado RP, de Buhr I, Heringer-Walther S, Pinheiro SV, Lopes MT, Bader M, Mendes EP, Lemos VS, Campagnole-Santos MJ, Schultheiss HP, Speth R, Walther T. Angiotensin-(1–7) is an endogenous ligand for the G protein-coupled receptor Mas. Proc Natl Acad Sci USA 100: 8258–8263, 2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Savergnini SQ, Beiman M, Lautner RQ, de Paula-Carvalho V, Allahdadi K, Pessoa DC, Costa-Fraga FP, Fraga-Silva RA, Cojocaru G, Cohen Y, Bader M, Pinto de Almeida A, Rotman G, Souza dos Santos RA. Vascular relaxation, antihypertensive effect, and cardioprotection of a novel peptide agonist of the Mas receptor. Hypertension 56: 1–7, 2010 [DOI] [PubMed] [Google Scholar]
- 34.Shenoy V, Ferreira AJ, Qi Y, Fraga-Silva RA, Díez-Freire C, Dooies A, Jun JY, Sriramula S, Mariappan N, Pourang D, Venugopal CS, Francis J, Reudelhuber T, Santos RA, Patel JM, Raizada MK, Katovich MJ. The angiotensin-converting enzyme 2/angiogenesis-(1–7)/Mas axis confers cardiopulmonary protection against lung fibrosis and pulmonary hypertension. Am J Respir Crit Care Med 182: 1065–1072, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Shinohara T, Harada M, Ogi K, Maruyama M, Fujii R, Tanaka H, Fukusumi S, Komatsu H, Hosoya M, Noguchi Y, Watanabe T, Moriya T, Itoh Y, Hinuma S. Identification of a G protein-coupled receptor specifically responsive to beta-alanine. J Biol Chem 279: 23559–235564, 2004 [DOI] [PubMed] [Google Scholar]
- 36.Steckelings UM, Widdop RE, Paulis L, Unger T. The angiotensin AT2 receptor in left ventricular hypertrophy. J Hypertens 28, Suppl 1: S50–S55, 2010 [DOI] [PubMed] [Google Scholar]
- 37.Steinhorn RH. Neonatal pulmonary hypertension. Pediatr Crit Care Med 11: S79–S84, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sun H, Choo-Wing R, Sureshbabu A, Fan J, Leng L, Yu S, Jiang D, Noble P, Homer RJ, Bucala R, Bhandari V. A critical regulatory role for macrophage migration inhibitory factor in hyperoxia-induced injury in the developing murine lung. PLoS One 8: e60560, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Trask AJ, Ferrario CM. Angiotensin-(1–7): pharmacology and new perspectives in cardiovascular treatments. Cardiovasc Drug Rev 25: 162–174, 2007 [DOI] [PubMed] [Google Scholar]
- 40.Tuder RM, Abman SH, Braun T, Capron F, Stevens T, Thistlethwaite PA, Haworth SG. Development and pathology of pulmonary hypertension. J Am Coll Cardiol 54: S3–S9, 2009 [DOI] [PubMed] [Google Scholar]
- 41.Uhal BD, Li X, Piasecki CC, Molina-Molina M. Angiotensin signalling in pulmonary fibrosis. Int J Biochem Cell Biol 44: 465–468, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Varagic J, Trask AJ, Jessup JA, Chappell MC, Ferrario CM. New angiotensins. J Mol Med 86: 663–671, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Verdonk K, Danser AH, van Esch JH. Angiotensin II type 2 receptor agonists: where should they be applied? Expert Opin Investig Drugs 21: 501–513, 2012 [DOI] [PubMed] [Google Scholar]
- 44.Wagenaar GT, ter Horst SA, van Gastelen MA, Leijser LM, Mauad T, van der Velden PA, de Heer E, Hiemstra PS, Poorthuis BJ, Walther FJ. Gene expression profile and histopathology of experimental bronchopulmonary dysplasia induced by prolonged oxidative stress. Free Radic Biol Med 36: 782–801, 2004 [DOI] [PubMed] [Google Scholar]
- 45.Wagenaar GT, Laghmani EH, Fidder M, Sengers RM, de Visser YP, de Vries L, Rink R, Roks AJ, Folkerts G, Walther FJ. Agonists of MAS oncogene and angiotensin II type 2 receptors attenuate cardiopulmonary disease in rats with neonatal hyperoxia-induced lung injury. Am J Physiol Lung Cell Mol Physiol 305: L341–L351, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Waseda Y, Yasui M, Nishizawa Y, Inuzuka K, Takato H, Ichikawa Y, Tagami A, Fujimura M, Nakao S. Angiotensin II type 2 receptor antagonist reduces bleomycin-induced pulmonary fibrosis in mice. Respir Res 9: 43, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Woyda K, Koebrich S, Reiss I, Rudloff S, Pullamsetti SS, Rühlmann A, Weissmann N, Ghofrani HA, Günther A, Seeger W, Grimminger F, Morty RE, Schermuly RT. Inhibition of phosphodiesterase 4 enhances lung alveolarisation in neonatal mice exposed to hyperoxia. Eur Respir J 33: 861–870, 2009 [DOI] [PubMed] [Google Scholar]
- 48.Wösten-van Asperen RM, Lutter R, Specht PA, Moll GN, van Woensel JB, van der Loos CM, van Goor H, Kamilic J, Florquin S, Bos AP. Acute respiratory distress syndrome leads to reduced ratio of ACE/ACE2 activities and is prevented by angiotensin-(1–7) or an angiotensin II receptor antagonist. J Pathol 225: 618–627, 2011 [DOI] [PubMed] [Google Scholar]
- 49.Yi M, Jankov RP, Belcastro R, Humes D, Copland I, Shek S, Sweezey NB, Post M, Albertine KH, Auten RL, Tanswell AK. Opposing effects of 60% oxygen and neutrophil influx on alveologenesis in the neonatal rat. Am J Respir Crit Care Med 170: 1188–1196, 2004 [DOI] [PubMed] [Google Scholar]