

Abstract
Rhizobia are symbiotic nitrogen-fixing soil bacteria that are associated with host legumes. The establishment of rhizobial symbiosis requires signal exchanges between partners in microaerobic environments that result in mutualism for the two partners. We developed a macroarray for Mesorhizobium loti MAFF303099, a microsymbiont of the model legume Lotus japonicus, and monitored the transcriptional dynamics of the bacterium during symbiosis, microaerobiosis, and starvation. Global transcriptional profiling demonstrated that the clusters of genes within the symbiosis island (611 kb), a transmissible region distinct from other chromosomal regions, are collectively expressed during symbiosis, whereas genes outside the island are downregulated. This finding implies that the huge symbiosis island functions as clustered expression islands to support symbiotic nitrogen fixation. Interestingly, most transposase genes on the symbiosis island were highly upregulated in bacteroids, as were nif, fix, fdx, and rpoN. The genome region containing the fixNOPQ genes outside the symbiosis island was markedly upregulated as another expression island under both microaerobic and symbiotic conditions. The symbiosis profiling data suggested that there was activation of amino acid metabolism, as well as nif-fix gene expression. In contrast, genes for cell wall synthesis, cell division, DNA replication, and flagella were strongly repressed in differentiated bacteroids. A highly upregulated gene in bacteroids, mlr5932 (encoding 1-aminocyclopropane-1-carboxylate deaminase), was disrupted and was confirmed to be involved in nodulation enhancement, indicating that disruption of highly expressed genes is a useful strategy for exploring novel gene functions in symbiosis.
Through the symbiotic nitrogen fixation process, bacteria belonging to the family Rhizobiaceae convert atmospheric dinitrogen (N2) to ammonia (NH3), which can be effectively used by host legume plants. The establishment of a rhizobiumlegume symbiosis requires induction of new developmental programs in the partners. The symbiotic interaction begins with signal exchanges of flavonoids and Nod factors (lipochitooligosaccharides) between the two partners (6). In legume nodules, microaerobic environments trigger the rhizobial expression of nitrogen-fixing genes, such as nif and fix, via an oxygen-sensing system (13). However, the establishment of nitrogen-fixing symbiosis probably requires more complex steps triggered by reciprocal signal exchanges that lead to the organogenesis of nodules, differentiation of microsymbionts, and efficacy of nodulation (27). In addition to this symbiotic lifestyle, rhizobia survive in soils with many environment stresses, such as nutrient starvation.
Lotus japonicus is a promising model legume for studying molecular interactions between symbiosis partners (20). Schauser et al. (40) first identified the plant regulatory gene nin, which is responsible for the nodule organogenesis program, in this legume. Recently, the receptor-like kinase genes have been discovered to be required for microsymbiont recognition (35) and nodule number regulation (22, 28).
Mesorhizobium loti is a nitrogen-fixing endosymbiont associated with L. japonicus. The entire genome structure of M. loti MAFF303099 (7.04 Mb) was recently reported (18), and this allowed us to construct a rhizobial array system by using libraries generated by the M. loti sequencing project and to profile the global transcription of rhizobia under various conditions. One of the notable features of the M. loti chromosome is the integration of a horizontally transferred DNA segment, the symbiosis island. It was first reported that a 500-kb DNA region on the chromosome of M. loti strain ICMP3153 is transmissible to nonsymbiotic Mesorhizobium species, is inserted into a phenylalanine tRNA gene in the recipient, and confers the ability to fix nitrogen symbiotically (42). A symbiosis island with a structure similar to that of ICMP3153 was found on the chromosome of M. loti MAFF303099, although the size and the genes of the MAFF303099 island (611 kb) differ from those of ICMP3153 (18, 43). The purposes of the present work were to address by using genome-wide monitoring the transcriptional dynamics of rhizobia throughout their life cycle and to examine the usefulness of transcriptional profiling as a screening method for identifying novel genes relevant to symbiosis. We found that the symbiosis island functions as clustered expression islands (EIs) to support symbiotic nitrogen fixation in M. loti MAFF303099.
MATERIALS AND METHODS
Preparation of genomic DNA macroarrays.
To cover the entire genome of M. loti MAFF303099 as widely as possible, we selected a minimally overlapping set of 3,832 clones from the 74,753 M13 clones of the RLB and RLE genomic libraries that were used for whole-genome sequencing (18). In practice, the end sequences of the library clones were fed to the Phred/Phrap program (11, 12) to automatically extract an initial set of about 3,700 candidates. The set was then manually optimized to minimize gaps and to exclude rRNA genes. The clones used consisted of 3,302 RLB and 530 RLE clones with average insert sizes of 2.6 and 1.0 kbp, respectively. The resultant coverage was more than 99.5% of the entire genome of M. loti MAFF303099.
M13 phage supernatants of the selected clones were used as templates for PCR amplification. Each PCR was performed by using M13 universal primers (5′-GGGTTTTCCCAGTCACGAC and 5′-TTATGCTTCCGGCTCGTAATGTTGTG) and 30 cycles of 97°C for 20 s and 68°C for 6 min, followed by 7 min of incubation at 72°C. The insert of each clone was amplified by PCR in a 100-μl reaction mixture, precipitated by addition of isopropyl alcohol, and then dissolved in 20 μl of 10 mM Tris-HCl buffer (pH 8.0) containing 1 mM EDTA. We then added 10 μl of a spotting dye solution containing 0.25% bromophenol blue and 60% glycerol, and the amplified inserts were singly spotted onto nylon filters (80 by 120 mm; Biodyne-A; Japan Pall Co. Ltd., Tokyo, Japan) by using a Biomek 2000 spotting machine (Bio-Rad, Tokyo, Japan). To normalize the signal intensities of the array spots, we spotted the PCR product of the mll2466 gene, which is the ortholog of the sigA gene of Bradyrhizobium japonicum. The sigA gene encodes the primary sigma factor of RNA polymerase and is essential for growth as a part of the housekeeping transcription machinery (2).
Strain and growth conditions.
Wild-type M. loti MAFF303099 (18, 37) was used throughout our experiments. For preparation of free-living cells, M. loti MAFF303099 was cultured in TY liquid medium (4) at 28°C. Mid-log-phase cultures (optical density at 660 nm [OD660], 0.2 to 0.3) were harvested by centrifugation at 4°C and then used for isolation of total RNA. For starvation experiments, M. loti MAFF303099 was cultured in B− minimal medium (29) to an OD660 of 0.2 to 0.3, and then the cells were harvested by centrifugation at 28°C, washed, and resuspended in B− minimal medium with or without 5 g of mannitol per liter for 20 min. B− minimum medium contained (per liter) 5 g of mannitol, 0.55 g of MgSO4 · 7H2O, 0.55 g of KNO3, 1.3 g of Ca(NO3)2 · 4H2O, 1.5 mg of MnSO4 · H2O, 0.25 mg of ZnSO4 · 7H2O, 3 mg of H3BO3, 1 mg of NaMoO4 · 2H2O, 0.1 mg of CuSO4 · 5H2O, 0.2 mg of biotin, 5 mg of thiamine-HCl, 33 mg of Fe(III)-Na-EDTA, 100 mg of K2HPO4, and 30 mg of KH2PO4 (29). The cells were harvested by centrifugation for isolation of total RNA.
For microaerobic conditions, 3 ml of an overnight culture was inoculated into 40 ml of TY liquid medium and then incubated at 28°C with reciprocal shaking at 180 rpm. When the OD660 of the culture reached 0.3, the gas phase was replaced with a gas mixture containing 1.5% (vol/vol) O2 and 98.5% (vol/vol) N2. After 30 min of incubation with reciprocal shaking at 100 rpm, the cells were harvested. The O2 concentration of the gas phase was monitored by using gas chromatography-mass spectrometry (QP-5000; Shimadzu, Kyoto, Japan).
Preparation of M. loti MAFF303099 bacteroids.
Washed sand was mixed with an equal volume of vermiculite and sterilized by autoclaving at 120°C. Surface-sterilized seeds of L. japonicus MG20 (21) were sown on the sand-vermiculite mixture. Plants were grown in a greenhouse and supplied with sterilized water containing M. loti MAFF303099. After 6 weeks of cultivation, nodules were detached from the roots and frozen immediately in liquid nitrogen. For each preparation, 1 g of frozen nodules was ground with a pestle in an ice-cold mortar. The nodule powder was transferred into a 50-ml tube containing zirconia balls (diameters, 3 to 5 mm; five particles of each size; Nikkato, Tokyo, Japan) and 6 ml of grinding buffer (0.3 M sucrose, 5 mM magnesium acetate, 0.2 M l-ascorbate, 2% polyvinylpyrrolidone, and 1.45 M 2-mercaptoethanol in 50 mM KH2PO4-Na2HPO4 buffer [pH 7.4]). After vigorous shaking with a vortex mixer, the nodule homogenate was filtered through three layers of Miracloth (Calbiochem, La Jolla, Calif.). The residue on the Miracloth was washed with 6 ml of extraction buffer (80 mM Tris-HCl [pH 7.5], 10 mM MgCl2, 10 mM 2-mercaptoethanol) containing 0.3 M sucrose. The combined filtrate was centrifuged at 10,000 × g for 3 min at 4°C, and the pellet was used as crude bacteroids.
Isolation of total RNA and preparation of labeled cDNA.
Total RNA of free-living cells (cultured in 100 ml of TY medium) and bacteroids (isolated from 1 g of nodules) were prepared by the method of Ditta et al. (7). Total RNA of cells grown under other conditions (starved, unstarved, aerobic, and microaerobic) were prepared by using RNAWiz (Ambion, Austin, Tex.). After removal of 16S and 23S rRNA from the total RNA with MicrobeExpress (Ambion) to decrease the background levels, the RNA was used as a template for synthesis of cDNA probes. Radioactive cDNA probes were prepared from the RNA by incorporation of [33P]dCTP during first-strand cDNA synthesis with reverse transcriptase. Each 30 μl of labeling mixture contained 40 μg of RNA, 250 ng of random hexamers (Invitrogen, Carlsbad, Calif.), 0.7 mM dATP, 0.7 mM dTTP, 0.7 mM dGTP, 4 μM dCTP, 40 U of RNase inhibitor (TaKaRa, Kyoto, Japan), 6 μl of 5× First Strand buffer (Invitrogen), 7 mM dithiothreitol, and 10 μl of [α-33P]dCTP. The mixture was incubated at 25°C for 10 min and then at 42°C for 2 min. We then added 200 U of SuperScript II reverse transcriptase (Invitrogen), and the reverse transcription reaction was performed at 42°C for 50 min.
Hybridization, image acquisition, and data analysis.
Prehybridization was carried out in 20 ml of Church's phosphate buffer (0.5 M Na2HPO4 [pH 7.2], 1 mM EDTA, 7% sodium dodecyl sulfate [SDS]) at 55°C for at least 3 h. Hybridization was performed in 10 ml of Church's phosphate buffer containing probe cDNA at 55°C for at least 15 h. Washing was done twice at room temperature in 2× SSC containing 0.1% SDS for 15 min, twice at room temperature in 0.2× SSC containing 0.1% SDS for 15 min, and twice at 55°C in 0.2× SSC containing 0.1% SDS for 15 min (1× SSC is 0.15 M NaCl plus 0.015 M sodium citrate). After the macroarray filters were washed, they were exposed to imaging plates (Fuji Film, Tokyo, Japan) for 15 h to 3 days, depending on the intensity of the signals, and the hybridized signals were captured as image files by using a BioImaging analyzer (BAS2000; Fuji Film). The primary processing of the data was performed with the ArrayVision software (Amersham Pharmacia, Uppsala, Sweden) to distinguish spots and to quantify the signal intensity after subtraction of the local background value.
Subsequently, the intensity data were processed by in-house programs written in Perl language. By using the programs, the data were sorted according to genome coordinate, used to determine averages and errors for repeated experiments, and normalized based on the total sum of the signal intensities or the signal intensity of sigA gene expression. The calculated data and comparison are available at a web page (http://orca10.bio.sci.osaka-u.ac.jp/array01/). Hybridization and image acquisition were performed in triplicate for different conditions.
To detect a genomic region in which clusters of genes are regulated collectively, we calculated the ratio (R) of the sum of signal intensities within a window range (w bases) around a genome position (n). The value was calculated for defined condition B versus control condition A with the following equation:
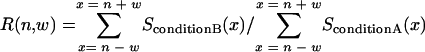 |
where SconditionA(x) and SconditionB(x) are the average signal intensities at position x under conditions A and B, respectively.
Quantification of gene expression by real-time RT-PCR.
The relative intensity of gene expression was estimated by quantitative real-time reverse transcription (RT)-PCR. The primers were designed by using the PrimerExpress software (Applied Biosystems, Tokyo, Japan). Each reaction mixture (total volume, 50 μl) containing 50 ng of purified RNA, 75 U of SuperScript II (Invitrogen), 20 U of RNase inhibitor (TaKaRa), and primers was prepared by using the SYBR Green PCR master mixture (Applied Biosystems). PCR amplification and detection of amplified DNA were performed with the ABI Prism 7700 sequence detection system and a GeneAmp 5700 (Applied Biosystems).
Gene disruption and phenotype assays.
The gentamicin resistance gene cassette (3) was inserted into the StuI (AatI) site, within mlr5932, in a 6.6-kb BamHI fragment from clone 233 of an ordered cosmid library (16) and cloned in pK18mob (39). The resulting plasmid was conjugated into M. loti MAFF303099, and gentamicin-resistant and neomycin-sensitive clones were selected. Correct insertion into the M. loti genome was confirmed by Southern hybridization. 1-Aminocyclopropane-1-carboxylic acid (ACC) deaminase activities of bacteroids and free-living cells of M. loti were determined by catalyzing the conversion of ACC to α-ketobutyrate (17). L. japonicus MG20 seeds inoculated with M. loti MAFF303099 and mutants were germinated in plastic growth pouches with nitrogen-free medium, and the numbers of nodules were determined (30).
RESULTS
Expression profiling under symbiotic, microaerobic, and starvation conditions.
We monitored genome-wide expression by using a macroarray that covers the entire genome of M. loti MAFF303099 during symbiosis, microaerobiosis, and carbon starvation; we then compared the data obtained with the data for appropriate controls and the data from three independent experiments. The hybridization signals of corresponding spots were automatically normalized with respect to the sigA signal on the same nylon membrane. In the comparative transcriptional profile analysis of bacteroids, the sum of the sigA-normalized intensities of bacteroids was similar to that obtained for free-living cells in TY medium. Because microaerobiosis and carbon starvation led to decreases in the sigA signal intensity relative to the sum of all signal intensities, further normalization was carried out based on the sum of all signal intensities in these comparisons. The profiling data were evaluated with in-house analysis programs (Fig. 1). The comparative expression data for the chromosome reflect quite different profiles during symbiosis (Fig. 1A), microaerobiosis (Fig. 1B), and carbon starvation (Fig. 1C).
FIG. 1.

Schematic representation of regions with up- and downregulated gene expression on the M. loti chromosome. (A to C) Upregulated (blue lines) and downregulated (red lines) clones in bacteroids (A) and under microaerobic growth conditions (B) and carbon starvation conditions (C). The line length indicates the logarithmic ratio of the hybridization signal to the signal obtained under reference conditions (for panel A, free-living cells; for panel B, cells grown under aerobic conditions; for panel C, unstarved cells). The innermost circle indicates fluctuations in the expression of transposase genes. (D) To compare regions of up- and downregulation (blue and red, respectively) at the genome level, we constructed expression profiles by using 100-kb windows. The arrowhead indicates a region that was upregulated both during symbiosis and under microaerobic conditions. The 2X and 10X bars indicate 2-fold and 10-fold differences, respectively, in expression levels. The symbiosis island (green bar) was located on the chromosome at coordinates 4644702 to 5255766 (18).
Giant expression region corresponds to the symbiosis island.
A comparison of the transcription profiles of bacteroids with those of free-living cells grown in TY medium showed that the region containing the symbiosis island was markedly upregulated under symbiotic conditions (Fig. 1A). To evaluate the genomic regions where clusters of genes are regulated collectively, we calculated a ratio of signal intensities by using a window size of 100 kb (Fig. 1D). The region where the enhancement of expression in bacteroids was greatest corresponded to the symbiosis island (Fig. 1A and D). However, transcription outside the island was strongly repressed in bacteroids compared with the transcription in free-living cells. The expression profile generated by comparing bacteroids with free-living cells grown in TY medium (Fig. 1A) was almost identical to the expression profile generated by comparing bacteroids with free-living cells cultured in B− medium either with or without a carbon source. The second-greatest enhancement of collective expression occurred under microaerobic conditions in a region downstream of the symbiosis island (Fig. 1B and D). In contrast to cells under symbiotic and microaerobic conditions, carbon-starved cells showed no regions where expression was remarkably upregulated (Fig. 1C and D).
Features of the giant expression region during symbiosis.
When we looked more closely at the giant expression region corresponding to the symbiosis island (Fig. 1D) by using smoothing calculations with a window size of 20 kb, seven independent EIs, designated EI-1 to EI-7, were identified (Fig. 2A). A comparison of the nucleotide sequences of the symbiosis islands of M. loti strains R7A and MAFF303099 revealed that the two islands share highly conserved colinear DNA regions (248 kb) with multiple deletions and insertions (43). To examine whether the conserved regions are upregulated in bacteroids, we compared the EIs in MAFF303099 and the conserved DNA region in the two strains (Fig. 2B).
FIG. 2.

EI on symbiosis island. (A) Expression profile constructed by using 20-kb windows to look more closely at the giant region upregulated during symbiosis that corresponded to the symbiosis island and contained seven EIs, which we designated EI-1 to EI-7. EIs in DNA regions specific for strain MAFF303099 are indicated by asterisks. The arrowheads indicate the positions of genes generally accepted to be involved in symbiotic nitrogen fixation. The 2X and 10X bars indicate 2-fold and 10-fold differences, respectively, in expression levels (upregulated, blue; downregulated, red). TTSS, type III secretion system. (B) Synteny between the symbiosis islands of M. loti strains MAFF303099 (18) and R7A (43). Clusters of red lines indicate conserved colinear DNA regions in the two strains. The yellow bars indicate the MAFF303099-specific regions. (C) Positions of the MAFF303099-specific region (yellow bars), the transposase gene (green arrowheads), EIs (blue bars), and active (red arrowheads) and inactive (black arrowheads) RpoN regulons in the symbiosis island of M. loti MAFF303099.
EI-1, EI-3, EI-6, and EI-7 contained the conserved DNA regions that are likely to be required for symbiotic nitrogen fixation, including genes for nitrogen fixation (nif, fix, fdx, and rpoN) (EI-1), cytochrome P450 (EI-6), and fixNOQPGHIS (EI-7). On the other hand, the MAFF303099-specific DNA regions were found in EI-2, EI-4, and EI-5, as well as the latter part of EI-1. The strain-specific expressed regions included seven small open reading frames that encode hypothetical or unknown proteins in EI-2, genes that encode 5-methyltetrahydrofolate-homocysteine S-methyltransferase, histidine decarboxylase, glutamine synthetase III, alanine racemase, and a probable dipeptidase in EI-4, and genes that encode phosphinothricin tripeptide synthetase B, l-proline 3-hydroxylase, a putative carboxylase, and γ-glutamyl kinase in EI-5.
Many transposase genes of insertion sequences (IS) are concentrated in the symbiosis island on the genome (18). When we plotted the positions of transposase genes in the symbiosis island, most of transposase genes were located in MAFF303099-specific DNA regions (Fig. 2C). In particular, there were transposase genes in and around the strain-specific EIs, EI-2, EI-4, EI-5, and the latter part of EI-1, suggesting that the strain-specific EIs are probably transferred and reconstructed by IS-mediated processes. Thus, we plotted the expression of clones containing the transposase genes, which are shown in the innermost circles in Fig. 1A through C. The expression of transposase genes on the symbiosis island was induced in bacteroids (Fig. 1A), and the IS families of these transposase genes were diverse and included IS3, IS4, IS5, IS6, IS21, IS91, and IS110. This suggests that the transposase genes are activated in symbiosis regardless of the IS family. Expression of transposase genes was found in the syrM-nodD3 intergenic region of Sinorhizobium meliloti bacteroids as well (1).
EI shared under microaerobic and symbiotic conditions.
M. loti cells cultured for 30 min under microaerobic conditions induced a strong EI downstream of the symbiosis island (Fig. 1B and D). This downstream island contained fixNOQP and other genes for the anaerobic cascade and anaerobic metabolism, such as the genes encoding the Fnr-type transcriptional regulator, coproporphyrinogen oxidase III, and aldehyde dehydrogenase (Fig. 3A). fixNOQP encodes a symbiotic cytochrome c oxidase complex that is specifically required for bacteroid respiration in the presence of very low O2 concentrations (13, 34). Like Rhizobium leguminosarum (41), Rhizobium etli (24), and S. meliloti (14), M. loti MAFF303099 possesses two sets of fixNOQP genes, one inside the symbiosis island and the other outside the symbiosis island. Interestingly, the DNA region around the copy of fixNOQP outside the symbiosis island is an EI even in bacteroids (Fig. 1D). Therefore, we directly compared the expression patterns of clones corresponding to the regions surrounding the copies of fixNOQP inside and outside the symbiosis island (fixNOPQsym and fixNOPQout). The regions of fixNOPQout and unknown genes (Fig. 3A) were expressed in both microaerobically grown cells and bacteroids, although other clones (Fig. 3A) were expressed exclusively during microaerobiosis. The expression patterns around fixNOQPsym were similar under microaerobic and symbiotic conditions (Fig. 3A). The expression of fixNsym gradually increased after the start of microaerobiosis, whereas fixNout was rapidly upregulated in a manner similar to that of other genes expressed in the presence of low O2 concentrations (Fig. 3B).
FIG. 3.

Expression around two different fixNOPQ regions inside and outside the symbiosis island of M. loti MAFF303099. (A) Profile of macroarray signals under microaerobic conditions and in bacteroids, on the basis of sigA normalization. (B) Time course of expression of selected genes after exposure to microaerobic conditions, obtained by using quantitative real-time RT-PCR assays. SYM and OUT indicate inside and outside the symbiosis island, respectively. The OUT region corresponds to the positions of the arrowheads in Fig. 1A, B, and D. M13 clones corresponded to SYM1 (RLB08201), to SYM15 (RLB21557) at 5220 to 5258 kb, and to OUT1 (RLB15504) to OUT33 (RLB03720) at 5379 to 5460 kb. Fnr, Fnr-type transcriptional regulator; Heme, coproporphyrinogen oxidase III; ALDH, aldehyde dehydrogenase.
Profiling of genes that were up- and downregulated during symbiosis, microaerobiosis, and starvation.
To identify differentially expressed genes with confidence, we adopted two stepwise strategies: (i) stringent selection of putative up- and downregulated genes from macroarray data and (ii) validation by quantitative real-time RT-PCR. First, we selected candidates according to the distribution profiles of scatter plots during symbiosis, microaerobic growth, and starvation (Fig. 4A through C). For example, putative upregulated clones were selected by using two thresholds; candidate upregulated clones had to have a signal intensity that was 20 times greater in bacteroids than in free-living cells, as well as a sigA-normalized signal intensity of >1.0 in bacteroids. The area corresponding to these thresholds is shown Fig. 4A. Figure 4D shows the results of an actual selection process for putative up- and downregulated genes in a nif region within the symbiosis island. Using the criteria described above, we selected eight genes, including nifDKE, as putative upregulated genes in this nif region. Sigma 54 consensus promoters were found upstream of six of the eight selected genes, suggesting that our gene selection process was valid.
FIG. 4.

Selection of putative upregulated genes in a nif region within the symbiosis island. (A to C) Scatter plots for bacteroids (A), microaerobiosis (B), and carbon starvation (C): log plots of the macroarray hybridization signals obtained under the different test conditions against the signals obtained under the reference conditions (for panel A, free-living cells; for panel B, aerobiosis; for panel C, unstarved cells). The x and y axes indicate the log values of the spot signals. (D) Bars indicate M13 clone inserts used for macroarray construction. The selected upregulated genes (red) include nifDKE, mlr5923 (lysine:N6-hydroxylase), mlr5924 (unknown protein), mlr5932 (ACC deaminase), mlr5943 (l-2,4-diaminobutyric acid transaminase), and mlr5944 (unknown protein). Arrows indicate the positions of the sigma 54 promoter consensus sequence (5′-TGGCAC[N5]TTGCT/A-3′) and the FixK box consensus sequence (5′-TTGA[N6]TCAA-3′) (9). (E) Differential expression of signals in bacteroids and free-living cells. Log (Bacteroids/Free-living cells) indicates the logarithmic ratio of the hybridization signal of bacteroids to the hybridization signal of free-living cells in TY medium. The solid and dotted boxes indicate clones that showed relatively strong (>1.0) and weak (<1.0) hybridization signals based on sigA normalization, respectively. We selected clones putatively upregulated in bacteroids that showed both a hybridization signal ratio of >20 and a signal intensity of >1.0 on the basis of sigA normalization (blue bars).
As a result, we selected (i) 99 genes that were upregulated and 26 genes that were downregulated in bacteroids compared with their expression in free-living cells; (ii) 83 genes that were upregulated and 22 genes that were downregulated during microaerobiosis compared to their expression under aerobic conditions; and (iii) 56 genes that were upregulated and 32 genes that were downregulated in starved cells compared to their expression in unstarved cells. All of the data are available at a website (http://orca10.bio.sci.osaka-u.ac.jp/array01/). To examine the validity of our selection, we determined the relative amounts of transcripts of the selected genes by using quantitative real-time RT-PCR based on the total amount of RNA prepared. The results supported the conclusion that our process for selecting putative up- and downregulated genes was valid (Table 1).
TABLE 1.
Validation of putative up- and downregulated genes by real-time RT-PCR
| Conditions | Gene | Gene annotation | Locationa | Relative expression
|
|
|---|---|---|---|---|---|
| Real-time RT-PCR | Array (clone) | ||||
| Bacteroid vs free-living | mlr5906 | Nitrogenase Mo-Fe protein alpha chain; NifD | SYM | 360 ± 56 | 698 (RLB09405) |
| mll9123 | Nitrilotriacetate monooxygenase component A | pMLa | 181 ± 28 | 27 (RLB12270) | |
| mlr6282 | Phosphinothricin tripeptide synthetase B | SYM | 116 ± 33 | 185 (RLB21690) | |
| msr5972 | Transposase (IS3 family orfA) | SYM | 26 ± 2 | 98 (RLB21047) | |
| mlr5876 | Cytochrome P450 | SYM | 25 ± 6 | 46 (RLB06802) | |
| mlr5943 | l-2,4-Diaminobutyric acid transaminase | SYM | 21 ± 4 | 223 (RLB13370) | |
| mlr5932 | ACC deaminase | SYM | 10.4 ± 0.3 | 53 (RLB15575) | |
| mll2921 | Flagellar L-ring protein; FlgH | OUT | 0.36 ± 0.10 | 0.02 (RLB24577) | |
| mlr9251 | Conjugal transfer protein | pMLa | 0.085 ± 0.015 | 0.01 (RLB09665) | |
| mll7254 | Glutamine synthetase (GS1) | OUT | 0.077 ± 0.024 | 0.05 (RLB05534) | |
| mlr7226 | Sugar ABC transporter, permease protein | OUT | 0.055 ± 0.008 | 0.043 (RLB23327) | |
| Microaerobic vs aerobic | mll6630 | Cytochrome c oxidase FixN chain | OUT | 86 ± 27 | 182 (RLB22846) |
| mlr6411 | Cytochrome c oxidase FixN chain | SYM | 11 ± 0.7 | 42 (RLB04306) | |
| mll6639 | Aldehyde dehydrogenase | OUT | 19 ± 2.2 | 37 (RLB12389) | |
| mlr6633 | Anaerbic coproporphyrinogen III oxidase | OUT | 83 ± 18 | 22 (RLB22521) | |
| mlr6580 | Serine/threonine kinase | OUT | 2.1 ± 0.2 | 13 (RLB17832) | |
| mll6600 | 5-Aminolevulinic acid synthase | OUT | 9.8 ± 0.3 | 7.7 (RLB08379) | |
| Carbon starvation vs no | mll1644 | Probable sterol methytransferase | OUT | >50b | 11 (RLB06335) |
| starvation | mll0710 | Glycerol-3-phosphate dehydrogenase | OUT | 20 ± 6.5 | 13 (RLB25054) |
| mlr2338 | Hypothetical protein | OUT | 14 ± 2.0 | 9.5 (RLB20249) | |
| mll1629 | Dihydropyrimidinase | OUT | 9.9 ± 1.0 | 6 (RLB06680) | |
| mlr4771 | Secreted sugar-binding protein | OUT | 8.1 ± 0.7 | 4.9 (RLB10318) | |
| mlr2341 | Hypothetical protein | OUT | 7.9 ± 1.3 | 9.5 (RLB20249) | |
| mll3075 | Amino acid/metabolite permease | OUT | 7.8 ± 1.1 | 5.4 (RLB08066) | |
| mll1631 | N-Carbamoyl-beta-alanine amidohydrolase | OUT | 4.8 ± 0.6 | 6 (RLB06680) | |
| mll3076 | Hypothetical protein | OUT | 3.6 ± 0.7 | 5.4 (RLB08066) | |
| mll4697 | Two-compoment system response regulator | OUT | 1.8 ± 0.1 | 2.2 (RLB15814) | |
| mlr1634 | Probable transcriptional regulator | OUT | 1.8 ± 0.3 | 2.7 (RLB17456) | |
| mll4698 | Two-compoment system histidine protein kinase | OUT | 1.8 ± 0.2 | 2.2 (RLB15814) | |
| mll6710 | Transcriptional regulator | OUT | 1.6 ± 0 | 2.9 (RLB23708) | |
Locations of replicons: chromosome, pMLa, and pMLb. OUT and SYM indicate outside and inside the symbiosis island on the chromosome at coordinates 4644702 to 5255766 (18).
The signal of unstarved cells was very weak.
RpoN (σ54) is a well-known σ factor that enables transcription of nitrogen fixation genes with the NifA protein in rhizobia. Thus, we compared the upregulated genes mentioned above with an in silico prediction of potential RpoN-dependent promoters on the genome of M. loti MAFF303099 (8). Of 99 genes upregulated in bacteroids, 24 (24%) were involved in the RpoN regulon (data are available at the http://orca10.bio.sci.osaka-u.ac.jp/array01/ website). Even in a symbiosis island, activation of 10 promoters (42%) among 24 predicted RpoN regulons was observed in bacteroids by this array analysis (Fig. 2C). In microaerobic conditions, only the exoP gene (mlr5276) was under the RpoN regulon. These results suggested that most of the upregulated genes in M. loti bacteroids were induced in a NifA-RpoN-independent manner.
Features of genes that were up- and downregulated during symbiosis.
When gene expression in bacteroids was examined, 90% of the 99 upregulated genes were located in the symbiosis island or the pMLa plasmid, whereas all 26 of the downregulated genes were located outside the symbiosis island. Genes related to symbiotic nitrogen fixation (nif and fix) exhibited greatly increased expression levels in bacteroids; rpoN, a σ factor that regulates the expression of the nif and fix genes, was also induced.
In addition, genes encoding enzymes involved in amino acid metabolism, such as aatA (mlr5883) and the glutamine synthetase GSIII (mlr6210), 2,4-diaminobutyric acid aminotransferase (mlr5943), and alanine racemase (mll6211) genes, were markedly upregulated in bacteroids compared with the expression in TY and B− minimum media, suggesting that activation of amino acid metabolism plays an important role in symbiotic nitrogen fixation (data are available at the http://orca10.bio.sci.osaka-u.ac.jp/array01/ website). Rhizobium has three different isozymes of glutamine synthetase, GSI, GSII, and GSIII (10). The GSI and GSIII genes were down- and upregulated in bacteroids of M. loti MAFF303099, respectively. Ethylene, a phytohormone generated from ACC in plants, is an inhibitor of nodulation, and application of ACC inhibits nodulation of legumes (30, 32). Interestingly, the gene for the enzyme that degrades the ethylene precursor, ACC deaminase, was induced in bacteroids (Table 1).
In contrast, the expression of clones carrying genes for the synthesis of fatty acids, flagella, and peptide glycans was thoroughly repressed during symbiosis. Other genes related to transcription, translation, DNA replication, aminoacyl-tRNA synthetase, and DNA polymerase also were repressed in bacteroids compared with their expression in vegetative cells. This repression seems to be reasonable in light of the biology of differentiated endosymbionts.
Features of genes that were up- and downregulated under microaerobic and starvation conditions.
Short exposure to microaerobic conditions induced the expression of genes for heme biosynthesis, such as the genes encoding corproporphyrinogen III dehydrogenase (mlr6392) and 5-aminolevulinic acid synthase (mll6600), as well as the two fixNOPQ operons (Fig. 2). Fnr-type transcription regulator (mll6632) and aldehyde dehydrogenase (mll6632) genes were upregulated, as were genes encoding unknown or hypothetical proteins. The genes that were upregulated in carbon-starved cells were involved in lipid metabolism and included genes that encode glycerol-3-phoshphate dehydrogenase (mll0710), sterol methyltransferase (mll1644), and coenzyme A biosynthesis (mll1631 and mll1629), suggesting that there was enhanced production of high-energy metabolites. In contrast, under microaerobic and starvation conditions cells commonly repressed genes for translation apparatus factors, including genes encoding many ribosomal proteins, peptidyl-tRNA hydrolase (mlr2694), and elongation factor Ts (mll0650).
Symbiotic phenotype of a mutant upregulated gene in bacteroids.
Rhizobial genes induced within nodules often affect the interactions between the two partners (31). Thus, we constructed a mutant that carried a defective mlr5932 gene, whose wild-type form (encoding ACC deaminase) was upregulated greatly during symbiosis (Fig. 4D and Table 1). The mlr5932 disruption mutant exhibited no ACC deaminase activity in bacteroids or free-living cells, whereas this activity was detected in the parent strain during symbiosis (Fig. 5A). This result demonstrates that mlr5932 encodes ACC deaminase, whose gene expression is induced exclusively during symbiosis, probably in a NifA-RpoN-dependent manner (Table 1 and Fig. 5A). The ACC deaminase knockout mutant D21 had a decreased ability to nodulate L. japonicus MG20 singly (Fig. 4C) and competitively (Fig. 4D), indicating that the induced ACC deaminase gene enhances nodulation and competitiveness in a manner similar to rhizobitoxine, a bradyrhizobial inhibitor of ACC synthase (26, 44, 45).
FIG. 5.

Phenotypic analyses of an insertion mutation in mlr5932, a gene that was upregulated during symbiosis. (A) Construction of the mlr5932 insertion mutant D21. A gentamicin cassette (Gm cassette) was inserted into the StuI site of mlr5932. The arrows and arrowhead indicate putative sigma 54 promoters and an upstream activator sequence (UAS), respectively. (B) ACC deaminase activities of wild-type strain MAFF303099 (WT) and mlr5932 insertion mutant D21 in bacteroids and free-living cells. The error bar indicates 1 standard deviation. ND, not detected. (C) Nodulation of L. japonicus MG20 inoculated with wild-type MAFF303099 or the mlr5932 insertion mutant D21. The error bars indicate standard errors. One asterisk and two asterisks indicate significant differences in mean numbers of nodules between the treatments at confidence levels of 0.05 and 0.01, respectively, as determined by Student's t test. (D) Nodulation competitiveness of wild-type strain MAFF303099 and the mlr5932 insertion mutant D21 when MAFF303099(pDG499) was used as a reference strain.
DISCUSSION
The symbiosis island of M. loti is a chromosomally integrated large element that can be transferred into the phenylalanine tRNA gene of nonsymbiotic mesorhizobia in the environment (41). This element resembles a pathogenicity island in terms of its tRNA gene target, different G+C content, and P4-type integrase-mediated transfer (15, 41). Pathogenicity islands of pathogenic bacteria code for toxins, adhesins, invasions, or other virulence factors, and their sizes range from 35 to 200 kb (15, 36). However, rhizobial symbiosis islands, which code for housekeeping functions, such as cellular processes, micro- and macromolecular metabolism other than nodulation, and nitrogen fixation, apparently are longer and have more complicated structures than the known pathogenesis islands. Symbiosis islands occur in the chromosomes of MAFF303099 (611 kb and phenylalanine-accepting tRNA gene target) (18) and B. japonicum USDA110 (681 kb and valine tRNA gene target) (19). Global expression analysis of bacteroids and free-living cells of M. loti MAFF303099 demonstrated that a symbiosis island is an extremely active region for gene expression during symbiosis (Fig. 1). One-half of the EIs clustered in the symbiosis island were located in M. loti strain MAFF303099-specific DNA regions, which are not found in strain R7A (Fig. 2A). It is unclear whether the strain-specific EIs contribute to the establishment and maintenance of symbiotic nitrogen fixation. However, one possible explanation is that the strain-specific EIs support host-specific nodulation and nitrogen fixation with L. japonicus, because MAFF303099 and R7A were isolated from different Lotus species, L. japonicus and L. corniculatus, growing in different countries (Japan and New Zealand, respectively).
Most of the upregulated genes in bacteroids are not in a conventional NifA-RpoN regulon and fixLJ-fixK circuit (Fig. 2C and Fig. 3) (for details see supplemental data at the website http://orca10.bio.sci.osaka-u.ac.jp/array01/). Thus, there might be other mechanisms of region expression, such as a change in the configuration of the chromosome, which is equivalent to the chromatin remodeling that occurs in eukaryotes (33), and histone-like proteins in bacteria (9). Hence, some sort of reconfiguration could allow expression from the symbiosis island and thus increased expression of transposase and nonsymbiotic genes. This high expression level not only could support symbiotic nitrogen fixation but also could give rise to DNA rearrangement by the activated transposase genes (Fig. 1A). This hypothesis could account for the rapid evolution of symbiosis islands in mesorhizobia. The two islands of M. loti strains R7A and MAFF303099 carry many strain-specific islets; only 40% (248 kb) of the backbone DNA region is conserved (Fig. 2) (43). MAFF303099 accumulated 19% (114 kb) of the transposase, integrase, and resolvase genes compared with the size of the symbiosis island (18). Lateral gene transfer plays a crucial role in the acquisition of symbiotic nitrogen fixation by rhizobia (5). In a sense, the rhizobial genome is merely a vehicle for the symbiosis island.
As in M. loti, duplicated fixNOPQ operons occur in R. leguminosarum (41), R. etli (24), and S. meliloti (14); this is probably a result of the evolution of symbiosis and oxygen respiration (13, 38). The array of fixNOPQ operons in M. loti MAFF303099 is unique in that both operons are located on a single chromosome (18). fixNOPQsym and fixNOPQout in M. loti MAFF303099 probably arose from lateral transfer with the symbiosis island and vertical inheritance, respectively, because of their locations and the relatively low levels of amino acid homology (79 to 88%) between the sets of gene. The rapid upregulation of fixNout compared with the expression of fixNsym (Fig. 2B) indicates that the fixNOPQout operon preferentially functions in microaerobic environments. However, bacteroids likely use both fixNOPQout and fixNOPQsym for symbiotic respiration (Fig. 2). In this context, a single fixNOPQ operon outside the putative symbiosis island functions in B. japonicum bacteroids (13, 19, 34). Therefore, rhizobia appear to have recruited a housekeeping fixNOPQ operon present in bacteria prior to transfer of the symbiosis island.
Although amino acid biosynthetic genes should be expressed more in minimal medium than in TY medium, the profiling data indicated that there was marked activation of amino acid metabolism in bacteroids compared with the metabolism in B− minimal medium, as well as TY medium. This implies that the genes are induced exclusively for symbiotic nitrogen fixation. Using R. leguminosarum bv. viciae and pea plants, Lodwig et al. recently proposed that amino acid cycling drives nitrogen fixation in legume-rhizobium symbioses (23). Our profiling data partially support this model. The aatA (mlr5883) and dctA (mll5840) homologs on the symbiosis island and the malate dehydrogenase gene (mlr9216) on pMLa were strongly expressed in M. loti MAFF303099 bacteroids, which are able to increase their oxaloacetate and aspartate supplies. M. loti MAFF303099 lacks homologs of the Aap/Bra transporters in R. leguminosarum but does have many genes for the amino acid ABC transporter, some of which perhaps function as exporters and importers of amino acids. 2,4-Diaminobutyric acid (DABA) aminotransferase synthesizes alanine by using DABA and pyruvate as substrates. Thus, the upregulation of genes for DABA aminotransferase (mlr5943) and alanine racemase (mll6211) suggests that there is activation of alanine metabolism in bacteroids as well, which might participate in the amino acid cycling between the two partners.
Our disruption analysis demonstrated that the mlr5932 gene, which is upregulated in bacteroids, encodes ACC deaminase and enhances nodulation and competitiveness in L. japonicus MG20 (Fig. 4). Recently, ACC deaminase in R. leguminosarum was reported to promote nodulation of pea plants as well (25). Moreover, rhizobitoxine synthesized by Bradyrhizobium elkanii enhances nodulation and the competitiveness of legumes by inhibiting endogenous ethylene production in host plants (44, 45). Therefore, a likely general strategy of rhizobial organisms is to reduce the amount of ethylene synthesized by their legume symbionts (26). Indeed, the genomic sequences of M. loti R7A (43) and B. japonicum USDA110 (19) both contain a nifA-dependent ACC deaminase gene.
In conclusion, our genome-wide transcriptional analysis revealed the presence of EIs and their clustering in and around the symbiosis island in the M. loti MAFF303099 genome. This finding suggests possible mechanisms for global gene regulation, symbiotic functions, and rhizobial evolution. Moreover, our profiling data should be useful as screening tools for identification of novel rhizobial genes relevant to symbiosis.
Acknowledgments
This work was supported in part by the Tokachi Federation of Agricultural Cooperatives, Obihiro, Hokkaido, Japan. We thank PROBRAIN (Japan) for supporting the research of K. Minamisawa and K. Saeki. Research by K. Saeki and H. Omori and by K. Minamisawa is also supported in part by Grants-in-Aid for Scientific Research 15013232 and 14360037, respectively.
We are grateful to Shin-ichrou Kitayama for his assistance with O2 tension determination.
REFERENCES
- 1.Ampe, F., E. Kiss, F. Sabourdy, and J. Batut. 2003. Transcriptome analysis of Sinorhizobium meliloti during symbiosis. Genome Biol. 4:R15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Beck, C., R. Marty, S. Klausli, H. Hennecke, and M. Gottfert. 1997. Dissection of the transcription machinery for housekeeping genes of Bradyrhizobium japonicum. J. Bacteriol. 179:364-369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Becker, A., M. Schmidt, W. Jäger, and A. Pühler. 1995. New gentamicin-resistance and lacZ promoter-probe cassettes suitable for insertion mutagenesis and generation of transcriptional fusions. Gene 162:37-39. [DOI] [PubMed] [Google Scholar]
- 4.Beringer, J. E. 1974. R-factor transfer in Rhizobium leguminosarum. J. Gen. Microbiol. 84:188-198. [DOI] [PubMed] [Google Scholar]
- 5.Broughton, W. G., and X. Perret. 1999. Genealogy of legume-Rhizobium symbioses. Curr. Opin. Plant Biol. 2:305-311. [DOI] [PubMed] [Google Scholar]
- 6.Denarie, J., F. Debeelle, and J. C. Prome. 1996. Rhizobium lipo-chitooligosaccharide nodulation factors: signaling molecules mediating recognition and morphogenesis. Annu. Rev. Biochem. 65:503-535. [DOI] [PubMed] [Google Scholar]
- 7.Ditta, G., E. Virts, A. Palomares, and C. Kin. 1987. The nifA gene of Rhizobium meliloti is oxygen regulated. J. Bacteriol. 169:3217-3223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dombrech, B., K. Marchal, J. Vanderleyden, and J. Michiels. 2002. Prediction and overview of the RpoN-regulon in closely related species of the Rhizobiales. Genome Biol. 3:0076.1-0076.11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dorman, C. J., and Deighan, P. 2003. Regulation of gene expression by histone-like proteins in bacteria. Curr. Opin. Genes Dev. 13:179-184. [DOI] [PubMed] [Google Scholar]
- 10.Encarnación, S., J. Calderón, A. S. Gelbard, A. J. L. Cooper, and J. Mora. 1998. Glutamine biosynthesis and the utilization of succinate and glutamine by Rhizobium etli and Sinorhizobium meliloti. Microbiology 144:2629-2638. [DOI] [PubMed] [Google Scholar]
- 11.Ewing, B., and P. Green. 1998. Base-calling of automated sequencer traces using phred. II. Error probabilities. Genome Res. 8:186-194. [PubMed] [Google Scholar]
- 12.Ewing, B., L. Hillier, M. C. Wendl, and P. Green. 1998. Base-calling of automated sequencer traces using phred. I. Accuracy assessment. Genome Res. 8:175-185. [DOI] [PubMed] [Google Scholar]
- 13.Fischer, H. M. 1994. Genetic regulation of nitrogen fixation in rhizobia. Microbiol. Rev. 58:352-386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Galibert F, T. M. Finan, S. R. Long, A. Puhler, P. Abola, F. Ampe, F. Barloy-Hubler, M. M. J. Barnett, A. Becker, P. Boistard, et al. 2001. The composite genome of the legume symbiont Sinorhizobium meliloti. Science 292:668-672. [DOI] [PubMed] [Google Scholar]
- 15.Hacker, J., G. Blum-Oeher, I. Muhldorfer, and H. Tschape. 1997. Pathogenecity islands of virulent bacteria: structure, function and impack on microbial evolution. Mol. Microbiol. 23:1089-1097. [DOI] [PubMed] [Google Scholar]
- 16.Hattori, Y., H. Omori, M. Hanyu, N. Kaseda, E. Mishima, T. Kaneko, S. Tabata. S., and K. Saeki. 2002. Ordered cosmid library of the Mesorhizobium loti MAFF303099 genome for systematic gene disruption and complementation analysis. Plant Cell Physiol. 43:1542-1557. [DOI] [PubMed] [Google Scholar]
- 17.Honma, M., and T. Shimomura, T. 1978. Metabolism of 1-aminocyclopropane-a-carboxylic acid. Agric. Biol. Chem. 42:1825-1831. [Google Scholar]
- 18.Kaneko, T., Y. Nakamura, S. Sato, E. Asamizu, T. Kato, S. Sasamoto, A. Watanabe, K. Idesawa, A. Ishikawa, K. Kawashima, T. Kimura, Y. Kishida, C. Kiyokawa, M. Kohara, M. Matsumoto, A. Matsuno, Y. Mochizuki, S. Nakayama, N. Nakazaki, S. Shimpo, M. Sugimoto, C. Takeuchi, M. Yamada, and S. Tabata. 2000. Complete genome structure of the nitrogen-fixing symbiotic bacterium Mesorhizobium loti. DNA Res. 7:331-338. [DOI] [PubMed] [Google Scholar]
- 19.Kaneko, T., Y. Nakamura, S. Sato, K. Minamisawa, T. Uchiumi, S. Sasamoto, A. Watanabe, K. Idesawa, M. Iriguchi, K. Kawashima, M. Kohara, M. Matsumoto, S. Shimpo, H. Tsuruoka, T. Wada, M. Yamada, and S. Tabata. 2002. Complete genomic sequence of nitrogen-fixing symbiotic bacterium Bradyrhizobium japonicum USDA110. DNA Res. 9:189-197. [DOI] [PubMed] [Google Scholar]
- 20.Kaneko. T., K. Saeki, and K. Minamisawa. 2003. Genome analysis of Mesorhizobium loti: a symbiotic partner to Lotus japonicus, p. 203-216. In T. Nagata and S. Tabata (ed.), Bio/technology in agriculture and forestry, vol. 52. Springer-Verlag, Berlin, Germany. [Google Scholar]
- 21.Kawaguchi, M. 2000. Lotus japonicus ′Miyakozima' MG-20: an early-flowering accession suitable for indoor handling. J. Plant Res. 113:507-509. [Google Scholar]
- 22.Krusell, L., L. H. Madsen, S. Sato, G. Aubert, A. Genua, K. Szczyglowski, G. Duc, T. Kaneko, S. Tabata, F. de Bruijin, E. Pajuelo, N. Sandal, and J. Stougaard. 2002. Shoot control of root development and nodulation is mediated by a receptor-like kinase. Nature 420:422-426. [DOI] [PubMed] [Google Scholar]
- 23.Lodwig, E. M., A. H. F. Hosie, A. Bourdes, K. Findlay, D. Allaway, R. Karunakaran, J. A. Downie, and P. S. Poole. 2003. Amino-acid cycling drives nitrogen fixation in the legume-Rhizobium symbiosis. Nature 422:722-726. [DOI] [PubMed] [Google Scholar]
- 24.Lopez, O., C. Morera, J. Miranda-Rios, L. Girard, D. Romero, and M. Soberon. 2001. Regulation of gene expression in response to oxygen in Rhizobium etli: role of FnrN in fixNOPQ expression and in symbiotic nitrogen fixation. J. Bacteriol. 183:6999-7006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ma, W., F. C. Guinel, and B. R. Glick. 2003. Rhizobium leguminosarum biovar viciae 1-aminocyclopropane-1-carboxylate deaminase promotes nodulation of pea plants. Appl. Environ. Microbiol. 69:4396-4402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ma, W., D. M. Penrose, and B. R. Glick. 2002. Strategies used by rhizobia to lower plant ethylene levels and increase nodulation. Can. J. Microbiol. 48:947-954. [DOI] [PubMed] [Google Scholar]
- 27.Niner, B. M., and A. M. Hirsch. 1998. How many Rhizobium genes, in addition to nod, nif/fix, and exo, are needed for nodule development and function? Symbiosis 24:51-102. [Google Scholar]
- 28.Nisimura, R., M. Hayashi, G. Wu, H. Kouchi, H. Imaizumi-Anraku, Y. Murakami, S. Kawasaki, S. Akao, M. Ohmori, M. Nagasawa, K. Harada, and M. Kawaguchi. 2002. HAR1 mediates systemic regulation of symbiotic organ development. Nature 420:426-429. [DOI] [PubMed] [Google Scholar]
- 29.Niwa, S., M. Kawaguchi, H. Imaizumi-Anraku, S. A. Chechetka, N. Ishizawa, A. Ikuta, and H. Kouchi. 2001. Responses of a model legume Lotus japonicus to lipochitin oligosaccharide nodulation factors purified from Mesorhizobium loti JRL501. Mol. Plant-Microbe Interact. 14:848-856. [DOI] [PubMed] [Google Scholar]
- 30.Nukui, N., H. Ezura, K. Yuhashi, T. Yasuta, and K. Minamisawa. 2000. Effects of ethylene precursor and inhibitors for ethylene biosynthesis and perception on nodulation in Lotus japonicus and Macroptilium atropurpureum. Plant Cell Physiol. 41:893-897. [DOI] [PubMed] [Google Scholar]
- 31.Oke, V., and S. R. Long. 1999. Bacterial genes induced within the nodule during the Rhziobium-legume symbiosis. Mol. Microbiol. 32:837-849. [DOI] [PubMed] [Google Scholar]
- 32.Penmetsa, R. V., and D. R. Cook. 1997. A legume ethylene-insensitive mutant hyperinfected by its rhizobial symbiont. Science 275:527-530. [DOI] [PubMed] [Google Scholar]
- 33.Perterson, C. L. 2003. Transcription activation: getting a dispatch grip on condensed chromatin. Curr. Biol. 13:R195-R197. [DOI] [PubMed] [Google Scholar]
- 34.Preisig, O., D. Anthamatten, and H. Hennecke. 1993. Genes for a microaerobically induced oxidase complex in Bradyrhizobium japonicum are essential for a nitrogen-fixing endosymbiosis. Proc. Natl. Acad. Sci. 90:3309-3313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Radutoiu, S., L. H. Madsen, E. B. Madsen, H. H. Felle, Y. Umehara, M. Gronkund, S. Sato, Y. Nakamura, S. Tabata, N. Sandal, and J. Stougaard. 2003. Plant recognition of symbiotic bacteria requires two LysM receptor-like kinases. Nature 425:585-593. [DOI] [PubMed] [Google Scholar]
- 36.Reid, S. D., C. J. Corinne, A. C. Bumbaugh, R. K. Selander, and T. S. Whittam. 2000. Parallel evolution of virulence in pathogenic Escherichia coli. Nature 406:64-67. [DOI] [PubMed] [Google Scholar]
- 37.Saeki, K., and H. Kouchi, H. 2000. The Lotus symbiont, Mesorhizobium loti: molecular genetic techniques and application. J. Plant Res. 113:457-465. [Google Scholar]
- 38.Saraste, M., and J. Castresana. 1994. Cytochrome oxidase evolved by tinkering with denitrification enzymes. FEBS Lett. 341:1-4. [DOI] [PubMed] [Google Scholar]
- 39.Schafer, A., A. Tauch, W. Jäger, J. Kalinowski, G. Thierbach, and A. Pühler. 1994. Small mobilizable multi-purpose cloning vectors derived from the Escherichia coli plasmids pK18 and pK19: selection of defined deletions in the chromosome of Corynebacterium glutamicum. Gene 145:69-73. [DOI] [PubMed] [Google Scholar]
- 40.Schauser, L., A. Roussis, J. Stiller, and J. Stougaard. 1999. A plant regulator controlling development of symbiotic root nodules. Nature 402:191-195. [DOI] [PubMed] [Google Scholar]
- 41.Schluter, A., T. Patschknowski, J. Quandet, L. B. Selinger, S. Weidner, M. Kramer, L. Zhou, M. F. Hynes, and U. B. Priefer. 1997. Functional and regulatory analysis of the two copies of the fixNOPQ operon of Rhziobium leguminosarum strain VF39. Mol. Plant-Microbe Interact. 10:605-616. [DOI] [PubMed] [Google Scholar]
- 42.Sullivan, J. T., and C. W. Ronson. 1998. Evolution of rhizobia by acquisition of a 500-kb symbiosis island that integrates into a phe-tRNA gene. Proc. Natl. Acad. Sci. 95:5145-5149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sullivan, J. T., J. R. Trzebiatowski, R. W. Cruickshank, J. Gouzy, S. D. Brown, R. M. Elliot, D. J. Fleetwood, N. G. McCallum, U. Rossbach, G. S. Stuart, J. E. Weaver. R. J. Webby, F. J. De Bruijn, and C. W. Ronson. 2002. Comparative sequence analysis of the symbiosis island of Mesorhizobium loti strain R7A. J. Bacteriol. 184:3086-3095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yasuta, T., S. Satoh, and K. Minamisawa. 1999. New assay for rhizobitoxine based on inhibition of 1-aminocyclopropane-1-carboxylate synthase. Appl. Environ. Microbiol. 65:849-852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Yuhashi, K., N. Ichikawa, H. Ezura, S. Akao, Y. Minakawa, N. Nukui, T. Yasuta, and K. Minamisawa. 2000. Rhizobitoxine production by Bradyrhizobium elkanii enhances nodulation and competitiveness on Macroptilium atropurpureum. Appl. Environ. Microbiol. 66:2658-2663. [DOI] [PMC free article] [PubMed] [Google Scholar]