

Abstract
Microglia are the first line of immune defense against central nervous system (CNS) injuries and disorders. These highly plastic cells play dualistic roles in neuronal injury and recovery and are known for their ability to assume diverse phenotypes. A broad range of surface receptors are expressed on microglia and mediate microglial ‘On’ or ‘Off’ responses to signals from other host cells as well as invading microorganisms. The integrated actions of these receptors result in tightly regulated biological functions, including cell mobility, phagocytosis, the induction of acquired immunity, and trophic factor/inflammatory mediator release. Over the last few years, significant advances have been made towards deciphering the signaling mechanisms related to these receptors and their specific cellular functions. In this review, we describe the current state of knowledge of the surface receptors involved in microglial activation, with an emphasis on their engagement of distinct functional programs and their roles in CNS injuries. It will become evident from this review that microglial homeostasis is carefully maintained by multiple counterbalanced strategies, including, but not limited to, ‘On’ and ‘Off’ receptor signaling. Specific regulation of theses microglial receptors may be a promising therapeutic strategy against CNS injuries.
1. Introduction
Microglia were first recognized as a distinct cellular entity in the central nervous system (CNS) by the German anatomist Franz Nissl and subsequently given their name by the Spanish neuroscientist Pío del Río-Hortega between 1919 and 1921. Over the course of the past century, much evidence has accumulated on the importance of this cell population in CNS homeostasis and its involvement in CNS pathologies. Similar to the role of peripheral macrophages, microglia are now known as the first line of defense against CNS injuries, including stroke, traumatic brain injury, and spinal cord injury. Following an insult, resident microglia rapidly mobilize to the injury site, where they play a role in acute damage and modulate the long-term progression of injury. Whether this microglial activation in the compromised CNS is helpful or destructive remains controversial. In support of a beneficial role of microglia, selective depletion of proliferative microglia is known to exacerbate ischemic brain injuries (Faustino et al., 2011; Lalancette-Hebert et al., 2007), whereas injection of microglia into the ischemic brain is known to ameliorate injuries (Imai et al., 2007). Microglial activation is thought to benefit the injured brain by removing cell debris and restoring tissue integrity (Hanisch and Kettenmann, 2007; Kwon et al., 2013; Miron et al., 2013; Thored et al., 2009). In contrast, mounting evidence reveals that inappropriate or excessive microglial activation may lead to secondary expansion of brain damage and deterioration of neurological outcomes (Barone and Feuerstein, 1999; Dirnagl et al., 1999). The toxicity of microglia is mediated by the release of a plethora of harmful substances, including nitric oxide (NO), reactive oxygen species (ROS), and proinflammatory cytokines (Block et al., 2007). Microglia can also impair neurogenesis (Ekdahl et al., 2003; Liu et al., 2007), oligodendrogenesis (Miron 2013) and prevent axon regeneration (Schwab and Bartholdi, 1996). As a result of findings such as these, it is increasingly well-accepted that microglia play dualistic roles in neural injury and recovery and can improve or destroy tissue integrity depending upon the cellular context. An improved understanding of the mechanisms underlying microglial activation and their functional modulation by the local microenvironment is likely to advance our knowledge of many CNS pathological states.
Because of the dual-faced nature of microglia in the CNS, their activity must be tightly regulated so that they can be promptly turned on as the first responders to noxious stimuli and then rapidly turned off to avoid unwanted side effects. In general, microglia communicate with other CNS cells in one of two ways. First, microglia are able to recognize the signals secreted and released by other CNS cells from a considerable distance. Second, microglia bind to surface molecules expressed on adjacent CNS cells. These microglia-regulating signals can be further divided into two broad categories, so-called ‘Off’ and ‘On’ signals (Biber et al., 2007). The ‘Off’ signals are usually constitutively expressed or released under physiological, resting conditions. Under pathological conditions, either the loss of the ‘Off’ signal or the gain of the ‘On’ signal initiates the rapid activation and mobilization of microglia. Microglia sense these signals through a wide array of recognition receptors. Specific regulation of these receptors with pharmacological and other tools thus represents a promising therapeutic strategy against CNS injuries.
In this review, we will describe the current state of knowledge on microglial surface receptors and their roles in CNS injuries, with a special emphasis on their engagement of distinct functional programs. This information leads to an improved understanding of how different receptors work in concert to maintain microglia in a state of balanced equilibrium. It will become evident from this review that microglial homeostasis is carefully maintained by multiple opposing and complementary switches, including the ‘On’ and ‘Off’ receptors and their downstream signaling events. These opposing signals appear to have evolved such that they are normally activated only when needed at the site of injury and subsequently inactivated to prevent excessive release of toxic mediators. Indeed, the counterbalancing of these opposing signals is a hallmark of a well-balanced and healthy CNS immune system (Figure 1). When this homeostatic equilibrium is disrupted, neurological injury may be exacerbated. Thus, the carefully timed modulation of microglial receptor activity is a rational therapeutic strategy.
Figure 1. Microglial receptors.
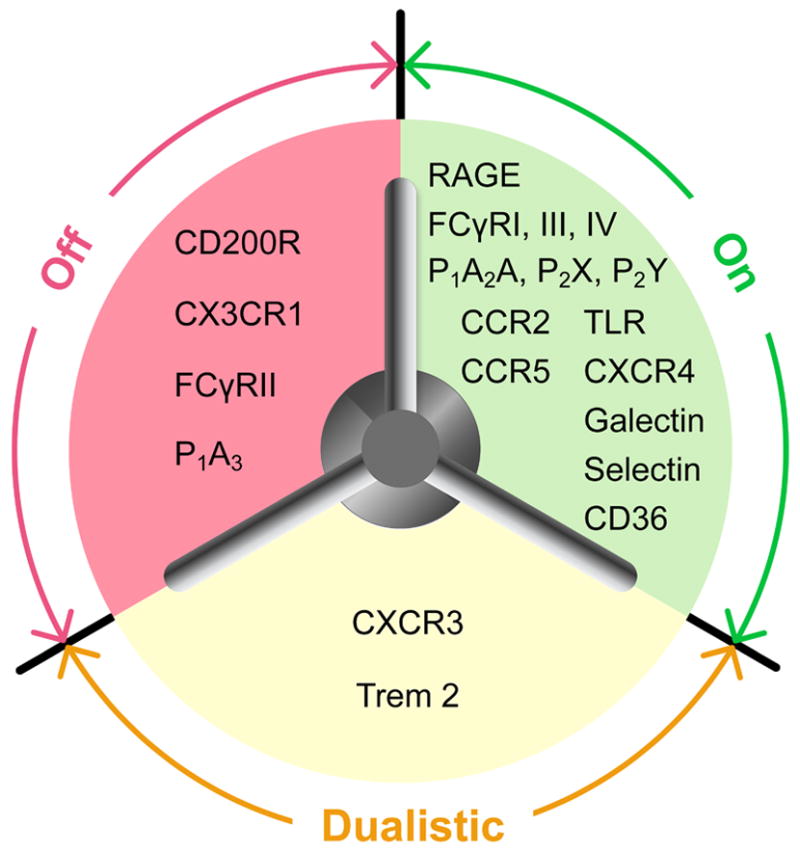
Various surface receptors act as opposing and complementary switches to maintain microglial homeostasis. “On” receptors: A large number of receptors have been identified to turn on microglial activation in response to different stimuli. “Off” receptors: Several receptors constitutively expressed on resting microglia help to maintain their quiescence. Dualistic receptors: The activation of these receptors induces both activating and inhibitory effects in microglia, allowing for a high degree of resolution in microglial responsiveness to differing stimuli. P1: P1 adenosine receptors; P2: P2 ATP receptors; RAGE: receptor for advanced glycation endproducts; TLR: toll-like receptor; TREM: triggering receptors expressed on myeloid cells.
2. Microglial receptors in the immunoglobulin superfamily
The receptors in the immunoglobulin superfamily (IgSF) consist of proteins with one or more IgSF structural domains. Recently, this family of receptors has been the focus of much attention because several of its members recognize the ‘Off’ signal and maintain microglia in a normal, surveillent status. In general, these ‘Off’ receptors signal through a cytoplasmic-domain immunoreceptor tyrosine-based inhibition motif (ITIM). In contrast, the ‘On’ receptors in this family sense pathological changes in neurons and other CNS components and signal through a cytoplasmic-domain immunoreceptor tyrosine-based activation motif (ITAM), thereby leading to microglial activation (Linnartz and Neumann, 2013). These opposing roles of ‘Off’ versus ‘On’ receptors and ITIM versus ITAM sigaling are typical of the counterbalancing strategies that determine the direction of microglial responses to fluctuations in the environmental milieu and maintain microglial equilibrium. Therefore, we will discuss several receptors in this family in the following section.
2.1. Triggering receptors expressed on myeloid cells (TREM)
TREM is a family of cell surface receptors specifically expressed on myeloid cells. The trem gene cluster is located on human chromosome 6p21 and mouse chromosome 17C3, and encodes TREM-1, TREM-2, TREM-3 (in the mouse), and other TREM-like genes (Klesney-Tait et al., 2006). The activation of TREM-1 enhances inflammatory responses while TREM-2 activation inhibits inflammation (Klesney-Tait et al., 2006), again exemplifying the principle of counterbalanced activating and inhibitory immune responses. TREM-2 is most relevant to the CNS by virtue of its constitutive expression on microglia and its proven role in microglia-neuron interactions.
2.1.1. TREM-2 mediates microglial clearance activity
TREM-2 has been detected in microglia both in vitro and in vivo by in situ hybridization and immunohistochemical staining (Kiialainen et al., 2005; Schmid et al., 2002; Sessa et al., 2004; Thrash et al., 2009). Microglial expression of TREM-2 is highly heterogeneous across brain regions, with high levels of TREM-2-expressing microglia in the lateral entorhinal cortex, cingulate cortex, and white matter, whereas few, if any, TREM-2-expressing microglia are found in the hypothalamus, circumventricular organs, or the median eminence. Even in areas with a relatively high proportion of TREM-2-expressing microglia, TREM-2-positive and TREM-2-negative microglia tend to be intermixed. These differences in TREM-2 expression in the healthy brain may reflect the diversity in microglial responsiveness and may contribute to regional variations in sensitivity to pathological signals.
The intracellular distribution of TREM-2 within microglia also shows distinct features. The majority of TREM-2 is located intracellularly in the Golgi complex and in exocytotic vesicles (Prada et al., 2006; Sessa et al., 2004). This intracellular pool of TREM-2 is dislodged and migrates to the cell surface following ionomycin or interferon-gamma (IFN-γ) exposure, resulting in transient elevation of surface TREM-2 (Sessa et al., 2004). Another study reported that microglial TREM-2 mRNA expression is rapidly decreased in response to lipopolysaccharide (LPS) or LPS+IFN-γ stimulation (Schmid et al., 2002). All these studies suggest that microglial TREM-2 responds rapidly to inflammatory signals by alterations in transcription and subcellular distribution, supporting the notion that TREM-2 is exquisitely sensitive and that it may regulate microglial activity. Indeed, TREM-2 overexpression and knockdown studies in microglia have demonstrated that TREM-2 stimulation promotes microglial migration and the phagocytosis of neuronal cell debris without inducing inflammatory cytokine expression (Takahashi et al., 2005). Such special ‘turning on’ properties render TREM-2 a reasonable candidate for directing the benefical activation of microglia.
The first exploration of the role of TREM-2 in CNS pathologies was initiated upon the genetic analysis of a recessively inherited disorder, polycystic lipomembranous osteodysplasia with sclerosing leukoencephalopathy (PLOSL, otherwise named Nasu-Hakola disease). This disease is characterized by insidious and progressive presenile dementia associated with multiple bone cysts. Disruption of TREM-2 or its adaptor protein DNAX-activating protein of molecular mass 12 kilodaltons (DAP12) was identified as the causal mutation underlying PLOSL (Paloneva et al., 2002). TREM-2 deficiency may result in insufficient clearance of apoptotic cells and exaggerated inflammation, which may elicit the frank CNS degeneration that is evident in PLOSL patients. Interestingly, the loss of TREM-2 seems to be important for white matter pathology because PLOSL patients exhibit sclerotic lesions that include activated microglia in the white matter and myelin pallor (Verloes et al., 1997). Consistent with this notion are the frequent close appositions of TREM-2-positive microglia with oligodendrocytes during development (Thrash et al., 2009) and the predominant distribution of TREM-2-positive microglia in the white matter of the adult brain (Schmid et al., 2002).
Recently, microglial TREM-2 expression has also been shown to be involved in other neurological disorders such as multiple sclerosis (MS) (Piccio et al., 2007) and Alzheimer’s disease (AD) (Frank et al., 2008). Some studies also reveal increased microglial expression of TREM-2 after ischemic stroke (Heldmann et al., 2011; Sugimoto et al., 2014). The function of TREM-2 in acute brain injuries, however, has not yet been defined.
2.1.2. TREM signaling
Despite an awareness of the importance of TREM-2 in microglial function, the ligands of TREM-2 have not yet been identified. This has hindered our understanding of microglial activation through this receptor. Using TREM-Fc fusion proteins, Hsieh and colleagues have demonstrated that multiple types of cultured neuronal cells bind to TREM2-Fc but not TREM1-Fc, suggesting that TREM-2 ligand(s) are constitutively expressed on healthy neurons (Hsieh et al., 2009). In addition, apoptotic signals were found to enhance the neuronal expression of TREM-2 ligand(s). The Hsieh study further demonstrated that TREM-2 and TREM-2 ligand(s) form a receptor-ligand pair connecting microglia with apoptotic neurons and directing the phagocytic removal of damaged cells. However, the specific identity of TREM-2 ligand(s) remains elusive. Another study has shown that heat shock protein (HSP) 60, a mitochondrial chaperone that can also be expressed at the cell surface, is a TREM-2-binding protein exposed on the surface of neurons as well as astrocytes (Stefano et al., 2009). Accordingly, treatment with HSP60 was found to stimulate microglial phagocytosis in a TREM-2-dependent manner.
In contrast to the idea of a single specific ligand for TREM-2, Takegahara and colleagues have documented that TREM-2 associates with another receptor on the cell surface of dendritic cells named Plexin-A1, and forms a higher order receptor complex containing TREM-2, plexin-A1, and DAP12 (Takegahara et al., 2006). The activation of plexin-A1 by its ligand, semaphorin 6D, can be blocked by decreasing either TREM-2 or DAP12 expression, suggesting a functional connection between plexin-A1/semaphorin 6D and TREM-2/DAP12 signaling. In short, the exploration of TREM-2 ligands may involve more than the identification of a single binding partner and may require an approach encompassing possible co-receptors. Furthermore, it still remains to be determined whether similar receptor complexes exist in microglia that regulate TREM-2–mediated communication of microglia with other CNS cells.
TREM-2 and TREM-1 both have very short cytoplasmic domains and lack intrinsic signaling capacity (Bouchon et al., 2000). Thus, both must signal through the transmembrane adaptor molecule DAP12. DAP12 contains an ITAM that can bind to the positively charged lysine residue in the transmembrane domain of TREM. Upon the engagement of TREM receptors, Src kinases phosphorylate the tyrosine residues in the DAP12 ITAM, forming a docking site for the binding of protein tyrosine kinase Syk (Ivashkiv, 2009; N’Diaye et al., 2009). Despite the similarity between TREM-2 and TREM-1 signaling, distinct downstream signaling pathways are activated by these two receptors beyond this point in order to maintain microglial equilibrium. Upon TREM-1 ligation in inflammatory phagocytes, Syk activates signaling cascades involving ERK1/2, PLCγ, the NFκB transcription factor, and MAPKs (Bouchon et al., 2000; Gibot et al., 2004), thereby leading to cell activation. In contrast to the putative activating effects of TREM-1/DAP12 signaling, TREM-2/DAP12 signaling results in diverse cellular responses ranging from increased cell activation to propagation of inhibitory signals within the cell. In microglia, stimulation of TREM-2 activates ERK1/2, leading to reorganization of the actin cytoskeleton and enhanced phagocytosis (Takahashi et al., 2005). TREM-2/DAP12 signaling may also stimulate the expression of chemokine receptors and promote microglial migration toward chemokine ligands CCL19 and CCL21. Interestingly, TREM-2 also induces inhibitory signals that ameliorate microglial inflammatory responses during the phagocytosis of apoptotic neurons (Takahashi et al., 2005).
TREM-2 can also relay ‘paradoxical’ signaling (activating and inhibitory) within microglia, although the mechanisms are unclear. Nonetheless, the ITAM-containing adapter DAP12 seems to be a critical player (Barrow and Trowsdale, 2006; Peng et al., 2010). A consensus ITIM sequence (SPYQEL) is embedded in the ITAM sequence of DAP12 (ESPYQELQGQRSDVYSDL) and is known as a ‘closet’ ITIM. This closet ITIM may recruit inhibitory phosphatases [eg. SH2 domain-containing inositol polyphosphate 5′ phosphatase (SHIP) and SH2 domain-containing phosphatase-1 (SHP1)] in order to attenuate DAP12 activation (Barrow and Trowsdale, 2006). This hypothesis has already found support in studies of macrophages, where SHIP1 is recruited to DAP12 in response to the ligation of TREM-2 and inhibits TREM-2/DAP12 signaling (Peng et al., 2010). The interaction between SHIP1 and DAP12 requires the binding of the SHIP1 SH2 domain with the phosphorylated tyrosine in the DAP12 closet ITIM. In the absence of SHIP1, ligation of TREM-2 recruits PI3K p85 subunit to DAP12 ITAM with the assistance of another adaptor protein, DAP10 (Peng et al., 2010). This, in turn, results in the activation of ERK1/2, Akt, and the guanine nucleotide exchange factor Vav3 (Figure 2). The factors that dictate the differential response of DAP12 to TREM-2 ligation remain to be elucidated. Barrow and colleagues have proposed that DAP12 might be either incompletely or completely phosphorylated depending on the binding affinity between the associated receptor and its ligands. Incomplete phosphorylation of DAP12 upon weak TREM-2 receptor binding may result in ITIM phosphorylation and lead to recruitment of inhibitory phosphatases. In contrast, high affinity receptor binding may result in complete ITAM phosphorylation and sustained activation (Figure 2). TREM-2 ligands will have to be identified to validate this hypothesis. An alternative possibility is that closet ITIM may extinguish ITAM signaling after the desired activation threshold is achieved, in order to prevent excessive activation.
Figure 2. TREM-2 signaling.
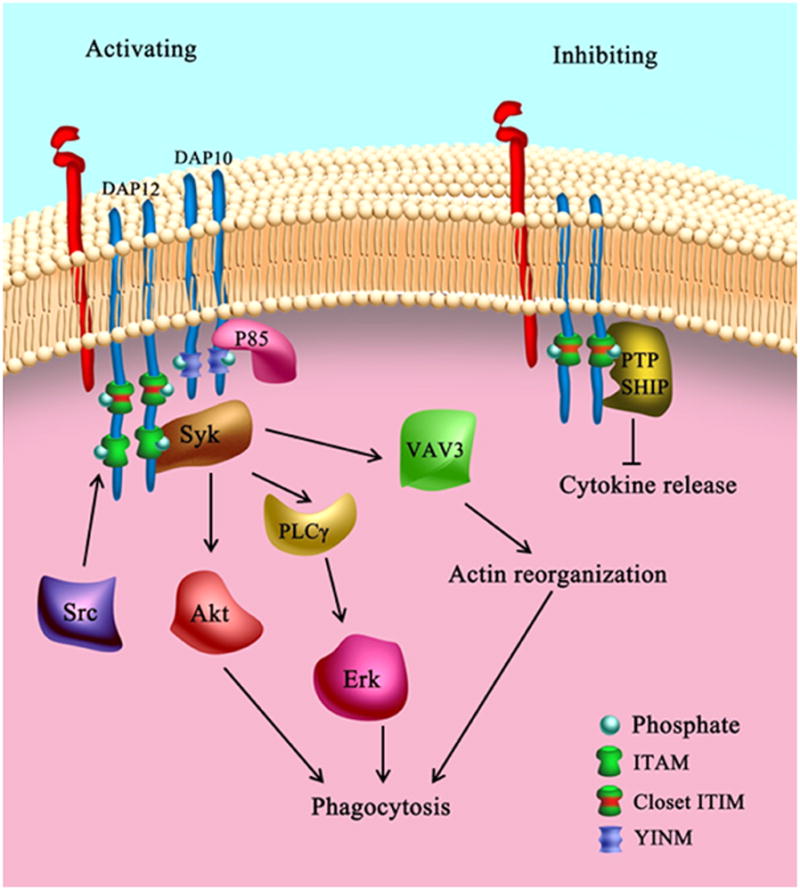
The ITAM-containing adapter DAP12 is important for both activating and inhibitory TREM-2 signaling. A consensus ITIM sequence is embedded in the ITAM sequence of DAP12 and is called a ‘closet’ ITIM. DAP12 might be incompletely or completely phosphorylated depending on the binding affinity between the associated receptor and its ligands. While incomplete phosphorylation of DAP12 upon weak TREM-2 receptor binding can result in ITIM phosphorylation, high affinity receptor binding may result in complete ITAM phosphorylation. The phosphorylated tyrosine in the DAP12 closet ITIM may recruit inhibitory phosphatases (SHP-1 or SHIP1) to attenuate TREM-2 activation. In the absence of SHIP1, ligation of TREM-2 recruits PI3K p85 subunit to the DAP12 ITAM with the assistance of another adaptor protein, DAP10. This, in turn, results in the activation of ERK1/2, Akt,, and the guanine nucleotide exchange factor Vav3.
The evident complexity of TREM signaling, as detailed above, may allow for a high degree of resolution in microglial responsiveness to differing stimuli and remains a fruitful and active area of investigation. However, there is still a dearth of information on the involvement of microglial TREM signaling in pathological states.
2.2. Fcγ Receptors (FcγRs)
FcγRs bind specifically to the Fc (fragment, crystallizable) region of IgG immunoglobulin antibodies and are therefore a gateway to responsiveness to various immune stimuli. As expected, FcγRs are involved in a variety of microglial biological functions that include phagocytosis, cytokine production, and oxidative bursts (Song et al., 2002; Ulvestad et al., 1994; Vedeler et al., 1994).
2.2.1. FcγRs differentially regulate microglial function
There are four principal types of FcγR: FcγRI (CD64), FcγRII (CD32), FcγRIII (CD16) and FcγRIV, which bind different isotypes of IgG with varying affinities (Nimmerjahn and Ravetch, 2008). FcγRs can induce both activating and inhibitory responses in microglia, as one might expect from the overarching need to maintain microglial homeostasis. For instance, FcγRI, FcγRIII and FcγRIV are activating receptors. In contrast, FcγRII (FcγRII in mice and FcγRIIb in humans) is an inhibitory feedback receptor that dampens responses triggered by activating receptors (Syam et al., 2010). Cultured human microglia have been shown to express mRNA for both activating (FcγRI and FcγRIII) and inhibitory (FcγRIIb) FcγR receptors (Lue and Walker, 2002). FcγR stimulation may regulate microglial phagocytosis of IgG-opsonized particles and enhance superoxide and nitric oxide production (Bard et al., 2000; Le et al., 2001; Ueyama et al., 2004). FcγRs are also important for microglial secretion of multiple chemokines, including macrophage inflammatory protein 1α (MIP-1α), monocyte chemotactic protein-1 β (MCP1β), and regulated on activation, normal T cell expressed and secreted (RANTES) in response to immobilized IgG antibodies (Lendvai et al., 2000). Studies using specific IgG antibody-coated Cryptococcus neoformans immune complexes have shown that FcγRI and FcγRIII are critical for MIP-1α production in microglia. Blockade or genetic deficiency of FcγRI and FcγRIII, but not FcγRII, significantly reduces microglial MIP-1α induction (Song et al., 2002). The function of inhibitory FcγRIIb in microglia still awaits identification.
The importance of FcγRs in the pathology of cerebral ischemia was shown by Komie-Kobayashi and colleagues in a mouse model of transient middle cerebral artery occlusion (MCAO, Komie-kobayashi, 2004). FcγR-deficient mice were protected from the progression and expansion of infarct volume after MCAO. This was accompanied by reductions in microglial activation, macrophage infiltration, and microglia/macrophage iNOS production. These findings are consistent with the abovementioned role of FcγRs in the secretion of multiple chemokines and the enhancement of superoxide and NO production. Another seminal study showed that intravenous infusions of immunoglobulin (IVIG), which contain pooled IgG immunoglobulins extracted from the plasma of multiple blood donors, protected against experimental stroke (Arumugam et al., 2007). Although such protection has been attributed to a direct effect of IVIG on neurons, the possibility that IVIG may also work through FcγRs on microglia as well as other immune cells cannot be excluded. Although the Arumugam study appears to contradict the Komie-Kobayashi study, one might speculate that IVIG preferentially engaged the inhibitory microglial FcγRs (eg. FcγRII) in the former study, thereby dampening immune overactivation. More studies are needed to test this hypothesis and to identify how engagement of FcγRs can lead to either beneficial or destructive responses in experimental stroke.
2.2.2. FcγR signaling
As mentioned above, the Fc regions of immunoglobulins are well-known ligands for FcRs. In addition, several non-immunoglobulin ligands for FcRs have also been identified. For example, Galectin-3, an animal lectin, is an endogenous ligand for inhibitory FcγRII (Cortegano et al., 2000). The interaction between Galectin-3 and FcγRII inhibits the production of interleukin-5 (IL-5) from peripheral blood mononuclear cells. Interestingly, soluble forms of FcγRs (sFcγRIII) have been detected in human plasma (Fleit et al., 1992; Huizinga et al., 1990). These sFcRs can bind to non-immunoglobulin ligands, such as complement receptors on various cell types and induce cytokine production (Galon et al., 1996; Heyman, 2000). The biological significance of these non-immunoglobulins ligands has not been addressed for microglia or neurological disorders and warrants further investigation.
Most FcγRs are transmembrane glycoproteins. Their common α chain contains an extracellular region with varying numbers of the IgSF domain. The IgSF domain includes a transmembrane region and a cytoplasmic region. Some FcγRs have only a single α chain (eg. FcγRIIa and FcγRIIb). Others are coupled to additional receptor subunits (eg. two γ chains for FcγRI and FcγRIIIa) involved in downstream signaling (Ortiz-Stern and Rosales, 2003) (Figure 3).
Figure 3. Fcγ receptors.
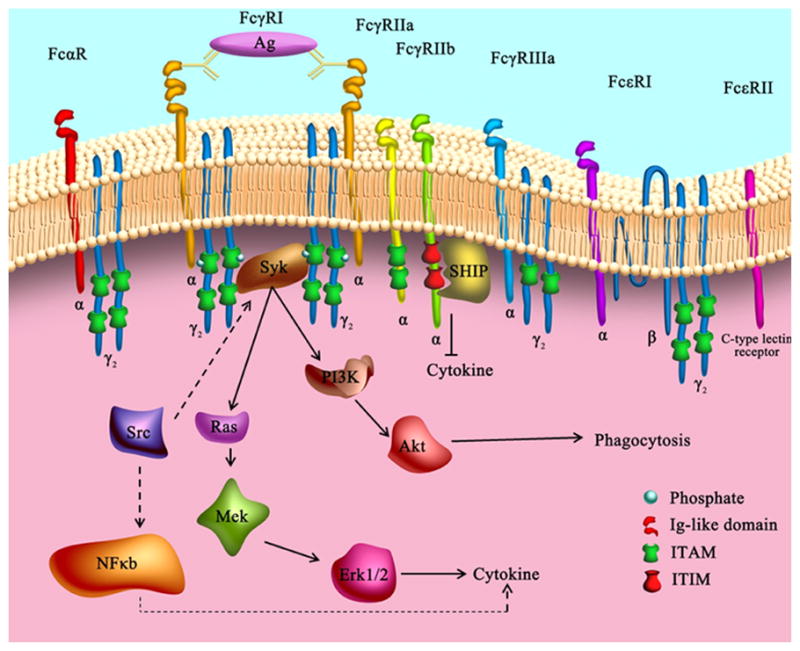
Most FcγRs have a common α chain that contains an extracellular region with varying numbers of the immunoglobulin (Ig)-like domain. FcγRIIa has only a single α chain. FcγRI and FcγRIIIa are coupled to additional receptor subunits (two γ chains) that are involved in downstream signaling. The signaling pathways initiated by the engagement of different activating FcγRs usually begin with tyrosine phosporylation of the conserved immunoreceptor tyrosine-based activation motif (ITAM) in the cytoplasmic tail of the receptors or their associated subunits by kinases in the Src family. The subsequent recruitment of Syk-family kinases in turn permits interaction with other signaling proteins, such as PI3K, Src, and Ras, and further activates downstream signaling pathways that lead to specific biological functions. The activation of NFκB or the Ras/MEK/ERK pathway is critical for cytokine/chemokine production whereas the PI3K-Akt pathway is important for microglial phagocytosis. In contrast to the ITAM-mediated activation of microglia, the engagement of inhibitory FcγRIIb triggers immunoreceptor tyrosine-based inhibition motif (ITIM) activation through its cytoplasmic domain, resulting in the recruitment of phosphatases such as SHIP and SHP1. This attenuates the signaling cascades engaged by activating FcRs and diminishes downstream biological effects by acting in counterpoint to ITAM.
The signaling pathways initiated by the engagement of different activating FcγRs usually begin with tyrosine phosporylation of the conserved ITAM in the cytoplasmic tail of the receptors or their associated subunits by kinases in the Src family. The subsequent recruitment of Syk-family and/or zeta-chain-associated protein kinase 70 (ZAP70) allows for interactions with other signaling proteins, such as linker for activation of T cells (LAT), phosphoinositide-3-kinase (PI3K), Src homology 2 (SH2) domain containing leukocyte protein of 76 kDa, and PLCγ2; these interactions activate further downstream signaling pathways that lead to specific and divergent biological functions (Crowley et al., 1997; Gross et al., 1999; Jakus et al., 2009; Park and Schreiber, 1995; Saitoh et al., 2000; Taylor et al., 1997; Tridandapani et al., 2000). For example, Src and Syk kinases have been shown to be important for microglial phagocytosis and release of the chemokine MIP-1α following FcγR engagement (Song et al., 2004). Furthermore, activation of NFκB or the Ras/MEK/ERK pathway is critical for MIP-1α production whereas the PI3K-Akt pathway is important for microglial phagocytosis, suggesting that multiple downstream signaling pathways are activated by FcγR to regulate different functions (Figure 3). Thus, a single FcγR can initiate distinct microglial functions (eg. phagocytosis and cytokine production), thereby illustrating that pathway divergence is another important principle of microglial receptor engagement.
In contrast to the ITAM-mediated activation of microglia, the engagement of inhibitory FcγRIIb triggers ITIM activation through its cytoplasmic domain, resulting in the recruitment of phosphatases such as SHIP and SHP1. This attenuates the signaling cascades engaged by activating FcγRs and diminishes downstream biological effects by acting in counterpoint to ITAM (Huang et al., 2003) (Figure 3).
The coexpression of activating and inhibitory FcγRs on microglia and their recruitment of opposing signaling cascades upon binding the same ligand reveal how FcγRs may contribute to the maintenance of microglial equilibrium and balance various immune functions. It may also underlie the contradictory effects of FcγR engagement in stroke models (see above).
2.3. CD200 receptor (CD200R)
Cluster of Differentiation 200 (CD200) receptor (CD200R, also named the OX2 receptor) and its ligand CD200 (OX2) are both cell surface glycoproteins that contain two immunoglobulin domains. While CD200 is broadly expressed on many types of cells including lymphoid cells, neurons and endothelial cells, CD200R is exclusively expressed on leukocytes, especially cells of the myeloid lineage (Wright et al., 2000).
2.3.1. CD200R maintains microglia in a quiescent state
CD200R was first identified on macrophages (Preston et al., 1997) and later detected on microglia (Wright et al., 2000). CD200R actively interacts with CD200 on healthy neurons and maintains microglia in a quiescent state. Disruption of CD200-CD200R binding with CD200 blocking antibodies increases microglial activation and exacerbates the signs of experimental autoimmune encephalomyelitis (EAE) (Wright et al., 2000) and experimental Parkinson’s disease (PD) (Zhang et al., 2011b) in rats. Studies in CD200 deficient mice further confirm that, in the absence of the inhibitory signaling from neuronal CD200, microglia spontaneously exhibit features of activation, including enlarged cell bodies, shortened and thickened processes, and increased expression of CD11b and CD45 (Hoek et al., 2000). Similar to the results with CD200 antibodies, CD200 deficiency also accelerates the onset of and enhanced microglia/macrophage activation in EAE mice (Copland et al., 2007; Hoek et al., 2000). These studies strongly suggest that CD200R is an important inhibitory receptor on microglia and that it plays a major role in maintaining immune balance in the CNS.
The involvement of CD200-CD200R in acute CNS injuries has also been studied. For example, in facial nerve axotomy, a traditional model for in situ microglial activation induced by damaged neurons, microglial activation is more apparent in CD200-null mice than in control mice (Hoek et al., 2000). This study shows that CD200 suppresses microglial activation also in response to acute neuronal injury. Furthermore, in the MCAO model of ischemic stroke, CD200 mRNA levels are significantly decreased as early as 1 day after ischemia (Matsumoto et al., 2007). Levels of CD200 mRNA plateau at 5 days and gradually increase back to control values thereafter. This acute post-ischemic drop in CD200 levels may contribute to short-term microglial activation after stroke. Interestingly, macrophage-like cells in the ischemic core have been shown to express CD200, suggesting that CD200-CD200R engagement may also be involved in the crosstalk between microglia and other immune cells.
2.3.2. CD200R signaling
CD200 is the only known ligand for CD200R. CD200R interacts with CD200 through its N-terminal Ig-like domain (Hatherley and Barclay, 2004). Unlike most other inhibitory receptors, CD200R does not contain any ITIM, nor does it recruit any tyrosine phosphatases upon engagement (Mihrshahi and Brown, 2010). Instead, CD200R has a cytoplasmic phosphotyrosine-binding (PTB) domain recognition motif (NPxY). Phosphorylation of the tyrosine residue within this motif leads to the direct binding of this motif to the PTB domain-containing adaptor downstream of tyrosine kinase 2 (Dok2) (Mihrshahi and Brown, 2010; Zhang et al., 2004; Zhang and Phillips, 2006). Further phosphorylation of CD200R-bound Dok2 recruits and activates Ras GTPase activating protein (RasGAP). RasGAP hydrolyzes RasGTP into the inactive form RasGDP, resulting in the subsequent inhibition of PI3K and ERK signaling (Mihrshahi et al., 2009). In addition to this main inhibitory pathway, CD200R ligation also causes a relatively delayed phosphorylation of Dok1 (Mihrshahi et al., 2009; Mihrshahi and Brown, 2010; Zhang et al., 2004). Upon phosphorylation, Dok1 activates other signaling molecules and finally inhibits the activation of Dok2 and RasGAP (Mihrshahi and Brown, 2010). Therefore, the activation of Dok1 initiates an elegant negative feedback cycle to fine-tune the activation of CD200R signaling pathway.
2.4. Receptor for advanced glycation endproducts (RAGE)
RAGE is named after its ability to bind advanced glycation end-products, which are proteins or lipids that are non-enzymatically glycated after exposure to sugars. RAGE exists in membrane-bound forms or, alternatively, in soluble forms that lack the transmembrane domain (Hudson et al., 2003). RAGE is highly expressed on immune cells and contributes to many pathological states with inflammatory components (Sparvero et al., 2009).
2.4.1. RAGE mediates microglia accumulation and activation in CNS injuries
RAGE is highly expressed in the CNS. During early development, RAGE is mainly found in neurons and contributes to neurite outgrowth (Hori et al., 1995). In the adult CNS, RAGE is expressed in various types of CNS cells including microglia, neurons, and endothelial cells (Giri et al., 2000; Yan et al., 1996). The function of microglial RAGE was initially characterized in models of AD. In postmortem tissue from AD victims, RAGE immunoreactivity is increased within microglia in close proximity to senile plaques (Lue et al., 2001; Yan et al., 1996). Further in vitro studies have confirmed that RAGE is present in cultured microglia and serves as a direct receptor for Aβ (Yan et al., 1996). The involvement of microglial RAGE in brain injuries has also been reported. In in vitro and in vivo models of ischemic stroke, high-mobility group box 1 (HMGB1), a ligand for RAGE, is released from injured neurons early after ischemia and contributes to ischemic brain injury through its interaction with RAGE (Muhammad et al., 2008). Further studies using chimeric mice generated by transplanting RAGE−/− bone marrow into wild-type mice confirmed that RAGE expression on immigrant macrophages was essential for post-stroke cerebral inflammation and brain damage. This study also suggests that HMGB1-RAGE signaling links neuronal necrosis with microglia/macrophage activation. Thus, RAGE signaling is a rational target for anti-inflammatory therapy in stroke. In animal models of TBI and in TBI patients, upregulation of RAGE in the brain has been reported, with a prominent increase in phagocytic microglia surrounding the lesion core at late stages of injury. Indeed, HMGB1 is released from cells early after TBI and primarily expressed in phagocytic microglia at late stages of TBI (Gao et al., 2012). However, further studies are needed to elucidate the biological significance of the temporal changes in RAGE and HMGB1 in TBI.
2.4.2. RAGE signaling
RAGE is an integral membrane protein composed of an extracellular domain, a single transmembrane-spanning domain, and a short, highly charged cytosolic domain (Hudson et al., 2008). RAGE recognizes many different binding partners through a common motif and is therefore also known as a pattern recognition receptor. Microglial RAGE has been documented to recognize ligands such as advanced glycation endproducts (AGEs) (Dukic-Stefanovic et al., 2003), Aβ (Yan et al., 1996), HMGB1 (Muhammad et al., 2008), and S100 proteins (Bianchi et al., 2007; Bianchi et al., 2010).
Ligand binding to microglial RAGE triggers many different intracellular signaling pathways. Microglial RAGE signaling following Aβ binding has been the most thoroughly investigated. Aβ-RAGE interactions elevate microglial macrophage colony-stimulating factor (m-csf) expression and initiate microglial chemotaxis (Lue et al., 2001). In addition, RAGE enhances secretion of several proinflammatory mediators (eg. TNF-α, IL-1β) from microglia, partly through the activation of p38 and ERK1/2. Such RAGE-induced neuroinflammation has the potential to greatly exaggerate Aβ neurotoxicity, leading to impaired learning and memory (Fang et al., 2010). Furthermore, IL-1β, p38MAPK and JNK are all involved in microglial RAGE signaling and mediate Aβ-induced synaptic dysfunction and impairments in long-term potentiation (Origlia et al., 2010).
RAGE activation in microglia by AGEs results in the upregulation of pro-inflammatory cytokines such as IL-6, TNF-α and iNOS through the activation of NFκB, MEK, and PI3K signaling pathways (Dukic-Stefanovic et al., 2003). AGE-RAGE interactions have been shown to induce microglia inducible nitric oxide synthase (iNOS) expression in a JNK-dependent manner and to elicit subsequent NO production (Khazaei et al., 2008). AGEs have also been found to stimulate the expression of RAGE in a human microglial cell line. AGE-RAGE interactions induce TNF-α and TNF-α receptor II expression on microglia, which in turn enhance the expression of the gap junction protein connexin 43 and thereby enhance communication between activated microglia (Shaikh et al., 2012).
S100B stimulates NO production from microglia in a manner dependent on RAGE extracellular domains but independent of RAGE transducing activity (Adami et al., 2004). These findings suggest that RAGE may concentrate S100B on the surface of microglia. In contrast, S100B-induced upregulation of cyclo-oxygenase-2 (COX2) expression in microglia seems to be RAGE-dependent through simultaneous stimulation of the Cdc42-Rac1-JNK and Ras-Rac1-NF-κB pathways (Bianchi et al., 2007). In addition, microglial RAGE engagement by S100B induces IL-1β and TNF-α expression by concurrent stimulation of NFκB and AP-1 transcriptional activities (Bianchi et al., 2010). A recent study also documented a role for S100B-engaged RAGE in microglial migration via activation of multiple signaling pathways and the resulting upregulation of several chemokines (CCL3, CCL5, and CXCL12) and chemokine receptors (CCR1 and CCR5) (Bianchi et al., 2011). These results suggest that S100B engages the RAGE receptor to turn on microglial function. However, the S100B-RAGE interaction does not always turn on microglial function. For example, in special pathological conditions such as tumor formation, S100B actually attenuates the activation of glioma associated microglia and macrophages, through the induction of STAT3 (Zhang et al., 2011a). As a result of these findings, blunt suppression of all the effects of RAGE in the clinic might lead to unwanted side effects and investigators will have to work towards a subtler fine-tuning of its pro- and anti-inflammatory properties.
3. Chemokine receptors
Chemokines belong to a family of small secreted proteins with molecular mass usually less than 10 kDa. Chemokines are so-named because they are leukocyte chemoattractants and also possess cytokine activities, in a portmanteau of the words ‘chemoattractant’ and ‘cytokine’ (Asensio and Campbell, 1999). Chemokines have four conserved cysteine residues that form disulfide bonds. The chemokine family is divided into four subclasses (CC, CXC, CX3C and C subclass) according to the position of the first two cytokines in the N-terminal region. Chemokines send signals through chemokine receptors, all of which are G-protein-coupled. The interaction between chemokines and chemokine receptors not only directs immune cell migration, but also controls a variety of other cellular functions. The receptors most often implicated in microglial functions in CNS injuries are CX3CR1, CCR2, CXCR4, CCR5, and CXCR3.
3.1. CX3CR1
3.1.1. CX3CR1 and its ligand CX3CL1 regulate microglia-neuron crosstalk
CX3CR1 is mainly expressed on glial cells in the CNS, especially microglia (Harrison et al., 1998; Meucci et al., 1998; Nishiyori et al., 1998). CX3CR1 exhibits a monogamous relationship (Imai et al., 1997) with its ligand CX3CL1 and plays an important role in neuron-microglial crosstalk. CX3CL1 (also named fractalkine) was the first chemokine found to be expressed in neurons (Harrison et al., 1998; Meucci et al., 1998; Nishiyori et al., 1998). Its expression in the brain is higher than in the periphery and mainly localized to the olfactory bulb, cerebral cortex, hippocampus, caudate, putamen, and nucleus accumbens (Nishiyori et al., 1998). CX3CL1 can either be membrane-bound or released from the cell surface after constitutive or inducible cleavage by proteases of the A Disintegrin And Metalloprotease (ADAM) family (Garton et al., 2001; Hundhausen et al., 2003). CX3CL1 is one of the ‘find me’ signals that apoptotic cells utilize to advertise their presence and to recruit local phagocytes (Ravichandran, 2011). Thus, CX3CL1 is important for the adaptive clearance and removal of dysfunctional, dying cells from the organism.
Mechanistic studies on CX3CR1 have revealed that it modulates multiple microglial activities, including adhesion, migration, proliferation, inflammatory cytokine production, and clearance capacity (Bhaskar et al., 2010; Cardona et al., 2006; Denes et al., 2008; Lyons et al., 2009). Importantly, CX3CR1 inhibits the microglial production of inflammatory cytokines following stimulation (Bhaskar et al., 2010; Cardona et al., 2006; Denes et al., 2008). The role of CX3CR1 in microglial phagocytosis is still controversial. Some studies have documented that CX3CR1 signaling inhibits or has no effect on microglial phagocytosis of β-amyloid in models of AD (Lee et al., 2010; Liu et al., 2010). However, other studies show that stimulation of CX3CR1 with its ligand increases microglial clearance of neuronal debris and induces neuroprotection (Noda et al., 2011). These differences might reflect the varying roles of CX3CR1 in the presence of distinct stimuli. Further work to distinguish how CX3CR1 exerts these potentially dualistic roles is warranted.
3.1.2. CX3CR1 and CX3CL1 in CNS injuries
The biological functions of CX3CL1 and CX3CR1 signaling in vivo have been investigated using CX3CR1-deficient mice. Under physiological conditions, disruptions in CX3CR1 signaling lead to impairments in cognitive function and synaptic plasticity via increased action of IL-1β (Rogers et al., 2011). These studies indicate an important role for CX3CR1 in maintaining CNS homeostasis and normal interneuronal communication. CX3CR1 deficiency augments microglial activation and neuronal damage in models of neurodegenerative diseases such as PD, AD, amyotrophic lateral sclerosis, and macular degeneration (Cardona et al., 2006; Chen et al., 2007; Cho et al., 2011; Pabon et al., 2011), suggesting an inhibitory effect of CX3CR1 on inappropriate microglial activation and subsequent neuronal toxicity. Paradoxically, however, CX3CR1 knockout mice display reduced neuroinflammation and mitigated neuronal damage after focal cerebral ischemia and SCI (Denes et al., 2008; Donnelly et al., 2011). A recent study provided a plausible explanation for this discrepancy (Donnelly et al., 2011):-in experimental paradigms where microglia are the predominant CNS macrophages (e.g., PD and amytrophic lateral sclerosis), deficient CX3CR1 signaling was proposed to enhance microglial neurotoxicity due to the loss of inhibitory signaling through CX3CR1-CX3CL1 interactions (Cardona et al., 2006). In contrast, in acute neurological injuries such as stroke or spinal cord injury, early microglial activation is followed by prominent macrophage infiltration, which contributes greatly to the expansion of primary injuries. A deficiency in CX3CR1 may limit the recruitment and activation of peripheral macrophages and thus exert neuroprotective effects. Therefore, CX3CL1-CX3CR1 signaling may exert opposite effects in different pathological conditions.
Another factor that may complicate the impact of CX3CL1-CX3CR1 is the timing of the immune response in relation to disease progression. For example, increased CX3CL1-CX3CR1 signaling may be beneficial in early AD by restraining microglial activity and preventing the exacerbation of phosphorylated tau-mediated damage to neurons (Bhaskar et al., 2010; Cho et al., 2011). However, sustained elevation of CX3CL1-CX3CR1 signaling may become detrimental by interfering with microglial phagocytosis in advanced stages when removal of extracellular Aβ deposits becomes necessary (Desforges et al., 2012; Lee et al., 2010). All of these complex issues need to be taken into full consideration when developing therapeutic interventions targeting CX3CR1.
Beyond the abovementioned regulation of inflammatory responses, CX3CL1 may sensitize microglia in the spinal cord and cause allodynia and thermal hyperalgesia. These additional functions of CX3CL1 have been proposed because antibodies against CX3CR1 are known to inhibit the development of neuropathic pain (Clark et al., 2007; Milligan et al., 2004; Zhuang et al., 2007). Further studies have also shown that membrane-bound CX3CL1 can be cleaved by microglial cathepsin S after SCI. The released soluble CX3CL1 then activates microglia through an interaction with CX3CR1 and causes neuropathic pain (Clark et al., 2007; Clark et al., 2009). These data provide potential therapeutic targets on microglia for conditions involving neuropathic pain.
3.1.3. CX3CL1-CX3CR1 signaling
CX3CR1 is a G-protein-coupled seven transmembrane domain receptor. Upon CX3CL1 recognition and binding, CX3CR1 undergoes a conformational change and activates associated G proteins by exchanging bound GDP for GTP, known as guanine nucleotide exchange. Following this exchange, the α subunit of the G-protein dissociates from the β/γ subunits to activate other intracellular signaling proteins, including phospholipase C (PLC), MAPK, and PI3K (Savarin-Vuaillat and Ransohoff, 2007). For example, the engagement of CX3CR1 on monocytes has been shown to induce the coordinated activation of ERK, p38, JNK1, and PI3K to efficiently mediate the CX3CR1-dependent cell adhesion to the extracellular glycoprotein fibronectin (Cambien et al., 2001). Thus, chemokine receptors play a role in cell adhesion to the extracellular matrix, which is known to regulate cell migration and is consistent with the chemoattractive properties of their cytokine ligands.
Despite the chemotactic properties of CX3CL1-CX3CR1 interactions in monocytes, CX3CL1-CX3CR1 interactions actually maintain microglia in a quiescent state by the activation of PI3K, as manifested by reduced IL-1 production after LPS exposure (Lyons et al., 2009). It has also been shown that the activation of the p38 pathway by CX3CR1 is important for pain processing after spinal cord injury or bone cancer (Hu et al., 2012; Zhuang et al., 2007). Apparently, multiple signaling pathways are activated downstream of CX3CL1-CX3CR1 engagement and are involved in a variety of cellular functions. The specific signaling pathways activated by CX3CR1 still need to be explored for each individual function, such as phagocytosis and cytokine production.
3.2. CCR2
3.2.1. CCR2 and its ligands direct microglial migration toward the site of damage
The chemokine receptor CCR2 is widely expressed on almost all types of immune cells including lymphocytes, neutrophils, macrophages, and natural killer cells (Savarin-Vuaillat and Ransohoff, 2007). As expected from its classification as a chemokine receptor, the main function of CCR2 is to direct immune cells to the site of inflammation, where the CCR2 ligands accumulate (Serbina and Pamer, 2006; Tsou et al., 2007). The chemoattractant functions of CCR2 are important for monocyte deployment from bone marrow or other lymph organs and for their subsequent infiltration into the CNS under pathological circumstances (Babcock et al., 2003; Bao et al., 2010; Chen et al., 2001; D’Mello et al., 2009; Eugenin et al., 2006; Naert and Rivest, 2012). It has been shown that circulating Ly-6Chigh ‘inflammatory’ monocytes equipped with CCR2 can penetrate the brain upon blood-brain barrier disruption and serve as migratory precursors of microglia (Getts et al., 2008; Mildner et al., 2011; Mildner et al., 2007). The constitutive expression of CCR2 on resident microglia is very low if present. However, LPS and other pathological stimuli dramatically induce the expression of this receptor on microglia (Banisadr et al., 2002b; Boddeke et al., 1999; Sivakumar et al., 2011; Zhang et al., 2007). In this manner, microglia might acquire migratory properties only in the presence of the appropriate inflammatory stimuli.
3.2.2. Microglial CCR2 and its ligands in CNS injuries
Unlike CX3CR1, CCR2 has multiple ligands, namely CCL2 (also named monocyte chemoattractant protein (MCP)1), CCL7 (MCP3), CCL8 (MCP2), CCL12 (MCP5), CCL13 (MCP4) and CCL16 (Charo et al., 1994; Franci et al., 1995; Garcia-Zepeda et al., 1996; Moore et al., 1997; Nomiyama et al., 2001). CCR2 and/or its ligands are upregulated in many types of CNS injury, including ischemia, hemorrhage, trauma, and hypoxia (Deng et al., 2009; Dimitrijevic et al., 2007; Kim et al., 2008). CCR2 activation on microglia after CNS injuries was initially reported to be destructive. This was based on findings that depletion of CCL2 or CCR2 reduces microglial activation and neuronal degeneration following aspiration lesions of the visual cortex in adult mice (Muessel et al., 2002). Similarly, it was shown that ischemia-induced infarcts are reduced in CCR2-deficient mice and that this is accompanied by reduced cerebral inflammation (Dimitrijevic et al., 2007). These studies suggest that the CCL2-CCR2 axis plays manifold roles in cerebral ischemia, including recruiting peripheral leukocytes and disrupting the blood-brain barrier. Subsequent studies using green fluorescent protein (GFP)-transgenic bone marrow chimeras further demonstrated that CCR2 deficiency reduces the influx of GFP-positive cells (macrophages and neutrophils) into the brain after ischemic stroke, whereas the activation and migration of resident microglia was not affected (Schilling et al., 2009)). It seems plausible that CCR2 is more important in peripheral immune cell trafficking than in local microglial responses following CNS injuries and that activation of CCR2 leads to the secondary expansion of primary damage.
In contrast, some studies suggest a possible protective role of the CCL2-CCR2 system in brain recovery. For example, although a lack of CCL2 or CCR2 decreases hematoma volume early after collagenase-induced intracerebral hemorrhage (ICH), it also delays the late phase resolution of the hematoma (Yao and Tsirka, 2012). This study also found that microglia activation/migration was attenuated early after ICH in CCR2 or CCL2 knockout mice and that this was followed by an exaggerated activation of microglia in later phases, possibly due to the activation of alternative signaling pathways. Therefore, any therapeutic strategies designed to target the CCR2-CCL2 complex after CNS injuries must be carefully timed to avoid adverse effects on long-term recovery and compensatory activation of alternative pro-inflammatory signalling.
Similar to CX3CR1, microglial CCR2 has been shown to be important in inflammatory pain and neuropathic pain states (Abbadie et al., 2003). To distinguish the roles of central and peripheral CCR2, bone marrow chimeric mice have been constructed to achieve selective knockout of CCR2 in resident microglia or in bone-marrow derived macrophages (BMDM). The results show that CCR2 expression in either resident microglia or BMDM is sufficient for the development of mechanical allodynia after peripheral nerve injury induced by partial ligation of the sciatic nerve (Zhang et al., 2007). These data support an essential role for microglial receptors in the development of neuropathic pain and reveal a potential therapeutic target for conditions associated with inflammation.
3.2.3. CCR2 signaling
A model of neuron-microglia interactions through the CCL2-CCR2 system has recently been proposed by Yao and colleagues (Yao and Tsirka, 2010). In their model, the C-terminus of CCL2, which is extensively decorated with O-linked carbohydrates, anchors the CCL2 molecule on neurons through its interaction with the glycosaminoglycans (GAGs) on neuronal cell membranes. This event increases the local CCL2 concentration, which in turn promotes the formation of CCL2 dimers or higher order aggregates. The C-terminal extension negatively regulates the chemotactic ability of CCL2 and hinders CCL2-CCR2 crosstalk (Sheehan et al., 2007). Upon neuronal injury or other noxious stimuli, plasmin is released from disturbed neurons and cleaves the C-terminal extension of CCL2 and abrogates CCL2 dimers. The truncated CCL2 N-terminal monomers then bind to CCR2 on microglia and initiate microglial intracellular signaling pathways. However, it was also noted that this model is based on mouse CCL2 structure. Human CCL2 does not have a glycosylated C-terminal, although its N-terminal is highly homologous to mouse CCL2. Therefore, other mechanisms might have evolved to regulate CCR2-CCL2 interactions in higher species (Yao and Tsirka, 2010).
The engagement of CCR2 induces GDP/GTP exchange on the α subunit (αi, αq or α16) of the associated G protein (Arai and Charo, 1996). The α–GTP complex then dissociates from the βγ heterodimer, and both complexes activate intracellular signaling cascades that differ depending on cell type. For example, the αi–GTP complex inhibits adenylate cyclase and the βγ complex activates phospholipase C to generate diacylglycerol and inositol 1,4,5-trisphosphate, leading to the release of intracellular calcium and activation of protein kinase C (PKC) (Myers et al., 1995). Activated CCR2 can also bind directly to FROUNT, a unique clathrin heavy-chain repeat homology protein, and form clusters at the leading edge of migrating cells (Terashima et al., 2005). Association of FROUNT with activated CCR2 leads to the activation of PI3K-Rac (a member of the Rho family of small GTPase signaling proteins) and the resultant protrusion of lamellipodia, the cytoskeletal actin projections observed at the leading edges of migrating cells. Although the involvement of FROUNT in microglial migration is not yet definitive, the activation of Rac is known to be important for the protrusion of lamellipodia and microglial migration upon CCR2 stimulation (Yao et al., 2010).
3.3. CCR5
3.3.1. Microglial CCR5 and its ligands in CNS injuries
CCR5 is expressed on many types of leukocytes involved in immune regulation. Like all the other chemokine receptors, CCR5 is important for immune cell migration toward specific ligands and thus results in recruitment of leukocytes to the site of inflammation or injury (Samson et al., 1996). The natural chemokine ligands that bind to CCR5 include CCL5 (also named regulated and normal T cell expressed and secreted, RANTES), CCL3, and CCL4. The importance of CCR5 in microglia was first reported more than a decade ago when it was identified as one of the most important co-receptors for HIV infection of microglia (He et al., 1997; Shieh et al., 1998). Subsequent studies revealed that CCR5 is expressed in many types of CNS cells (microglia, neurons, astrocytes, and vascular endothelial cells) and plays multifarious roles mediating glial-glial and glial-neuronal interactions (Cartier et al., 2003; Gamo et al., 2008; Rottman et al., 1997; Skuljec et al., 2011).
CCR5 is expressed at low levels by microglial cells throughout normal CNS tissue. In contrast, high expression of microglial CCR5 has been observed in several neurological injuries including excitotoxic brain injury (Galasso et al., 1998), perinatal hypoxia-ischemia (Cowell et al., 2006) and TBI (Carbonell et al., 2005). The primary function of upregulated CCR5 in neurodegenerative diseases is to direct microglial migration (Marella and Chabry, 2004). In contrast, the functions of microglial CCR5 are more complicated in acute CNS injuries. For example, in an experimental model of focal brain injury, a specific CCR5 antagonist, TAK-779, was able to reduce migration velocity and the number of perilesional migratory microglia (Carbonell et al., 2005) highlighting an additional trafficking role for CCR5. However, the upregulation of CCR5 following acute motor nerve injury is not essential for the recruitment of microglia. This is evidenced by the normal migration of microglia toward injured motor neurons in CCR5 deficient mice (Babcock et al., 2003; Gamo et al., 2008). An alternative function of CCR5 may be to protect neurons by ameliorating the production of NO and inflammatory cytokines from microglia following initial nerve damage (Gamo et al., 2008). However, conflicting in vitro findings also show that the activation of CCR5 induces a pro-inflammatory profile in microglia, as manifested by increased NO and reduced IL-10 production following CCL5 exposure (Skuljec et al., 2011). Further studies to explain the distinct functions of CCR5 on microglial behavior in different experimental paradigms are warranted.
3.3.2. CCR5 signaling
Signaling pathways initiated by G protein coupled chemical receptors are frequently related to ion channel regulation (Dascal, 2001). It is therefore not surprising that some microglial receptors open ion channels. For example, CCR5 is coupled to pertussis toxin (PTX)-sensitive inhibitory G protein (GαIβγ) and exerts its cellular function by regulating intracellular Ca2+ concentrations (Shideman et al., 2006). The engagement of CCR5 results in the dissociation of βγ subunits from the Gi heterotrimer. The liberated βγ subunits activate multiple signaling pathways, including PI3K/PLC/Bruton’s tyrosine kinase (Btk)/Cyclic ADP-ribose (cADPR)-mediated Ca2+ release from intracellular stores and ADP-ribose (ADPR)-dependent Ca2+ influx through nimodipine-sensitive Ca2+ channels on the plasma membrane.
3.4. CXCR3
mRNA expression of CXCR3 was first reported in cultured microglia by Biber and colleagues using RT-PCR (Biber et al., 2002) and was confirmed with in situ hybridization, immunohistochemistry, and flow cytometry (Biber et al., 2001; Flynn et al., 2003). CXCR3 binds to multiple CXC chemokines including CXCL4 (also named Platelet factor 4 (PF4)), CXCL9 (also named monokine induced by gamma interferon (MIG)), CXCL10 (also named interferon gamma-induced protein 10 (IP10)), CXCL11 (also named IP9); and CXCL21 (also named secondary lymphoid-tissue chemokine (SLC)) (Clark-Lewis et al., 2003; de Jong et al., 2008; Rappert et al., 2002; van Weering et al., 2011). Interestingly, many of these chemokines, such as CXCL21 and CXCL10, are markedly induced in injured neurons (Biber et al., 2001). The interaction between microglial CXCR3 and its ligands on endangered neurons is important for neuron-microglia crosstalk and the regulation of neuroinflammation in response to neuronal damage. Furthermore, these neuronal chemokines can be packed into vesicles and transported throughout neuronal processes to reach distant presynaptic structures (de Jong et al., 2005). This feature may explain the activation of microglia at locations remote from primary lesion sites. The major biological effect of microglial CXCR3 is to direct microglial migration towards CXCR3 ligands (Biber et al., 2001; Dijkstra et al., 2004; Rappert et al., 2004). In addition, microglial CXCR3 activation may attenuate microglial phagocytosis and production of NO upon LPS stimulation (de Jong et al., 2008). It is not known whether the activation of microglial CXCR3 leads to neuronal rescue or further neuronal damage. Nevertheless, a recent study suggests that the CXCR3+ microglia is neurosupportive, displaying high sensitivity to apoptosis and permitting the production of high levels of IL-10 and TGF-β (Li et al., 2006). Of note, CXCR3 function also depends on the cellular background on which the receptor is expressed because human CCL21 does not show any effects in CXCR3-transfected HEK293 cells, which are of human origin (Dijkstra et al., 2004).
CXCR3-mediated microglia-neuron interactions are known to be involved in brain injuries. Neuronal expression of CXCL21 is induced in the brain soon after the onset of ischemia, suggesting an involvement of CXCL21 and CXCR3 in early neuroimmunological events after stroke (Biber et al., 2001). Rappert and colleagues have examined the impact of CXCR3 on microglial migration and proliferation using the entorhinal cortex lesion and facial nerve axotomy models, respectively (Rappert et al., 2004). The authors found that CXCR3 signaling was critical for the migration but not for the proliferation of microglia after brain injuries (Biber et al., 2001; Rappert et al., 2002). These findings were consistent with their in vitro studies using microglial cultures. Interestingly, the impaired microglial migration after entorhinal cortex lesions in CXCR3 knockout mice was accompanied by the preservation of denervated dendrites. Denervated dendrites that have lost their afferent synaptic contacts are ordinarily cleared by microglia. Thus, the studies by Rappert and colleagues suggest that CXCR3 is essential for microglial clearance of dysfunctional dendrites. A recent study further suggests that microglial CXCR3 may regulate neuronal death in a brain region-specific or topographic manner (van Weering et al., 2011).
Very little information is available on the signal transduction pathways activated by microglial CXCR3 and their possible correlation with the biologic actions elicited by agonists. The engagement of CXCR3 on different types of cells has been associated with the activation of a variety of signaling pathways, including the p38, ERK, Src, PI3K/Akt, and PLC pathways (Bonacchi et al., 2001; Shahabuddin et al., 2006; Smit et al., 2003). The involvement of these pathways in microglial CXCR3 signaling, however, has not been investigated. Nevertheless, activation of microglial CXCR3 is known to be associated with the opening of ion channels. Thus, brief exposure of microglia to CCL21 is known to trigger a long lasting elevation in Cl− conductance, which is critical for CCL21-induced chemotaxis (Rappert et al., 2002). Further identification of CXCR3 signaling will shed more light on the function of this chemokine receptor in microglia.
3.5. CXCR4 and CXCL12
CXCR4 is constitutively expressed in the CNS in a variety of cell types including microglia, astrocytes, neurons, oligodendrocyte progenitors, and vascular endothelial cells (Banisadr et al., 2002a; Ohtani et al., 1998; Patel et al., 2010). The CXCR4 receptor is coupled with GαIβγ protein. PTX, which specifically uncouples the Gαi subunit from G protein, can inhibit CXCL12-induced calcium mobilization in microglia (Tanabe et al., 1997). CXCL12 (also named stromal-derived-factor-1, SDF-1) was initially identified as the only ligand for CXCR4. However, recent studies demonstrate that ubiquitin is also a natural ligand of CXCR4 (Saini et al., 2010). Although many studies on CXCR4 function in CNS focus on its involvement in HIV infection, accumulating evidence suggests that the CXCR4-CXCL12 axis has important functions in normal physiology in addition to an important role in disease states (Limatola et al., 2000; Zou et al., 1998). In support of this notion, mice with either CXCR4 or CXCL12 gene deletions die soon after birth with severe abnormalities affecting many organs, including the brain. Studies by Bezzi and colleagues further demonstrate that the CXCR4-CXCL12 system is important for communication between astrocytes and neurons via astrocytic glutamate release (Bezzi et al., 2001). However, excessive CXCL12 in the injured or infected brain activates CXCR4 on microglia, which then release large amounts of TNFα. This massive release of TNFα results in turn in a surge of glutamate release from astrocytes. These responses switch astrocyte function from normal physiology to excitotoxicity, perturbing the transmission of information from glial networks to neuronal circuits. CXCR4 engagement has also been shown to regulate IL-6 production from microglia in an ERK, PI3K, and NFκB-dependent manner (Lu et al., 2009). As expected from its classification as a chemokine receptor, CXCR4 also mediates microglial migration toward CXCL12 (Wang et al., 2008).
CXCL12 and CXCR4 are implicated in many CNS injuries. For example, CXCL12 and CXCR4 are involved in the pathology of ischemic stroke. Cerebral ischemia results in a prompt and long-lasting elevation of CXCL12 in the ischemic penumbra, particularly in association with reactive astrocytes in perivascular areas (Hill et al., 2004). Interestingly, transplanted GFP+ bone marrow cells are recruited in proximity to these CXCL12+ vessels and display the characteristics of activated microglial cells. These results suggest that CXCL12 is important in the homing of bone marrow-derived monocytes, which transform into microglia at the site of ischemic injury.
An in vitro study with microglial cultures suggests that exposure to hypoxia significantly enhances CXCR4 expression on microglia in a hypoxia inducible factor-1alpha (HIF-1alpha)-dependent manner, resulting in the accelerated migration toward CXCL12 (Wang et al., 2008). Such accumulation of monocyte-derived and local microglia may contribute to severe neuroinflammation after brain injury. However, there is also evidence that CXCL12 and CXCR4 might be important for neurorepair processes after brain ischemia by regulating neurogenesis and angiogenesis within the vascular niche (Merino et al., 2011; Wang et al., 2012). Finally, chronic intrathecal delivery of CXCL12 after spinal cord contusion has been shown to reduce cell apoptosis, boost astroglial and microglial responses, enhance angiogenesis, and ultimately improve long-term functional recovery after SCI (Zendedel et al., 2012). Such dualistic roles of CXCL12/CXCR4 in neuroinflammation and neurovascular repair need to be carefully considered when targeting this molecular pair with rational drug design.
4. Purinergic receptors
Purinergic receptors (purinoreceptors) are divided into two broad classes, P1 and P2, based on their binding properties. P1 receptors bind adenosine whereas P2 receptors bind ATP.
4.1. P1 adenosine receptors
P1 adenosine receptors are a class of G protein coupled seven-transmembrane proteins. Four different subtypes of adenosine receptors have been identified: A1, A2A, A2B, and A3. Each receptor subtype is encoded by a separate gene and exerts both distinct and overlapping functions. In general, A1 and A3 receptors are coupled to Gi and mediate biological functions distinct from those mediated by Gs protein-coupled A2A or A2B receptors. Purine nucleoside adenosine is the natural ligand of P1 adenosine receptors. The concentration of extracellular adenosine is very low under physiological conditions. This small amount of adenosine nonetheless exerts important roles in the modulation of many brain functions, including sleep and arousal, locomotion, anxiety, cognition and memory (Hasko et al., 2005). Under pathological CNS conditions such as hypoxia, ischemia, or trauma, adenosine may be released from injured neurons and other types of cells as a consequence of energy metabolism failure (Headrick et al., 1994; Koos et al., 1997; Latini et al., 1996). This release results in both protective and injurious effects on tissue via the stimulation of various P1 adenosine receptors.
4.1.1. A2A receptor
All four subtypes of adenosine receptors have been detected on microglia (Hasko and Pacher, 2012; Hasko et al., 2005; van der Putten et al., 2009) and were shown to individually or cooperatively modulate microglial activities such as proliferation, apoptosis, protein production, and chemotaxis. Among these receptors, A2A appears to be actively involved in the secretion of inflammatory factors from stimulated microglia. The expression of microglial A2A receptors is enhanced with inflammation, such as following stimulation with LPS or other toll-like receptor (TLR) ligands (van der Putten et al., 2009; Wittendorp et al., 2004). Microglial A2A receptors are also increased in pathological conditions such as AD and brain ischemia (Angulo et al., 2003; Trincavelli et al., 2008). In vitro experiments reveal that A2A receptor activation potentiates the release of NO from LPS-stimulated microglia (Saura et al., 2005) and leads to the upregulation of COX2 and the release of prostaglandin E2 (PGE2) from cultured microglia (Fiebich et al., 1996). These results may explain the anti-inflammatory and neuroprotective effects of A2A antagonists or gene knockout in animal models of cerebral ischemia, TBI, and SCI (Chen et al., 1999; Genovese et al., 2009; Yu et al., 2004).
A recent elegant study proposed that local glutamate levels may dictate the function of A2A receptors in microglia. In the presence of low concentrations of glutamate, an A2A receptor agonist inhibited LPS-induced microglial activation. High concentrations of glutamate, however, redirected A2A receptor signaling from the PKA to the PKC pathway, resulting in a switch from anti-inflammatory to pro-inflammatory effects (Dai et al., 2010). Therefore, the neuroprotective effect of A2A antagonists and agonists may well depend on the extent of brain injury and disease stage. Therapies targeted at adenosine pathways must take this into full consideration.
In addition to protein factor secretion, the A2A receptor is also involved in other microglial functions. For example, the A2A receptor is important for the retraction of microglial processes, which results in the amoeboid morphology assumed by activated microglia during in vivo LPS exposure or brain injury. Finally, the A2A receptor has also been shown to promote microglial proliferation and inhibit phagocytosis by LPS-activated microglia (Gebicke-Haerter et al., 1996). The contribution of these A2A functions to CNS pathologies awaits further investigation.
4.1.2. A3 receptor
The function of A3 receptors on microglia is not clear. Conflicting results have been presented by different groups. The early studies of microglial A3 receptors documented that stimulation of A3 receptors activates Gi protein-dependent phosphorylation of ERK1/2 and p38. However, the role of the A3 receptor in microglial function was not addressed (Hammarberg et al., 2004; Hammarberg et al., 2003). A subsequent report argued that the A3 receptor had no impact on the phosphorylation of p38 and ERK1/2. Instead, the A3 receptor was reported to inhibit LPS-induced PI3K/Akt and NFκB activation, thereby reducing TNF-α production (Lee et al., 2006). Similarly, Choi and colleagues have documented that an A3 receptor agonist, LJ529, significantly reduces the release of IL-1β, TNF-α, and MCP-1 in LPS-treated microglial cells. However, they also observed that the IC50 values of LJ529 for the inhibition of cytokine/chemokine release were much higher (in the micromolar range) than the nanomolar Ki values for the A3 receptor (Tchilibon et al., 2005). These observations suggest that the A3 receptor may not play a major role in cytokine release in microglia. Choi and colleagues further demonstrated that A3 receptor engagement inhibits microglial migration toward chemoattractants by downregulating the spatiotemporal expression of Rho GTPases (Choi et al., 2011). As a consequence, LJ529 suppresses microglia/monocyte accumulation into the ischemic brain and protects the brain in an in vivo model of cerebral ischemia. In contrast to this conclusion, a recent publication showed that A3 receptor signaling may promote the ADP-induced process extension and migration of microglia (Ohsawa et al., 2012).
Interactions between the A3 receptor and other P1 adenosine receptors have also been reported. The A3 receptor and A2A receptor recipically inhibit each other and thereby orchestrate cellular responses to stressors (van der Putten et al., 2009). Specifically, the A2A receptor mediates inhibitory signaling and the A3 receptor mediates activation in microglia. In unstimulated microglia, these two receptors counterbalance each other to maintain homeostatic equilibrium. However, under conditions of inflammatory stimulation, the decrease in A3 receptor-mediated signaling sensitizes microglia to A2A receptor-mediated inhibitory signaling. Thus, the A3 receptor may be a dynamically regulated suppressor of the A2A receptor.
In short, there is a dearth of conclusive knowledge on the function of A3 receptors. Further studies are required in order to clarify how this receptor affects microglial functions.
4.2. P2 ATP
P2 receptors are divided into five subclasses: P2X, P2Y, P2Z, P2U, and P2T. P2Y, P2U, and P2T purinergic receptors are metabotropic whereas P2X and P2Z receptors are ionotropic. Microglia express multiple types of these receptors, including P2X and P2Y (James and Butt, 2002). P2 receptors bind to ATP and UTP/UDP. Many CNS cells, including neurons and astrocytes, release ATP as a transmitter to mediate intercellular communication (Butt, 2011). During acute brain injuries such as ischemic stroke, neurons enter rapid energy failure, resulting in loss of ionic gradients and consequent depolarization. This is followed by intracellular ATP depletion and outflow of ATP from cells (Juranyi et al., 1999; Latini and Pedata, 2001). In addition to neurons, ATP is also released from vascular cells and blood cells (Bune et al., 2010). Furthermore, the release of UTP/UDP from damaged neurons has been observed after brain injury (Koizumi et al., 2007). These purines trigger various responses in microglia through corresponding P2 receptors. Interestingly, the impact of ATP may depend on the model and/or ATP concentration. Microglia exposed to 3 mM ATP acquire an amoeboid phenotype (Butchi et al., 2010) whereas exposure to 0.6 – 1 mM ATP has the opposite effect and induces the ramified phenotype (Wollmer et al., 2001). Microglia can also release ATP, suggesting the presence of an autocrine loop (Ferrari et al., 1997c).
4.2.1. P2X4
P2X4 is the most abundant P2X receptor in the CNS (Buell et al., 1996). Direct application of ATP or LPS increases P2X4 receptor expression in cultured microglia (Gong et al., 2009; Raouf et al., 2007). Functionally, microglial P2X4 receptors are involved in chemotaxis by the modulation of PI3K signaling (Ohsawa et al., 2007). An interaction between P2X4 and mu-opioid receptors has been shown to enhance microglial migration in response to morphine in a PI3K/Akt-dependent manner (Horvath and DeLeo, 2009). P2X4 also mediates the production of proinflammatory cytokines from microglia upon exposure to hypoxia (Li et al., 2011).
An induction of microglial P2X4 has been observed in CNS disease models that involve inflammation, such as SCI, cerebral ischemia, and preterm hypoxia-ischemia (Cavaliere et al., 2003; Li et al., 2011; Schwab et al., 2005; Tsuda et al., 2003; Ulmann et al., 2008; Wixey et al., 2009). Specifically, microglial P2X4 receptor expression is essential for spinal inflammatory pain processes (Schwab et al., 2005; Tsuda et al., 2003; Ulmann et al., 2008). Pharmacological blockade or molecular suppression of the P2X4 receptor leads to a reduction in tactile allodynia after SCI, whereas intraspinal administration of microglia in which P2X4 receptors has been pre-activated produces tactile allodynia in normal rats (Tsuda et al., 2003). Mechanistically, stimulation of the P2X4 receptor by ATP causes the SNARE-mediated release and synthesis of BDNF, which is dependent on extracellular Ca2+ influx and activation of p38-MAPK (Ulmann et al., 2008) (Figure 4). The elevation in BDNF in turn weakens the tonic inhibition of lamina I GABAergic interneurons (Coull et al., 2005) and results in the development of allodynia.
Figure 4. P2X4 receptors in tactile allodynia after spinal nerve injury.
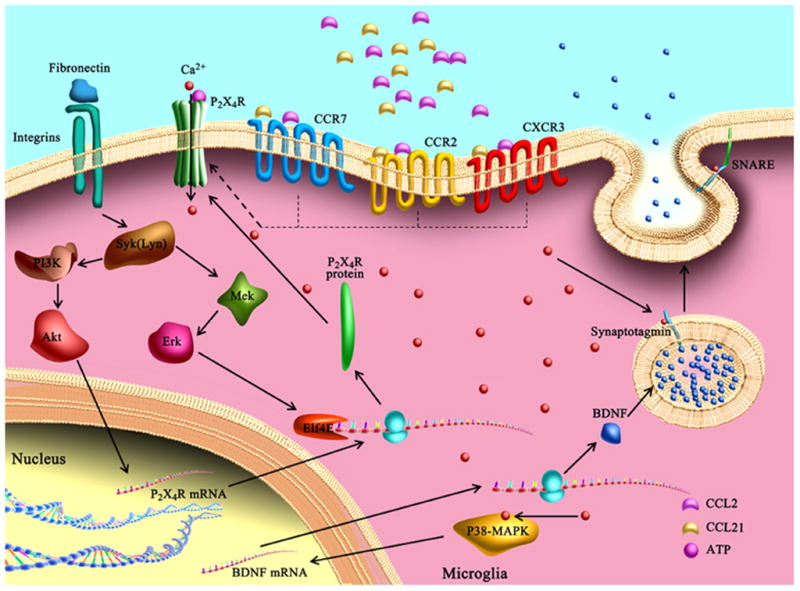
Spinal nerve injury may induce P2X4 receptor upregulation in microglia through the activation of fibronectin/integrin, Lyn kinase, PI3K/Akt, and MEK/ERK-dependent signaling pathways. Chemokine CCL2 and CCL21 are also involved in transforming quiescent microglia into the P2X4-expressing phenotype through interaction with microglial CCR2, CXCR3, and CCR7 receptors. Stimulation of the P2X4 receptor by ATP causes the SNARE-mediated release and synthesis of BDNF, which is dependent on extracellular Ca2+ influx and activation of p38-MAPK. The elevated BDNF in turn weakens the tonic inhibition of lamina I GABAergic interneurons and results in the development of allodynia.
The molecular mechanisms mediating the upregulation of P2X4 receptors on microglia have been actively studied. Spinal nerve injury may induce P2X4 receptor upregulation and neuropathic pain through fibronectin/integrin, Lyn kinase, PI3K/Akt, and MEK/ERK-dependent mechanisms (Tsuda et al., 2008a; Tsuda et al., 2009b; Tsuda et al., 2008b) (Figure 4). Another study demonstrated that CCL21 is rapidly expressed in injured small-sized primary sensory neurons and transported to their central terminals in the dorsal horn after SCI. The upregulation of neuronal CCL21 enhances microglial P2X4 receptor expression and leads to tactile allodynia (Biber et al., 2011). Chemokine CCL2 and cytokine IFNγ have also been shown to transform quiescent microglia into the P2X4-expressing phenotype (Beggs et al., 2012; Toyomitsu et al., 2012; Tsuda et al., 2009a). Further elucidation of microglial P2X4 receptor upregulation and function following SCI may help identify new therapeutic targets for neuropathic pain.
4.2.2. P2X7
Microglial P2X7 receptor stimulation by exogenous ATP triggers a rapid and sustained transmembrane influx of extracellular Ca2+ as well as an efflux of intracellular K+ (Ferrari et al., 1996; Gudipaty et al., 2003; Kahlenberg and Dubyak, 2004). Such dramatic changes in intracellular ion homeostasis activate multiple signaling molecules, including NFκB, ERK1/2, p38, and JNK1/2 (Ferrari et al., 1997d; Parvathenani et al., 2003; Shiratori et al., 2010; Takenouchi et al., 2007). Several transcription factors whose activation are involved in the expression of inflammatory genes, such as NFκB, nuclear factor of activated T cells (NAFT), and cyclic AMP response element-binding (CREB), are also activated by P2X7 receptors in microglia (Ferrari et al., 1999; Kataoka et al., 2009; Shiratori et al., 2010). Consequently, P2X7 engagement induces the secretion of pro-inflammatory mediators such as TNF-α, IL-1β, superoxide, NO, CXCL2, and CCL3 in microglial cells (Gendron et al., 2003; Hide et al., 2000; Kataoka et al., 2009; Parvathenani et al., 2003; Shiratori et al., 2010). In particular, the P2X7 receptor has been shown to be critical for IL-1β production in microglial cells (Ferrari et al., 1997b; Takenouchi et al., 2009). LPS or other inflammatory stimulants induce the synthesis and cytoplasmic accumulation of microglial pro-IL-1β, which is biologically inactive. The P2X7 receptor, in response to relatively high concentrations of ATP, induces the rapid processing and massive release of mature IL-1β in LPS-primed microglia (Sanz and Di Virgilio, 2000). Thus, purinoreceptors mediate the release of pro-inflammatory cytokines from microglia under conditions of high ATP.
Interestingly, mechanically stimulated astrocytes also release ATP. Astrocyte-derived ATP induces the shedding of IL-1β-containing vesicles from nearby microglia in a P2X7-dependent manner, suggesting that astrocyte-microglia crosstalk through ATP/P2X7 interactions plays a critical role in the release of IL-1β from microglia (Bianco et al., 2005). Mechanistic studies further reveal that P2X7 receptor engagement activates IL-1β-converting enzyme/caspase 1 in microglia (Sanz and Di Virgilio, 2000; Takenouchi et al., 2008), which in turn induces actin filament reorganization and the shedding of IL-1β-containing vesicles (Takenouchi et al., 2008).
In addition to regulating inflammatory cytokine secretion, the P2X7 receptor might be important for microglial cell death and proliferation in a dose- or duration-dependent manner. Prolonged activation of the P2X7 receptor leads to the formation of membrane pores in microglia and results in the induction of apoptotic/necrotic cell death (Ferrari et al., 1997a; Ferrari et al., 1999; Takenouchi et al., 2008). In contrast, moderate P2X7 receptor activation demonstrates trophic effects and increases microglial proliferation, especially under pathological conditions associated with increased TNFα (Bianco et al., 2006; Monif et al., 2009).
Disease states appear to upregulate P2X7 expression on microglia. For example, microglial P2X7 receptors are upregulated in response to injuries such as cerebral ischemia (Franke et al., 2004). Such receptor upregulation enhances microglial activation and might be a therapeutic target for brain injury. For instance, P2X7 receptor antagonists have been shown to protect against transient global cerebral ischemia reperfusion injury by reducing inflammation (Chu et al., 2012). In addition, P2X7 receptor antagonists suppress seizures, confer neuroprotection during status epilepticus, and reduce neuropathic pain (Engel et al., 2012; He et al., 2012).
4.2.3. P2Y receptors
P2Y receptors are a family of G-protein coupled receptors that bind ATP, ADP, UTP, and UDP. The expression and functions of P2Y6 and P2Y12 in microglia have been studied. For example, the P2Y6 receptor is known to be important for microglial phagocytic activity (Koizumi et al., 2007). Koizumi and colleagues first reported that P2Y6 receptors are upregulated in microglia following kainic acid-induced neuronal injury and act as a sensor to detect the UTP/UDP signals released from damaged neurons. The stimulation of P2Y6 by UTP/UDP enhances microglial phagocytosis and therefore facilitates the removal of damaged cells and cellular debris. P2Y6 receptors also regulate other aspects of microglial activity. For example, an in vitro study with glial cell and slice cultures documented that UDP-P2Y6 interactions resulted in elevated expression of CCL3 and CCL2 in microglia and astrocytes, suggesting a possible role of the P2Y6 receptor in chemokine production from glial cells (Kim et al., 2011).
In contrast to the function of P2Y6 in microglial phagocytosis, P2Y12 has been shown to be important for microglial motility (Haynes et al., 2006). P2Y12 is expressed predominantly in microglia in the brain (Sasaki et al., 2003), but is dramatically reduced after microglial activation (Haynes et al., 2006; Orr et al., 2009). Microglia from P2Y12 deficient mice exhibited normal baseline motility but are unable to extend processes or migrate toward ATP/ADP in vitro or in vivo, resulting in reduced directional process extension toward sites of cortical damage (Haynes et al., 2006). Further studies confirm that the P2Y12 receptor is critical for microglial chemotaxis and have identified several signaling molecules downstream of P2Y12 activation, such as integrin β1, PLC, and PI3K/Akt (Irino et al., 2008; Ohsawa et al., 2007; Ohsawa et al., 2010)
In summary, purinergic receptors are involved in multiple microglial functions, including process motility, chemotaxis, phagocytosis, and cytokine/chemokine production. A close collaboration between these receptors is critical in mediating efficient neuron-microglia interactions. For instance, damaged neurons as well as other CNS cells release ATP and UTP. Whereas ATP serves as a ‘find me’ signal to activate P2Y12-mediated cell motility, UTP and its degradated product UDP serve as ‘eat-me’ signals to trigger P2Y6-mediated phagocytosis. It is also notable that the expression of purine receptors on microglia is dynamic and activation state-specific (Orr et al., 2009; van der Putten et al., 2009). For example, LPS-treated microglia exhibit marked A2A receptor upregulation along with a loss of P2Y12 receptors. Such changes switch microglial motility from chemoattraction to repulsion, suggesting that a context-dependent shift in purinergic receptor signaling may orchestrate microglial responses to stressors (Orr et al., 2009).
5. Phosphatidylserine receptors and bridging proteins
5.1 Phosphatidylserine receptors and bridging proteins in neuron-microglia interactions
Phosphatidylserine is normally expressed on the inner leaflet of the plasma membrane in healthy cells. However, phosphatidylserine exposure on the exoplasmic leaflet is a characteristic feature of cell apoptosis (Fadok et al., 1998). The recognition of phosphatidylserine by its receptors on phagocytes is believed to be an essential ‘eat me’ signal for the clearance of dying cells (Ravichandran, 2011). A variety of engulfment receptors on phagocytes are capable of binding to phosphatidylserine. These phosphatidylserine receptors function in two modes: direct recognition and bridged recognition. The receptors directly recognizing phosphatidylserine include the phosphatidylserine receptor (PSR) (Fadok et al., 2000), the T cell immunoglobulin and mucin (TIM) family members (Kobayashi et al., 2007; Miyanishi et al., 2007), Stabilin2 (Park et al., 2008) and brain angiogenesis inhibitor I (BAI-I) (Park et al., 2007). For indirect phosphatidylserine recognition, bridging proteins such as milk fat globule-EGF factor 8 (MFG-E8) and growth arrest specific gene 6 (Gas6) are essential (Hanayama et al., 2002; Leonardi-Essmann et al., 2005). These proteins usually have two binding sites; one site binds to the phosphatidylserine and the other binds to receptors on phagocytes. Together, these sites mediate phosphatidylserine-dependent phagocytosis of apoptotic cells.
Phosphatidylserine receptors are critical for the microglial clearance of apoptotic neurons (Hirt and Leist, 2003; Witting et al., 2000). As one might expect, O-phospho-L-serine, a molecule that mimics the phosphatidylserine head group and blocks microglial phosphatidylserine receptors, interferes with the uptake of apoptotic neurons by microglia (Witting et al., 2000). Specifically, the expression of PSR has been detected on quiescent microglia and is enhanced by LPS stimulation (De et al., 2002). The engagement of PSR may promote the phagocytosis of apoptotic neurons and shift microglial activation toward an anti-inflammatory phenotype (De et al., 2002; Hirt and Leist, 2003).
Bridging proteins are also important for microglial function. MFG-E8 is expressed in microglia and secreted in response to soluble factors, such as CX3CL1, which is released from degenerating neurons (Fuller and Van Eldik, 2008; Leonardi-Essmann et al., 2005; Li et al., 2012; Noda et al., 2011) (Figure 5). MFG-E8 can bind to phosphatidylserine on apoptotic neurons and simultaneously engage integrin αvβ3 on microglia. Elevated MFG-E8 expression therefore results in accelerated microglial clearance activity. Moreover, MFG-E8 enhances production of heme oxygenase-1 (HO-1) and reduces neurodegeneration.
Figure 5. Phosphatidylserine receptors and bridging proteins are critical for the microglial clearance of apoptotic neurons.
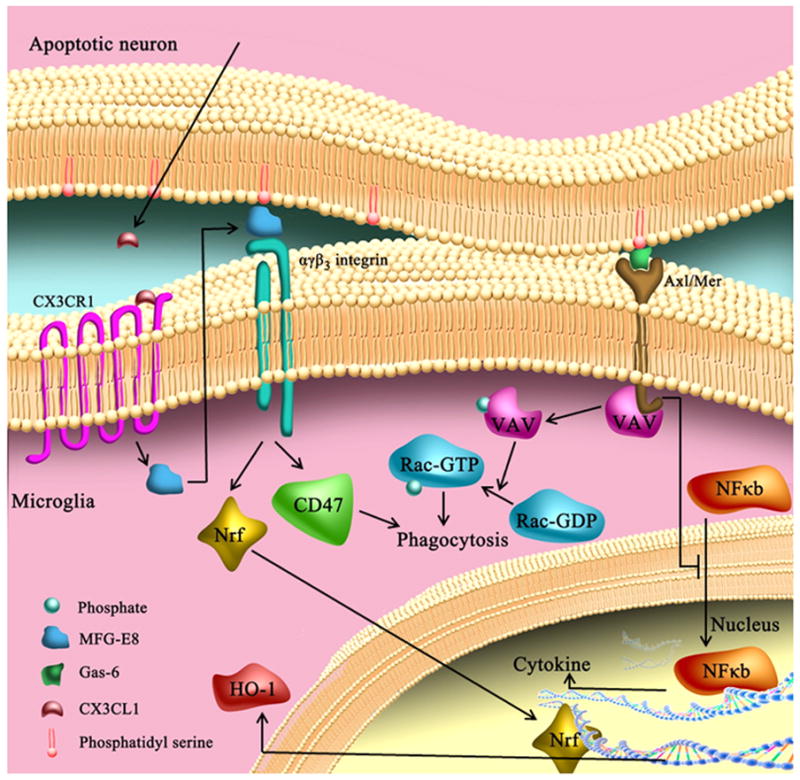
MFG-E8 is expressed in microglia and secreted in response to soluble factors, such as CX3CL1, which is released from degenerating neurons. MFG-E8 can bind to phosphatidylserine on apoptotic neurons and simultaneously engage integrin αvβ3 on microglia. Elevated MFG-E8 expression therefore results in accelerated microglial clearance activity. Moreover, MFG-E8 enhances production of heme oxygenase-1 (HO-1) through Nrf2 activation. Another bridging protein, Gas6, bridges phosphatidylserine residues on the surface of apoptotic cells to the Axl/Mer family of tyrosine kinases (Mer, Axl, and Tyro3) on microglia and stimulates microglial phagocytosis of apoptotic neurons in a vav/Rac-dependent manner. Gas6 receptors mediate an anti-inflammatory response to apoptotic cells by inhibiting NF-κB activation.
The bridging protein Gas6 has also been detected on microglia (Grommes et al., 2008) (Figure 5). Gas6 bridges phosphatidylserine residues on the surface of apoptotic cells to the Axl/Mer family of tyrosine kinases (Mer, Axl, and Tyro3) on microglia and stimulates microglial phagocytosis of apoptotic neurons in a vav/Rac-dependent manner. Similar to MFG-E8, Gas6 receptors mediate an anti-inflammatory response to apoptotic cells in addition to boosting phagocytosis.
A question that has undergone years of debate is whether phosphatidylserine exposure alone is sufficient to induce phagocytosis. Although early studies claim that phosphatidylserine by itself could send a signal strong enough to elicit phagocytic responses in macrophages, subsequent evidence challenged this conclusion by showing that many cells are not taken up by phagocytes despite the presence of phosphatidylserine on their outer leaflet (Borisenko et al., 2003; Suzuki et al., 2010). A second signal has therefore been proposed that may work in concert with phosphatidylserine to send an efficient ‘eat-me’ signal to phagocytes (Gardai et al., 2005). Other possible explanations include the requirement of a threshold number of exposed phosphatidylserine molecules (Borisenko et al., 2003) and the preference for modified phosphatidylserine (Tyurin et al., 2008) for phagocytosis induction. However, all these hypotheses still remain to be tested.
5.2 Phosphatidylserine receptors and bridging proteins in CNS injuries
As mentioned above, bridging proteins such as MFG-E8 and Gas6 mediate indirect phosphatidylserine recognition. A recent study demonstrates neuroprotective and anti-inflammatory effects of recombinant MFG-E8 protein in a mouse model of stroke, indicating the potentially therapeutic value of this protein in neurological disorders (Cheyuo et al., 2012). Gas6 deficiency exacerbates microglial activation as well as oligodendrocyte loss after cuprizone-induced demyelination (Binder et al., 2008). Consistent with its role as a microglial tyrosine kinase that is bridged to phosphatidylserine residues on apoptotic cells, Axl deficiency reduces microglia/macrophage recruitment and impairs myelin debris clearance following demyelination during EAE (Weinger et al., 2011). In contrast, treatment with exogenous Gas6 ameliorates microglial activation and enhances remyelination following cuprizone-induced demyelination (Binder et al., 2011; Tsiperson et al., 2010). Interestingly, Gas6 not only modulates microglial activity, but also independently influences the oligodendrocyte response after CNS damage.
6. Pattern recognition receptors (PRR)
It is well known that microglial activation can be mediated by a superfamily of PRR that recognize molecular motifs comprised of pathogen associated molecular patterns (PAMPs) and danger associated molecular patterns (DAMPs). PAMPs are a set of molecular determinants derived from invading pathogenic bacteria, fungi, or viruses that command immediate neutralization and subsequent removal by the innate immune system. DAMPs, on the other hand, are derived from cellular debris, intracellular proteins/enzymes released from necrosis following catastrophic events such as ischemic injury and spinal cord injury, as well as neurological disorders such as MS, AD, and PD.
Different classes of PRR such as Toll like receptors, carbohydrate binding receptors, Scavenger receptors, Rig like receptors (Rig like helicases) (Lech et al., 2010), Nod like receptors (Rosenzweig et al., 2011) and Aim2 like receptors (Brunette et al., 2012) with distinct recognition motifs and subcellular localizations work in concert to neutralize exogenous pathogens and toxic material. Among them, Toll like receptors, carbohydrate binding receptors, and scavenger receptors have all been implicated in CNS injuries. The function of PRR in microglial activation has been extensively reviewed elsewhere. We will only discuss them briefly here.
6.1. Toll like receptors (TLRs)
Toll like receptors are mammalian homologues of the Toll gene identified in Drosophila nearly three decades ago. In mammals, 13 isoforms designated as TLR1 to TLR13 have been identified. Only 11 of them are expressed in human cells (Hanke and Kielian, 2011) and 9 in microglia (Bsibsi et al., 2002; Lee and Lee, 2002). In the brain, TLR signaling is a part of the innate immune system. TLRs undergo homodimerization to achieve specific ligand or agonist recognition, with the exception of TLR2, which associates with TLR1 and TLR6 in order to recognize bacterial lipopeptides (Kawai and Akira, 2007).
Induction of several TLRs has been strongly associated with activation of microglial cells in response to inflammation (Lehnardt et al., 2007; Olson and Miller, 2004; Trinchieri and Sher, 2007). Among those TLRs, TLR2 and 4 have been shown to modulate the extent of the inflammatory response and neuronal demise. Immediately after stroke, neuronal degeneration-induced inflammatory responses lead to the production and release of pro-inflammatory cytokines and excitotoxins via activation of TLR2 and TLR4 in microglia (Caso et al., 2007; Hua et al., 2007; Kilic et al., 2008; Lehnardt et al., 2007). Indeed, stroke-induced injury, infarct size and neurological deficits are all significantly reduced in mice deficient in TLR2 and TLR4 (Tang et al., 2007). Inhibition of neuronal TLR2 and TLR4 prevents JNK activation in ischemia (Tang et al., 2007). In addition, TLR4 deficiency decreases TNFα, iNOS and COX2 production (Pradillo et al., 2009), potentially resulting in greater tolerance towards ischemic injury. In contrast, TLR3 or TLR9 activation has no impact on stroke outcome.
6.2. Carbohydrate binding receptors
Recent reports suggest that carbohydrate structures attached onto proteins during post-translational modification can interact with microglia to modulate their activation status and function. Understanding the relationship between carbohydrate moieties and carbohydrate binding receptors on microglia may provide a novel means of mitigating inflammation-induced damage in various neurological disorders.
6.2.1. Galectins
In mammals, there are 15 isoforms of galectins that are further subdivided into galectin1 and galectin3 based on sequence homologies and functions (Liu, 2002). Typically, galectins contain a highly conserved motif known as the carbohydrate recognition domain (CRD) that enables their direct association with carbohydrates, specifically, β-galactosides. It has been speculated that galectins recognize galactose-containing oligosaccharides on a group of glycoproteins and glycolipids in vivo (Liu, 2002). Microglial galectin1 and galectin3 are functional counterparts in modulating inflammatory responses. The former facilitates anti-inflammatory events and the latter promotes pro-inflammatory cascades (Rabinovich and Toscano, 2009). Therefore, a tightly temporally titrated expression of specific subsets of galectins on microglia helps ensure a balanced and productive inflammatory response.
An increase in galectin3 has been correlated with microglial activation in ischemic injury (Lalancette-Hebert et al., 2012; Rotshenker, 2003), leading to the demise of hippocampal neurons (Satoh et al., 2011). Indeed, preconditioning strategies or hypothermia both result in the downregulation of microglial galectin3 and attenuate these deleterious events (Lalancette-Hebert et al., 2012; Satoh et al., 2011). Alternatively, upregulation of microglial galectin1 has been shown to suppress pro-inflammatory responses in MS (Starossom et al., 2012), supporting the counter functions of galectin1 and galectin3 that were originally proposed by Rabinovich and Toscano (Rabinovich and Toscano, 2009). In addition, supplementation with recombinant galectin1 enhances astrocytic BDNF production and reduced neuronal apoptosis in ischemic injury (Qu et al., 2010). These responses suggest an inherently protective function of microglial galectin1 and a potentially toxic role for galectin3. In non-neuronal systems, galectins may engage multiple signaling pathways such as Akt, p38, ERK, NF βB, and JNK (Chung et al., 2012; Koh et al., 2008; Pineda et al., 2012). The impact of galectin on signal transduction in the brain has yet to be defined.
6.2.2. Selectins
Selectins are part of a receptor family that associates with sialyated and fucosylated glycan structures (Varki, 2007). Among them, E- and P-selectins are expressed in endothelium and platelets cells, respectively. In contrast, exclusive expression of L-selectins has been reported in astrocytes and microglia (Jeon et al., 2008). Selectins are involved in cell-cell interactions during the initiation of inflammatory responses (Varki, 2007). P- and L-selectins are bound by sulfatides, a class of sulfated glycosylceramides. Thus, sulfatide-induced inflammatory responses in the brain may be mediated via microglial selectins. The concomitant reduction of pro-inflammatory cytokines by microglial L-selectins also suggests that these carbohydrate-binding receptors modulate neuroinflammation (Jeon et al., 2008).
Our understanding of the roles of selectins in various neurological disorders lies in its infancy. In stroke, E- and P-selectin have been shown to affect platelet and endothelial activities (Beer et al., 2011; Jaremo et al., 2012), which in turn modulate inflammatory responses and degeneration (Ma et al., 2012). Interestingly, polymorphisms in L-selectin appear to increase the risk of stroke (Wei et al., 2011). Nevertheless, whether microglial selectins are mechanistically involved in this disease still remains elusive because epidemiological studies are correlational in nature.
6.3. CD36
Scavenger receptors are a set of PRR that have been used to identify modified lipoproteins liberated during cellular damage and degeneration. Scavenger receptors facilitate the removal of these lipoproteins. To date, over 20 structurally distinct scavenger receptors with overlapping ligands have been identified (Husemann et al., 2002). Nevertheless, the exact structural recognition motifs for all scavenger receptors have not been elucidated. Scavenger receptors can be classified into 6 categories (A, B, C, D, E and F). Among them, CD36, which belongs to SCARB, is closedly related to CNS injuries.
CD36 participates in HDL-mediated transport of lipids and cholesterol to the liver (Krieger, 1999). This transport process, termed “reverse cholesterol transport,” is the principal mechanism by which HDL prevents the cholesterol buildup that leads to atherosclerosis (Trigatti et al., 1999). In addition, CD36 is found on macrophages and dendritic cells derived from monocytes and may mediate the uptake of degenerating neutrophils, lymphocytes, and fibroblasts (Albert et al., 1998; Puente Navazo et al., 1996; Ren and Savill, 1995; Savill et al., 1992). CD36 is highly expressed in mouse neonatal microglia but is expressed at lower levels in adult microglia (Coraci et al., 2002). The function of microglial CD36 is not yet clear. However, it is known that CD36 protein expression is increased in the ischemic brain, predominantly in microglia/macrophages. CD36-null mice exhibit reduced infarct volumes after MCAO with attenuated ROS production in the early stages after reperfusion. These findings support a role for CD36 in ischemia-induced oxidative stress and brain injury. Despite these toxic roles for CD36, CD36 has also been shown to promote hematoma absorption via microglial phagocytosis, thereby protecting brain cells from intracerebral hemorrhage-induced damage (Zhao et al., 2009; Zhao et al., 2007). A recent study further reveals that CD36 deficiency worsens short-term outcomes from focal stroke in neonates due to enhanced inflammation and diminished removal of apoptotic neuronal debris (Woo et al., 2012). These studies suggest that the function of CD36 in brain injuries depends on the nature of the injury, the stage of damage, or the age of the victims.
7. Future directions
In summary, a broad range of surface receptors are expressed on microglia and mediate microglial recognition of ‘On’ or ‘Off’ signals on other host cells as well as invading microorganisms. Integrated actions of these receptors result in tightly-regulated biological functions, including cell mobility, phagocytosis, inflammatory mediator release, and the induction of acquired immunity. Over the last few years, significant advances have been made towards deciphering the signaling mechanisms related to these receptors and identifying their contribution to specific cellular functions. As described above, a recurring theme in the mechanism of action of microglial receptors is the orchestration of opposing actions. The juxtaposition of ITAM against ITIM, SHP-1 against SHP-2, galectin1 against galectin3, and numerous other examples reveal an elegant means of fine-tuning microglial responses in response to different challenges so that homeostasis of the CNS can be achieved. Also as a result of these juxtrapositions, microglial sensitivity to different stimuli has an unexpectedly high degree of resolution. The complexity of microglial responses to environmental challenges is also reflected in their dual-faced nature, where they can switch from a protective to a toxic phenotype in ways that are not completely understood. Indeed, there remain many unresolved challenges in this field, some of which will be discussed below.
First and foremost, there remain serious technical obstacles in studies of microglia. For example, the in vivo studies of microglial receptors have been largely dependent on pharmacological inhibition and/or genetic deletion of the receptor of interest. However, most microglial receptors are also expressed on other types of cells, posing a potential confound to the interpretation of pharmacological or gene deletion studies. In other words, the effects of global inhibition or ablation of these receptors may also reflect cells other than microglia. Second, the characterization of microglial responses is usually based on immunostaining or biochemical methods using microglia-specific markers. However, the lack of an antibody specific to either microglia or macrophages precludes our ability to distinguish between local microglia and circulating macrophages that were recruited to the brain following injuries. Third, because microglia react to even minute changes in brain milieu, it is critical to examine these cells in their natural environment as far as possible. Recently, novel techniques such as microglia-specific conditional knockout, transgenic cell labeling, in vivo microglia imaging/tracking have been developed and optimized to help overcome some of these difficulties (Sieger and Peri, 2013; Xiao et al., 2011). However, new quantitative imaging probes are still necessary to permit researchers to monitor signaling events within living cells. In combination with live cell imaging of microglia, such a technique would greatly improve our understanding of microglia-specific signaling dynamics in different microenvironments (Sieger and Peri, 2013).
Besides technical challenges, another obstacle to studying microglia is the relative lack of information on the interactions between different microglial receptors and their combined downstream effects. It is known that multiple distinct receptors on microglia are activated simultaneously or sequentially in response to the same pathological challenge. Despite our knowledge of individual receptors, the interplay between these receptors remains uncharted territory. To address this issue, we need to describe the spatiotemporal kinetics and mode of action for each specific receptor in isolation and in combination with other receptors, the sequence of receptor activation and deactivation, and, finally, how the coordinated actions of multiple receptors fulfill the subtle biological roles of microglia. Recent research has made some progress toward this goal. For example, accumulating evidence suggests that multiple microglia receptors, including CD36, CD47, integrinα6β1, TLR4, TLR2 and scavenger receptor A, are co-activated in response to Aβ and form a large receptor complex to mediate microglial phagocytosis of Aβ fibrils and the subsequent activation of pro-inflammatory intracellular signaling pathways (Bamberger et al., 2003; Reed-Geaghan et al., 2009; Wilkinson et al., 2006). In contrast to such cooperative functions, FcγR activation has been shown to stimulate inhibitory signaling through another microglial receptor, signal regulatory protein α (SIRPα), which in turn inhibits FcγR- and complementary receptor-mediated phagocytosis (Gresham et al., 2000; Oldenborg et al., 2001). Again, advances in real time imaging technologies may offer a better experimental platform to further clarify our understanding of how different surface receptors work together in both time and space in order to modulate microglial functions in CNS injuries.
It must also be borne in mind that the expression and function of many microglial receptors are sexually dimorphic and age-dependent. For example, the expression of several ‘Off’ receptors, including CX3CR1 and CD200, decreases with age (Lyons et al., 2007; Wynne et al., 2010), which might contribute to the age-related dysregulation of microglial responses to CNS injuries (Wong, 2013). In addition, the expression of several P2 nucleotide receptors on microglia has been shown to vary with age and gender (Crain et al., 2009). For example, the microglial expression of P2X4 is age-dependent and significantly lower in females than in males. Furthermore, the expression of P2X7 or P2Y6 is low in neonates but significantly increases from 21 days of age onwards, with no obvious gender difference. Many CNS diseases are more common in one gender and increase in occurance in elderly populations. Thus, elucidating how these gender-specific and age-related differences influence the function of microglia will provide valuable insights on the contribution of these receptors to the aging process and their roles in CNS diseases.
Many experimental strategies that target microglial receptors have been used for the treatment of neurological disorders in animal models (Binder et al., 2011; Clark et al., 2007; Pertwee, 2009; Tsiperson et al., 2010; Zhuang et al., 2007). Translating this research into the clinic will be far more challenging because we do not yet have a comprehensive view of microglial receptors. For example, more information on the topographic distribution of receptors on non-microglial cells under normal and pathological conditions would be useful to predict possible off-target effects. Furthermore, better strategies to selectively target microglia will be essential to achieve drug specificity (Cucchiarini et al., 2003; Minami et al., 2012). The temporal kinetics of microglial behavior will be particularly critical in deciding the treatment regimen. As one example, inhibition of CXCL12-CXCR4 signaling in early phases of stroke may restrain microglial activity. However, interference with microglial activity must be avoided in advanced stages when neurovascular remodeling becomes active. Finally, researchers must also bear in mind that the long-term modulation of microglial activities may negatively affect homeostasis and immune defense and result in undesired side effects such as opportunistic brain infections and the buildup of necrotic cellular debris. Fortunately, the pace of research on microglia has increased exponentially in the past decade, not to mention since del-Hortega seminal observations of microglia/macrophages almost a hundred years ago. If we continue to advance our understanding of microglial receptors at this more recent pace, new strategies to modulate microglial activity without adversely impacting immune status may lie on the horizon.
Highlights.
Microglia is the first line of immune defense in the central nervous system
Integrated actions of microglial receptors result in tightly regulated biological functions.
Microglial homeostasis is carefully maintained by multiple counterbalanced strategies, including ‘On’ and ‘Off’ receptor signaling.
Understanding of the signaling mechanisms related to these receptors and identifying their contributions to specific cellular functions will advance our knowledge of many CNS injuries.
Acknowledgments
Dr. Xiaoming Hu is supported by the American Heart Association (13SDG14570025) and the Ethyl Vincent pilot grant in multiple sclerosis from Department of Neurology, University of Pittsburgh. Dr. Rehana K. Leak is supported by a Commonwealth Universal Research Enhancement (C.U.R.E.) Award from the Department of Health and a Michael J. Fox Foundation Innovation Award. Dr. Yanqin Gao is supported by the Chinese Natural Science Foundation grants (81171149 and 81371306). Dr. Jun Chen is supported by the National Institutes of Health Grants (NS45048, NS62157, NS59806, and NS36736), Chinese Natural Science Foundation grants (81228008) and a VA Merit Review.
Glossary
- Aβ
amyloidβ
- AD
Alzheimer’s disease
- AGEs
advanced glycation endproducts
- COX2
cyclo-oxygenase-2
- CNS
central nervous system
- DAP12
DNAX-activating protein of molecular mass 12 kilodaltons
- Dok
downstream of tyrosine kinase
- ERK
Extracellular signal-regulated kinases
- Gas6
growth arrest specific gene 6
- HMGB1
high-mobility group box 1
- HO-1
heme oxygenase-1
- IFN-γ
interferon-gamma
- IgSF
immunoglobulin superfamily
- IL
interleukin
- JNK
c-Jun-N-terminal protein kinase
- iNOS
inducible nitric oxide synthase
- ITAM
immunoreceptor tyrosine-based activation motif
- ITIM
immunoreceptor tyrosine-based inhibition motif
- LPS
lipopolysaccharide
- MAPK
mitogen-activated protein kinase
- m-csf
macrophage colony-stimulating factor
- MFG-E8
milk fat globule-EGF factor 8
- MMP
matrix metalloproteinase
- MCP-1
monocyte chemotactic protein-1
- MIP
macrophage inflammatory protein
- MS
multiple sclerosis
- NFκB
nuclear factor-κB
- NO
nitric oxide
- PD
Parkinson’s disease
- PI3K
phosphoinositide-3-kinase
- PKC
protein kinase C
- PLCγ
phospholipase Cγ
- RAGE
Receptor for advanced glycation endproducts
- PSR
phosphatidylserine receptor
- RANTES
regulated on activation, normal T cell expressed and secreted
- ROS
reactive oxygen species
- SCI
spinal cord injury
- SHIP
SH2 domain-containing inositol polyphosphate 5′ phosphatase
- SHP1
SH2 domain-containing phosphatase-1
- TBI
traumatic brain injury
- TGF-β
transforming growth factor-β
- TLR
toll-like receptor
- TNFα
tumor necrosis factor α
- TREM
Triggering receptors expressed on myeloid cells
Footnotes
Author Disclosure Statement
No competing financial interests exist.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Abbadie C, Lindia JA, Cumiskey AM, Peterson LB, Mudgett JS, Bayne EK, DeMartino JA, MacIntyre DE, Forrest MJ. Impaired neuropathic pain responses in mice lacking the chemokine receptor CCR2. Proc Natl Acad Sci U S A. 2003;100:7947–7952. doi: 10.1073/pnas.1331358100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adami C, Bianchi R, Pula G, Donato R. S100B-stimulated NO production by BV-2 microglia is independent of RAGE transducing activity but dependent on RAGE extracellular domain. Biochim Biophys Acta. 2004;1742:169–177. doi: 10.1016/j.bbamcr.2004.09.008. [DOI] [PubMed] [Google Scholar]
- Albert ML, Pearce SF, Francisco LM, Sauter B, Roy P, Silverstein RL, Bhardwaj N. Immature dendritic cells phagocytose apoptotic cells via alphavbeta5 and CD36, and cross-present antigens to cytotoxic T lymphocytes. The Journal of experimental medicine. 1998;188:1359–1368. doi: 10.1084/jem.188.7.1359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Angulo E, Casado V, Mallol J, Canela EI, Vinals F, Ferrer I, Lluis C, Franco R. A1 adenosine receptors accumulate in neurodegenerative structures in Alzheimer disease and mediate both amyloid precursor protein processing and tau phosphorylation and translocation. Brain Pathol. 2003;13:440–451. doi: 10.1111/j.1750-3639.2003.tb00475.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arai H, Charo IF. Differential regulation of G-protein-mediated signaling by chemokine receptors. J Biol Chem. 1996;271:21814–21819. doi: 10.1074/jbc.271.36.21814. [DOI] [PubMed] [Google Scholar]
- Arumugam TV, Tang SC, Lathia JD, Cheng A, Mughal MR, Chigurupati S, Magnus T, Chan SL, Jo DG, Ouyang X, Fairlie DP, Granger DN, Vortmeyer A, Basta M, Mattson MP. Intravenous immunoglobulin (IVIG) protects the brain against experimental stroke by preventing complement-mediated neuronal cell death. Proc Natl Acad Sci U S A. 2007;104:14104–14109. doi: 10.1073/pnas.0700506104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asensio VC, Campbell IL. Chemokines in the CNS: plurifunctional mediators in diverse states. Trends Neurosci. 1999;22:504–512. doi: 10.1016/s0166-2236(99)01453-8. [DOI] [PubMed] [Google Scholar]
- Babcock AA, Kuziel WA, Rivest S, Owens T. Chemokine expression by glial cells directs leukocytes to sites of axonal injury in the CNS. J Neurosci. 2003;23:7922–7930. doi: 10.1523/JNEUROSCI.23-21-07922.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bamberger ME, Harris ME, McDonald DR, Husemann J, Landreth GE. A cell surface receptor complex for fibrillar beta-amyloid mediates microglial activation. J Neurosci. 2003;23:2665–2674. doi: 10.1523/JNEUROSCI.23-07-02665.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banisadr G, Fontanges P, Haour F, Kitabgi P, Rostene W, Melik Parsadaniantz S. Neuroanatomical distribution of CXCR4 in adult rat brain and its localization in cholinergic and dopaminergic neurons. Eur J Neurosci. 2002a;16:1661–1671. doi: 10.1046/j.1460-9568.2002.02237.x. [DOI] [PubMed] [Google Scholar]
- Banisadr G, Queraud-Lesaux F, Boutterin MC, Pelaprat D, Zalc B, Rostene W, Haour F, Parsadaniantz SM. Distribution, cellular localization and functional role of CCR2 chemokine receptors in adult rat brain. J Neurochem. 2002b;81:257–269. doi: 10.1046/j.1471-4159.2002.00809.x. [DOI] [PubMed] [Google Scholar]
- Bao Y, Kim E, Bhosle S, Mehta H, Cho S. A role for spleen monocytes in post-ischemic brain inflammation and injury. J Neuroinflammation. 2010;7:92. doi: 10.1186/1742-2094-7-92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bard F, Cannon C, Barbour R, Burke RL, Games D, Grajeda H, Guido T, Hu K, Huang J, Johnson-Wood K, Khan K, Kholodenko D, Lee M, Lieberburg I, Motter R, Nguyen M, Soriano F, Vasquez N, Weiss K, Welch B, Seubert P, Schenk D, Yednock T. Peripherally administered antibodies against amyloid beta-peptide enter the central nervous system and reduce pathology in a mouse model of Alzheimer disease. Nat Med. 2000;6:916–919. doi: 10.1038/78682. [DOI] [PubMed] [Google Scholar]
- Barone FC, Feuerstein GZ. Inflammatory mediators and stroke: new opportunities for novel therapeutics. J Cereb Blood Flow Metab. 1999;19:819–834. doi: 10.1097/00004647-199908000-00001. [DOI] [PubMed] [Google Scholar]
- Barrow AD, Trowsdale J. You say ITAM and I say ITIM, let’s call the whole thing off: the ambiguity of immunoreceptor signalling. Eur J Immunol. 2006;36:1646–1653. doi: 10.1002/eji.200636195. [DOI] [PubMed] [Google Scholar]
- Beer C, Blacker D, Hankey GJ, Puddey IB. Association of clinical and aetiologic subtype of acute ischaemic stroke with inflammation, oxidative stress and vascular function: a cross-sectional observational study. Medical science monitor : international medical journal of experimental and clinical research. 2011;17:CR467–473. doi: 10.12659/MSM.881931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beggs S, Trang T, Salter MW. P2X4R+ microglia drive neuropathic pain. Nat Neurosci. 2012;15:1068–1073. doi: 10.1038/nn.3155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bezzi P, Domercq M, Brambilla L, Galli R, Schols D, De Clercq E, Vescovi A, Bagetta G, Kollias G, Meldolesi J, Volterra A. CXCR4-activated astrocyte glutamate release via TNFalpha: amplification by microglia triggers neurotoxicity. Nat Neurosci. 2001;4:702–710. doi: 10.1038/89490. [DOI] [PubMed] [Google Scholar]
- Bhaskar K, Konerth M, Kokiko-Cochran ON, Cardona A, Ransohoff RM, Lamb BT. Regulation of tau pathology by the microglial fractalkine receptor. Neuron. 2010;68:19–31. doi: 10.1016/j.neuron.2010.08.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bianchi R, Adami C, Giambanco I, Donato R. S100B binding to RAGE in microglia stimulates COX-2 expression. J Leukoc Biol. 2007;81:108–118. doi: 10.1189/jlb.0306198. [DOI] [PubMed] [Google Scholar]
- Bianchi R, Giambanco I, Donato R. S100B/RAGE-dependent activation of microglia via NF-kappaB and AP-1 Co-regulation of COX-2 expression by S100B, IL-1beta and TNF-alpha. Neurobiol Aging. 2010;31:665–677. doi: 10.1016/j.neurobiolaging.2008.05.017. [DOI] [PubMed] [Google Scholar]
- Bianchi R, Kastrisianaki E, Giambanco I, Donato R. S100B protein stimulates microglia migration via RAGE-dependent up-regulation of chemokine expression and release. J Biol Chem. 2011;286:7214–7226. doi: 10.1074/jbc.M110.169342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bianco F, Ceruti S, Colombo A, Fumagalli M, Ferrari D, Pizzirani C, Matteoli M, Di Virgilio F, Abbracchio MP, Verderio C. A role for P2X7 in microglial proliferation. J Neurochem. 2006;99:745–758. doi: 10.1111/j.1471-4159.2006.04101.x. [DOI] [PubMed] [Google Scholar]
- Bianco F, Pravettoni E, Colombo A, Schenk U, Moller T, Matteoli M, Verderio C. Astrocyte-derived ATP induces vesicle shedding and IL-1 beta release from microglia. J Immunol. 2005;174:7268–7277. doi: 10.4049/jimmunol.174.11.7268. [DOI] [PubMed] [Google Scholar]
- Biber K, Dijkstra I, Trebst C, De Groot CJ, Ransohoff RM, Boddeke HW. Functional expression of CXCR3 in cultured mouse and human astrocytes and microglia. Neuroscience. 2002;112:487–497. doi: 10.1016/s0306-4522(02)00114-8. [DOI] [PubMed] [Google Scholar]
- Biber K, Neumann H, Inoue K, Boddeke HW. Neuronal ‘On’ and ‘Off’ signals control microglia. Trends in neurosciences. 2007;30:596–602. doi: 10.1016/j.tins.2007.08.007. [DOI] [PubMed] [Google Scholar]
- Biber K, Sauter A, Brouwer N, Copray SC, Boddeke HW. Ischemia-induced neuronal expression of the microglia attracting chemokine Secondary Lymphoid-tissue Chemokine (SLC) Glia. 2001;34:121–133. doi: 10.1002/glia.1047. [DOI] [PubMed] [Google Scholar]
- Biber K, Tsuda M, Tozaki-Saitoh H, Tsukamoto K, Toyomitsu E, Masuda T, Boddeke H, Inoue K. Neuronal CCL21 up-regulates microglia P2X4 expression and initiates neuropathic pain development. EMBO J. 2011;30:1864–1873. doi: 10.1038/emboj.2011.89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Binder MD, Cate HS, Prieto AL, Kemper D, Butzkueven H, Gresle MM, Cipriani T, Jokubaitis VG, Carmeliet P, Kilpatrick TJ. Gas6 deficiency increases oligodendrocyte loss and microglial activation in response to cuprizone-induced demyelination. J Neurosci. 2008;28:5195–5206. doi: 10.1523/JNEUROSCI.1180-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Binder MD, Xiao J, Kemper D, Ma GZ, Murray SS, Kilpatrick TJ. Gas6 increases myelination by oligodendrocytes and its deficiency delays recovery following cuprizone-induced demyelination. PLoS One. 2011;6:e17727. doi: 10.1371/journal.pone.0017727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Block ML, Zecca L, Hong JS. Microglia-mediated neurotoxicity: uncovering the molecular mechanisms. Nature reviews Neuroscience. 2007;8:57–69. doi: 10.1038/nrn2038. [DOI] [PubMed] [Google Scholar]
- Boddeke EW, Meigel I, Frentzel S, Gourmala NG, Harrison JK, Buttini M, Spleiss O, Gebicke-Harter P. Cultured rat microglia express functional beta-chemokine receptors. J Neuroimmunol. 1999;98:176–184. doi: 10.1016/s0165-5728(99)00096-x. [DOI] [PubMed] [Google Scholar]
- Bonacchi A, Romagnani P, Romanelli RG, Efsen E, Annunziato F, Lasagni L, Francalanci M, Serio M, Laffi G, Pinzani M, Gentilini P, Marra F. Signal transduction by the chemokine receptor CXCR3: activation of Ras/ERK, Src, and phosphatidylinositol 3-kinase/Akt controls cell migration and proliferation in human vascular pericytes. J Biol Chem. 2001;276:9945–9954. doi: 10.1074/jbc.M010303200. [DOI] [PubMed] [Google Scholar]
- Borisenko GG, Matsura T, Liu SX, Tyurin VA, Jianfei J, Serinkan FB, Kagan VE. Macrophage recognition of externalized phosphatidylserine and phagocytosis of apoptotic Jurkat cells--existence of a threshold. Arch Biochem Biophys. 2003;413:41–52. doi: 10.1016/s0003-9861(03)00083-3. [DOI] [PubMed] [Google Scholar]
- Bouchon A, Dietrich J, Colonna M. Cutting edge: inflammatory responses can be triggered by TREM-1, a novel receptor expressed on neutrophils and monocytes. J Immunol. 2000;164:4991–4995. doi: 10.4049/jimmunol.164.10.4991. [DOI] [PubMed] [Google Scholar]
- Brunette RL, Young JM, Whitley DG, Brodsky IE, Malik HS, Stetson DB. Extensive evolutionary and functional diversity among mammalian AIM2-like receptors. The Journal of experimental medicine. 2012 doi: 10.1084/jem.20121960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bsibsi M, Ravid R, Gveric D, van Noort JM. Broad expression of Toll-like receptors in the human central nervous system. J Neuropathol Exp Neurol. 2002;61:1013–1021. doi: 10.1093/jnen/61.11.1013. [DOI] [PubMed] [Google Scholar]
- Buell G, Lewis C, Collo G, North RA, Surprenant A. An antagonist-insensitive P2X receptor expressed in epithelia and brain. EMBO J. 1996;15:55–62. [PMC free article] [PubMed] [Google Scholar]
- Bune LT, Thaning P, Johansson PI, Bochsen L, Rosenmeier JB. Effects of nucleotides and nucleosides on coagulation. Blood Coagul Fibrinolysis. 2010;21:436–441. doi: 10.1097/MBC.0b013e328338db27. [DOI] [PubMed] [Google Scholar]
- Butchi NB, Du M, Peterson KE. Interactions between TLR7 and TLR9 agonists and receptors regulate innate immune responses by astrocytes and microglia. Glia. 2010;58:650–664. doi: 10.1002/glia.20952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butt AM. ATP: a ubiquitous gliotransmitter integrating neuron-glial networks. Semin Cell Dev Biol. 2011;22:205–213. doi: 10.1016/j.semcdb.2011.02.023. [DOI] [PubMed] [Google Scholar]
- Cambien B, Pomeranz M, Schmid-Antomarchi H, Millet MA, Breittmayer V, Rossi B, Schmid-Alliana A. Signal transduction pathways involved in soluble fractalkine-induced monocytic cell adhesion. Blood. 2001;97:2031–2037. doi: 10.1182/blood.v97.7.2031. [DOI] [PubMed] [Google Scholar]
- Carbonell WS, Murase S, Horwitz AF, Mandell JW. Migration of perilesional microglia after focal brain injury and modulation by CC chemokine receptor 5: an in situ time-lapse confocal imaging study. J Neurosci. 2005;25:7040–7047. doi: 10.1523/JNEUROSCI.5171-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cardona AE, Pioro EP, Sasse ME, Kostenko V, Cardona SM, Dijkstra IM, Huang D, Kidd G, Dombrowski S, Dutta R, Lee JC, Cook DN, Jung S, Lira SA, Littman DR, Ransohoff RM. Control of microglial neurotoxicity by the fractalkine receptor. Nat Neurosci. 2006;9:917–924. doi: 10.1038/nn1715. [DOI] [PubMed] [Google Scholar]
- Cartier L, Dubois-Dauphin M, Hartley O, Irminger-Finger I, Krause KH. Chemokine-induced cell death in CCR5-expressing neuroblastoma cells. J Neuroimmunol. 2003;145:27–39. doi: 10.1016/j.jneuroim.2003.09.008. [DOI] [PubMed] [Google Scholar]
- Caso JR, Pradillo JM, Hurtado O, Lorenzo P, Moro MA, Lizasoain I. Toll-like receptor 4 is involved in brain damage and inflammation after experimental stroke. Circulation. 2007;115:1599–1608. doi: 10.1161/CIRCULATIONAHA.106.603431. [DOI] [PubMed] [Google Scholar]
- Cavaliere F, Florenzano F, Amadio S, Fusco FR, Viscomi MT, D’Ambrosi N, Vacca F, Sancesario G, Bernardi G, Molinari M, Volonte C. Up-regulation of P2X2, P2X4 receptor and ischemic cell death: prevention by P2 antagonists. Neuroscience. 2003;120:85–98. doi: 10.1016/s0306-4522(03)00228-8. [DOI] [PubMed] [Google Scholar]
- Charo IF, Myers SJ, Herman A, Franci C, Connolly AJ, Coughlin SR. Molecular cloning and functional expression of two monocyte chemoattractant protein 1 receptors reveals alternative splicing of the carboxyl-terminal tails. Proc Natl Acad Sci U S A. 1994;91:2752–2756. doi: 10.1073/pnas.91.7.2752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen BP, Kuziel WA, Lane TE. Lack of CCR2 results in increased mortality and impaired leukocyte activation and trafficking following infection of the central nervous system with a neurotropic coronavirus. J Immunol. 2001;167:4585–4592. doi: 10.4049/jimmunol.167.8.4585. [DOI] [PubMed] [Google Scholar]
- Chen J, Connor KM, Smith LE. Overstaying their welcome: defective CX3CR1 microglia eyed in macular degeneration. J Clin Invest. 2007;117:2758–2762. doi: 10.1172/JCI33513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen JF, Huang Z, Ma J, Zhu J, Moratalla R, Standaert D, Moskowitz MA, Fink JS, Schwarzschild MA. A(2A) adenosine receptor deficiency attenuates brain injury induced by transient focal ischemia in mice. J Neurosci. 1999;19:9192–9200. doi: 10.1523/JNEUROSCI.19-21-09192.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho SH, Sun B, Zhou Y, Kauppinen TM, Halabisky B, Wes P, Ransohoff RM, Gan L. CX3CR1 protein signaling modulates microglial activation and protects against plaque-independent cognitive deficits in a mouse model of Alzheimer disease. J Biol Chem. 2011;286:32713–32722. doi: 10.1074/jbc.M111.254268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi IY, Lee JC, Ju C, Hwang S, Cho GS, Lee HW, Choi WJ, Jeong LS, Kim WK. A3 adenosine receptor agonist reduces brain ischemic injury and inhibits inflammatory cell migration in rats. Am J Pathol. 2011;179:2042–2052. doi: 10.1016/j.ajpath.2011.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu K, Yin B, Wang J, Peng G, Liang H, Xu Z, Du Y, Fang M, Xia Q, Luo B. Inhibition of P2X7 receptor ameliorates transient global cerebral ischemia/reperfusion injury via modulating inflammatory responses in the rat hippocampus. J Neuroinflammation. 2012;9:69. doi: 10.1186/1742-2094-9-69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung LY, Tang SJ, Sun GH, Chou TY, Yeh TS, Yu SL, Sun KH. Galectin-1 promotes lung cancer progression and chemoresistance by upregulating p38 MAPK, ERK, and cyclooxygenase-2. Clinical cancer research : an official journal of the American Association for Cancer Research. 2012;18:4037–4047. doi: 10.1158/1078-0432.CCR-11-3348. [DOI] [PubMed] [Google Scholar]
- Clark-Lewis I, Mattioli I, Gong JH, Loetscher P. Structure-function relationship between the human chemokine receptor CXCR3 and its ligands. J Biol Chem. 2003;278:289–295. doi: 10.1074/jbc.M209470200. [DOI] [PubMed] [Google Scholar]
- Clark AK, Yip PK, Grist J, Gentry C, Staniland AA, Marchand F, Dehvari M, Wotherspoon G, Winter J, Ullah J, Bevan S, Malcangio M. Inhibition of spinal microglial cathepsin S for the reversal of neuropathic pain. Proc Natl Acad Sci U S A. 2007;104:10655–10660. doi: 10.1073/pnas.0610811104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark AK, Yip PK, Malcangio M. The liberation of fractalkine in the dorsal horn requires microglial cathepsin S. J Neurosci. 2009;29:6945–6954. doi: 10.1523/JNEUROSCI.0828-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Copland DA, Calder CJ, Raveney BJ, Nicholson LB, Phillips J, Cherwinski H, Jenmalm M, Sedgwick JD, Dick AD. Monoclonal antibody-mediated CD200 receptor signaling suppresses macrophage activation and tissue damage in experimental autoimmune uveoretinitis. Am J Pathol. 2007;171:580–588. doi: 10.2353/ajpath.2007.070272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coraci IS, Husemann J, Berman JW, Hulette C, Dufour JH, Campanella GK, Luster AD, Silverstein SC, El-Khoury JB. CD36, a class B scavenger receptor, is expressed on microglia in Alzheimer’s disease brains and can mediate production of reactive oxygen species in response to beta-amyloid fibrils. Am J Pathol. 2002;160:101–112. doi: 10.1016/s0002-9440(10)64354-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cortegano I, Pozo V, Cardaba B, Arrieta I, Gallardo S, Rojo M, Aceituno E, Takai T, Verbeek S, Palomino P, Liu FT, Lahoz C. Interaction between galectin-3 and FcgammaRII induces down-regulation of IL-5 gene: implication of the promoter sequence IL-5REIII. Glycobiology. 2000;10:237–242. doi: 10.1093/glycob/10.3.237. [DOI] [PubMed] [Google Scholar]
- Coull JA, Beggs S, Boudreau D, Boivin D, Tsuda M, Inoue K, Gravel C, Salter MW, De Koninck Y. BDNF from microglia causes the shift in neuronal anion gradient underlying neuropathic pain. Nature. 2005;438:1017–1021. doi: 10.1038/nature04223. [DOI] [PubMed] [Google Scholar]
- Cowell RM, Xu H, Parent JM, Silverstein FS. Microglial expression of chemokine receptor CCR5 during rat forebrain development and after perinatal hypoxia-ischemia. J Neuroimmunol. 2006;173:155–165. doi: 10.1016/j.jneuroim.2006.01.005. [DOI] [PubMed] [Google Scholar]
- Crain JM, Nikodemova M, Watters JJ. Expression of P2 nucleotide receptors varies with age and sex in murine brain microglia. J Neuroinflammation. 2009;6:24. doi: 10.1186/1742-2094-6-24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crowley MT, Costello PS, Fitzer-Attas CJ, Turner M, Meng F, Lowell C, Tybulewicz VL, DeFranco AL. A critical role for Syk in signal transduction and phagocytosis mediated by Fcgamma receptors on macrophages. J Exp Med. 1997;186:1027–1039. doi: 10.1084/jem.186.7.1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cucchiarini M, Ren XL, Perides G, Terwilliger EF. Selective gene expression in brain microglia mediated via adeno-associated virus type 2 and type 5 vectors. Gene Ther. 2003;10:657–667. doi: 10.1038/sj.gt.3301925. [DOI] [PubMed] [Google Scholar]
- D’Mello C, Le T, Swain MG. Cerebral microglia recruit monocytes into the brain in response to tumor necrosis factoralpha signaling during peripheral organ inflammation. J Neurosci. 2009;29:2089–2102. doi: 10.1523/JNEUROSCI.3567-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai SS, Li W, An JH, Wang H, Yang N, Chen XY, Zhao Y, Li P, Liu P, Chen JF, Zhou YG. Adenosine A2A receptors in both bone marrow cells and non-bone marrow cells contribute to traumatic brain injury. J Neurochem. 2010;113:1536–1544. doi: 10.1111/j.1471-4159.2010.06716.x. [DOI] [PubMed] [Google Scholar]
- Dascal N. Ion-channel regulation by G proteins. Trends Endocrinol Metab. 2001;12:391–398. doi: 10.1016/s1043-2760(01)00475-1. [DOI] [PubMed] [Google Scholar]
- de Jong EK, de Haas AH, Brouwer N, van Weering HR, Hensens M, Bechmann I, Pratley P, Wesseling E, Boddeke HW, Biber K. Expression of CXCL4 in microglia in vitro and in vivo and its possible signaling through CXCR3. J Neurochem. 2008;105:1726–1736. doi: 10.1111/j.1471-4159.2008.05267.x. [DOI] [PubMed] [Google Scholar]
- de Jong EK, Dijkstra IM, Hensens M, Brouwer N, van Amerongen M, Liem RS, Boddeke HW, Biber K. Vesicle-mediated transport and release of CCL21 in endangered neurons: a possible explanation for microglia activation remote from a primary lesion. J Neurosci. 2005;25:7548–7557. doi: 10.1523/JNEUROSCI.1019-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De SR, Ajmone-Cat MA, Nicolini A, Minghetti L. Expression of phosphatidylserine receptor and down-regulation of pro-inflammatory molecule production by its natural ligand in rat microglial cultures. J Neuropathol Exp Neurol. 2002;61:237–244. doi: 10.1093/jnen/61.3.237. [DOI] [PubMed] [Google Scholar]
- Denes A, Ferenczi S, Halasz J, Kornyei Z, Kovacs KJ. Role of CX3CR1 (fractalkine receptor) in brain damage and inflammation induced by focal cerebral ischemia in mouse. Journal of cerebral blood flow and metabolism : official journal of the International Society of Cerebral Blood Flow and Metabolism. 2008;28:1707–1721. doi: 10.1038/jcbfm.2008.64. [DOI] [PubMed] [Google Scholar]
- Deng YY, Lu J, Ling EA, Kaur C. Monocyte chemoattractant protein-1 (MCP-1) produced via NF-kappaB signaling pathway mediates migration of amoeboid microglia in the periventricular white matter in hypoxic neonatal rats. Glia. 2009;57:604–621. doi: 10.1002/glia.20790. [DOI] [PubMed] [Google Scholar]
- Desforges NM, Hebron ML, Algarzae NK, Lonskaya I, Moussa CE. Fractalkine Mediates Communication between Pathogenic Proteins and Microglia: Implications of Anti-Inflammatory Treatments in Different Stages of Neurodegenerative Diseases. Int J Alzheimers Dis. 2012;2012:345472. doi: 10.1155/2012/345472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dijkstra IM, Hulshof S, van der Valk P, Boddeke HW, Biber K. Cutting edge: activity of human adult microglia in response to CC chemokine ligand 21. J Immunol. 2004;172:2744–2747. doi: 10.4049/jimmunol.172.5.2744. [DOI] [PubMed] [Google Scholar]
- Dimitrijevic OB, Stamatovic SM, Keep RF, Andjelkovic AV. Absence of the chemokine receptor CCR2 protects against cerebral ischemia/reperfusion injury in mice. Stroke. 2007;38:1345–1353. doi: 10.1161/01.STR.0000259709.16654.8f. [DOI] [PubMed] [Google Scholar]
- Dirnagl U, Iadecola C, Moskowitz MA. Pathobiology of ischaemic stroke: an integrated view. Trends in neurosciences. 1999;22:391–397. doi: 10.1016/s0166-2236(99)01401-0. [DOI] [PubMed] [Google Scholar]
- Donnelly DJ, Longbrake EE, Shawler TM, Kigerl KA, Lai W, Tovar CA, Ransohoff RM, Popovich PG. Deficient CX3CR1 signaling promotes recovery after mouse spinal cord injury by limiting the recruitment and activation of Ly6Clo/iNOS+ macrophages. J Neurosci. 2011;31:9910–9922. doi: 10.1523/JNEUROSCI.2114-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dukic-Stefanovic S, Gasic-Milenkovic J, Deuther-Conrad W, Munch G. Signal transduction pathways in mouse microglia N-11 cells activated by advanced glycation endproducts (AGEs) J Neurochem. 2003;87:44–55. doi: 10.1046/j.1471-4159.2003.01988.x. [DOI] [PubMed] [Google Scholar]
- Ekdahl CT, Claasen JH, Bonde S, Kokaia Z, Lindvall O. Inflammation is detrimental for neurogenesis in adult brain. Proceedings of the National Academy of Sciences of the United States of America. 2003;100:13632–13637. doi: 10.1073/pnas.2234031100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engel T, Gomez-Villafuertes R, Tanaka K, Mesuret G, Sanz-Rodriguez A, Garcia-Huerta P, Miras-Portugal MT, Henshall DC, Diaz-Hernandez M. Seizure suppression and neuroprotection by targeting the purinergic P2X7 receptor during status epilepticus in mice. FASEB J. 2012;26:1616–1628. doi: 10.1096/fj.11-196089. [DOI] [PubMed] [Google Scholar]
- Eugenin EA, Osiecki K, Lopez L, Goldstein H, Calderon TM, Berman JW. CCL2/monocyte chemoattractant protein-1 mediates enhanced transmigration of human immunodeficiency virus (HIV)-infected leukocytes across the blood-brain barrier: a potential mechanism of HIV-CNS invasion and NeuroAIDS. J Neurosci. 2006;26:1098–1106. doi: 10.1523/JNEUROSCI.3863-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fadok VA, Bratton DL, Frasch SC, Warner ML, Henson PM. The role of phosphatidylserine in recognition of apoptotic cells by phagocytes. Cell Death Differ. 1998;5:551–562. doi: 10.1038/sj.cdd.4400404. [DOI] [PubMed] [Google Scholar]
- Fadok VA, Bratton DL, Rose DM, Pearson A, Ezekewitz RA, Henson PM. A receptor for phosphatidylserine-specific clearance of apoptotic cells. Nature. 2000;405:85–90. doi: 10.1038/35011084. [DOI] [PubMed] [Google Scholar]
- Fang F, Lue LF, Yan S, Xu H, Luddy JS, Chen D, Walker DG, Stern DM, Schmidt AM, Chen JX, Yan SS. RAGE-dependent signaling in microglia contributes to neuroinflammation, Abeta accumulation, and impaired learning/memory in a mouse model of Alzheimer’s disease. FASEB J. 2010;24:1043–1055. doi: 10.1096/fj.09-139634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faustino JV, Wang X, Johnson CE, Klibanov A, Derugin N, Wendland MF, Vexler ZS. Microglial cells contribute to endogenous brain defenses after acute neonatal focal stroke. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2011;31:12992–13001. doi: 10.1523/JNEUROSCI.2102-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrari D, Chiozzi P, Falzoni S, Dal Susino M, Collo G, Buell G, Di Virgilio F. ATP-mediated cytotoxicity in microglial cells. Neuropharmacology. 1997a;36:1295–1301. doi: 10.1016/s0028-3908(97)00137-8. [DOI] [PubMed] [Google Scholar]
- Ferrari D, Chiozzi P, Falzoni S, Dal Susino M, Melchiorri L, Baricordi OR, Di Virgilio F. Extracellular ATP triggers IL-1 beta release by activating the purinergic P2Z receptor of human macrophages. J Immunol. 1997b;159:1451–1458. [PubMed] [Google Scholar]
- Ferrari D, Chiozzi P, Falzoni S, Hanau S, Di Virgilio F. Purinergic modulation of interleukin-1 beta release from microglial cells stimulated with bacterial endotoxin. J Exp Med. 1997c;185:579–582. doi: 10.1084/jem.185.3.579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrari D, Los M, Bauer MK, Vandenabeele P, Wesselborg S, Schulze-Osthoff K. P2Z purinoreceptor ligation induces activation of caspases with distinct roles in apoptotic and necrotic alterations of cell death. FEBS Lett. 1999;447:71–75. doi: 10.1016/s0014-5793(99)00270-7. [DOI] [PubMed] [Google Scholar]
- Ferrari D, Villalba M, Chiozzi P, Falzoni S, Ricciardi-Castagnoli P, Di Virgilio F. Mouse microglial cells express a plasma membrane pore gated by extracellular ATP. J Immunol. 1996;156:1531–1539. [PubMed] [Google Scholar]
- Ferrari D, Wesselborg S, Bauer MK, Schulze-Osthoff K. Extracellular ATP activates transcription factor NF-kappaB through the P2Z purinoreceptor by selectively targeting NF-kappaB p65. J Cell Biol. 1997d;139:1635–1643. doi: 10.1083/jcb.139.7.1635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fiebich BL, Biber K, Lieb K, van Calker D, Berger M, Bauer J, Gebicke-Haerter PJ. Cyclooxygenase-2 expression in rat microglia is induced by adenosine A2a-receptors. Glia. 1996;18:152–160. doi: 10.1002/(SICI)1098-1136(199610)18:2<152::AID-GLIA7>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- Fleit HB, Kobasiuk CD, Daly C, Furie R, Levy PC, Webster RO. A soluble form of Fc gamma RIII is present in human serum and other body fluids and is elevated at sites of inflammation. Blood. 1992;79:2721–2728. [PubMed] [Google Scholar]
- Flynn G, Maru S, Loughlin J, Romero IA, Male D. Regulation of chemokine receptor expression in human microglia and astrocytes. J Neuroimmunol. 2003;136:84–93. doi: 10.1016/s0165-5728(03)00009-2. [DOI] [PubMed] [Google Scholar]
- Franci C, Wong LM, Van Damme J, Proost P, Charo IF. Monocyte chemoattractant protein-3, but not monocyte chemoattractant protein-2, is a functional ligand of the human monocyte chemoattractant protein-1 receptor. J Immunol. 1995;154:6511–6517. [PubMed] [Google Scholar]
- Frank S, Burbach GJ, Bonin M, Walter M, Streit W, Bechmann I, Deller T. TREM2 is upregulated in amyloid plaque-associated microglia in aged APP23 transgenic mice. Glia. 2008;56:1438–1447. doi: 10.1002/glia.20710. [DOI] [PubMed] [Google Scholar]
- Franke H, Gunther A, Grosche J, Schmidt R, Rossner S, Reinhardt R, Faber-Zuschratter H, Schneider D, Illes P. P2X7 receptor expression after ischemia in the cerebral cortex of rats. J Neuropathol Exp Neurol. 2004;63:686–699. doi: 10.1093/jnen/63.7.686. [DOI] [PubMed] [Google Scholar]
- Fuller AD, Van Eldik LJ. MFG-E8 regulates microglial phagocytosis of apoptotic neurons. J Neuroimmune Pharmacol. 2008;3:246–256. doi: 10.1007/s11481-008-9118-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galasso JM, Harrison JK, Silverstein FS. Excitotoxic brain injury stimulates expression of the chemokine receptor CCR5 in neonatal rats. Am J Pathol. 1998;153:1631–1640. doi: 10.1016/S0002-9440(10)65752-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galon J, Gauchat JF, Mazieres N, Spagnoli R, Storkus W, Lotze M, Bonnefoy JY, Fridman WH, Sautes C. Soluble Fcgamma receptor type III (FcgammaRIII, CD16) triggers cell activation through interaction with complement receptors. J Immunol. 1996;157:1184–1192. [PubMed] [Google Scholar]
- Gamo K, Kiryu-Seo S, Konishi H, Aoki S, Matsushima K, Wada K, Kiyama H. G-protein-coupled receptor screen reveals a role for chemokine receptor CCR5 in suppressing microglial neurotoxicity. J Neurosci. 2008;28:11980–11988. doi: 10.1523/JNEUROSCI.2920-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao TL, Yuan XT, Yang D, Dai HL, Wang WJ, Peng X, Shao HJ, Jin ZF, Fu ZJ. Expression of HMGB1 and RAGE in rat and human brains after traumatic brain injury. J Trauma Acute Care Surg. 2012;72:643–649. doi: 10.1097/TA.0b013e31823c54a6. [DOI] [PubMed] [Google Scholar]
- Garcia-Zepeda EA, Combadiere C, Rothenberg ME, Sarafi MN, Lavigne F, Hamid Q, Murphy PM, Luster AD. Human monocyte chemoattractant protein (MCP)-4 is a novel CC chemokine with activities on monocytes, eosinophils, and basophils induced in allergic and nonallergic inflammation that signals through the CC chemokine receptors (CCR)-2 and -3. J Immunol. 1996;157:5613–5626. [PubMed] [Google Scholar]
- Gardai SJ, McPhillips KA, Frasch SC, Janssen WJ, Starefeldt A, Murphy-Ullrich JE, Bratton DL, Oldenborg PA, Michalak M, Henson PM. Cell-surface calreticulin initiates clearance of viable or apoptotic cells through trans-activation of LRP on the phagocyte. Cell. 2005;123:321–334. doi: 10.1016/j.cell.2005.08.032. [DOI] [PubMed] [Google Scholar]
- Garton KJ, Gough PJ, Blobel CP, Murphy G, Greaves DR, Dempsey PJ, Raines EW. Tumor necrosis factor-alpha-converting enzyme (ADAM17) mediates the cleavage and shedding of fractalkine (CX3CL1) J Biol Chem. 2001;276:37993–38001. doi: 10.1074/jbc.M106434200. [DOI] [PubMed] [Google Scholar]
- Gebicke-Haerter PJ, Christoffel F, Timmer J, Northoff H, Berger M, Van Calker D. Both adenosine A1- and A2-receptors are required to stimulate microglial proliferation. Neurochem Int. 1996;29:37–42. [PubMed] [Google Scholar]
- Gendron FP, Chalimoniuk M, Strosznajder J, Shen S, Gonzalez FA, Weisman GA, Sun GY. P2X7 nucleotide receptor activation enhances IFN gamma-induced type II nitric oxide synthase activity in BV-2 microglial cells. J Neurochem. 2003;87:344–352. doi: 10.1046/j.1471-4159.2003.01995.x. [DOI] [PubMed] [Google Scholar]
- Genovese T, Melani A, Esposito E, Mazzon E, Di Paola R, Bramanti P, Pedata F, Cuzzocrea S. The selective adenosine A2A receptor agonist CGS 21680 reduces JNK MAPK activation in oligodendrocytes in injured spinal cord. Shock. 2009;32:578–585. doi: 10.1097/SHK.0b013e3181a20792. [DOI] [PubMed] [Google Scholar]
- Getts DR, Terry RL, Getts MT, Muller M, Rana S, Shrestha B, Radford J, Van Rooijen N, Campbell IL, King NJ. Ly6c+ “inflammatory monocytes” are microglial precursors recruited in a pathogenic manner in West Nile virus encephalitis. J Exp Med. 2008;205:2319–2337. doi: 10.1084/jem.20080421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibot S, Kolopp-Sarda MN, Bene MC, Bollaert PE, Lozniewski A, Mory F, Levy B, Faure GC. A soluble form of the triggering receptor expressed on myeloid cells-1 modulates the inflammatory response in murine sepsis. J Exp Med. 2004;200:1419–1426. doi: 10.1084/jem.20040708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giri R, Shen Y, Stins M, Du Yan S, Schmidt AM, Stern D, Kim KS, Zlokovic B, Kalra VK. beta-amyloid-induced migration of monocytes across human brain endothelial cells involves RAGE and PECAM-1. Am J Physiol Cell Physiol. 2000;279:C1772–1781. doi: 10.1152/ajpcell.2000.279.6.C1772. [DOI] [PubMed] [Google Scholar]
- Gong QJ, Li YY, Xin WJ, Zang Y, Ren WJ, Wei XH, Zhang T, Liu XG. ATP induces long-term potentiation of C-fiber-evoked field potentials in spinal dorsal horn: the roles of P2X4 receptors and p38 MAPK in microglia. Glia. 2009;57:583–591. doi: 10.1002/glia.20786. [DOI] [PubMed] [Google Scholar]
- Gresham HD, Dale BM, Potter JW, Chang PW, Vines CM, Lowell CA, Lagenaur CF, Willman CL. Negative regulation of phagocytosis in murine macrophages by the Src kinase family member, Fgr. J Exp Med. 2000;191:515–528. doi: 10.1084/jem.191.3.515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grommes C, Lee CY, Wilkinson BL, Jiang Q, Koenigsknecht-Talboo JL, Varnum B, Landreth GE. Regulation of microglial phagocytosis and inflammatory gene expression by Gas6 acting on the Axl/Mer family of tyrosine kinases. J Neuroimmune Pharmacol. 2008;3:130–140. doi: 10.1007/s11481-007-9090-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gross BS, Lee JR, Clements JL, Turner M, Tybulewicz VL, Findell PR, Koretzky GA, Watson SP. Tyrosine phosphorylation of SLP-76 is downstream of Syk following stimulation of the collagen receptor in platelets. J Biol Chem. 1999;274:5963–5971. doi: 10.1074/jbc.274.9.5963. [DOI] [PubMed] [Google Scholar]
- Gudipaty L, Munetz J, Verhoef PA, Dubyak GR. Essential role for Ca2+ in regulation of IL-1beta secretion by P2X7 nucleotide receptor in monocytes, macrophages, and HEK-293 cells. Am J Physiol Cell Physiol. 2003;285:C286–299. doi: 10.1152/ajpcell.00070.2003. [DOI] [PubMed] [Google Scholar]
- Hammarberg C, Fredholm BB, Schulte G. Adenosine A3 receptor-mediated regulation of p38 and extracellular-regulated kinase ERK1/2 via phosphatidylinositol-3′-kinase. Biochem Pharmacol. 2004;67:129–134. doi: 10.1016/j.bcp.2003.08.031. [DOI] [PubMed] [Google Scholar]
- Hammarberg C, Schulte G, Fredholm BB. Evidence for functional adenosine A3 receptors in microglia cells. J Neurochem. 2003;86:1051–1054. doi: 10.1046/j.1471-4159.2003.01919.x. [DOI] [PubMed] [Google Scholar]
- Hanayama R, Tanaka M, Miwa K, Shinohara A, Iwamatsu A, Nagata S. Identification of a factor that links apoptotic cells to phagocytes. Nature. 2002;417:182–187. doi: 10.1038/417182a. [DOI] [PubMed] [Google Scholar]
- Hanisch UK, Kettenmann H. Microglia: active sensor and versatile effector cells in the normal and pathologic brain. Nature neuroscience. 2007;10:1387–1394. doi: 10.1038/nn1997. [DOI] [PubMed] [Google Scholar]
- Hanke ML, Kielian T. Toll-like receptors in health and disease in the brain: mechanisms and therapeutic potential. Clin Sci (Lond) 2011;121:367–387. doi: 10.1042/CS20110164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrison JK, Jiang Y, Chen S, Xia Y, Maciejewski D, McNamara RK, Streit WJ, Salafranca MN, Adhikari S, Thompson DA, Botti P, Bacon KB, Feng L. Role for neuronally derived fractalkine in mediating interactions between neurons and CX3CR1-expressing microglia. Proc Natl Acad Sci U S A. 1998;95:10896–10901. doi: 10.1073/pnas.95.18.10896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasko G, Pacher P. Regulation of macrophage function by adenosine. Arterioscler Thromb Vasc Biol. 2012;32:865–869. doi: 10.1161/ATVBAHA.111.226852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasko G, Pacher P, Vizi ES, Illes P. Adenosine receptor signaling in the brain immune system. Trends Pharmacol Sci. 2005;26:511–516. doi: 10.1016/j.tips.2005.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hatherley D, Barclay AN. The CD200 and CD200 receptor cell surface proteins interact through their N-terminal immunoglobulin-like domains. Eur J Immunol. 2004;34:1688–1694. doi: 10.1002/eji.200425080. [DOI] [PubMed] [Google Scholar]
- Haynes SE, Hollopeter G, Yang G, Kurpius D, Dailey ME, Gan WB, Julius D. The P2Y12 receptor regulates microglial activation by extracellular nucleotides. Nat Neurosci. 2006;9:1512–1519. doi: 10.1038/nn1805. [DOI] [PubMed] [Google Scholar]
- He J, Chen Y, Farzan M, Choe H, Ohagen A, Gartner S, Busciglio J, Yang X, Hofmann W, Newman W, Mackay CR, Sodroski J, Gabuzda D. CCR3 and CCR5 are co-receptors for HIV-1 infection of microglia. Nature. 1997;385:645–649. doi: 10.1038/385645a0. [DOI] [PubMed] [Google Scholar]
- He WJ, Cui J, Du L, Zhao YD, Burnstock G, Zhou HD, Ruan HZ. Spinal P2X(7) receptor mediates microglia activation-induced neuropathic pain in the sciatic nerve injury rat model. Behav Brain Res. 2012;226:163–170. doi: 10.1016/j.bbr.2011.09.015. [DOI] [PubMed] [Google Scholar]
- Headrick JP, Bendall MR, Faden AI, Vink R. Dissociation of adenosine levels from bioenergetic state in experimental brain trauma: potential role in secondary injury. J Cereb Blood Flow Metab. 1994;14:853–861. doi: 10.1038/jcbfm.1994.107. [DOI] [PubMed] [Google Scholar]
- Heldmann U, Mine Y, Kokaia Z, Ekdahl CT, Lindvall O. Selective depletion of Mac-1-expressing microglia in rat subventricular zone does not alter neurogenic response early after stroke. Experimental neurology. 2011;229:391–398. doi: 10.1016/j.expneurol.2011.03.005. [DOI] [PubMed] [Google Scholar]
- Heyman B. Regulation of antibody responses via antibodies, complement, and Fc receptors. Annu Rev Immunol. 2000;18:709–737. doi: 10.1146/annurev.immunol.18.1.709. [DOI] [PubMed] [Google Scholar]
- Hide I, Tanaka M, Inoue A, Nakajima K, Kohsaka S, Inoue K, Nakata Y. Extracellular ATP triggers tumor necrosis factor-alpha release from rat microglia. J Neurochem. 2000;75:965–972. doi: 10.1046/j.1471-4159.2000.0750965.x. [DOI] [PubMed] [Google Scholar]
- Hill WD, Hess DC, Martin-Studdard A, Carothers JJ, Zheng J, Hale D, Maeda M, Fagan SC, Carroll JE, Conway SJ. SDF-1 (CXCL12) is upregulated in the ischemic penumbra following stroke: association with bone marrow cell homing to injury. J Neuropathol Exp Neurol. 2004;63:84–96. doi: 10.1093/jnen/63.1.84. [DOI] [PubMed] [Google Scholar]
- Hirt UA, Leist M. Rapid, noninflammatory and PS-dependent phagocytic clearance of necrotic cells. Cell Death Differ. 2003;10:1156–1164. doi: 10.1038/sj.cdd.4401286. [DOI] [PubMed] [Google Scholar]
- Hoek RM, Ruuls SR, Murphy CA, Wright GJ, Goddard R, Zurawski SM, Blom B, Homola ME, Streit WJ, Brown MH, Barclay AN, Sedgwick JD. Down-regulation of the macrophage lineage through interaction with OX2 (CD200) Science. 2000;290:1768–1771. doi: 10.1126/science.290.5497.1768. [DOI] [PubMed] [Google Scholar]
- Hori O, Brett J, Slattery T, Cao R, Zhang J, Chen JX, Nagashima M, Lundh ER, Vijay S, Nitecki D, et al. The receptor for advanced glycation end products (RAGE) is a cellular binding site for amphoterin. Mediation of neurite outgrowth and co-expression of rage and amphoterin in the developing nervous system. J Biol Chem. 1995;270:25752–25761. doi: 10.1074/jbc.270.43.25752. [DOI] [PubMed] [Google Scholar]
- Horvath RJ, DeLeo JA. Morphine enhances microglial migration through modulation of P2X4 receptor signaling. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2009;29:998–1005. doi: 10.1523/JNEUROSCI.4595-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsieh CL, Koike M, Spusta SC, Niemi EC, Yenari M, Nakamura MC, Seaman WE. A role for TREM2 ligands in the phagocytosis of apoptotic neuronal cells by microglia. J Neurochem. 2009;109:1144–1156. doi: 10.1111/j.1471-4159.2009.06042.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu JH, Yang JP, Liu L, Li CF, Wang LN, Ji FH, Cheng H. Involvement of CX3CR1 in bone cancer pain through the activation of microglia p38 MAPK pathway in the spinal cord. Brain Res. 2012;1465:1–9. doi: 10.1016/j.brainres.2012.05.020. [DOI] [PubMed] [Google Scholar]
- Hua F, Ma J, Ha T, Xia Y, Kelley J, Williams DL, Kao RL, Browder IW, Schweitzer JB, Kalbfleisch JH, Li C. Activation of Toll-like receptor 4 signaling contributes to hippocampal neuronal death following global cerebral ischemia/reperfusion. Journal of neuroimmunology. 2007;190:101–111. doi: 10.1016/j.jneuroim.2007.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang ZY, Hunter S, Kim MK, Indik ZK, Schreiber AD. The effect of phosphatases SHP-1 and SHIP-1 on signaling by the ITIM- and ITAM-containing Fcgamma receptors FcgammaRIIB and FcgammaRIIA. J Leukoc Biol. 2003;73:823–829. doi: 10.1189/jlb.0902454. [DOI] [PubMed] [Google Scholar]
- Hudson BI, Bucciarelli LG, Wendt T, Sakaguchi T, Lalla E, Qu W, Lu Y, Lee L, Stern DM, Naka Y, Ramasamy R, Yan SD, Yan SF, D’Agati V, Schmidt AM. Blockade of receptor for advanced glycation endproducts: a new target for therapeutic intervention in diabetic complications and inflammatory disorders. Arch Biochem Biophys. 2003;419:80–88. doi: 10.1016/j.abb.2003.08.030. [DOI] [PubMed] [Google Scholar]
- Hudson BI, Carter AM, Harja E, Kalea AZ, Arriero M, Yang H, Grant PJ, Schmidt AM. Identification, classification, and expression of RAGE gene splice variants. FASEB J. 2008;22:1572–1580. doi: 10.1096/fj.07-9909com. [DOI] [PubMed] [Google Scholar]
- Huizinga TW, de Haas M, Kleijer M, Nuijens JH, Roos D, von dem Borne AE. Soluble Fc gamma receptor III in human plasma originates from release by neutrophils. J Clin Invest. 1990;86:416–423. doi: 10.1172/JCI114727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hundhausen C, Misztela D, Berkhout TA, Broadway N, Saftig P, Reiss K, Hartmann D, Fahrenholz F, Postina R, Matthews V, Kallen KJ, Rose-John S, Ludwig A. The disintegrin-like metalloproteinase ADAM10 is involved in constitutive cleavage of CX3CL1 (fractalkine) and regulates CX3CL1-mediated cell-cell adhesion. Blood. 2003;102:1186–1195. doi: 10.1182/blood-2002-12-3775. [DOI] [PubMed] [Google Scholar]
- Husemann J, Loike JD, Anankov R, Febbraio M, Silverstein SC. Scavenger receptors in neurobiology and neuropathology: their role on microglia and other cells of the nervous system. Glia. 2002;40:195–205. doi: 10.1002/glia.10148. [DOI] [PubMed] [Google Scholar]
- Imai F, Suzuki H, Oda J, Ninomiya T, Ono K, Sano H, Sawada M. Neuroprotective effect of exogenous microglia in global brain ischemia. Journal of cerebral blood flow and metabolism : official journal of the International Society of Cerebral Blood Flow and Metabolism. 2007;27:488–500. doi: 10.1038/sj.jcbfm.9600362. [DOI] [PubMed] [Google Scholar]
- Imai T, Hieshima K, Haskell C, Baba M, Nagira M, Nishimura M, Kakizaki M, Takagi S, Nomiyama H, Schall TJ, Yoshie O. Identification and molecular characterization of fractalkine receptor CX3CR1, which mediates both leukocyte migration and adhesion. Cell. 1997;91:521–530. doi: 10.1016/s0092-8674(00)80438-9. [DOI] [PubMed] [Google Scholar]
- Irino Y, Nakamura Y, Inoue K, Kohsaka S, Ohsawa K. Akt activation is involved in P2Y12 receptor-mediated chemotaxis of microglia. J Neurosci Res. 2008;86:1511–1519. doi: 10.1002/jnr.21610. [DOI] [PubMed] [Google Scholar]
- Ivashkiv LB. Cross-regulation of signaling by ITAM-associated receptors. Nat Immunol. 2009;10:340–347. doi: 10.1038/ni.1706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jakus Z, Simon E, Frommhold D, Sperandio M, Mocsai A. Critical role of phospholipase Cgamma2 in integrin and Fc receptor-mediated neutrophil functions and the effector phase of autoimmune arthritis. J Exp Med. 2009;206:577–593. doi: 10.1084/jem.20081859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- James G, Butt AM. P2Y and P2X purinoceptor mediated Ca2+ signalling in glial cell pathology in the central nervous system. Eur J Pharmacol. 2002;447:247–260. doi: 10.1016/s0014-2999(02)01756-9. [DOI] [PubMed] [Google Scholar]
- Jaremo P, Eriksson M, Lindahl TL, Nilsson S, Milovanovic M. Platelets and acute cerebral infarction. Platelets. 2012 doi: 10.3109/09537104.2012.712168. [DOI] [PubMed] [Google Scholar]
- Jeon SB, Yoon HJ, Park SH, Kim IH, Park EJ. Sulfatide, a major lipid component of myelin sheath, activates inflammatory responses as an endogenous stimulator in brain-resident immune cells. J Immunol. 2008;181:8077–8087. doi: 10.4049/jimmunol.181.11.8077. [DOI] [PubMed] [Google Scholar]
- Juranyi Z, Sperlagh B, Vizi ES. Involvement of P2 purinoceptors and the nitric oxide pathway in [3H]purine outflow evoked by short-term hypoxia and hypoglycemia in rat hippocampal slices. Brain Res. 1999;823:183–190. doi: 10.1016/s0006-8993(99)01169-5. [DOI] [PubMed] [Google Scholar]
- Kahlenberg JM, Dubyak GR. Mechanisms of caspase-1 activation by P2X7 receptor-mediated K+ release. Am J Physiol Cell Physiol. 2004;286:C1100–1108. doi: 10.1152/ajpcell.00494.2003. [DOI] [PubMed] [Google Scholar]
- Kataoka A, Tozaki-Saitoh H, Koga Y, Tsuda M, Inoue K. Activation of P2X7 receptors induces CCL3 production in microglial cells through transcription factor NFAT. J Neurochem. 2009;108:115–125. doi: 10.1111/j.1471-4159.2008.05744.x. [DOI] [PubMed] [Google Scholar]
- Kawai T, Akira S. TLR signaling. Seminars in immunology. 2007;19:24–32. doi: 10.1016/j.smim.2006.12.004. [DOI] [PubMed] [Google Scholar]
- Khazaei MR, Habibi-Rezaei M, Karimzadeh F, Moosavi-Movahedi AA, Sarrafnejhad AA, Sabouni F, Bakhti M. Microglial cell death induced by glycated bovine serum albumin: nitric oxide involvement. J Biochem. 2008;144:197–206. doi: 10.1093/jb/mvn059. [DOI] [PubMed] [Google Scholar]
- Kiialainen A, Hovanes K, Paloneva J, Kopra O, Peltonen L. Dap12 and Trem2, molecules involved in innate immunity and neurodegeneration, are co-expressed in the CNS. Neurobiol Dis. 2005;18:314–322. doi: 10.1016/j.nbd.2004.09.007. [DOI] [PubMed] [Google Scholar]
- Kilic U, Kilic E, Matter CM, Bassetti CL, Hermann DM. TLR-4 deficiency protects against focal cerebral ischemia and axotomy-induced neurodegeneration. Neurobiol Dis. 2008;31:33–40. doi: 10.1016/j.nbd.2008.03.002. [DOI] [PubMed] [Google Scholar]
- Kim B, Jeong HK, Kim JH, Lee SY, Jou I, Joe EH. Uridine 5′-diphosphate induces chemokine expression in microglia and astrocytes through activation of the P2Y6 receptor. J Immunol. 2011;186:3701–3709. doi: 10.4049/jimmunol.1000212. [DOI] [PubMed] [Google Scholar]
- Kim GH, Kellner CP, Hahn DK, Desantis BM, Musabbir M, Starke RM, Rynkowski M, Komotar RJ, Otten ML, Sciacca R, Schmidt JM, Mayer SA, Connolly ES., Jr Monocyte chemoattractant protein-1 predicts outcome and vasospasm following aneurysmal subarachnoid hemorrhage. J Neurosurg. 2008;109:38–43. doi: 10.3171/JNS/2008/109/7/0038. [DOI] [PubMed] [Google Scholar]
- Klesney-Tait J, Turnbull IR, Colonna M. The TREM receptor family and signal integration. Nat Immunol. 2006;7:1266–1273. doi: 10.1038/ni1411. [DOI] [PubMed] [Google Scholar]
- Kobayashi N, Karisola P, Pena-Cruz V, Dorfman DM, Jinushi M, Umetsu SE, Butte MJ, Nagumo H, Chernova I, Zhu B, Sharpe AH, Ito S, Dranoff G, Kaplan GG, Casasnovas JM, Umetsu DT, Dekruyff RH, Freeman GJ. TIM-1 and TIM-4 glycoproteins bind phosphatidylserine and mediate uptake of apoptotic cells. Immunity. 2007;27:927–940. doi: 10.1016/j.immuni.2007.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koh HS, Lee C, Lee KS, Ham CS, Seong RH, Kim SS, Jeon SH. CD7 expression and galectin-1-induced apoptosis of immature thymocytes are directly regulated by NF-kappaB upon T-cell activation. Biochemical and biophysical research communications. 2008;370:149–153. doi: 10.1016/j.bbrc.2008.03.049. [DOI] [PubMed] [Google Scholar]
- Koizumi S, Shigemoto-Mogami Y, Nasu-Tada K, Shinozaki Y, Ohsawa K, Tsuda M, Joshi BV, Jacobson KA, Kohsaka S, Inoue K. UDP acting at P2Y6 receptors is a mediator of microglial phagocytosis. Nature. 2007;446:1091–1095. doi: 10.1038/nature05704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koos BJ, Kruger L, Murray TF. Source of extracellular brain adenosine during hypoxia in fetal sheep. Brain Res. 1997;778:439–442. doi: 10.1016/s0006-8993(97)01207-9. [DOI] [PubMed] [Google Scholar]
- Krieger M. Charting the fate of the “good cholesterol”: identification and characterization of the high-density lipoprotein receptor SR-BI. Annual review of biochemistry. 1999;68:523–558. doi: 10.1146/annurev.biochem.68.1.523. [DOI] [PubMed] [Google Scholar]
- Kwon MJ, Kim J, Shin H, Jeong SR, Kang YM, Choi JY, Hwang DH, Kim BG. Contribution of macrophages to enhanced regenerative capacity of dorsal root ganglia sensory neurons by conditioning injury. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2013;33:15095–15108. doi: 10.1523/JNEUROSCI.0278-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lalancette-Hebert M, Gowing G, Simard A, Weng YC, Kriz J. Selective ablation of proliferating microglial cells exacerbates ischemic injury in the brain. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2007;27:2596–2605. doi: 10.1523/JNEUROSCI.5360-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lalancette-Hebert M, Swarup V, Beaulieu JM, Bohacek I, Abdelhamid E, Weng YC, Sato S, Kriz J. Galectin-3 is required for resident microglia activation and proliferation in response to ischemic injury. J Neurosci. 2012;32:10383–10395. doi: 10.1523/JNEUROSCI.1498-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Latini S, Corsi C, Pedata F, Pepeu G. The source of brain adenosine outflow during ischemia and electrical stimulation. Neurochem Int. 1996;28:113–118. doi: 10.1016/0197-0186(95)00062-d. [DOI] [PubMed] [Google Scholar]
- Latini S, Pedata F. Adenosine in the central nervous system: release mechanisms and extracellular concentrations. J Neurochem. 2001;79:463–484. doi: 10.1046/j.1471-4159.2001.00607.x. [DOI] [PubMed] [Google Scholar]
- Le W, Rowe D, Xie W, Ortiz I, He Y, Appel SH. Microglial activation and dopaminergic cell injury: an in vitro model relevant to Parkinson’s disease. J Neurosci. 2001;21:8447–8455. doi: 10.1523/JNEUROSCI.21-21-08447.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lech M, Avila-Ferrufino A, Skuginna V, Susanti HE, Anders HJ. Quantitative expression of RIG-like helicase, NOD-like receptor and inflammasome-related mRNAs in humans and mice. International immunology. 2010;22:717–728. doi: 10.1093/intimm/dxq058. [DOI] [PubMed] [Google Scholar]
- Lee JY, Jhun BS, Oh YT, Lee JH, Choe W, Baik HH, Ha J, Yoon KS, Kim SS, Kang I. Activation of adenosine A3 receptor suppresses lipopolysaccharide-induced TNF-alpha production through inhibition of PI 3-kinase/Akt and NF-kappaB activation in murine BV2 microglial cells. Neurosci Lett. 2006;396:1–6. doi: 10.1016/j.neulet.2005.11.004. [DOI] [PubMed] [Google Scholar]
- Lee S, Varvel NH, Konerth ME, Xu G, Cardona AE, Ransohoff RM, Lamb BT. CX3CR1 deficiency alters microglial activation and reduces beta-amyloid deposition in two Alzheimer’s disease mouse models. Am J Pathol. 2010;177:2549–2562. doi: 10.2353/ajpath.2010.100265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee SJ, Lee S. Toll-like receptors and inflammation in the CNS. Current drug targets Inflammation and allergy. 2002;1:181–191. doi: 10.2174/1568010023344698. [DOI] [PubMed] [Google Scholar]
- Lehnardt S, Lehmann S, Kaul D, Tschimmel K, Hoffmann O, Cho S, Krueger C, Nitsch R, Meisel A, Weber JR. Toll-like receptor 2 mediates CNS injury in focal cerebral ischemia. J Neuroimmunol. 2007;190:28–33. doi: 10.1016/j.jneuroim.2007.07.023. [DOI] [PubMed] [Google Scholar]
- Lendvai N, Qu XW, Hsueh W, Casadevall A. Mechanism for the isotype dependence of antibody-mediated toxicity in Cryptococcus neoformans-infected mice. J Immunol. 2000;164:4367–4374. doi: 10.4049/jimmunol.164.8.4367. [DOI] [PubMed] [Google Scholar]
- Leonardi-Essmann F, Emig M, Kitamura Y, Spanagel R, Gebicke-Haerter PJ. Fractalkine-upregulated milk-fat globule EGF factor-8 protein in cultured rat microglia. J Neuroimmunol. 2005;160:92–101. doi: 10.1016/j.jneuroim.2004.11.012. [DOI] [PubMed] [Google Scholar]
- Li E, Noda M, Doi Y, Parajuli B, Kawanokuchi J, Sonobe Y, Takeuchi H, Mizuno T, Suzumura A. The neuroprotective effects of milk fat globule-EGF factor 8 against oligomeric amyloid beta toxicity. J Neuroinflammation. 2012;9:148. doi: 10.1186/1742-2094-9-148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li F, Wang L, Li JW, Gong M, He L, Feng R, Dai Z, Li SQ. Hypoxia induced amoeboid microglial cell activation in postnatal rat brain is mediated by ATP receptor P2X4. BMC Neurosci. 2011;12:111. doi: 10.1186/1471-2202-12-111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Gang Z, Yuling H, Luokun X, Jie X, Hao L, Li W, Chunsong H, Junyan L, Mingshen J, Youxin J, Feili G, Boquan J, Jinquan T. Different neurotropic pathogens elicit neurotoxic CCR9- or neurosupportive CXCR3-expressing microglia. J Immunol. 2006;177:3644–3656. doi: 10.4049/jimmunol.177.6.3644. [DOI] [PubMed] [Google Scholar]
- Limatola C, Giovannelli A, Maggi L, Ragozzino D, Castellani L, Ciotti MT, Vacca F, Mercanti D, Santoni A, Eusebi F. SDF-1alpha-mediated modulation of synaptic transmission in rat cerebellum. Eur J Neurosci. 2000;12:2497–2504. doi: 10.1046/j.1460-9568.2000.00139.x. [DOI] [PubMed] [Google Scholar]
- Linnartz B, Neumann H. Microglial activatory (immunoreceptor tyrosine-based activation motif)- and inhibitory (immunoreceptor tyrosine-based inhibition motif)-signaling receptors for recognition of the neuronal glycocalyx. Glia. 2013;61:37–46. doi: 10.1002/glia.22359. [DOI] [PubMed] [Google Scholar]
- Liu FT. Galectins: novel anti-inflammatory drug targets. Expert opinion on therapeutic targets. 2002;6:461–468. doi: 10.1517/14728222.6.4.461. [DOI] [PubMed] [Google Scholar]
- Liu Z, Condello C, Schain A, Harb R, Grutzendler J. CX3CR1 in microglia regulates brain amyloid deposition through selective protofibrillar amyloid-beta phagocytosis. J Neurosci. 2010;30:17091–17101. doi: 10.1523/JNEUROSCI.4403-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Z, Fan Y, Won SJ, Neumann M, Hu D, Zhou L, Weinstein PR, Liu J. Chronic treatment with minocycline preserves adult new neurons and reduces functional impairment after focal cerebral ischemia. Stroke; a journal of cerebral circulation. 2007;38:146–152. doi: 10.1161/01.STR.0000251791.64910.cd. [DOI] [PubMed] [Google Scholar]
- Lu DY, Tang CH, Yeh WL, Wong KL, Lin CP, Chen YH, Lai CH, Chen YF, Leung YM, Fu WM. SDF-1alpha up-regulates interleukin-6 through CXCR4, PI3K/Akt, ERK, and NF-kappaB-dependent pathway in microglia. Eur J Pharmacol. 2009;613:146–154. doi: 10.1016/j.ejphar.2009.03.001. [DOI] [PubMed] [Google Scholar]
- Lue LF, Walker DG. Modeling Alzheimer’s disease immune therapy mechanisms: interactions of human postmortem microglia with antibody-opsonized amyloid beta peptide. J Neurosci Res. 2002;70:599–610. doi: 10.1002/jnr.10422. [DOI] [PubMed] [Google Scholar]
- Lue LF, Walker DG, Brachova L, Beach TG, Rogers J, Schmidt AM, Stern DM, Yan SD. Involvement of microglial receptor for advanced glycation endproducts (RAGE) in Alzheimer’s disease: identification of a cellular activation mechanism. Exp Neurol. 2001;171:29–45. doi: 10.1006/exnr.2001.7732. [DOI] [PubMed] [Google Scholar]
- Lyons A, Downer EJ, Crotty S, Nolan YM, Mills KH, Lynch MA. CD200 ligand receptor interaction modulates microglial activation in vivo and in vitro: a role for IL-4. J Neurosci. 2007;27:8309–8313. doi: 10.1523/JNEUROSCI.1781-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyons A, Lynch AM, Downer EJ, Hanley R, O’Sullivan JB, Smith A, Lynch MA. Fractalkine-induced activation of the phosphatidylinositol-3 kinase pathway attentuates microglial activation in vivo and in vitro. J Neurochem. 2009;110:1547–1556. doi: 10.1111/j.1471-4159.2009.06253.x. [DOI] [PubMed] [Google Scholar]
- Ma XJ, Cheng JW, Zhang J, Liu AJ, Liu W, Guo W, Shen FM, Lu GC. E-selectin deficiency attenuates brain ischemia in mice. CNS neuroscience & therapeutics. 2012;18:903–908. doi: 10.1111/cns.12000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marella M, Chabry J. Neurons and astrocytes respond to prion infection by inducing microglia recruitment. J Neurosci. 2004;24:620–627. doi: 10.1523/JNEUROSCI.4303-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsumoto H, Kumon Y, Watanabe H, Ohnishi T, Takahashi H, Imai Y, Tanaka J. Expression of CD200 by macrophage-like cells in ischemic core of rat brain after transient middle cerebral artery occlusion. Neurosci Lett. 2007;418:44–48. doi: 10.1016/j.neulet.2007.03.027. [DOI] [PubMed] [Google Scholar]
- Merino JJ, Gutierrez-Fernandez M, Rodriguez-Frutos B, Alvarez-Grech J, Alcalde ME, Vallejo-Cremades MT, Diez-Tejedor E. CXCR4/SDF-1alpha-chemokine regulates neurogenesis and/or angiogenesis within the vascular niche of ischemic rats; however, does SDF-1alpha play a role in repair? Int J Stroke. 2011;6:466–467. doi: 10.1111/j.1747-4949.2011.00651.x. [DOI] [PubMed] [Google Scholar]
- Meucci O, Fatatis A, Simen AA, Bushell TJ, Gray PW, Miller RJ. Chemokines regulate hippocampal neuronal signaling and gp120 neurotoxicity. Proc Natl Acad Sci U S A. 1998;95:14500–14505. doi: 10.1073/pnas.95.24.14500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mihrshahi R, Barclay AN, Brown MH. Essential roles for Dok2 and RasGAP in CD200 receptor-mediated regulation of human myeloid cells. J Immunol. 2009;183:4879–4886. doi: 10.4049/jimmunol.0901531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mihrshahi R, Brown MH. Downstream of tyrosine kinase 1 and 2 play opposing roles in CD200 receptor signaling. J Immunol. 2010;185:7216–7222. doi: 10.4049/jimmunol.1002858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mildner A, Schlevogt B, Kierdorf K, Bottcher C, Erny D, Kummer MP, Quinn M, Bruck W, Bechmann I, Heneka MT, Priller J, Prinz M. Distinct and non-redundant roles of microglia and myeloid subsets in mouse models of Alzheimer’s disease. J Neurosci. 2011;31:11159–11171. doi: 10.1523/JNEUROSCI.6209-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mildner A, Schmidt H, Nitsche M, Merkler D, Hanisch UK, Mack M, Heikenwalder M, Bruck W, Priller J, Prinz M. Microglia in the adult brain arise from Ly-6ChiCCR2+ monocytes only under defined host conditions. Nat Neurosci. 2007;10:1544–1553. doi: 10.1038/nn2015. [DOI] [PubMed] [Google Scholar]
- Milligan ED, Zapata V, Chacur M, Schoeniger D, Biedenkapp J, O’Connor KA, Verge GM, Chapman G, Green P, Foster AC, Naeve GS, Maier SF, Watkins LR. Evidence that exogenous and endogenous fractalkine can induce spinal nociceptive facilitation in rats. Eur J Neurosci. 2004;20:2294–2302. doi: 10.1111/j.1460-9568.2004.03709.x. [DOI] [PubMed] [Google Scholar]
- Minami SS, Sun B, Popat K, Kauppinen T, Pleiss M, Zhou Y, Ward ME, Floreancig P, Mucke L, Desai T, Gan L. Selective targeting of microglia by quantum dots. J Neuroinflammation. 2012;9:22. doi: 10.1186/1742-2094-9-22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miron VE, Boyd A, Zhao JW, Yuen TJ, Ruckh JM, Shadrach JL, van Wijngaarden P, Wagers AJ, Williams A, Franklin RJ, ffrench-Constant C. M2 microglia and macrophages drive oligodendrocyte differentiation during CNS remyelination. Nature neuroscience. 2013;16:1211–1218. doi: 10.1038/nn.3469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyanishi M, Tada K, Koike M, Uchiyama Y, Kitamura T, Nagata S. Identification of Tim4 as a phosphatidylserine receptor. Nature. 2007;450:435–439. doi: 10.1038/nature06307. [DOI] [PubMed] [Google Scholar]
- Monif M, Reid CA, Powell KL, Smart ML, Williams DA. The P2X7 receptor drives microglial activation and proliferation: a trophic role for P2X7R pore. J Neurosci. 2009;29:3781–3791. doi: 10.1523/JNEUROSCI.5512-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore UM, Kaplow JM, Pleass RD, Castro SW, Naik K, Lynch CN, Daly S, Roach AG, Jaye M, Williams RJ. Monocyte chemoattractant protein-2 is a potent agonist of CCR2B. J Leukoc Biol. 1997;62:911–915. doi: 10.1002/jlb.62.6.911. [DOI] [PubMed] [Google Scholar]
- Muessel MJ, Klein RM, Wilson AM, Berman NE. Ablation of the chemokine monocyte chemoattractant protein-1 delays retrograde neuronal degeneration, attenuates microglial activation, and alters expression of cell death molecules. Brain Res Mol Brain Res. 2002;103:12–27. doi: 10.1016/s0169-328x(02)00158-4. [DOI] [PubMed] [Google Scholar]
- Muhammad S, Barakat W, Stoyanov S, Murikinati S, Yang H, Tracey KJ, Bendszus M, Rossetti G, Nawroth PP, Bierhaus A, Schwaninger M. The HMGB1 receptor RAGE mediates ischemic brain damage. J Neurosci. 2008;28:12023–12031. doi: 10.1523/JNEUROSCI.2435-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myers SJ, Wong LM, Charo IF. Signal transduction and ligand specificity of the human monocyte chemoattractant protein-1 receptor in transfected embryonic kidney cells. J Biol Chem. 1995;270:5786–5792. doi: 10.1074/jbc.270.11.5786. [DOI] [PubMed] [Google Scholar]
- N’Diaye EN, Branda CS, Branda SS, Nevarez L, Colonna M, Lowell C, Hamerman JA, Seaman WE. TREM-2 (triggering receptor expressed on myeloid cells 2) is a phagocytic receptor for bacteria. J Cell Biol. 2009;184:215–223. doi: 10.1083/jcb.200808080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naert G, Rivest S. Hematopoietic CC-chemokine receptor 2 (CCR2) competent cells are protective for the cognitive impairments and amyloid pathology in a transgenic mouse model of Alzheimer’s disease. Mol Med. 2012;18:297–313. doi: 10.2119/molmed.2011.00306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nimmerjahn F, Ravetch JV. Fcgamma receptors as regulators of immune responses. Nat Rev Immunol. 2008;8:34–47. doi: 10.1038/nri2206. [DOI] [PubMed] [Google Scholar]
- Nishiyori A, Minami M, Ohtani Y, Takami S, Yamamoto J, Kawaguchi N, Kume T, Akaike A, Satoh M. Localization of fractalkine and CX3CR1 mRNAs in rat brain: does fractalkine play a role in signaling from neuron to microglia? FEBS Lett. 1998;429:167–172. doi: 10.1016/s0014-5793(98)00583-3. [DOI] [PubMed] [Google Scholar]
- Noda M, Doi Y, Liang J, Kawanokuchi J, Sonobe Y, Takeuchi H, Mizuno T, Suzumura A. Fractalkine attenuates excito-neurotoxicity via microglial clearance of damaged neurons and antioxidant enzyme heme oxygenase-1 expression. J Biol Chem. 2011;286:2308–2319. doi: 10.1074/jbc.M110.169839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nomiyama H, Mera A, Ohneda O, Miura R, Suda T, Yoshie O. Organization of the chemokine genes in the human and mouse major clusters of CC and CXC chemokines: diversification between the two species. Genes Immun. 2001;2:110–113. doi: 10.1038/sj.gene.6363742. [DOI] [PubMed] [Google Scholar]
- Ohsawa K, Irino Y, Nakamura Y, Akazawa C, Inoue K, Kohsaka S. Involvement of P2X4 and P2Y12 receptors in ATP-induced microglial chemotaxis. Glia. 2007;55:604–616. doi: 10.1002/glia.20489. [DOI] [PubMed] [Google Scholar]
- Ohsawa K, Irino Y, Sanagi T, Nakamura Y, Suzuki E, Inoue K, Kohsaka S. P2Y12 receptor-mediated integrin-beta1 activation regulates microglial process extension induced by ATP. Glia. 2010;58:790–801. doi: 10.1002/glia.20963. [DOI] [PubMed] [Google Scholar]
- Ohsawa K, Sanagi T, Nakamura Y, Suzuki E, Inoue K, Kohsaka S. Adenosine A3 receptor is involved in ADP-induced microglial process extension and migration. J Neurochem. 2012;121:217–227. doi: 10.1111/j.1471-4159.2012.07693.x. [DOI] [PubMed] [Google Scholar]
- Ohtani Y, Minami M, Kawaguchi N, Nishiyori A, Yamamoto J, Takami S, Satoh M. Expression of stromal cell-derived factor-1 and CXCR4 chemokine receptor mRNAs in cultured rat glial and neuronal cells. Neurosci Lett. 1998;249:163–166. doi: 10.1016/s0304-3940(98)00425-x. [DOI] [PubMed] [Google Scholar]
- Oldenborg PA, Gresham HD, Lindberg FP. CD47-signal regulatory protein alpha (SIRPalpha) regulates Fcgamma and complement receptor-mediated phagocytosis. J Exp Med. 2001;193:855–862. doi: 10.1084/jem.193.7.855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olson JK, Miller SD. Microglia initiate central nervous system innate and adaptive immune responses through multiple TLRs. J Immunol. 2004;173:3916–3924. doi: 10.4049/jimmunol.173.6.3916. [DOI] [PubMed] [Google Scholar]
- Origlia N, Bonadonna C, Rosellini A, Leznik E, Arancio O, Yan SS, Domenici L. Microglial receptor for advanced glycation end product-dependent signal pathway drives beta-amyloid-induced synaptic depression and long-term depression impairment in entorhinal cortex. J Neurosci. 2010;30:11414–11425. doi: 10.1523/JNEUROSCI.2127-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orr AG, Orr AL, Li XJ, Gross RE, Traynelis SF. Adenosine A(2A) receptor mediates microglial process retraction. Nat Neurosci. 2009;12:872–878. doi: 10.1038/nn.2341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ortiz-Stern A, Rosales C. Cross-talk between Fc receptors and integrins. Immunol Lett. 2003;90:137–143. doi: 10.1016/j.imlet.2003.08.004. [DOI] [PubMed] [Google Scholar]
- Pabon MM, Bachstetter AD, Hudson CE, Gemma C, Bickford PC. CX3CL1 reduces neurotoxicity and microglial activation in a rat model of Parkinson’s disease. J Neuroinflammation. 2011;8:9. doi: 10.1186/1742-2094-8-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paloneva J, Manninen T, Christman G, Hovanes K, Mandelin J, Adolfsson R, Bianchin M, Bird T, Miranda R, Salmaggi A, Tranebjaerg L, Konttinen Y, Peltonen L. Mutations in two genes encoding different subunits of a receptor signaling complex result in an identical disease phenotype. Am J Hum Genet. 2002;71:656–662. doi: 10.1086/342259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park D, Tosello-Trampont AC, Elliott MR, Lu M, Haney LB, Ma Z, Klibanov AL, Mandell JW, Ravichandran KS. BAI1 is an engulfment receptor for apoptotic cells upstream of the ELMO/Dock180/Rac module. Nature. 2007;450:430–434. doi: 10.1038/nature06329. [DOI] [PubMed] [Google Scholar]
- Park JG, Schreiber AD. Determinants of the phagocytic signal mediated by the type IIIA Fc gamma receptor, Fc gamma RIIIA: sequence requirements and interaction with protein-tyrosine kinases. Proc Natl Acad Sci U S A. 1995;92:7381–7385. doi: 10.1073/pnas.92.16.7381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park SY, Jung MY, Kim HJ, Lee SJ, Kim SY, Lee BH, Kwon TH, Park RW, Kim IS. Rapid cell corpse clearance by stabilin-2, a membrane phosphatidylserine receptor. Cell Death Differ. 2008;15:192–201. doi: 10.1038/sj.cdd.4402242. [DOI] [PubMed] [Google Scholar]
- Parvathenani LK, Tertyshnikova S, Greco CR, Roberts SB, Robertson B, Posmantur R. P2X7 mediates superoxide production in primary microglia and is up-regulated in a transgenic mouse model of Alzheimer’s disease. J Biol Chem. 2003;278:13309–13317. doi: 10.1074/jbc.M209478200. [DOI] [PubMed] [Google Scholar]
- Patel JR, McCandless EE, Dorsey D, Klein RS. CXCR4 promotes differentiation of oligodendrocyte progenitors and remyelination. Proc Natl Acad Sci U S A. 2010;107:11062–11067. doi: 10.1073/pnas.1006301107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng Q, Malhotra S, Torchia JA, Kerr WG, Coggeshall KM, Humphrey MB. TREM2- and DAP12-dependent activation of PI3K requires DAP10 and is inhibited by SHIP1. Sci Signal. 2010;3:ra38. doi: 10.1126/scisignal.2000500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pertwee RG. Emerging strategies for exploiting cannabinoid receptor agonists as medicines. Br J Pharmacol. 2009;156:397–411. doi: 10.1111/j.1476-5381.2008.00048.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piccio L, Buonsanti C, Mariani M, Cella M, Gilfillan S, Cross AH, Colonna M, Panina-Bordignon P. Blockade of TREM-2 exacerbates experimental autoimmune encephalomyelitis. Eur J Immunol. 2007;37:1290–1301. doi: 10.1002/eji.200636837. [DOI] [PubMed] [Google Scholar]
- Pineda D, Ampurdanes C, Medina MG, Serratosa J, Tusell JM, Saura J, Planas AM, Navarro P. Tissue plasminogen activator induces microglial inflammation via a noncatalytic molecular mechanism involving activation of mitogen-activated protein kinases and Akt signaling pathways and AnnexinA2 and Galectin-1 receptors. Glia. 2012;60:526–540. doi: 10.1002/glia.22284. [DOI] [PubMed] [Google Scholar]
- Prada I, Ongania GN, Buonsanti C, Panina-Bordignon P, Meldolesi J. Triggering receptor expressed in myeloid cells 2 (TREM2) trafficking in microglial cells: continuous shuttling to and from the plasma membrane regulated by cell stimulation. Neuroscience. 2006;140:1139–1148. doi: 10.1016/j.neuroscience.2006.03.058. [DOI] [PubMed] [Google Scholar]
- Pradillo JM, Fernandez-Lopez D, Garcia-Yebenes I, Sobrado M, Hurtado O, Moro MA, Lizasoain I. Toll-like receptor 4 is involved in neuroprotection afforded by ischemic preconditioning. Journal of neurochemistry. 2009;109:287–294. doi: 10.1111/j.1471-4159.2009.05972.x. [DOI] [PubMed] [Google Scholar]
- Preston S, Wright GJ, Starr K, Barclay AN, Brown MH. The leukocyte/neuron cell surface antigen OX2 binds to a ligand on macrophages. Eur J Immunol. 1997;27:1911–1918. doi: 10.1002/eji.1830270814. [DOI] [PubMed] [Google Scholar]
- Puente Navazo MD, Daviet L, Ninio E, McGregor JL. Identification on human CD36 of a domain (155-183) implicated in binding oxidized low-density lipoproteins (Ox-LDL) Arteriosclerosis, thrombosis, and vascular biology. 1996;16:1033–1039. doi: 10.1161/01.atv.16.8.1033. [DOI] [PubMed] [Google Scholar]
- Qu WS, Wang YH, Wang JP, Tang YX, Zhang Q, Tian DS, Yu ZY, Xie MJ, Wang W. Galectin-1 enhances astrocytic BDNF production and improves functional outcome in rats following ischemia. Neurochemical research. 2010;35:1716–1724. doi: 10.1007/s11064-010-0234-z. [DOI] [PubMed] [Google Scholar]
- Rabinovich GA, Toscano MA. Turning ‘sweet’ on immunity: galectin-glycan interactions in immune tolerance and inflammation. Nat Rev Immunol. 2009;9:338–352. doi: 10.1038/nri2536. [DOI] [PubMed] [Google Scholar]
- Raouf R, Chabot-Dore AJ, Ase AR, Blais D, Seguela P. Differential regulation of microglial P2X4 and P2X7 ATP receptors following LPS-induced activation. Neuropharmacology. 2007;53:496–504. doi: 10.1016/j.neuropharm.2007.06.010. [DOI] [PubMed] [Google Scholar]
- Rappert A, Bechmann I, Pivneva T, Mahlo J, Biber K, Nolte C, Kovac AD, Gerard C, Boddeke HW, Nitsch R, Kettenmann H. CXCR3-dependent microglial recruitment is essential for dendrite loss after brain lesion. J Neurosci. 2004;24:8500–8509. doi: 10.1523/JNEUROSCI.2451-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rappert A, Biber K, Nolte C, Lipp M, Schubel A, Lu B, Gerard NP, Gerard C, Boddeke HW, Kettenmann H. Secondary lymphoid tissue chemokine (CCL21) activates CXCR3 to trigger a Cl-current and chemotaxis in murine microglia. J Immunol. 2002;168:3221–3226. doi: 10.4049/jimmunol.168.7.3221. [DOI] [PubMed] [Google Scholar]
- Ravichandran KS. Beginnings of a good apoptotic meal: the find-me and eat-me signaling pathways. Immunity. 2011;35:445–455. doi: 10.1016/j.immuni.2011.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reed-Geaghan EG, Savage JC, Hise AG, Landreth GE. CD14 and toll-like receptors 2 and 4 are required for fibrillar A{beta}-stimulated microglial activation. J Neurosci. 2009;29:11982–11992. doi: 10.1523/JNEUROSCI.3158-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren Y, Savill J. Proinflammatory cytokines potentiate thrombospondin-mediated phagocytosis of neutrophils undergoing apoptosis. J Immunol. 1995;154:2366–2374. [PubMed] [Google Scholar]
- Rogers JT, Morganti JM, Bachstetter AD, Hudson CE, Peters MM, Grimmig BA, Weeber EJ, Bickford PC, Gemma C. CX3CR1 deficiency leads to impairment of hippocampal cognitive function and synaptic plasticity. J Neurosci. 2011;31:16241–16250. doi: 10.1523/JNEUROSCI.3667-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenzweig HL, Planck SR, Rosenbaum JT. NLRs in immune privileged sites. Current opinion in pharmacology. 2011;11:423–428. doi: 10.1016/j.coph.2011.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rotshenker S. Microglia and macrophage activation and the regulation of complement-receptor-3 (CR3/MAC-1)-mediated myelin phagocytosis in injury and disease. J Mol Neurosci. 2003;21:65–72. doi: 10.1385/JMN:21:1:65. [DOI] [PubMed] [Google Scholar]
- Rottman JB, Ganley KP, Williams K, Wu L, Mackay CR, Ringler DJ. Cellular localization of the chemokine receptor CCR5. Correlation to cellular targets of HIV-1 infection. Am J Pathol. 1997;151:1341–1351. [PMC free article] [PubMed] [Google Scholar]
- Saini V, Marchese A, Majetschak M. CXC chemokine receptor 4 is a cell surface receptor for extracellular ubiquitin. J Biol Chem. 2010;285:15566–15576. doi: 10.1074/jbc.M110.103408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saitoh S, Arudchandran R, Manetz TS, Zhang W, Sommers CL, Love PE, Rivera J, Samelson LE. LAT is essential for Fc(epsilon)RI-mediated mast cell activation. Immunity. 2000;12:525–535. doi: 10.1016/s1074-7613(00)80204-6. [DOI] [PubMed] [Google Scholar]
- Samson M, Libert F, Doranz BJ, Rucker J, Liesnard C, Farber CM, Saragosti S, Lapoumeroulie C, Cognaux J, Forceille C, Muyldermans G, Verhofstede C, Burtonboy G, Georges M, Imai T, Rana S, Yi Y, Smyth RJ, Collman RG, Doms RW, Vassart G, Parmentier M. Resistance to HIV-1 infection in caucasian individuals bearing mutant alleles of the CCR-5 chemokine receptor gene. Nature. 1996;382:722–725. doi: 10.1038/382722a0. [DOI] [PubMed] [Google Scholar]
- Sanz JM, Di Virgilio F. Kinetics and mechanism of ATP-dependent IL-1 beta release from microglial cells. J Immunol. 2000;164:4893–4898. doi: 10.4049/jimmunol.164.9.4893. [DOI] [PubMed] [Google Scholar]
- Sasaki Y, Hoshi M, Akazawa C, Nakamura Y, Tsuzuki H, Inoue K, Kohsaka S. Selective expression of Gi/o-coupled ATP receptor P2Y12 in microglia in rat brain. Glia. 2003;44:242–250. doi: 10.1002/glia.10293. [DOI] [PubMed] [Google Scholar]
- Satoh K, Niwa M, Goda W, Binh NH, Nakashima M, Takamatsu M, Hara A. Galectin-3 expression in delayed neuronal death of hippocampal CA1 following transient forebrain ischemia, and its inhibition by hypothermia. Brain research. 2011;1382:266–274. doi: 10.1016/j.brainres.2011.01.049. [DOI] [PubMed] [Google Scholar]
- Saura J, Angulo E, Ejarque A, Casado V, Tusell JM, Moratalla R, Chen JF, Schwarzschild MA, Lluis C, Franco R, Serratosa J. Adenosine A2A receptor stimulation potentiates nitric oxide release by activated microglia. J Neurochem. 2005;95:919–929. doi: 10.1111/j.1471-4159.2005.03395.x. [DOI] [PubMed] [Google Scholar]
- Savarin-Vuaillat C, Ransohoff RM. Chemokines and chemokine receptors in neurological disease: raise, retain, or reduce? Neurotherapeutics. 2007;4:590–601. doi: 10.1016/j.nurt.2007.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savill J, Hogg N, Ren Y, Haslett C. Thrombospondin cooperates with CD36 and the vitronectin receptor in macrophage recognition of neutrophils undergoing apoptosis. The Journal of clinical investigation. 1992;90:1513–1522. doi: 10.1172/JCI116019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schilling M, Strecker JK, Ringelstein EB, Schabitz WR, Kiefer R. The role of CC chemokine receptor 2 on microglia activation and blood-borne cell recruitment after transient focal cerebral ischemia in mice. Brain Res. 2009;1289:79–84. doi: 10.1016/j.brainres.2009.06.054. [DOI] [PubMed] [Google Scholar]
- Schmid CD, Sautkulis LN, Danielson PE, Cooper J, Hasel KW, Hilbush BS, Sutcliffe JG, Carson MJ. Heterogeneous expression of the triggering receptor expressed on myeloid cells-2 on adult murine microglia. J Neurochem. 2002;83:1309–1320. doi: 10.1046/j.1471-4159.2002.01243.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwab JM, Guo L, Schluesener HJ. Spinal cord injury induces early and persistent lesional P2X4 receptor expression. J Neuroimmunol. 2005;163:185–189. doi: 10.1016/j.jneuroim.2005.02.016. [DOI] [PubMed] [Google Scholar]
- Schwab ME, Bartholdi D. Degeneration and regeneration of axons in the lesioned spinal cord. Physiological reviews. 1996;76:319–370. doi: 10.1152/physrev.1996.76.2.319. [DOI] [PubMed] [Google Scholar]
- Serbina NV, Pamer EG. Monocyte emigration from bone marrow during bacterial infection requires signals mediated by chemokine receptor CCR2. Nat Immunol. 2006;7:311–317. doi: 10.1038/ni1309. [DOI] [PubMed] [Google Scholar]
- Sessa G, Podini P, Mariani M, Meroni A, Spreafico R, Sinigaglia F, Colonna M, Panina P, Meldolesi J. Distribution and signaling of TREM2/DAP12, the receptor system mutated in human polycystic lipomembraneous osteodysplasia with sclerosing leukoencephalopathy dementia. Eur J Neurosci. 2004;20:2617–2628. doi: 10.1111/j.1460-9568.2004.03729.x. [DOI] [PubMed] [Google Scholar]
- Shahabuddin S, Ji R, Wang P, Brailoiu E, Dun N, Yang Y, Aksoy MO, Kelsen SG. CXCR3 chemokine receptor-induced chemotaxis in human airway epithelial cells: role of p38 MAPK and PI3K signaling pathways. Am J Physiol Cell Physiol. 2006;291:C34–39. doi: 10.1152/ajpcell.00441.2005. [DOI] [PubMed] [Google Scholar]
- Shaikh SB, Uy B, Perera A, Nicholson LF. AGEs-RAGE mediated up-regulation of connexin43 in activated human microglial CHME-5 cells. Neurochem Int. 2012;60:640–651. doi: 10.1016/j.neuint.2012.02.023. [DOI] [PubMed] [Google Scholar]
- Sheehan JJ, Zhou C, Gravanis I, Rogove AD, Wu YP, Bogenhagen DF, Tsirka SE. Proteolytic activation of monocyte chemoattractant protein-1 by plasmin underlies excitotoxic neurodegeneration in mice. J Neurosci. 2007;27:1738–1745. doi: 10.1523/JNEUROSCI.4987-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shideman CR, Hu S, Peterson PK, Thayer SA. CCL5 evokes calcium signals in microglia through a kinase-, phosphoinositide-, and nucleotide-dependent mechanism. J Neurosci Res. 2006;83:1471–1484. doi: 10.1002/jnr.20839. [DOI] [PubMed] [Google Scholar]
- Shieh JT, Albright AV, Sharron M, Gartner S, Strizki J, Doms RW, Gonzalez-Scarano F. Chemokine receptor utilization by human immunodeficiency virus type 1 isolates that replicate in microglia. J Virol. 1998;72:4243–4249. doi: 10.1128/jvi.72.5.4243-4249.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shiratori M, Tozaki-Saitoh H, Yoshitake M, Tsuda M, Inoue K. P2X7 receptor activation induces CXCL2 production in microglia through NFAT and PKC/MAPK pathways. J Neurochem. 2010;114:810–819. doi: 10.1111/j.1471-4159.2010.06809.x. [DOI] [PubMed] [Google Scholar]
- Sieger D, Peri F. Animal models for studying microglia: The first, the popular, and the new. Glia. 2013;61:3–9. doi: 10.1002/glia.22385. [DOI] [PubMed] [Google Scholar]
- Sivakumar V, Foulds WS, Luu CD, Ling EA, Kaur C. Retinal ganglion cell death is induced by microglia derived pro-inflammatory cytokines in the hypoxic neonatal retina. J Pathol. 2011;224:245–260. doi: 10.1002/path.2858. [DOI] [PubMed] [Google Scholar]
- Skuljec J, Sun H, Pul R, Benardais K, Ragancokova D, Moharregh-Khiabani D, Kotsiari A, Trebst C, Stangel M. CCL5 induces a pro-inflammatory profile in microglia in vitro. Cell Immunol. 2011;270:164–171. doi: 10.1016/j.cellimm.2011.05.001. [DOI] [PubMed] [Google Scholar]
- Smit MJ, Verdijk P, van der Raaij-Helmer EM, Navis M, Hensbergen PJ, Leurs R, Tensen CP. CXCR3-mediated chemotaxis of human T cells is regulated by a Gi- and phospholipase C-dependent pathway and not via activation of MEK/p44/p42 MAPK nor Akt/PI-3 kinase. Blood. 2003;102:1959–1965. doi: 10.1182/blood-2002-12-3945. [DOI] [PubMed] [Google Scholar]
- Song X, Shapiro S, Goldman DL, Casadevall A, Scharff M, Lee SC. Fcgamma receptor I- and III-mediated macrophage inflammatory protein 1alpha induction in primary human and murine microglia. Infect Immun. 2002;70:5177–5184. doi: 10.1128/IAI.70.9.5177-5184.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song X, Tanaka S, Cox D, Lee SC. Fcgamma receptor signaling in primary human microglia: differential roles of PI-3K and Ras/ERK MAPK pathways in phagocytosis and chemokine induction. J Leukoc Biol. 2004;75:1147–1155. doi: 10.1189/jlb.0403128. [DOI] [PubMed] [Google Scholar]
- Sparvero LJ, Asafu-Adjei D, Kang R, Tang D, Amin N, Im J, Rutledge R, Lin B, Amoscato AA, Zeh HJ, Lotze MT. RAGE (Receptor for Advanced Glycation Endproducts), RAGE ligands, and their role in cancer and inflammation. J Transl Med. 2009;7:17. doi: 10.1186/1479-5876-7-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Starossom SC, Mascanfroni ID, Imitola J, Cao L, Raddassi K, Hernandez SF, Bassil R, Croci DO, Cerliani JP, Delacour D, Wang Y, Elyaman W, Khoury SJ, Rabinovich GA. Galectin-1 deactivates classically activated microglia and protects from inflammation-induced neurodegeneration. Immunity. 2012;37:249–263. doi: 10.1016/j.immuni.2012.05.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stefano L, Racchetti G, Bianco F, Passini N, Gupta RS, Panina Bordignon P, Meldolesi J. The surface-exposed chaperone, Hsp60, is an agonist of the microglial TREM2 receptor. J Neurochem. 2009;110:284–294. doi: 10.1111/j.1471-4159.2009.06130.x. [DOI] [PubMed] [Google Scholar]
- Sugimoto K, Nishioka R, Ikeda A, Mise A, Takahashi H, Yano H, Kumon Y, Ohnishi T, Tanaka J. Activated microglia in a rat stroke model express NG2 proteoglycan in peri-infarct tissue through the involvement of TGF-beta1. Glia. 2014;62:185–198. doi: 10.1002/glia.22598. [DOI] [PubMed] [Google Scholar]
- Suzuki J, Umeda M, Sims PJ, Nagata S. Calcium-dependent phospholipid scrambling by TMEM16F. Nature. 2010;468:834–838. doi: 10.1038/nature09583. [DOI] [PubMed] [Google Scholar]
- Syam S, Mero P, Pham T, McIntosh CA, Bruhns P, Booth JW. Differential recruitment of activating and inhibitory Fc gamma RII during phagocytosis. J Immunol. 2010;184:2966–2973. doi: 10.4049/jimmunol.0900016. [DOI] [PubMed] [Google Scholar]
- Takahashi K, Rochford CD, Neumann H. Clearance of apoptotic neurons without inflammation by microglial triggering receptor expressed on myeloid cells-2. J Exp Med. 2005;201:647–657. doi: 10.1084/jem.20041611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takegahara N, Takamatsu H, Toyofuku T, Tsujimura T, Okuno T, Yukawa K, Mizui M, Yamamoto M, Prasad DV, Suzuki K, Ishii M, Terai K, Moriya M, Nakatsuji Y, Sakoda S, Sato S, Akira S, Takeda K, Inui M, Takai T, Ikawa M, Okabe M, Kumanogoh A, Kikutani H. Plexin-A1 and its interaction with DAP12 in immune responses and bone homeostasis. Nat Cell Biol. 2006;8:615–622. doi: 10.1038/ncb1416. [DOI] [PubMed] [Google Scholar]
- Takenouchi T, Iwamaru Y, Sugama S, Sato M, Hashimoto M, Kitani H. Lysophospholipids and ATP mutually suppress maturation and release of IL-1 beta in mouse microglial cells using a Rho-dependent pathway. J Immunol. 2008;180:7827–7839. doi: 10.4049/jimmunol.180.12.7827. [DOI] [PubMed] [Google Scholar]
- Takenouchi T, Sato M, Kitani H. Lysophosphatidylcholine potentiates Ca2+ influx, pore formation and p44/42 MAP kinase phosphorylation mediated by P2X7 receptor activation in mouse microglial cells. J Neurochem. 2007;102:1518–1532. doi: 10.1111/j.1471-4159.2007.04570.x. [DOI] [PubMed] [Google Scholar]
- Takenouchi T, Sugama S, Iwamaru Y, Hashimoto M, Kitani H. Modulation of the ATP-lnduced release and processing of IL-1beta in microglial cells. Crit Rev Immunol. 2009;29:335–345. doi: 10.1615/critrevimmunol.v29.i4.40. [DOI] [PubMed] [Google Scholar]
- Tanabe S, Heesen M, Yoshizawa I, Berman MA, Luo Y, Bleul CC, Springer TA, Okuda K, Gerard N, Dorf ME. Functional expression of the CXC-chemokine receptor-4/fusin on mouse microglial cells and astrocytes. J Immunol. 1997;159:905–911. [PubMed] [Google Scholar]
- Tang SC, Arumugam TV, Xu X, Cheng A, Mughal MR, Jo DG, Lathia JD, Siler DA, Chigurupati S, Ouyang X, Magnus T, Camandola S, Mattson MP. Pivotal role for neuronal Toll-like receptors in ischemic brain injury and functional deficits. Proceedings of the National Academy of Sciences of the United States of America. 2007;104:13798–13803. doi: 10.1073/pnas.0702553104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor N, Jahn T, Smith S, Lamkin T, Uribe L, Liu Y, Durden DL, Weinberg K. Differential activation of the tyrosine kinases ZAP-70 and Syk after Fc gamma RI stimulation. Blood. 1997;89:388–396. [PubMed] [Google Scholar]
- Tchilibon S, Joshi BV, Kim SK, Duong HT, Gao ZG, Jacobson KA. (N)-methanocarba 2,N6-disubstituted adenine nucleosides as highly potent and selective A3 adenosine receptor agonists. J Med Chem. 2005;48:1745–1758. doi: 10.1021/jm049580r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terashima Y, Onai N, Murai M, Enomoto M, Poonpiriya V, Hamada T, Motomura K, Suwa M, Ezaki T, Haga T, Kanegasaki S, Matsushima K. Pivotal function for cytoplasmic protein FROUNT in CCR2-mediated monocyte chemotaxis. Nat Immunol. 2005;6:827–835. doi: 10.1038/ni1222. [DOI] [PubMed] [Google Scholar]
- Thored P, Heldmann U, Gomes-Leal W, Gisler R, Darsalia V, Taneera J, Nygren JM, Jacobsen SE, Ekdahl CT, Kokaia Z, Lindvall O. Long-term accumulation of microglia with proneurogenic phenotype concomitant with persistent neurogenesis in adult subventricular zone after stroke. Glia. 2009;57:835–849. doi: 10.1002/glia.20810. [DOI] [PubMed] [Google Scholar]
- Thrash JC, Torbett BE, Carson MJ. Developmental regulation of TREM2 and DAP12 expression in the murine CNS: implications for Nasu-Hakola disease. Neurochem Res. 2009;34:38–45. doi: 10.1007/s11064-008-9657-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toyomitsu E, Tsuda M, Yamashita T, Tozaki-Saitoh H, Tanaka Y, Inoue K. CCL2 promotes P2X4 receptor trafficking to the cell surface of microglia. Purinergic Signal. 2012;8:301–310. doi: 10.1007/s11302-011-9288-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tridandapani S, Lyden TW, Smith JL, Carter JE, Coggeshall KM, Anderson CL. The adapter protein LAT enhances fcgamma receptor-mediated signal transduction in myeloid cells. J Biol Chem. 2000;275:20480–20487. doi: 10.1074/jbc.M909462199. [DOI] [PubMed] [Google Scholar]
- Trigatti B, Rayburn H, Vinals M, Braun A, Miettinen H, Penman M, Hertz M, Schrenzel M, Amigo L, Rigotti A, Krieger M. Influence of the high density lipoprotein receptor SR-BI on reproductive and cardiovascular pathophysiology. Proceedings of the National Academy of Sciences of the United States of America. 1999;96:9322–9327. doi: 10.1073/pnas.96.16.9322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trincavelli ML, Melani A, Guidi S, Cuboni S, Cipriani S, Pedata F, Martini C. Regulation of A(2A) adenosine receptor expression and functioning following permanent focal ischemia in rat brain. J Neurochem. 2008;104:479–490. doi: 10.1111/j.1471-4159.2007.04990.x. [DOI] [PubMed] [Google Scholar]
- Trinchieri G, Sher A. Cooperation of Toll-like receptor signals in innate immune defence. Nat Rev Immunol. 2007;7:179–190. doi: 10.1038/nri2038. [DOI] [PubMed] [Google Scholar]
- Tsiperson V, Li X, Schwartz GJ, Raine CS, Shafit-Zagardo B. GAS6 enhances repair following cuprizone-induced demyelination. PLoS One. 2010;5:e15748. doi: 10.1371/journal.pone.0015748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsou CL, Peters W, Si Y, Slaymaker S, Aslanian AM, Weisberg SP, Mack M, Charo IF. Critical roles for CCR2 and MCP-3 in monocyte mobilization from bone marrow and recruitment to inflammatory sites. J Clin Invest. 2007;117:902–909. doi: 10.1172/JCI29919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsuda M, Masuda T, Kitano J, Shimoyama H, Tozaki-Saitoh H, Inoue K. IFN-gamma receptor signaling mediates spinal microglia activation driving neuropathic pain. Proc Natl Acad Sci U S A. 2009a;106:8032–8037. doi: 10.1073/pnas.0810420106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsuda M, Shigemoto-Mogami Y, Koizumi S, Mizokoshi A, Kohsaka S, Salter MW, Inoue K. P2X4 receptors induced in spinal microglia gate tactile allodynia after nerve injury. Nature. 2003;424:778–783. doi: 10.1038/nature01786. [DOI] [PubMed] [Google Scholar]
- Tsuda M, Toyomitsu E, Komatsu T, Masuda T, Kunifusa E, Nasu-Tada K, Koizumi S, Yamamoto K, Ando J, Inoue K. Fibronectin/integrin system is involved in P2X(4) receptor upregulation in the spinal cord and neuropathic pain after nerve injury. Glia. 2008a;56:579–585. doi: 10.1002/glia.20641. [DOI] [PubMed] [Google Scholar]
- Tsuda M, Toyomitsu E, Kometani M, Tozaki-Saitoh H, Inoue K. Mechanisms underlying fibronectin-induced up-regulation of P2X4R expression in microglia: distinct roles of PI3K-Akt and MEK-ERK signalling pathways. J Cell Mol Med. 2009b;13:3251–3259. doi: 10.1111/j.1582-4934.2009.00719.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsuda M, Tozaki-Saitoh H, Masuda T, Toyomitsu E, Tezuka T, Yamamoto T, Inoue K. Lyn tyrosine kinase is required for P2X(4) receptor upregulation and neuropathic pain after peripheral nerve injury. Glia. 2008b;56:50–58. doi: 10.1002/glia.20591. [DOI] [PubMed] [Google Scholar]
- Tyurin VA, Tyurina YY, Feng W, Mnuskin A, Jiang J, Tang M, Zhang X, Zhao Q, Kochanek PM, Clark RS, Bayir H, Kagan VE. Mass-spectrometric characterization of phospholipids and their primary peroxidation products in rat cortical neurons during staurosporine-induced apoptosis. J Neurochem. 2008;107:1614–1633. doi: 10.1111/j.1471-4159.2008.05728.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ueyama T, Lennartz MR, Noda Y, Kobayashi T, Shirai Y, Rikitake K, Yamasaki T, Hayashi S, Sakai N, Seguchi H, Sawada M, Sumimoto H, Saito N. Superoxide production at phagosomal cup/phagosome through beta I protein kinase C during Fc gamma R-mediated phagocytosis in microglia. J Immunol. 2004;173:4582–4589. doi: 10.4049/jimmunol.173.7.4582. [DOI] [PubMed] [Google Scholar]
- Ulmann L, Hatcher JP, Hughes JP, Chaumont S, Green PJ, Conquet F, Buell GN, Reeve AJ, Chessell IP, Rassendren F. Up-regulation of P2X4 receptors in spinal microglia after peripheral nerve injury mediates BDNF release and neuropathic pain. J Neurosci. 2008;28:11263–11268. doi: 10.1523/JNEUROSCI.2308-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ulvestad E, Williams K, Matre R, Nyland H, Olivier A, Antel J. Fc receptors for IgG on cultured human microglia mediate cytotoxicity and phagocytosis of antibody-coated targets. J Neuropathol Exp Neurol. 1994;53:27–36. doi: 10.1097/00005072-199401000-00004. [DOI] [PubMed] [Google Scholar]
- van der Putten C, Zuiderwijk-Sick EA, van Straalen L, de Geus ED, Boven LA, Kondova I, APIJ, Bajramovic JJ. Differential expression of adenosine A3 receptors controls adenosine A2A receptor-mediated inhibition of TLR responses in microglia. J Immunol. 2009;182:7603–7612. doi: 10.4049/jimmunol.0803383. [DOI] [PubMed] [Google Scholar]
- van Weering HR, Boddeke HW, Vinet J, Brouwer N, de Haas AH, van Rooijen N, Thomsen AR, Biber KP. CXCL10/CXCR3 signaling in glia cells differentially affects NMDA-induced cell death in CA and DG neurons of the mouse hippocampus. Hippocampus. 2011;21:220–232. doi: 10.1002/hipo.20742. [DOI] [PubMed] [Google Scholar]
- Varki A. Glycan-based interactions involving vertebrate sialic-acid-recognizing proteins. Nature. 2007;446:1023–1029. doi: 10.1038/nature05816. [DOI] [PubMed] [Google Scholar]
- Vedeler C, Ulvestad E, Grundt I, Conti G, Nyland H, Matre R, Pleasure D. Fc receptor for IgG (FcR) on rat microglia. J Neuroimmunol. 1994;49:19–24. doi: 10.1016/0165-5728(94)90176-7. [DOI] [PubMed] [Google Scholar]
- Verloes A, Maquet P, Sadzot B, Vivario M, Thiry A, Franck G. Nasu-Hakola syndrome: polycystic lipomembranous osteodysplasia with sclerosing leucoencephalopathy and presenile dementia. J Med Genet. 1997;34:753–757. doi: 10.1136/jmg.34.9.753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Li C, Chen Y, Hao Y, Zhou W, Chen C, Yu Z. Hypoxia enhances CXCR4 expression favoring microglia migration via HIF-1alpha activation. Biochem Biophys Res Commun. 2008;371:283–288. doi: 10.1016/j.bbrc.2008.04.055. [DOI] [PubMed] [Google Scholar]
- Wang Y, Huang J, Li Y, Yang GY. Roles of chemokine CXCL12 and its receptors in ischemic stroke. Curr Drug Targets. 2012;13:166–172. doi: 10.2174/138945012799201603. [DOI] [PubMed] [Google Scholar]
- Wei YS, Lan Y, Meng LQ, Nong LG. The association of L-selectin polymorphisms with L-selectin serum levels and risk of ischemic stroke. Journal of thrombosis and thrombolysis. 2011;32:110–115. doi: 10.1007/s11239-011-0587-4. [DOI] [PubMed] [Google Scholar]
- Weinger JG, Brosnan CF, Loudig O, Goldberg MF, Macian F, Arnett HA, Prieto AL, Tsiperson V, Shafit-Zagardo B. Loss of the receptor tyrosine kinase Axl leads to enhanced inflammation in the CNS and delayed removal of myelin debris during experimental autoimmune encephalomyelitis. J Neuroinflammation. 2011;8:49. doi: 10.1186/1742-2094-8-49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilkinson B, Koenigsknecht-Talboo J, Grommes C, Lee CY, Landreth G. Fibrillar beta-amyloid-stimulated intracellular signaling cascades require Vav for induction of respiratory burst and phagocytosis in monocytes and microglia. J Biol Chem. 2006;281:20842–20850. doi: 10.1074/jbc.M600627200. [DOI] [PubMed] [Google Scholar]
- Wittendorp MC, Boddeke HW, Biber K. Adenosine A3 receptor-induced CCL2 synthesis in cultured mouse astrocytes. Glia. 2004;46:410–418. doi: 10.1002/glia.20016. [DOI] [PubMed] [Google Scholar]
- Witting A, Muller P, Herrmann A, Kettenmann H, Nolte C. Phagocytic clearance of apoptotic neurons by Microglia/Brain macrophages in vitro: involvement of lectin-, integrin-, and phosphatidylserine-mediated recognition. J Neurochem. 2000;75:1060–1070. doi: 10.1046/j.1471-4159.2000.0751060.x. [DOI] [PubMed] [Google Scholar]
- Wixey JA, Reinebrant HE, Carty ML, Buller KM. Delayed P2X4R expression after hypoxia-ischemia is associated with microglia in the immature rat brain. J Neuroimmunol. 2009;212:35–43. doi: 10.1016/j.jneuroim.2009.04.016. [DOI] [PubMed] [Google Scholar]
- Wollmer MA, Lucius R, Wilms H, Held-Feindt J, Sievers J, Mentlein R. ATP and adenosine induce ramification of microglia in vitro. J Neuroimmunol. 2001;115:19–27. doi: 10.1016/s0165-5728(01)00257-0. [DOI] [PubMed] [Google Scholar]
- Wong WT. Microglial aging in the healthy CNS: phenotypes, drivers, and rejuvenation. Front Cell Neurosci. 2013;7:22. doi: 10.3389/fncel.2013.00022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woo MS, Wang X, Faustino JV, Derugin N, Wendland MF, Zhou P, Iadecola C, Vexler ZS. Genetic deletion of CD36 enhances injury after acute neonatal stroke. Ann Neurol. 2012;72:961–970. doi: 10.1002/ana.23727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright GJ, Puklavec MJ, Willis AC, Hoek RM, Sedgwick JD, Brown MH, Barclay AN. Lymphoid/neuronal cell surface OX2 glycoprotein recognizes a novel receptor on macrophages implicated in the control of their function. Immunity. 2000;13:233–242. doi: 10.1016/s1074-7613(00)00023-6. [DOI] [PubMed] [Google Scholar]
- Wynne AM, Henry CJ, Huang Y, Cleland A, Godbout JP. Protracted downregulation of CX3CR1 on microglia of aged mice after lipopolysaccharide challenge. Brain Behav Immun. 2010;24:1190–1201. doi: 10.1016/j.bbi.2010.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao H, Li Y, Du J, Mosig A. Ct3d: tracking microglia motility in 3D using a novel cosegmentation approach. Bioinformatics. 2011;27:564–571. doi: 10.1093/bioinformatics/btq691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan SD, Chen X, Fu J, Chen M, Zhu H, Roher A, Slattery T, Zhao L, Nagashima M, Morser J, Migheli A, Nawroth P, Stern D, Schmidt AM. RAGE and amyloid-beta peptide neurotoxicity in Alzheimer’s disease. Nature. 1996;382:685–691. doi: 10.1038/382685a0. [DOI] [PubMed] [Google Scholar]
- Yao H, Yang Y, Kim KJ, Bethel-Brown C, Gong N, Funa K, Gendelman HE, Su TP, Wang JQ, Buch S. Molecular mechanisms involving sigma receptor-mediated induction of MCP-1: implication for increased monocyte transmigration. Blood. 2010;115:4951–4962. doi: 10.1182/blood-2010-01-266221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao Y, Tsirka SE. The C terminus of mouse monocyte chemoattractant protein 1 (MCP1) mediates MCP1 dimerization while blocking its chemotactic potency. J Biol Chem. 2010;285:31509–31516. doi: 10.1074/jbc.M110.124891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao Y, Tsirka SE. The CCL2-CCR2 system affects the progression and clearance of intracerebral hemorrhage. Glia. 2012;60:908–918. doi: 10.1002/glia.22323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu L, Huang Z, Mariani J, Wang Y, Moskowitz M, Chen JF. Selective inactivation or reconstitution of adenosine A2A receptors in bone marrow cells reveals their significant contribution to the development of ischemic brain injury. Nat Med. 2004;10:1081–1087. doi: 10.1038/nm1103. [DOI] [PubMed] [Google Scholar]
- Zendedel A, Nobakht M, Bakhtiyari M, Beyer C, Kipp M, Baazm M, Joghataie MT. Stromal cell-derived factor-1 alpha (SDF-1alpha) improves neural recovery after spinal cord contusion in rats. Brain Res. 2012;1473:214–226. doi: 10.1016/j.brainres.2012.07.037. [DOI] [PubMed] [Google Scholar]
- Zhang J, Shi XQ, Echeverry S, Mogil JS, De Koninck Y, Rivest S. Expression of CCR2 in both resident and bone marrow-derived microglia plays a critical role in neuropathic pain. J Neurosci. 2007;27:12396–12406. doi: 10.1523/JNEUROSCI.3016-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L, Liu W, Alizadeh D, Zhao D, Farrukh O, Lin J, Badie SA, Badie B. S100B attenuates microglia activation in gliomas: possible role of STAT3 pathway. Glia. 2011a;59:486–498. doi: 10.1002/glia.21118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang S, Cherwinski H, Sedgwick JD, Phillips JH. Molecular mechanisms of CD200 inhibition of mast cell activation. J Immunol. 2004;173:6786–6793. doi: 10.4049/jimmunol.173.11.6786. [DOI] [PubMed] [Google Scholar]
- Zhang S, Phillips JH. Identification of tyrosine residues crucial for CD200R-mediated inhibition of mast cell activation. J Leukoc Biol. 2006;79:363–368. doi: 10.1189/jlb.0705398. [DOI] [PubMed] [Google Scholar]
- Zhang S, Wang XJ, Tian LP, Pan J, Lu GQ, Zhang YJ, Ding JQ, Chen SD. CD200-CD200R dysfunction exacerbates microglial activation and dopaminergic neurodegeneration in a rat model of Parkinson’s disease. J Neuroinflammation. 2011b;8:154. doi: 10.1186/1742-2094-8-154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao X, Grotta J, Gonzales N, Aronowski J. Hematoma resolution as a therapeutic target: the role of microglia/macrophages. Stroke. 2009;40:S92–94. doi: 10.1161/STROKEAHA.108.533158. [DOI] [PubMed] [Google Scholar]
- Zhao X, Sun G, Zhang J, Strong R, Song W, Gonzales N, Grotta JC, Aronowski J. Hematoma resolution as a target for intracerebral hemorrhage treatment: role for peroxisome proliferator-activated receptor gamma in microglia/macrophages. Annals of neurology. 2007;61:352–362. doi: 10.1002/ana.21097. [DOI] [PubMed] [Google Scholar]
- Zhuang ZY, Kawasaki Y, Tan PH, Wen YR, Huang J, Ji RR. Role of the CX3CR1/p38 MAPK pathway in spinal microglia for the development of neuropathic pain following nerve injury-induced cleavage of fractalkine. Brain Behav Immun. 2007;21:642–651. doi: 10.1016/j.bbi.2006.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou YR, Kottmann AH, Kuroda M, Taniuchi I, Littman DR. Function of the chemokine receptor CXCR4 in haematopoiesis and in cerebellar development. Nature. 1998;393:595–599. doi: 10.1038/31269. [DOI] [PubMed] [Google Scholar]