

Abstract
Stroke is a devastating neurological disorder and one of the leading causes of death and serious disability. After cerebral ischemia, revascularization in the ischemic boundary zone provides nutritive blood flow as well as various growth factors to promote the survival and activity of neurons and neural progenitor cells. Enhancement of angiogenesis and the resulting improvement of cerebral microcirculation are key restorative mechanisms and represent an important therapeutic strategy for ischemic stroke. In the present study, we tested the hypothesis that post-stroke angiogenesis would be enhanced by omega-3 polyunsaturated fatty acids (n-3 PUFAs), a major component of dietary fish oil. To this end, we found that transgenic fat-1 mice that overproduce n-3 PUFAs exhibited long-term behavioral and histological protection against transient focal cerebral ischemia (tFCI). Importantly, fat-1 transgenic mice also exhibited robust improvements in revascularization and angiogenesis compared to wild type littermates, suggesting a potential role for n-3 fatty acids in post-stroke cerebrovascular remodeling. Mechanistically, n-3 PUFAs induced upregulation of angiopoietin 2 (Ang 2) in astrocytes after tFCI and stimulated extracellular Ang 2 release from cultured astrocytes after oxygen and glucose deprivation. Ang 2 facilitated endothelial proliferation and barrier formation in vitro by potentiating the effects of VEGF on phospholipase Cγ1 and Src signaling. Consistent with these findings, blockade of Src activity in post-stroke fat-1 mice impaired n-3 PUFA-induced angiogenesis and exacerbated long-term neurological outcomes. Taken together, our findings strongly suggest that n-3 PUFA supplementation is a potential angiogenic treatment capable of augmenting brain repair and improving long-term functional recovery after cerebral ischemia.
Keywords: angiogenesis, angiopoietin 2, astrocyte, neuroprotection, omega-3 polyunsaturated fatty acids, stroke, VEGF
INTRODUCTION
Stroke is the fourth leading cause of death and the leading cause of serious long-term disability in adults in the United States (Towfighi and Saver, 2011). Intravenous thrombolysis therapy with tissue plasminogen activator (tPA), the only FDA-approved treatment for ischemic strokes, is severely limited by its short therapeutic time window of less than 4.5 h (Hacke et al., 2008). To date, neuroprotective strategies that only target injured tissue during acute stages of stroke have not been successfully translated to the clinic. In contrast, neurorestorative therapies targeting the tissue spared from direct injury have recently gained attention for their potential to enhance post-stroke brain repair processes and to promote functional recovery with a longer therapeutic time window (Zhang and Chopp, 2009).
Vascular remodeling triggered by reduction of blood flow is a major endogenous defense mechanism in the brain following stroke. Revascularization is induced by shear fluid stress and consists of arteriogenesis, or an improvement in collateral blood flow through pre-existing vasculature in the early phase after ischemic injury. In the late stages after ischemic injury, revascularization also consists of a surge in angiogenesis through endothelial cell proliferation and the subsequent formation of new blood vessels (Liu et al., 2014). Post-stroke angiogenesis greatly improves tissue perfusion and endothelial cells release a plethora of neurotrophic factors, supporting the activity of neurons and neural progenitor cells and promoting long-term functional recovery (Zhang and Chopp, 2009). Thus, post-stroke angiogenesis is an important target for therapeutic interventions. Numerous experiments have attempted to enhance post-stroke angiogenesis, such as the transplantation of endothelial progenitor cells (EPCs), which are of great therapeutic value and already under clinical investigation (Yin et al., 2013; Fan et al., 2010; Ishikawa et al., 2013; Liu et al., 2014).
Omega-3 polyunsaturated fatty acids (n-3 PUFAs) are known to protect against ischemic brain injury in several stroke models (Hu et al., 2013; Belayev et al., 2009; Zhang et al., 2010). However, the underlying mechanisms are not fully understood. Multiple mechanisms have been proposed for n-3 PUFA-mediated protection, including reduction of oxidative stress (Bazan, 2005), anti-inflammatory effects (Zhang et al., 2010; Musiek et al., 2008), induction of heme oxygenase 1 by nuclear factor E2-related factor 2 (Zhang et al., 2014), and potentiation of neurogenesis and oligodendrogenesis (Hu et al., 2013). As neurogenesis is thought to only occur within an angiogenic microenvironment (Palmer et al., 2000), it seems likely that n-3 PUFAs also promote the formation of new blood vessels. However, the effect of n-3 PUFAs on vascular remodeling after acute ischemic stroke remains to be explored.
In order to fill this gap in the field, the present study examined the impact of stroke in transgenic (Tg) mice expressing the C. elegans fat-1 gene, which encodes an n-3 fatty acid desaturase. We demonstrated that n-3 PUFA levels were elevated in these mice through increased conversion from their endogenous n-6 forms. Overproduction n-3 PUFAs in fat-1 Tg mice provided remarkable protection against focal cerebral ischemia compared to wild type littermates. Interestingly, endogenous post-stroke angiogenesis was robustly enhanced by n-3 PUFAs, in a process involving angiopoietin 2 (Ang 2) and vascular endothelial growth factor (VEGF) signaling. Our results strongly support the view that n-3 PUFA supplementation is a potential prophylactic treatment to improve tissue repair and enhance long-term functional recovery after stroke.
MATERIALS AND METHODS
Animals
A transgenic mouse line expressing the C. elegans fat-1 gene was created as described previously (Kang et al., 2004). The fat-1 gene encodes an n-3 fatty acid desaturase that adds an extra n-3 double bond to n-6 fatty acids, thereby converting n-6 PUFAs to their corresponding n-3 forms. The coding region of C. elegans fat-1 gene was optimized for expression in mammalian cells (Wei et al., 2010), and the resultant fat-1 cDNA was driven by a cytomegalovirus (CMV) enhancer and a chicken β-actin promoter. The chimeric transgene was introduced into C57/B6 mice (The Jackson Laboratory, Bar Harbor, Maine, USA) by pronuclear microinjection. The fat-1 heterozygote and wild type (Wt) C57/B6 mice were interbred to produce fat-1 Tg mice and Wt littermates. Tail biopsies were performed and DNA was extracted for genotyping. The fat-1 genotype was confirmed with the forward primer 5′-CGG TTT CTG CGA TGG ATC CCA C-3′ and reverse primer 5′-CCG GTG AAA ACG CAG AAG TTG TTG-3′ amplifying a 631-bp band. Mice were backbred to Wt C57/B6 mice to minimize the potential impact of genetic heterogeneity on the susceptibility to cerebral ischemia. Both fat-1 and Wt mice were maintained on a normal lab-rodent diet (5% fat, n-3:n-6 ratio = 1:5, ProLab® IsoPro® RMH 3000, LabDiet, St. Louis, Missouri, USA). Animals were housed in a temperature- and humidity-controlled animal facility with a 12-hour light-dark cycle. Food and water were available ad libitum. All animal procedures were approved by the University of Pittsburgh Institutional Animal Care and Use Committee and performed in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals.
Lipid extraction and gas chromatography analysis of fatty acid composition
Lipid extraction from tissues was performed as described previously using a modified Folch extraction method (Hu et al., 2013). Fatty acid compositions were determined by capillary gas chromatography using a Clarus 500 Gas Chomatograph (Perkin Elmer, Waltham, Massachusetts, USA). Components were identified by comparing the peak retention times to a 30-component methyl ester standard (Sigma-Aldrich, St. Louis, Missouri, USA). Fatty acid concentrations were determined by calculation of the areas of peaks and expressed as pmol per mg of extracted lipid. Five animals were examined in each group (Wt and fat-1).
Transient focal cerebral ischemia model
Transient focal cerebral ischemia (tFCI) was induced in adult male mice (10–12 week old, 25–30g) by intraluminal occlusion of the left middle cerebral artery (MCA) for 60 min as described previously (Stetler et al., 2008). Experimental procedures were performed following Stroke Therapy Academic Industry Roundtable (STAIR) guidelines (Fisher et al., 2009). Mice were anesthetized with 3% isoflurane vaporized in 67%:30% N2O/O2 until they were unresponsive to the tail pinch test. Animals were then fitted with a nose cone blowing 1.5% isoflurane for anesthesia maintenance. A monofilament (8-0) with a silicon-coated tip was introduced into the common carotid artery, advanced to the origin of the MCA, and left undisturbed for 60 min. Rectal temperature was maintained at 37.0 ± 0.5°C during surgery with a temperature-controlled heating pad. Mean arterial blood pressure was monitored during surgery by a tail cuff, and arterial blood gas was analyzed at 10 min after the onset of ischemia and 10 min after reperfusion. Regional cerebral blood flow (rCBF) was measured using laser-Doppler flowmetry before, during and after transient MCA occlusion (tMCAO). Animals that did not show a CBF reduction of at least 75% of baseline levels or that died after ischemia induction were excluded from further experimentation. Sham-operated animals underwent the same anesthesia and surgical procedures with the exception of MCA occlusion. For the Src inhibitor treatment, AZD0530 was freshly prepared by dissolving the stock solution (5 mg/mL in DMSO) in corn oil at a ratio of 95% corn oil: 5% DMSO. Stroke mice were orally fed with AZD0530 (20 mg/kg) gavage solution or the same volume of vehicle gavage solution on a daily basis starting from 3 d after tMCAO until the day before sacrifice (Figure S1). After the surgery, body weight was measured daily. Surgeries and all outcome assessments listed below were performed by investigators blinded to mouse genotype and experimental groups. Measurements of rCBF and body weight loss were made in 8 animals per group. For survival rates, 92 Wt mice and 75 fat-1 mice were assessed. Eleven Wt mice and 12 fat-1 mice were analyzed for physiological parameters. Overall, 90% of the animals showed greater than 75% reduction in CBF during MCAO, survived beyond 24 h after MCAO and entered the study. Six Wt mice and 5 fat-1 mice that failed to show a CBF reduction greater than 75% during MCAO were excluded from the study. Of the included animals, 37 Wt mice and 19 fat-1 mice died during the long term studies (21 d after MCAO).
Neurobehavioral tests
Neurobehavioral tests were performed 1 d before and 3–21 d after MCAO. Sensorimotor deficits were assessed by the rotarod and cylinder tests. The rotarod test was carried out as described previously (Stetler et al., 2008). Briefly, mice were placed on a rotating drum with a speed accelerating from 4 to 40 rpm during a 5-min period. The time at which the animal fell off the drum was recorded. The test began 1 d before surgery and consisted of 2 trials. On the day of surgery the animals underwent 5 trials, the mean of which was used as the presurgery baseline value for each animal. After surgery, animals were tested for 5 trials on a daily basis up to 14 d. The cylinder test was performed as described previously (Gan et al., 2012) to assess asymmetries in forepaw use. The mouse was placed in a transparent cylinder (diameter: 9 cm; height: 15cm), and videotaped for 5 min. Videotapes were analyzed in slow motion, and forepaw (left/right/both) use during the first contact against the cylinder wall after rearing and during lateral exploration was recorded. Nonimpaired forepaw (left) preference was expressed as a relative proportion of right forepaw contacts, and calculated as: (left−right)/(left+right+both)×100%. Noninjured animals show no preference for either forepaw, while a left forepaw preference will increase in injured animals depending on the severity of the insult. Each neurobehavioral test was conducted on 8 animals per group.
Examination of recently proliferated cells by BrdU labeling
The S-phase marker 5-bromo-2-deoxyuridine (BrdU) was used to label recently proliferated cells. BrdU (50 mg/kg body weight, Sigma-Aldrich) was injected intraperitoneally twice per day with an interval of 8 h at 3–6 d and again at 10–13 d after MCAO (Figure S1). At 7 or 14 d after MCAO, animals were deeply anesthetized and transcardially perfused with 0.9% NaCl followed by 4% paraformaldehyde in PBS. Brains were cryoprotected in 30% sucrose in PBS, and frozen serial coronal brain sections (30μm) were prepared on a cryostat (CM1900, Leica, Bensheim, Germany). Sections were pretreated with 1 N HCl for 1 hour at 37°C, followed by 0.1 mol/L boric acid (pH 8.5) for 10 min at room temperature. Sections were then blocked with the M.O.M kit (Vector, Burlingame, CA, USA), followed by incubation with mouse anti-BrdU antibody (1:1000; BD Biosciences, San Jose, CA, USA) at 4°C overnight. After washing, sections were incubated with the Cy3-AffiniPure donkey anti-mouse IgG (1:1500, Jackson ImmunoResearch Laboratories, Inc., West Grove, PA, USA) for 1 hour at room temperature. Fluorescence images were captured with an Olympus Fluoview FV1000 confocal microscope with the FV10-ASW 2.0 software (Olympus America, Center Valley, PA, USA). Unbiased stereological principles were used to quantify BrdU immunopositive cells along the microvessels. BrdU+ cells were expressed as the number of cells in the designated fields divided by the area (mm2) using the MCID image analysis system (Imaging Research, G.E. Healthcare Biosciences, Pittsburgh, Pennsylvania, USA). At least 10 microscopic fields were randomly sampled in each section and 7 to 8 animals were analyzed in each group.
Vascular labeling and three-dimensional analysis of vascular density
Lectin is known to selectively bind to endothelial glycocalyx only within perfused vessels, and was thus used to identify functional vessels in the brain. Animals were transcardially perfused with FITC-conjugated tomato lectin (Sigma-Aldrich) at a dose of 100 μg/μL, 5 min before euthanasia. Coronal brain sections were prepared and imaged as described above. Six sections with 0.5-mm intervals were analyzed for each brain, and six regions of interest (ROIs) in the ischemic boundary zone (IBZ) were selected from each section. The ROIs were scanned in 512 × 512 pixel (233 × 233 μm2) format in the x-y direction, and 1-μm-step-size optical sections along the z-axis were acquired with a 40× objective lens. Three-dimensional reconstruction was performed using an image analysis software package (3D Doctor 3.5, Able software, USA). The vascular surface area (mm2) and the total vascular length (mm) per volume of tissue (mm3) were calculated by the software, and the number of vascular branch points was counted in the three-dimensional images by a blinded investigator. Six to 8 animals per group were used for this analysis.
Immunohistochemistry and infarct volume measurements
Free-floating coronal brain sections were prepared as described above. Sections were blocked with 10% donkey serum in PBS for 1 h, followed by overnight incubation (4°C) with the following primary antibodies: goat polyclonal anti-glial fibrillary acidic protein (GFAP) antibody (1:1000; Abcam, Cambridge, MA, USA) and rabbit polyclonal anti-angiopoietin 2 (Ang 2) antibody (1:200, Abcam). After washing, sections were incubated for 1 h at room temperature with donkey anti-goat secondary antibody conjugated with DyLight 488 (1:1000, Jackson ImmunoResearch Laboratories, Inc.) and donkey anti-rabbit secondary antibody conjugated with Cy3 (1:1000, Jackson ImmunoResearch Laboratories, Inc.). Alternate sections from each experimental condition were incubated in all solutions except the primary antibody to assess nonspecific staining. Sections were then counterstained with 4′,6-diamidino-2-phenylindole (DAPI; Thermo Scientific, Pittsburgh, PA, USA) for 2 min at room temperature, mounted and coverslipped with Fluoromount-G (Southern Biotech, Birmingham, AL, USA). Fluorescence images were captured as described above.
For infarct volume measurements, 7 equally-spaced coronal sections encompassing the middle cerebral artery territory were incubated in rabbit polyclonal anti-microtubule-associated protein 2 (MAP2) antibody (1:200; Santa Cruz Biotechnology, Dallas, TX, USA) followed by donkey anti-rabbit secondary antibody conjugated with DyLight 488 (1:1000, Jackson ImmunoResearch Laboratories, Inc.). Images were acquired as described above, and infarct volume was determined with NIH Image J analysis by an observer blinded to the experimental group assignment. The actual infarct volume with edema correction was calculated as the total volume of the contralateral hemisphere minus the non-infarct volume of the ipsilateral hemisphere. Each experimental group had 8 animals.
Primary astrocyte cultures and oxygen-glucose deprivation
Primary mouse astrocytes were prepared from 1-day-old mouse brains as described previously (Hu et al., 2012). Cells were seeded on poly-D-lysine coated glass coverslips in 24-well plates at a density of 5 × 104 cells per well. To model ischemia in vitro, cultured astrocytes were exposed to transient oxygen and glucose deprivation (OGD) for 60 min as described previously (Stetler et al., 2008). Culture medium was replaced with glucose-free medium, and cultures were placed in a Billups-Rothenberg modular incubator chamber (Del Mar, San Diego, CA, USA), which was flushed with 95% argon and 5% CO2 for 5 min and then sealed. The chamber was placed in a water-jacketed incubator (Forma, Thermo Fisher Scientific, Waltham, MA, USA) at 37°C for 60 min and then returned to 95% air, 5% CO2, and glucose-containing medium for a period of time indicated in each experiment. Control glucose-containing cultures were incubated for the same period of time at 37°C in humidified 95% air and 5% CO2. A docosahexaenoic/eicosapentaenoic acid (DHA/EPA) stock mixture was prepared freshly in ethanol at 100 mM and then diluted in culture medium containing bovine serum albumin (BSA) (5 μM). The ratio of the DHA (3 μM)/EPA (7 μM) mixture was consistent with the concentration ratio of DHA/EPA in commercially available fish oils and was added into the culture 48 h prior to OGD. Culture medium containing ethanol (0.01%) and BSA (5 μM) was used as the control.
Enzyme-linked immunosorbent assay (ELISA)
At 24 h after OGD or control conditions, the concentrations of Ang 2 and VEGF in the supernatants from astrocyte cell cultures were measured using the mouse VEGF quantikine ELISA kit and mouse angiopoietin-2 quantikine ELISA kit (R & D Systems, Minneapolis, MN, USA), respectively. All experimental procedures and data analyses were performed according to the manufacturer’s instructions.
Endothelial and astrocyte cocultures
Mouse primary brain microvascular endothelial cells (ECs) were purchased from CellBiologics (Cat. No. C57-6023, Chicago, IL, USA). ECs and astrocytes were cultured in a transwell coculture system that allowed contact-independent communication through diffusible factors (Figure 4B). The transwell PET membranes (0.4 μm pore, 11 mm diameter; Corning, Lowell, MA, USA) were treated with a gelatin-based coating solution (Cat. No. 6950, CellBiologics). Primary mouse ECs were seeded onto the luminal side of the membrane at a density of 2.5 × 105 cells per membrane and grown to complete confluence within 4–5 d. Astrocytes grown on glass coverslips were placed into the bottom of the abluminal chamber for the duration indicated in individual experiments. In some experiments, the coculture was maintained in VEGF-free media, or treated with the VEGF receptor (VEGFR) antagonist CPO-P11 (10 μM).
Figure 4. DHA/EPA promote astrocyte angiopoietin 2 release and facilitate endothelial proliferation and barrier formation.

A. Mouse primary astrocytes were treated with DHA/EPA (3μM/7μM) or vehicle for 48 h and then subjected to 60-min OGD. The concentrations of Ang 2 and VEGF in the culture medium were measured quantitatively 24 h after OGD using ELISA. Data are presented as mean ± SEM from three independent experiments, ***p≤0.001 vs. control. #p≤0.05, ##p≤0.01 vs. OGD. B. Illustration of the transwell coculture system allowing cell contact-independent interactions between endothelial cells (ECs) and astrocytes through diffusible factors. Mouse primary ECs were cultured in the luminal chamber of the transwell inserts in the presence or absence of primary astrocytes in the abluminal chamber. Astrocytes grown on a coverslip were treated with DHA/EPA (3μM/7μM) or vehicle for 48 h, followed by 60-min OGD, and then placed into the coculture abluminal chamber. Paracellular permeability of ECs was determined by measuring the diffusion coefficient of FITC-Dextran (40 kDa) from the luminal to the abluminal chamber, and data are expressed as percentage of paracellular permeability in controls. The paracellular permeability of vehicle-treated ECs in the absence of astrocytes at 1 d of coculture was expressed as 100%. C–D. ECs were treated with DHA/EPA (3μM/7μM) or vehicle for 48 h, or cocultured with OGD-treated astrocytes as described above, and then processed for immunostaining. Representative immunofluorescent staining for Ki67 (C) and VE-cadherin (D) in ECs 1–5 d after coculture (red). Cells were counterstained with DAPI (blue) for nuclear labeling. E–F. Immunostained Ki67+ cells were counted as a measure of active cell proliferation (E), and paracellular permeability was calculated as described above (F). Data are presented as mean ± SEM, *p≤0.05, **p≤0.01 vs. vehicle. #p≤0.05 vs. EC+astrocyte (Astro) from 3–4 independent experiments for each condition. G–I. Primary astrocytes were transfected with an empty vector (Lenti), vector with scrambled control sequence (Lenti-Sc), or lentiviral vectors containing shRNA targeting Ang 2 (Lenti-Ang 2). Cells were treated with lentiviral vectors and DHA/EPA (3μM/7μM) or vehicle for 48 h and then subjected to 60-min OGD. G. The release of Ang 2 from astrocytes was measured by ELISA 1 d after OGD. H–I. Transfected astrocytes were cocultured with ECs as described above. H. Ki67+ ECs 3 d after coculture. I. EC paracellular permeability 5 d after coculture. Data are presented as mean ± SEM, *p≤<0.05, **p≤0.01 vs. Lenti from 3–4 independent experiments for each condition.
To assess EC barrier function formation in vitro, FITC-Dextran (40 kDa) was added into the luminal chamber at a concentration of 2 mg/mL in 500 μL media. After various time intervals, fluorescence intensity was measured with a fluorescence reader by removing 30 μL media from the lower chamber. Thirty μL fresh media was added after each reading. Paracellular permeability coefficient was calculated using the method described previously (Dimitrijevic et al., 2006), based on the concentrations of the tracer in the luminal (donating) and the abluminal (receiving) chamber, respectively. The concentration of FITC-Dextran in samples was calculated from a standard curve generated using the tracer. Data were normalized and expressed as percentage of the level of vehicle-treated ECs at 1 d.
Construction of viral vectors and gene transfection
Lentiviral vectors were constructed expressing short hairpin interfering RNA (shRNA) against murine Ang 2 (Lenti-Ang 2, Sequence 1: 5′-GGC TGA TGA AGC TGG AGA A -3′; Sequence 2: 5′-GTA CTA AAC CAG ACG ACA A -3′). The gene-specific targeting sequence or its counterpart scramble sequence (Lenti-Sc, 5′-TGC AGG TAG CGA AAG AGT G-3′) was inserted into the transfer vector FSW under the control of the U6 promoter. The constructed transfer vectors were transformed into Stbl3 Escherichia coli, and then isolated using the EndoFree Plasmid Maxi Kit (Qiagen, Valencia, CA, USA). Large-scale production of the virus was achieved as described previously (Stetler et al., 2008). Briefly, a plasmid mixture containing 435 μg of pCMV ΔR8.9 (packaging construct), 237 μg of pVSVG (envelope plasmid), and 675 μg of FSW (transfer vector) was suspended in 34.2 mL of CaCl2 (250 mM) and then added volume for volume into 2× BES buffer, pH 6.95. The DNA-CaCl2 precipitate was added to human kidney 293 FT cells (on 15-cm plates at a density of 1.1×107/plate) drop by drop (1.125 mL for each plate), and allowed to incubate for 12 h before switching to fresh culture medium. The supernatant was collected 72 h after transfection, filtered through a 0.45 μm filter and centrifuged at 21,000 rpm for 2 h using an SW28 rotor (Beckman Coulter, Indianapolis, IN, USA). Viruses were further purified by sucrose gradient ultracentrifugation. The pellet was suspended in 3 mL of PBS, loaded on the top of 2 mL of 20% sucrose solution, and centrifuged at 22,000 rpm for 2 h using the SW50.1 rotor (Beckman Coulter). The resulting pellet was resuspended in 200 μL of DMEM, aliquoted, and stored at −70°C. The titer of the vector stock was determined using ELISA. The average titer is typically ~5–10 × 1010 particle units/mL. Primary mouse astrocyte cultures were infected for 3 d with Lenti-Ang 2, Lenti-Sc, or the control empty vector (Lenti). The knockdown of Ang 2 in astrocytes was confirmed by ELISA (Figure 4G).
Western blots
Protein isolation from brain tissues or cultured cells was performed as described previously (Cao et al., 2001). Western blot was performed using the standard SDS-PAGE method and enhanced chemiluminescence detection reagents (G.E. Healthcare Biosciences). Immunoreactivity was semi-quantitatively measured by gel densitometric scanning and analyzed with the MCID image analysis system (Imaging Research, Inc.). Antibodies against the following proteins were used: rabbit polyclonal anti-angiopoietin 1 (1:500, Abcam), rabbit polyclonal anti-angiopoietin 2 (1:500, Abcam), rabbit polyclonal anti-meteorin (1:250, Abcam), rabbit monoclonal anti-phospho-VEGFR2 (Tyr1059; 1:1000, Cell Signaling, Danvers, MA, USA), rabbit monoclonal antibody against total-VEGFR2 (1:1000, Cell Signaling), rabbit polyclonal anti-phospho-Src (Tyr416; 1:1000, Cell Signaling), rabbit polyclonal anti-total-Src (1:1000, Cell Signaling), rabbit monoclonal anti-phospho-c-Abl (Tyr89; 1:1000, Cell Signaling), rabbit polyclonal anti-c-Abl (1:1000, Abcam), rabbit polyclonal anti-phosphoPLCγ1 (Tyr783, 1:1000, Cell Signaling), rabbit polyclonal anti-PLCγ1 (1:1000, Cell Signaling), rabbit polyclonal anti-vascular endothelial (VE)-cadherin (1:500, Abcam) and mouse monoclonal anti-β-actin antibody (1:2000, Sigma-Aldrich). Data were quantified from 4–8 animals/group for brain tissue analysis, and from three independent experiments for in vitro studies.
Immunostaining of endothelial cells
Primary mouse ECs grown on collagen-coated coverslips in 24-well culture dishes were fixed in 4% paraformaldehyde and then blocked with donkey serum in 0.3 M glycine in PBS for 1 h at room temperature. The cells were then incubated with rabbit monoclonal anti-Ki67 (1:200, Abcam) or rabbit polyclonal anti-VE-cadherin primary antibodies (1:200, Abcam) overnight at 4°C, followed by incubation with donkey anti-rabbit secondary antibody conjugated with Cy3 (1:1000, Jackson ImmunoResearch Laboratories, Inc.). The cells were then counterstained with DAPI for 2 min at room temperature. After washing in PBS, the coverslips were mounted on glass slides with antifade Vectashield solution (Vector Laboratories). Fluorescence images were captured with an Olympus Fluoview FV1000 confocal microscope with FV10-ASW 2.0 software (Olympus America).
Statistical analysis
All data are expressed as mean ± SEM. The statistical difference between means of two groups was analyzed by the Student’s t-test. The differences between means of multiple groups were assessed by ANOVA followed by the Bonferroni/Dunn post hoc test. Comparisons of animal survival rates were performed by Kaplan-Meier survival analysis. A p value less than 0.05 was considered statistically significant.
RESULTS
Transgenic overproduction of n-3 PUFAs improves neurological functions and confers long-term protection against cerebral ischemia
Expression of the fat-1 transgene resulted in a shift in the total lipid profiles from n-6 to n-3 PUFAs in all the tissues tested (tail, liver, and brain; Figure S2A). The concentrations of three major n-3 PUFAs, eicosapentaenoic acid (EPA), docosapentaenoic acid (DPA), and docosahexaenoic acid (DHA) were significantly elevated in the brains of fat-1 transgenic (Tg) mice (Figure S2C). To examine the effects of n-3 PUFA overproduction on ischemic brain injury, adult male fat-1 mice and their Wt littermates were subjected to tMCAO (Figure S1). As shown in Figure 1B, fat-1 mice developed a significantly smaller infarct (40.2% reduction) than their Wt littermates 21 d after tMCAO. This protection could not be attributed to changes in rCBF (Figure 1A) or alterations in other physiological parameters that could affect stroke outcomes (Table 1). In addition, fat-1 mice experienced far less loss of body weight during 3–21 d of reperfusion (Figure 1C) and their survival rate was higher than Wt littermates (Figure 1D, p=0.044 by Kaplan-Meier survival analysis). To investigate the impact of n-3 PUFAs on functional outcomes after tMCAO, the rotarod and cylinder tests were performed up to 21 d of reperfusion. Animals with sham operations displayed no significant difference in neurobehavioral performance regardless of their genotype (Figure 1E–F, left panels). Following tMCAO, both fat-1 and Wt mice showed spontaneous recovery in sensorimotor functions during 3–21 d of reperfusion (Figure 1E–F, right panels). However, in fat-1 mice, neurological functions were further improved compared to Wt littermates, as evaluated by their performance in the rotarod and cylinder tests. In summary, transgenic overproduction of n-3 PUFAs in fat-1 mice provides long-term improvement of neurological functions after ischemia.
Figure 1. Improved neurological outcomes and long-term protection after tMCAO in fat-1 transgenic (Tg) mice.

A. Wt and fat-1 mice exhibited similar reductions in regional cerebral blood flow (rCBF) in the ischemic cortex during the 60-min MCAO and the subsequent reperfusion period (Rep). B. Brain infarct volume at 21 d after tMCAO in Wt and fat-1 mice. Data are presented as mean ± SEM, n=8 animals/group. **p<0.01 fat-1 vs. Wt. C. Loss of body weight was measured in Wt and fat-1 mice during 1–21 d of post-ischemia reperfusion and expressed as percentage of the body weight before MCAO. Data are presented as mean ± SEM, n=8 animals/group. *p≤0.05 fat-1 vs. Wt. D. Survival rate of animals after 1–21 d of reperfusion. n=75 for fat-1 mice; n=92 for wild type mice. p=0.044 between Wt and fat-1 groups by Kaplan-Meier survival analysis. E. Rotarod test before and 3–14 d after sham operation or tMCAO. F. Cylinder test 3–21 d after sham operation or tMCAO. Data are presented as mean ± SEM, n=8 animals/group. *p≤0.05, **p≤0.01, ***p≤0.001 vs. pre-MCAO. #p≤0.05, ##p≤.01 fat-1 vs. Wt.
Table 1.
Physiological parameters during and after transient focal cerebral ischemia
| Time | BP | pH | pO2 | pCO2 | Glucose | |
|---|---|---|---|---|---|---|
| Wild type | During tFCI | 90.2 ± 2.6 | 7.32 ± 0.01 | 118.4 ± 3.6 | 39.6 ± 2.4 | 125.6 ± 4.2 |
| After tFCI | 86.4 ± 3.4 | 7.34 ± 0.02 | 121.3 ± 4.1 | 41.5 ± 2.3 | 135.1 ± 3.3 | |
| Tg-Fat1 | During tFCI | 87.4 ± 2.5 | 7.34 ± 0.02 | 124.2 ± 3.8 | 40.8 ± 1.9 | 130.2 ± 3.6 |
| After tFCI | 85.6 ± 3.8 | 7.33 ± 0.01 | 122.5 ± 3.4 | 42.0 ± 2.5 | 136.7 ± 3.4 |
Comparison of physiological parameters during and after transient focal cerebral ischemia (tFCI) in Tg-Fat1 mice and wild type littermates. Physiological parameters include: BP, mean blood pressure in mmHg; pO2, arterial O2 pressure in mmHg; pCO2, arterial CO2 pressure in mmHg; glucose, blood glucose levels in mg/dL. Data were obtained 10 min after the onset of tFCI (during) and 10 min of reperfusion (after), and expressed as mean ± SEM. n = 11 for wild type mice; n = 12 for Tg-Fat-1 mice.
n-3 PUFAs enhance post-stroke revascularization in the IBZ
The long-term protection provided by n-3 PUFAs was suggestive of true neurorestoration, which is likely to be associated with angiogenesis. To test the hypothesis that post-stroke angiogenesis is enhanced by n-3 PUFAs, we next examined whether the overproduction of n-3 PUFAs affected revascularization in the IBZ (Figure 2A), a region with active vascular remodeling during the post-stroke recovery stage (Krupinski et al., 1994). As shown in Figure 2B, the number of functional vessels in the IBZ was decreased 1 d after tMCAO, as reflected by reduced fluorescent lectin signal, which reflects endothelial cells within properly perfused vessels. Revascularization developed gradually during the first 21 d after tMCAO in both Wt and fat-1 brains, but was significantly enhanced in fat-1 brains compared to Wt brains. Further analyses demonstrated that vascular surface area, branch points, and length were all increased in fat-1 mice for the long term (Figure 2C–E). No significant difference in vasculature formation was observed between fat-1 and Wt brains in non-injured sham animals (Figure 2B–E). These data suggest that n-3 PUFA overproduction promotes robust post-stroke revascularization, which may partially account for its long-term protective effects.
Figure 2. Enhanced revascularization after tMCAO in fat-1 Tg mice.
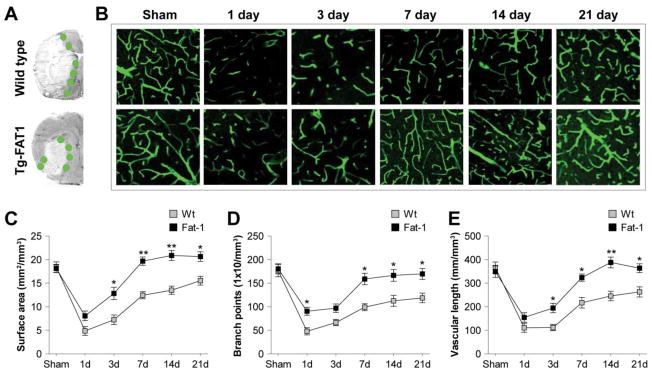
A. Brain infarct formation and the ischemic boundary zone (IBZ, green circles) in Wt and fat-1 Tg mice, as visualized by MAP2 staining. B. Representative images of lectin fluorescent signal in the IBZ of Wt and fat-1 brains at 1–21 d after tMCAO. C–E. Post-stroke revascularization was assessed by measurement of vascular surface area (C), branch points (D) and length (E) at 1–21 d after tMCAO. Data are presented as mean ± SEM, n=6 animals/group. *p≤0.05, **p≤0.01 fat-1 vs. Wt.
n-3 PUFAs elevate angiopoietin 2 expression in astrocytes in vivo
The enhanced post-stroke revascularization in fat-1 Tg mice indicates that n-3 PUFAs may promote brain repair by stimulating endogenous cerebral angiogenesis after ischemia. To begin to test this hypothesis, we measured the expression of three proteins that are critical for angiogenesis. Angiopoietin (Ang) 1 is important for EC sprouting and recruitment of periendothelial cells (Kim et al., 2000; Su et al., 2009). In contrast, Ang 2 serves as an endogenous antagonist to Ang 1, and at the same time promotes detachment of periendothelial cells and loosens the surrounding matrix (Maisonpierre et al., 1997; Holopainen et al., 2012). The brain-specific protein meteorin, secreted by perivascular astrocytes, also regulates angiogenesis by promoting vascular maturation (Park et al., 2008). As expected, we observed significant elevation of all three proteins in the ipsilateral hemisphere after stroke (Figure 3A–B), indicating a natural activation of the endogenous angiogenesis process. Interestingly, Ang 2 and meteorin levels were robustly elevated in fat-1 mice compared to Wt mice, suggesting a potential role of these proteins in the effects of n-3 PUFA on post-stroke revascularization. Moreover, tMCAO-induced Ang 2 upregulation was largely found in GFAP+ astrocytes (Figure 3C, arrow). Overproduction of n-3 PUFAs further elicited Ang 2 expression, not only in astrocytes, but also along the microvessels (Figure 3C, arrowhead). The colocalization of Ang 2 with GFAP and the lack of colocalization between Ang 2 and the endothelial marker lectin suggest that Ang 2 may be released by astrocytes and exert its function on nearby microvessels through extracellular diffusion.
Figure 3. n-3 PUFA overproduction elevates angiopoietin 2 expression after tMCAO.

A. Representative Western blots of angiopoietin 1 (Ang 1), angiopoietin 2 (Ang 2), and meteorin in the contralateral and ipsilateral hemispheres of Wt and fat-1 brains after 3 or 7 d of reperfusion. β-actin was used as an internal loading control. B. Protein expression of Ang 1, Ang 2 and meteorin was quantified and expressed relative to the contralateral side (Con) of Wt mice. Data are presented as mean ± SEM, n=4–5 animals/group. *p≤0.05, **p≤0.01 vs. Con. #p≤0.05 fat-1 vs. Wt. C. Representative images of Ang 2 immunofluorescent signal (red), double labeled with GFAP or lectin (green), in the IBZ of Wt and fat-1 brains 7 d after tMCAO. Sections were counterstained with DAPI (blue) for nuclear labeling. Arrow: Increased Ang 2 expression in GFAP+ astrocytes. Arrowhead: Enhanced Ang 2 signal along the microvessels. Scale bar: 20 μm.
DHA/EPA stimulate astrocyte angiopoietin 2 release and promote endothelial proliferation and barrier formation
The colocalization of Ang 2 with the astrocyte marker GFAP in the brain suggested that astrocytes may secrete Ang 2 after stroke and promote angiogenesis. We hypothesized that n-3 PUFAs stimulate astrocytic Ang 2 release to facilitate EC proliferation and barrier formation, both of which are important steps during angiogenesis (Liu et al., 2014). To test this hypothesis, we cultured primary mouse astrocytes and treated the cells with 60 min of OGD. OGD induced a significant increase in extracellular release of Ang 2 and VEGF into the culture media (Figure 4A). Interestingly, 48 h of treatment with DHA/EPA prior to OGD led to further increases in Ang 2 but a dramatic decrease in VEGF levels (Figure 4A). To further elucidate the role of astrocytes and Ang 2 in n-3 PUFA-mediated angiogenesis, we utilized a transwell system of cocultured primary mouse ECs and astrocytes (Figure 4B). DHA/EPA treatment inhibited EC proliferation in the absence of astrocytes as reflected by a reduction in Ki67+ cells (Figure S3A), a marker for active cell cycling (Scholzen and Gerdes, 2000). Astrocytes promoted EC proliferation in a contact-independent manner (Figure 4 C and E), and this effect was potentiated by DHA/EPA. In addition, astrocytes facilitated the function of the EC barrier. Tight junctions and adherens junctions together form the junctional complex between adjacent ECs and both types of junctions regulate endothelial paracellular permeability (Dejana et al., 2008; Petty and Lo, 2002). To assess the formation of EC junctional complexes and barrier function, we examined the expression of VE-cadherin, the main adherens junctional protein in ECs. The expression of VE-cadherin was substantially increased in ECs in the presence of astrocytes (Figure 4D), leading to improved barrier function and reduced paracellular permeability (Figure 4F). DHA/EPA treatment of astrocytes prior to coculture further enhanced these effects (Figure 4D and F) but had no direct effect on EC barrier formation in the absence of astrocytes (Figure S3B).
The above results indicate that DHA/EPA stimulated astrocytes to release soluble factors that promote EC proliferation and barrier formation. We suspected that Ang 2 played a critical role in this process, given that DHA/EPA significantly increased astrocytic Ang 2 production following OGD (Figure 4A). To test this hypothesis, we knocked down the expression of Ang 2 in astrocytes by shRNA (Figure 4G). The knockdown of Ang 2 in astrocytes significantly inhibited the effects of astrocytes and DHA/EPA on EC proliferation and barrier formation (Figure 4H and I). Taken together, these results demonstrate that DHA/EPA stimulates astrocytes to produce Ang 2, and that this response plays a crucial role in EC proliferation and barrier formation.
Angiopoietin 2 potentiates astrocyte VEGF signaling and mediates DHA/EPA-induced EC proliferation and barrier formation
To further investigate how Ang 2 promoted EC proliferation and barrier formation, we next examined the interactions between Ang 2 and VEGF signaling, a process known to enhance angiogenesis after cerebral ischemia (Zhang et al., 2000). Proliferation of ECs was dependent on VEGF, as absence of VEGF from the culture medium hampered EC proliferation and blunted the effects of astrocytes and DHA/EPA (Figure 5A, left panel). An even more potent inhibitory effect was observed when ECs were treated with the VEGFR antagonist CBO-P11, with almost complete prevention of astrocyte- and DHA/EPA-induced EC proliferation (Figure 5A, right panel). This may be due to the blockade of both endogenous and exogenous VEGF (Bozoyan et al., 2012; Lee et al., 2011).
Figure 5. Angiopoietin 2 potentiates VEGF-induced EC proliferation and barrier formation.

A. Primary mouse astrocytes were treated with DHA/EPA (3μM/7μM) or vehicle for 48 h and subjected to 60-min OGD. Astrocytes were then cocultured with ECs in the transwell system, and Ki67+ ECs were counted 3 d later as described in the text. Left: The coculture was maintained in medium with 1 ng/mL VEGF (Control) or VEGF-free medium. Right: The co-culture was treated with the VEGFR antagonist CPO-P11 (10 μM) or vehicle. Data are presented as mean ± SEM from 3 independent experiments, *p≤0.05, **p≤0.01 vs. control. B. Recombinant Ang 2 (20 ng/mL) was added to primary EC cultures without or with VEGF (10 ng/mL). Western blotting was performed 30 min after the treatment to detect phosphorylated Src, total Src, phosphorylated PLCγ1 and total PLCγ1. β-actin was used as an internal loading control. C. Protein expression of p-Src and p-PLCγ1 in ECs was quantified and expressed relative to the control group. Data are presented as mean ± SEM, n=3 independent cultures/group. *p≤0.05, **p≤0.01 vs. control. #p≤0.05 vs. VEGF. D–H. ECs were cultured in the transwell system with DHA/EPA- and OGD-treated astrocytes, as described above. The PLCγ1 inhibitor U73122 or Src inhibitor AZD0530 (3 or 10 μM) were added to the coculture for 3 d. D. The number of Ki67+ ECs was quantified. E–F. Western blotting was performed to detect VE-cadherin in ECs. Expression of VE-cadherin was quantified relative to the vehicle control. G. Representative fluorescent images of VE-cadherin (red) and DAPI (blue) in immunostained ECs. H. Paracellular permeability was measured and expressed as a percentage of vehicle-treated ECs in the absence of astrocytes at 1 d of coculture. Data are presented as mean ± SEM,*p≤0.05, **p≤0.01 vs. vehicle, from three independent experiments.
The downstream signaling of VEGF includes phosphorylation-mediated activation of Src and phospholipase Cγ (PLCγ), two major molecules that mediate angiogenesis (Zachary, 2001). VEGF induced phosphorylation of Src in EC cultures, but not significant phosphorylation of PLCγ1 (Figure 5B and C). Application of Ang 2 onto ECs failed to affect either p-Src or p-PLCγ1. Importantly, Ang 2 and VEGF together caused a dramatic activation of both Src and PLCγ1 (Figure 5B and C), suggesting that Ang 2 can potentiate the effects of VEGF.
We further examined the role of PLCγ1 and Src in VEGF-induced EC proliferation and barrier formation by blocking these two molecules. Blockade of PLCγ1 with U73122 reduced Ki67+ ECs in a concentration-dependent manner, while the Src inhibitor AZD0530 did not have significant inhibitory effects on this measure (Figure 5D). However, inhibition of Src with AZD0530 inhibited EC barrier formation by reducing the expression of the VE-cadherin in ECs in a concentration-dependent manner (Figure 5E and F). Importantly, the enhancing effect of DHA/EPA on EC VE-cadherin expression was almost completely abolished by Src inhibition (Figure 5E–G). As a result, EC paracellular permeability was significantly increased 1–5 d after coculture with DHA/EPA-treated astrocytes (Figure 5H). Taken together, these results suggest that VEGF-PLCγ1/Src signaling is important in EC proliferation and barrier formation, and that Ang 2 sensitizes ECs to the effects of VEGF.
n-3 PUFAs promote post-stroke angiogenesis via VEGF-Src signaling
We further examined the role of VEGF signaling in post-stroke angiogenesis in vivo. Stroke induced activation of VEGF signaling, as reflected by increased phosphorylation of VEGFR2 and the downstream molecule Src (Figure 6A and B). In fat-1 mice, VEGF-Src signaling was further intensified by n-3 PUFA overproduction. Blocking Src with AZD0530 drastically reduced p-Src level but had no significant effect on p-VEGFR2 (Figure 6A and B), confirming that Src is downstream of VEGF and receptor binding. Ischemia caused long-term angiogenesis in the IBZ, as illustrated by the presence of BrdU+ cells along newly-formed microvessels (Figure 6C, h and i). Post-stroke angiogenesis was dramatically enhanced in fat-1 brains and this enhancement was attenuated by Src inhibition (Figure 6C and D).
Figure 6. Omega-3 PUFAs enhance post-stroke angiogenesis via VEGF-Src signaling.

A. Representative Western blots of phosphorylated VEGFR2 (Tyr 1059), total VEGFR2, phosphorylated Src (Tyr 416) and total Src in the ipsilateral hemispheres of Wt and fat-1 brains after 7 and 14 d of reperfusion. The Src inhibitor AZD0530 (20 mg/kg) was administered daily by gavage at 3 to 6 d (for the 7-day outcome study) or 3 to 13 d (for the 14-day outcome study) after tMCAO. β-actin was used as an internal loading control. B. Protein levels of p-VEGFR2 and p-Src were quantified and expressed relative to Sham controls. Data are presented as mean ± SEM, n=6–8 animals/group. *p≤0.05, **p≤0.01 vs. Sham. #p≤0.05, ##p≤0.01 vs. Wt. @@p≤0.01 Fat-1+AZD vs. Fat-1. C. Representative fluorescent images of BrdU (red) and lectin (green) labeling in the IBZ following tMCAO. a. Three-dimensional confocal image showing BrdU and lectin double-labeled fluorescence. b-i. BrdU and lectin fluorescent signal in Wt and fat-1 brains, or brains treated with AZD0530 after 7 and 14 d of reperfusion. The BrdU+/Lectin+ dual labeled cells exhibit yellow fluorescence. Dashed boxes in d and e indicate areas that were magnified in h and i, respectively. Note that the BrdU+ cells distribute along the lectin+ vessels. D. Quantification of the BrdU+ cells that are associated with microvessels in the contralateral hemispheres (Con), or ipsilateral hemispheres (Ipsi) of animals with or without AZD0530 treatment at 14 d after tMCAO. Data are presented as mean ± SEM, n=7–8 animals/group. *p≤0.05, **p≤0.01 Ipsi+AZD vs. Ipsi.
Next we confirmed the role of Src in post-stroke recovery by examining revascularization. As shown in Figure 7A and B, post-stroke revascularization was significantly reduced by Src inhibition, and n-3 PUFA-afforded angiogenesis was largely abolished with AZD0530 treatment. More importantly, Src inhibition hampered the long-term functional improvements observed in fat-1 mice, as determined by the rotarod and cylinder tests (Figure 7C). There was also a statistical trend towards increased infarct volume with AZD0530 treatment (Figure 7D). In summary, these data strongly suggest that Src plays a critical role in post-stroke angiogenesis. That is, stimulation of VEGF-Src signaling mediates the enhancement of revascularization by n-3 PUFA production, thereby contributing to long-term functional improvement after stroke.
Figure 7. Src inhibition attenuates revascularization and exacerbates neurological outcomes after tCMAO.

A. Representative fluorescent lectin signal in the IBZ of wild type or fat-1 brains after 7 d of reperfusion, with or without Src inhibitor AZD0530. B. Quantification of revascularization in the IBZ by measurement of vascular surface area, branch points or vascular length after 7 d of reperfusion. Data are presented as mean ± SEM, n=7–8 animals/group; *p≤0.05, **p≤0.01 AZD-treated vs. untreated animals. C. Rotarod and cylinder tests during 3–14 d of reperfusion in fat-1 mice or fat-1 treated with AZD0530. Data are presented as mean ± SEM, n=8 animals/group; *p≤<0.05, **p≤0.01 AZD-treated vs. untreated animals. D. Infarct volume in fat-1 mice with or without AZD0530 treatment after 14 d of reperfusion. Data are presented mean ± SEM, n=8 animals/group; p=0.067 AZD-treated vs. untreated animals.
DISCUSSION
The present report is the first mechanistic study of the role of n-3 PUFAs in post-stroke revascularization and angiogenesis. The main findings include the following: 1) transgenic overproduction of n-3 PUFAs improved post-stroke revascularization and enhanced endogenous angiogenesis; 2) n-3 PUFAs induced Ang 2 production in astrocytes, which subsequently promoted EC proliferation and barrier formation; 3) Ang 2 potentiated VEGF-mediated angiogenic effects through the downstream molecules PLCγ1 and Src.
Although the impact of n-3 PUFAs on angiogenesis has been examined under various pathological conditions, their roles are still quite controversial. For example, n-3 PUFAs inhibit tumor vasculature formation (Spencer et al., 2009; Rose and Connolly, 1999; Mukutmoni-Norris et al., 2000) through multiple mechanisms, including the reduction of VEGF (Rose and Connolly, 1999), platelet-derived growth factor (PDGF) (Fox and DiCorleto, 1988) and matrix metalloproteinases (MMPs) (Tsuzuki et al., 2007). In the context of proliferative retinopathy, increased dietary intake of n-3 PUFAs has been shown to significantly reduce retinal neovascularization (Connor et al., 2007). In contrast to these findings, n-3 PUFAs were recently reported to promote angiogenesis after hindlimb ischemia (Turgeon et al., 2013). The distinct properties of different organs and their microenvironments in various pathological states are likely to contribute to these discrepancies. In various in vitro experiments, n-3 PUFAs were demonstrated to suppress EC proliferation and invasion, and induce cell apoptosis (Kim et al., 2005; Yang et al., 1998; Szymczak et al., 2008). These results are supported by our observation that direct treatment of cultured ECs with DHA/EPA had a negative impact on cell proliferation (Figure S3A). However, the strong angiogenic effects detected in fat-1 mice after stroke suggest that n-3 PUFAs may exert proangiogenic functions through other components in the neurovascular niche. Interestingly, the upregulation of Ang 2 after ischemia was not colocalized with ECs but with GFAP+ astrocytes. Similar results were reported in a previous study, where Ang 2 was induced in astrocytes by spinal cord injury, and contributed to improvements in functional recovery (Durham-Lee et al., 2012). Further investigation indicated that DHA/EPA stimulated cultured astrocytes to release Ang 2, which subsequently facilitated EC proliferation and barrier formation. These results may explain the enhancement of angiogenesis by n-3 PUFAs in vivo, and underscore the critical role of the neurovascular niche in post-stroke vascular remodeling.
Angiopoietins are a family of growth factors regulating vessel formation and vascular function (Augustin et al., 2009). Ang 1 exerts angiogenic functions through the EC-specific tyrosine kinase receptor Tie2. Ang 2 was initially described as an endogenous antagonist of Ang 1 acting on the Tie2 receptor, which disrupts blood vessel formation in the embryo when overexpressed in transgenic mice (Maisonpierre et al., 1997). Subsequent studies suggested that the promotion or inhibition of angiogenesis by Ang 2 is cellular context-dependent (Lobov et al., 2002; Daly et al., 2013). For example, Ang 2 destabilizes the vasculature by disengaging ECs from surrounding cells and matrix, making ECs more accessible to additional angiogenic factors, including VEGF (Jones et al., 2001). In vivo Ang 2 treatment alone does not increase microvessel density, but dramatically enhances VEGF-mediated angiogenesis in the adult mouse brain (Zhu et al., 2005). In the current study, Ang 2 appeared to sensitize ECs to VEGF signaling, promoting EC proliferation and coordinating EC barrier formation and the maturation of vessels.
VEGF is the most potent stimulator of EC proliferation and angiogenesis. The strong angiogenic ability and vascular permeating effects of VEGF allow it to exert dualistic roles in stroke pathogenesis (Jones et al., 2001). Administration of VEGF at late stages of ischemic stroke improves cerebral vascular perfusion, enhances angiogenesis, and ameliorates neurological deficits (Zhang et al., 2000). However, in the acute stages of stroke, VEGF also contributes to increased vascular permeability and blood-brain barrier (BBB) breakdown, leading to hemorrhagic transformation and exacerbating ischemic injury (Zhang et al., 2000). In our study, DHA/EPA treatment stimulated astrocytes to release Ang 2 but inhibited VEGF release. This dual action may help compensate against the deleterious effects of VEGF on the BBB while at the same time potentiating angiogenic effects. The major downstream signaling molecules of VEGF include PLCγ1 and Src (Zachary, 2001), blockade of which negatively impact EC proliferation and functional barrier formation. It has been reported that Src inhibition reduces ischemic BBB damage and infarct volume at early stages after injury (Liang et al., 2009; Zan et al., 2014). This protective effect of Src inhibition may reflect blockade of the deleterious effects of VEGF. In contrast, at later stages after ischemia, Src inhibition significantly reduced angiogenesis and hindered the recovery of neurological functions. Furthermore, our results are consistent with previous reports that Src inhibition reduces angiogenesis in tumor models (Summy et al., 2005). In the present study, the potent Src inhibitor AZD0530 was used to investigate the role of Src in vitro and in vivo. The compound is a relatively selective inhibitor for Src, although it is also a weaker inhibitor of Abl kinase (10 times of concentration are needed to inhibit Abl) (Green et al., 2009). Nevertheless, to determine if Abl is involved in the effects of n-3 PUFAs on ischemia-induced angiogenesis, the phosphorylation of Abl was investigated in brain extracts from Wt and fat-1 mice treated with or without AZD0530. Phospho-Abl levels were not significantly altered at either 7 d or 14 d after MCAO, and AZD treatment did not affect the phospho-Abl levels (Figure S4). Taken together, these results do not support a role for Abl signaling in n-3 PUFA-promoted angiogenesis, at least during the time frame examined in the current study. Notably, we observed discrepant effects of Src signaling on EC proliferation in vitro and in vivo. In other words, Src inhibition reduced BrdU+ ECs in vivo but did not have significant effects on Ki67+ ECs in vitro. One possible explanation is that Src signaling participates in the migration of EPCs along vessels, an essential step for angiogenesis. Thus, inhibition of Src may reduce BrdU+ ECs by inhibiting EPC migration, rather than directly suppressing EC proliferation. Further investigations will be needed to elucidate the precise role of Src in EC development.
To date, therapeutic interventions against cerebral ischemia that only target a single pathogenic component have been largely unsuccessful. Strategies targeting multiple events in the ischemic cascade and various components of the neurovascular unit may be more effective. In line with the latter view, n-3 PUFAs are thought to confer protection against cerebral ischemia through multiple mechanisms. In addition to the acute effects, such as the attenuation of oxidative stress (Bazan, 2005), anti-inflammatory effects (Zhang et al., 2010), induction of heme oxygenase 1 (Zhang et al., 2014), and pro-survival functions (Eady et al., 2012), we recently reported potent neurorestorative properties of n-3 PUFAs, such as the promotion of endogenous neurogenesis and oligodendrogenesis (Hu et al., 2013). Transgenic overexpression of n-3 PUFAs significantly increased neural stem cell proliferation and differentiation, and increased neuroblast migration to the injury site (Hu et al., 2013). In the current study, we showed that n-3 PUFAs also enhance endogenous angiogenesis after stroke. These newly formed vessels probably play an important supportive role for the survival of both neurons and glia, thereby leading to long-term functional improvements in neurological outcomes. Thus, multiple studies to date demonstrate that n-3 PUFAs exhibit the potential to be highly effective in the clinic. Fish oil capsule intake is a major source of n-3 PUFAs in humans in developed countries and can be safely ingested over the long term. However, further investigations are needed on how to combine long-term fish oil intake in high-risk populations with acute treatment of single n-3 PUFAs in stroke patients. An additional concern that needs to be addressed experimentally is the reliance on young and otherwise healthy animals in the majority of studies of n-3 PUFAs, including in the present report. As the majority of stroke patients are aged, further research on the effects of n-3 PUFAs as a function of age is warranted.
CONCLUSIONS
In summary, the present study reveals novel neurorestorative mechanisms underlying the long-term protective effects of n-3 PUFAs against cerebral ischemia. Endogenous post-stroke angiogenesis was promoted by n-3 PUFAs and the effect depended on the neurovascular niche. Our findings indicate that n-3 PUFA supplementation is a potential angiogenic treatment to enhance endogenous tissue repair and improve long-term functional recovery after stroke.
Supplementary Material
Mice were subjected to 60 minute-long transient middle cerebral artery occlusion (tMCAO). Neurobehavioral tests were performed up to 21 d after tMCAO and brain tissues were harvested for various analyses at indicated times. For AZD0530 treatments, two endpoints (7 and 14 d) were used. The drug was given daily starting from 3 d after tMCAO until the day before sacrifice, as described in Materials and Methods. To label recently proliferated cells, BrdU was injected intraperitoneally twice per day with an interval of 8 h at 3–6 d and again at 10–13 d after MCAO.
Fatty acid analyses were performed on 3–5 month-old wild type (Wt) and fat-1 transgenic (Tg) mice with gas chromatography. A. The overall ratio of n-3 to n-6 fatty acids was calculated from total n-3 and n-6 fatty acid content in tail, liver and brain samples. Tg fat-1 mice exhibited a substantial increase in the n-3/n-6 ratio in all three tissues compared to their Wt littermates. B–C. The individual content of five major n-6 PUFAs (B) and five major n-3 PUFAs (C) was measured in brain samples from Wt and fat-1 Tg mice. Data are presented as mean ± SEM, n=5 animals/group. *p≤0.05, **p≤0.01, ***p≤0.001 fat-1 vs. Wt. Of the five major n-6 PUFAs examined individually in the brain, the content of three fatty acids was decreased significantly in fat-1 mice, including arachidonic acid (AA, C20:4), adrenic acid (C22:4) and docosapentaenoic acid (DPA, C22:5). No difference was detected for the content of linoleic acid (LA, C18:2) and eicosadienoic acid (EDA, C20:2) between Wt and fat-1 mice. On the other hand, n-3 PUFA production was significantly elevated in fat-1 mice, accounting for the increase in the overall n-3/n-6 PUFA ratio. The content of three major n-3 fatty acids was elevated in fat-1 mouse brains, including eicosapentaenoic acid (EPA, C20:5), docosapentaenoic acid (DPA, C22:5) and docosahexaenoic acid (DHA, C22:6), while no significant difference was detected for two other n-3 fatty acids, alpha-linolenic acid (ALA, C18:3) and eicosatrienoic acid (ETE, C20:3). In summary, these lipid profiles reflect a shift from n-6 to n-3 PUFA production in fat-1 Tg mice, as a result of high expression of the n-3 fatty acid desaturase.
Mouse primary brain microvascular endothelial cells (ECs) were treated with DHA/EPA (3μM/7μM) or vehicle for 48 h. A. EC proliferation was assessed after 1–5 d by counting immunostained Ki67+ cells, as described in Materials and Methods. B. EC barrier formation was evaluated by measurement of paracellular permeability after 1–5 d. Paracellular permeability was calculated from the diffusion coefficient of the 40 kDa FITC-Dextran and expressed as a percentage of vehicle-treated cultures at 1 d. Data are presented as mean ± SEM. n=3 independent cultures/group. *p≤0.05 vs. vehicle control.
A. Representative Western blots of phosphorylated Abl (Tyr 89), and total Abl in the ipsilateral hemispheres of Wt and fat-1 brains after sham operation (S), or at 7 and 14 d after tMCAO. AZD0530 (20 mg/kg) was administered as described in Materials and Methods. β-actin was used as an internal loading control. B. Protein level of p-Abl was quantified and expressed relative to Sham controls. No significant difference in p-Abl levels was observed among groups at 7 d or 14 d after tMCAO. Data are presented as mean ± SEM, n=5 animals/group.
HIGHLIGHTS.
n-3 PUFAs elicit long-term protection against cerebral ischemic injury
n-3 PUFAs enhance post-stroke angiogenesis
n-3 PUFAs promote astrocyte-mediated endothelial proliferation/barrier formation
Angiopoietin 2 potentiates VEGF-induced endothelial proliferation/barrier formation
VEGF-Src signaling participates in n-3 PUFA-mediated revascularization
Acknowledgments
This project was supported by NIH grants NS036736, NS045048, and NS056118 (to J.C.), the Research Career Scientist Award from Department of Veterans Affairs and the VA RR & D Merit Review (to J.C.). L.C. was supported by the High Level Talent Fund of the Beijing Healthcare System (Grant No. 2011-3-093) and the Program for New Century Excellent Talents in University (Grant No. NCET-12-0612). J.W. was supported by the National Natural Science Foundation of China (Grant No. 81301066) and Beijing Nova Program (Grant No. XX2013019). The authors are indebted to Carol Culver for excellent editorial assistance and Pat Strickler for excellent administrative support.
LIST OF ABBREVIATIONS
- AA
Arachidonic acid
- ALA
Alpha-linolenic acid
- Ang
Angiopoietin
- BBB
Blood-brain barrier
- BrdU
5-bromo-2-deoxyuridine
- DAPI
4′,6-diamidino-2-phenylindole
- DHA
Docosahexaenoic acid
- DPA
Docosapentaenoic acid
- EC
Endothelial cell
- EDA
Eicosadienoic acid
- ELISA
Enzyme-linked immunosorbent assay
- EPA
Eicosapentaenoic acid
- EPC
Endothelial progenitor cell
- ETE
Eicosatrienoic acid
- GFAP
Glial fibrillary acidic protein
- IBZ
Ischemic boundary zone
- LA
Linoleic acid
- MCA
Middle cerebral artery
- OGD
Oxygen and glucose deprivation
- PLC
Phospholipase C
- PUFA
Polyunsaturated fatty acid
- rCBF
Regional cerebral blood flow
- ROI
Region of interest
- shRNA
Small hairpin RNA
- Tg
Transgenic
- tFCI
Transient focal cerebral ischemia
- tMCAO
Transient middle cerebral artery occlusion
- VE-cadherin
Vascular endothelial cadherin
- VEGF
Vascular endothelial growth factor
- VEGFR
Vascular endothelial growth factor receptor
- Wt
Wild type
Footnotes
CONFLICT OF INTEREST
None.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Augustin HG, Koh GY, Thurston G, Alitalo K. Control of vascular morphogenesis and homeostasis through the angiopoietin-Tie system. Nat Rev Mol Cell Biol. 2009;10:165–177. doi: 10.1038/nrm2639. [DOI] [PubMed] [Google Scholar]
- Bazan NG. Neuroprotectin D1 (NPD1): a DHA-derived mediator that protects brain and retina against cell injury-induced oxidative stress. Brain Pathol. 2005;15:159–166. doi: 10.1111/j.1750-3639.2005.tb00513.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belayev L, Khoutorova L, Atkins KD, Bazan NG. Robust docosahexaenoic acid-mediated neuroprotection in a rat model of transient, focal cerebral ischemia. Stroke. 2009;40:3121–3126. doi: 10.1161/STROKEAHA.109.555979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bozoyan L, Khlghatyan J, Saghatelyan A. Astrocytes control the development of the migration-promoting vasculature scaffold in the postnatal brain via VEGF signaling. J Neurosci. 2012;32:1687–1704. doi: 10.1523/JNEUROSCI.5531-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao G, Minami M, Pei W, Yan C, Chen D, O’Horo C, Graham SH, Chen J. Intracellular Bax translocation after transient cerebral ischemia: implications for a role of the mitochondrial apoptotic signaling pathway in ischemic neuronal death. J Cereb Blood Flow Metab. 2001;21:321–333. doi: 10.1097/00004647-200104000-00001. [DOI] [PubMed] [Google Scholar]
- Connor KM, SanGiovanni JP, Lofqvist C, Aderman CM, Chen J, Higuchi A, Hong S, Pravda EA, Majchrzak S, Carper D, Hellstrom A, Kang JX, Chew EY, Salem N, Jr, Serhan CN, Smith LE. Increased dietary intake of omega-3-polyunsaturated fatty acids reduces pathological retinal angiogenesis. Nat Med. 2007;13:868–873. doi: 10.1038/nm1591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daly C, Eichten A, Castanaro C, Pasnikowski E, Adler A, Lalani AS, Papadopoulos N, Kyle AH, Minchinton AI, Yancopoulos GD, Thurston G. Angiopoietin-2 functions as a Tie2 agonist in tumor models, where it limits the effects of VEGF inhibition. Cancer Res. 2013;73:108–118. doi: 10.1158/0008-5472.CAN-12-2064. [DOI] [PubMed] [Google Scholar]
- Dejana E, Orsenigo F, Lampugnani MG. The role of adherens junctions and VE-cadherin in the control of vascular permeability. J Cell Sci. 2008;121:2115–2122. doi: 10.1242/jcs.017897. [DOI] [PubMed] [Google Scholar]
- Dimitrijevic OB, Stamatovic SM, Keep RF, Andjelkovic AV. Effects of the chemokine CCL2 on blood-brain barrier permeability during ischemia-reperfusion injury. J Cereb Blood Flow Metab. 2006;26:797–810. doi: 10.1038/sj.jcbfm.9600229. [DOI] [PubMed] [Google Scholar]
- Durham-Lee JC, Wu Y, Mokkapati VU, Paulucci-Holthauzen AA, Nesic O. Induction of angiopoietin-2 after spinal cord injury. Neuroscience. 2012;202:454–464. doi: 10.1016/j.neuroscience.2011.09.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eady TN, Belayev L, Khoutorova L, Atkins KD, Zhang C, Bazan NG. Docosahexaenoic acid signaling modulates cell survival in experimental ischemic stroke penumbra and initiates long-term repair in young and aged rats. PLoS One. 2012;7:e46151. doi: 10.1371/journal.pone.0046151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan Y, Shen F, Frenzel T, Zhu W, Ye J, Liu J, Chen Y, Su H, Young WL, Yang GY. Endothelial progenitor cell transplantation improves long-term stroke outcome in mice. Ann Neurol. 2010;67:488–497. doi: 10.1002/ana.21919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fisher M, Feuerstein G, Howells DW, Hurn PD, Kent TA, Savitz SI, Lo EH. Update of the stroke therapy academic industry roundtable preclinical recommendations. Stroke. 2009;40:2244–2250. doi: 10.1161/STROKEAHA.108.541128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fox PL, DiCorleto PE. Fish oils inhibit endothelial cell production of platelet-derived growth factor-like protein. Science. 1988;241:453–456. doi: 10.1126/science.3393911. [DOI] [PubMed] [Google Scholar]
- Gan Y, Ji X, Hu X, Luo Y, Zhang L, Li P, Liu X, Yan F, Vosler P, Gao Y, Stetler RA, Chen J. Transgenic overexpression of peroxiredoxin-2 attenuates ischemic neuronal injury via suppression of a redox-sensitive pro-death signaling pathway. Antioxid Redox Signal. 2012;17:719–732. doi: 10.1089/ars.2011.4298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green TP, Fennell M, Whittaker R, Curwen J, Jacobs V, Allen J, Logie A, Hargreaves J, Hickinson DM, Wilkinson RW, Elvin P, Boyer B, Carragher N, Ple PA, Bermingham A, Holdgate GA, Ward WH, Hennequin LF, Davies BR, Costello GF. Preclinical anticancer activity of the potent, oral Src inhibitor AZD0530. Mol Oncol. 2009;3:248–261. doi: 10.1016/j.molonc.2009.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hacke W, Kaste M, Bluhmki E, Brozman M, Davalos A, Guidetti D, Larrue V, Lees KR, Medeghri Z, Machnig T, Schneider D, von KR, Wahlgren N, Toni D. Thrombolysis with alteplase 3 to 4.5 hours after acute ischemic stroke. N Engl J Med. 2008;359:1317–1329. doi: 10.1056/NEJMoa0804656. [DOI] [PubMed] [Google Scholar]
- Holopainen T, Saharinen P, D’Amico G, Lampinen A, Eklund L, Sormunen R, Anisimov A, Zarkada G, Lohela M, Helotera H, Tammela T, Benjamin LE, Yla-Herttuala S, Leow CC, Koh GY, Alitalo K. Effects of angiopoietin-2-blocking antibody on endothelial cell-cell junctions and lung metastasis. J Natl Cancer Inst. 2012;104:461–475. doi: 10.1093/jnci/djs009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu X, Li P, Guo Y, Wang H, Leak RK, Chen S, Gao Y, Chen J. Microglia/macrophage polarization dynamics reveal novel mechanism of injury expansion after focal cerebral ischemia. Stroke. 2012;43:3063–3070. doi: 10.1161/STROKEAHA.112.659656. [DOI] [PubMed] [Google Scholar]
- Hu X, Zhang F, Leak RK, Zhang W, Iwai M, Stetler RA, Dai Y, Zhao A, Gao Y, Chen J. Transgenic overproduction of omega-3 polyunsaturated fatty acids provides neuroprotection and enhances endogenous neurogenesis after stroke. Curr Mol Med. 2013;13:1465–1473. doi: 10.2174/15665240113139990075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishikawa H, Tajiri N, Shinozuka K, Vasconcellos J, Kaneko Y, Lee HJ, Mimura O, Dezawa M, Kim SU, Borlongan CV. Vasculogenesis in experimental stroke after human cerebral endothelial cell transplantation. Stroke. 2013;44:3473–3481. doi: 10.1161/STROKEAHA.113.001943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones N, Iljin K, Dumont DJ, Alitalo K. Tie receptors: new modulators of angiogenic and lymphangiogenic responses. Nat Rev Mol Cell Biol. 2001;2:257–267. doi: 10.1038/35067005. [DOI] [PubMed] [Google Scholar]
- Kang JX, Wang J, Wu L, Kang ZB. Transgenic mice: fat-1 mice convert n-6 to n-3 fatty acids. Nature. 2004;427:504. doi: 10.1038/427504a. [DOI] [PubMed] [Google Scholar]
- Kim HJ, Vosseler CA, Weber PC, Erl W. Docosahexaenoic acid induces apoptosis in proliferating human endothelial cells. J Cell Physiol. 2005;204:881–888. doi: 10.1002/jcp.20351. [DOI] [PubMed] [Google Scholar]
- Kim I, Kim HG, Moon SO, Chae SW, So JN, Koh KN, Ahn BC, Koh GY. Angiopoietin-1 induces endothelial cell sprouting through the activation of focal adhesion kinase and plasmin secretion. Circ Res. 2000;86:952–959. doi: 10.1161/01.res.86.9.952. [DOI] [PubMed] [Google Scholar]
- Krupinski J, Kaluza J, Kumar P, Kumar S, Wang JM. Role of angiogenesis in patients with cerebral ischemic stroke. Stroke. 1994;25:1794–1798. doi: 10.1161/01.str.25.9.1794. [DOI] [PubMed] [Google Scholar]
- Lee B, Clarke D, Al AA, Kahle M, Parham C, Auckland L, Shaw C, Fidanboylu M, Orr AW, Ogunshola O, Fertala A, Thomas SA, Bix GJ. Perlecan domain V is neuroprotective and proangiogenic following ischemic stroke in rodents. J Clin Invest. 2011;121:3005–3023. doi: 10.1172/JCI46358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang S, Pong K, Gonzales C, Chen Y, Ling HP, Mark RJ, Boschelli F, Boschelli DH, Ye F, Barrios Sosa AC, Mansour TS, Frost P, Wood A, Pangalos MN, Zaleska MM. Neuroprotective profile of novel SRC kinase inhibitors in rodent models of cerebral ischemia. J Pharmacol Exp Ther. 2009;331:827–835. doi: 10.1124/jpet.109.156562. [DOI] [PubMed] [Google Scholar]
- Liu J, Wang Y, Akamatsu Y, Lee CC, Stetler RA, Lawton MT, Yang GY. Vascular remodeling after ischemic stroke: Mechanisms and therapeutic potentials. Prog Neurobiol. 2014;115C:138–156. doi: 10.1016/j.pneurobio.2013.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lobov IB, Brooks PC, Lang RA. Angiopoietin-2 displays VEGF-dependent modulation of capillary structure and endothelial cell survival in vivo. Proc Natl Acad Sci U S A. 2002;99:11205–11210. doi: 10.1073/pnas.172161899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maisonpierre PC, Suri C, Jones PF, Bartunkova S, Wiegand SJ, Radziejewski C, Compton D, McClain J, Aldrich TH, Papadopoulos N, Daly TJ, Davis S, Sato TN, Yancopoulos GD. Angiopoietin-2, a natural antagonist for Tie2 that disrupts in vivo angiogenesis. Science. 1997;277:55–60. doi: 10.1126/science.277.5322.55. [DOI] [PubMed] [Google Scholar]
- Mukutmoni-Norris M, Hubbard NE, Erickson KL. Modulation of murine mammary tumor vasculature by dietary n-3 fatty acids in fish oil. Cancer Lett. 2000;150:101–109. doi: 10.1016/s0304-3835(99)00380-8. [DOI] [PubMed] [Google Scholar]
- Musiek ES, Brooks JD, Joo M, Brunoldi E, Porta A, Zanoni G, Vidari G, Blackwell TS, Montine TJ, Milne GL, McLaughlin B, Morrow JD. Electrophilic cyclopentenone neuroprostanes are anti-inflammatory mediators formed from the peroxidation of the omega-3 polyunsaturated fatty acid docosahexaenoic acid. J Biol Chem. 2008;283:19927–19935. doi: 10.1074/jbc.M803625200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palmer TD, Willhoite AR, Gage FH. Vascular niche for adult hippocampal neurogenesis. J Comp Neurol. 2000;425:479–494. doi: 10.1002/1096-9861(20001002)425:4<479::aid-cne2>3.0.co;2-3. [DOI] [PubMed] [Google Scholar]
- Park JA, Lee HS, Ko KJ, Park SY, Kim JH, Choe G, Kweon HS, Song HS, Ahn JC, Yu YS, Kim KW. Meteorin regulates angiogenesis at the gliovascular interface. Glia. 2008;56:247–258. doi: 10.1002/glia.20600. [DOI] [PubMed] [Google Scholar]
- Petty MA, Lo EH. Junctional complexes of the blood-brain barrier: permeability changes in neuroinflammation. Prog Neurobiol. 2002;68:311–323. doi: 10.1016/s0301-0082(02)00128-4. [DOI] [PubMed] [Google Scholar]
- Rose DP, Connolly JM. Antiangiogenicity of docosahexaenoic acid and its role in the suppression of breast cancer cell growth in nude mice. Int J Oncol. 1999;15:1011–1015. doi: 10.3892/ijo.15.5.1011. [DOI] [PubMed] [Google Scholar]
- Scholzen T, Gerdes J. The Ki-67 protein: from the known and the unknown. J Cell Physiol. 2000;182:311–322. doi: 10.1002/(SICI)1097-4652(200003)182:3<311::AID-JCP1>3.0.CO;2-9. [DOI] [PubMed] [Google Scholar]
- Spencer L, Mann C, Metcalfe M, Webb M, Pollard C, Spencer D, Berry D, Steward W, Dennison A. The effect of omega-3 FAs on tumour angiogenesis and their therapeutic potential. Eur J Cancer. 2009;45:2077–2086. doi: 10.1016/j.ejca.2009.04.026. [DOI] [PubMed] [Google Scholar]
- Stetler RA, Cao G, Gao Y, Zhang F, Wang S, Weng Z, Vosler P, Zhang L, Signore A, Graham SH, Chen J. Hsp27 protects against ischemic brain injury via attenuation of a novel stress-response cascade upstream of mitochondrial cell death signaling. J Neurosci. 2008;28:13038–13055. doi: 10.1523/JNEUROSCI.4407-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su H, Takagawa J, Huang Y, Arakawa-Hoyt J, Pons J, Grossman W, Kan YW. Additive effect of AAV-mediated angiopoietin-1 and VEGF expression on the therapy of infarcted heart. Int J Cardiol. 2009;133:191–197. doi: 10.1016/j.ijcard.2007.12.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Summy JM, Trevino JG, Lesslie DP, Baker CH, Shakespeare WC, Wang Y, Sundaramoorthi R, Metcalf CA, III, Keats JA, Sawyer TK, Gallick GE. AP23846, a novel and highly potent Src family kinase inhibitor, reduces vascular endothelial growth factor and interleukin-8 expression in human solid tumor cell lines and abrogates downstream angiogenic processes. Mol Cancer Ther. 2005;4:1900–1911. doi: 10.1158/1535-7163.MCT-05-0171. [DOI] [PubMed] [Google Scholar]
- Szymczak M, Murray M, Petrovic N. Modulation of angiogenesis by omega-3 polyunsaturated fatty acids is mediated by cyclooxygenases. Blood. 2008;111:3514–3521. doi: 10.1182/blood-2007-08-109934. [DOI] [PubMed] [Google Scholar]
- Towfighi A, Saver JL. Stroke declines from third to fourth leading cause of death in the United States: historical perspective and challenges ahead. Stroke. 2011;42:2351–2355. doi: 10.1161/STROKEAHA.111.621904. [DOI] [PubMed] [Google Scholar]
- Tsuzuki T, Shibata A, Kawakami Y, Nakagawa K, Miyazawa T. Conjugated eicosapentaenoic acid inhibits vascular endothelial growth factor-induced angiogenesis by suppressing the migration of human umbilical vein endothelial cells. J Nutr. 2007;137:641–646. doi: 10.1093/jn/137.3.641. [DOI] [PubMed] [Google Scholar]
- Turgeon J, Dussault S, Maingrette F, Groleau J, Haddad P, Perez G, Rivard A. Fish oil-enriched diet protects against ischemia by improving angiogenesis, endothelial progenitor cell function and postnatal neovascularization. Atherosclerosis. 2013;229:295–303. doi: 10.1016/j.atherosclerosis.2013.05.020. [DOI] [PubMed] [Google Scholar]
- Wei D, Li J, Shen M, Jia W, Chen N, Chen T, Su D, Tian H, Zheng S, Dai Y, Zhao A. Cellular production of n-3 PUFAs and reduction of n-6-to-n-3 ratios in the pancreatic beta-cells and islets enhance insulin secretion and confer protection against cytokine-induced cell death. Diabetes. 2010;59:471–478. doi: 10.2337/db09-0284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang SP, Morita I, Murota SI. Eicosapentaenoic acid attenuates vascular endothelial growth factor-induced proliferation via inhibiting Flk-1 receptor expression in bovine carotid artery endothelial cells. J Cell Physiol. 1998;176:342–349. doi: 10.1002/(SICI)1097-4652(199808)176:2<342::AID-JCP12>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]
- Yin KJ, Hamblin M, Chen YE. Angiogenesis-Regulating microRNAs and Ischemic Stroke. Curr Vasc Pharmacol. 2013 doi: 10.2174/15701611113119990016. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zachary I. Signaling mechanisms mediating vascular protective actions of vascular endothelial growth factor. Am J Physiol Cell Physiol. 2001;280:C1375–C1386. doi: 10.1152/ajpcell.2001.280.6.C1375. [DOI] [PubMed] [Google Scholar]
- Zan L, Zhang X, Xi Y, Wu H, Song Y, Teng G, Li H, Qi J, Wang J. Src regulates angiogenic factors and vascular permeability after focal cerebral ischemia-reperfusion. Neuroscience. 2014;262:118–128. doi: 10.1016/j.neuroscience.2013.12.060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang M, Wang S, Mao L, Leak RK, Shi Y, Zhang W, Hu X, Sun B, Cao G, Gao Y, Xu Y, Chen J, Zhang F. Omega-3 Fatty Acids Protect the Brain against Ischemic Injury by Activating Nrf2 and Upregulating Heme Oxygenase 1. J Neurosci. 2014;34:1903–1915. doi: 10.1523/JNEUROSCI.4043-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang W, Hu X, Yang W, Gao Y, Chen J. Omega-3 polyunsaturated fatty acid supplementation confers long-term neuroprotection against neonatal hypoxic-ischemic brain injury through anti-inflammatory actions. Stroke. 2010;41:2341–2347. doi: 10.1161/STROKEAHA.110.586081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang ZG, Chopp M. Neurorestorative therapies for stroke: underlying mechanisms and translation to the clinic. Lancet Neurol. 2009;8:491–500. doi: 10.1016/S1474-4422(09)70061-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang ZG, Zhang L, Jiang Q, Zhang R, Davies K, Powers C, Bruggen N, Chopp M. VEGF enhances angiogenesis and promotes blood-brain barrier leakage in the ischemic brain. J Clin Invest. 2000;106:829–838. doi: 10.1172/JCI9369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu Y, Lee C, Shen F, Du R, Young WL, Yang GY. Angiopoietin-2 facilitates vascular endothelial growth factor-induced angiogenesis in the mature mouse brain. Stroke. 2005;36:1533–1537. doi: 10.1161/01.STR.0000170712.46106.2e. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Mice were subjected to 60 minute-long transient middle cerebral artery occlusion (tMCAO). Neurobehavioral tests were performed up to 21 d after tMCAO and brain tissues were harvested for various analyses at indicated times. For AZD0530 treatments, two endpoints (7 and 14 d) were used. The drug was given daily starting from 3 d after tMCAO until the day before sacrifice, as described in Materials and Methods. To label recently proliferated cells, BrdU was injected intraperitoneally twice per day with an interval of 8 h at 3–6 d and again at 10–13 d after MCAO.
Fatty acid analyses were performed on 3–5 month-old wild type (Wt) and fat-1 transgenic (Tg) mice with gas chromatography. A. The overall ratio of n-3 to n-6 fatty acids was calculated from total n-3 and n-6 fatty acid content in tail, liver and brain samples. Tg fat-1 mice exhibited a substantial increase in the n-3/n-6 ratio in all three tissues compared to their Wt littermates. B–C. The individual content of five major n-6 PUFAs (B) and five major n-3 PUFAs (C) was measured in brain samples from Wt and fat-1 Tg mice. Data are presented as mean ± SEM, n=5 animals/group. *p≤0.05, **p≤0.01, ***p≤0.001 fat-1 vs. Wt. Of the five major n-6 PUFAs examined individually in the brain, the content of three fatty acids was decreased significantly in fat-1 mice, including arachidonic acid (AA, C20:4), adrenic acid (C22:4) and docosapentaenoic acid (DPA, C22:5). No difference was detected for the content of linoleic acid (LA, C18:2) and eicosadienoic acid (EDA, C20:2) between Wt and fat-1 mice. On the other hand, n-3 PUFA production was significantly elevated in fat-1 mice, accounting for the increase in the overall n-3/n-6 PUFA ratio. The content of three major n-3 fatty acids was elevated in fat-1 mouse brains, including eicosapentaenoic acid (EPA, C20:5), docosapentaenoic acid (DPA, C22:5) and docosahexaenoic acid (DHA, C22:6), while no significant difference was detected for two other n-3 fatty acids, alpha-linolenic acid (ALA, C18:3) and eicosatrienoic acid (ETE, C20:3). In summary, these lipid profiles reflect a shift from n-6 to n-3 PUFA production in fat-1 Tg mice, as a result of high expression of the n-3 fatty acid desaturase.
Mouse primary brain microvascular endothelial cells (ECs) were treated with DHA/EPA (3μM/7μM) or vehicle for 48 h. A. EC proliferation was assessed after 1–5 d by counting immunostained Ki67+ cells, as described in Materials and Methods. B. EC barrier formation was evaluated by measurement of paracellular permeability after 1–5 d. Paracellular permeability was calculated from the diffusion coefficient of the 40 kDa FITC-Dextran and expressed as a percentage of vehicle-treated cultures at 1 d. Data are presented as mean ± SEM. n=3 independent cultures/group. *p≤0.05 vs. vehicle control.
A. Representative Western blots of phosphorylated Abl (Tyr 89), and total Abl in the ipsilateral hemispheres of Wt and fat-1 brains after sham operation (S), or at 7 and 14 d after tMCAO. AZD0530 (20 mg/kg) was administered as described in Materials and Methods. β-actin was used as an internal loading control. B. Protein level of p-Abl was quantified and expressed relative to Sham controls. No significant difference in p-Abl levels was observed among groups at 7 d or 14 d after tMCAO. Data are presented as mean ± SEM, n=5 animals/group.