

Abstract
Cells of Flavobacterium johnsoniae glide rapidly over surfaces by an unknown mechanism. Seven genes (gldA, gldB, gldD, gldF, gldG, gldH, and ftsX) that are required for gliding motility have been described. Complementation of the nonmotile mutants UW102-41, UW102-85, and UW102-92 identified another gene, gldI, that is required for gliding motility. gldI mutants formed nonspreading colonies, and individual cells were completely nonmotile. They were also resistant to bacteriophages that infect wild-type cells, and they failed to digest chitin. Introduction of wild-type gldI on a plasmid restored colony spreading, cell motility, phage sensitivity, and the ability to digest chitin to the gldI mutants. gldI encodes a predicted 199-amino-acid protein that localized to the membrane fraction. Labeling studies with [3H]palmitate indicated that GldI is a lipoprotein. GldI is similar to peptidyl-prolyl cis/trans-isomerases of the FK506-binding protein family and may be involved in folding cell envelope protein components of the motility machinery.
Many members of the Cytophaga-Flavobacterium-Bacteroides group exhibit rapid gliding motility. Cells of Flavobacterium johnsoniae glide over surfaces at speeds of up to 10 μm/s. Cells suspended in liquid do not swim, but their motility apparatus continues to function, since latex spheres that bind to a cell are rapidly propelled along multiple paths on the cell surface (27). The mechanism of F. johnsoniae gliding motility is not known, but several models have been proposed. Lapidus and Berg (22) suggested that outer membrane components are driven along tracks by periplasmic and cytoplasmic membrane proteins that obtain energy from the proton motive force. McBride et al. (24) postulated that coordinated export and import of polysaccharide, protein, or other macromolecules may form “conveyor belts” along the cell surface which propel cells.
Techniques to genetically manipulate F. johnsoniae have been developed (25), and a number of genes and proteins that are required for gliding have been described (1, 18-20, 24). Strains with mutations in gldA, gldB, gldD, gldF, gldG, or gldH form nonspreading colonies, and individual cells lack the gliding movements and ability to propel latex spheres along their surfaces that are characteristic of wild-type cells. Mutations in any of these genes also result in resistance to bacteriophages that infect wild-type cells (1, 18-20, 24) and loss of the ability to utilize the insoluble polysaccharide chitin (24). GldA, GldF, and GldG are thought to form an ATP-binding cassette transporter (18), and GldB, GldD, and GldH are lipoproteins that are required for gliding (24).
This paper describes the identification of gldI, which encodes another lipoprotein that is required for F. johnsoniae gliding motility, chitin utilization, and bacteriophage sensitivity. The GldI sequence is similar to the sequences of members of the FK506-binding protein (FKBP) family of peptidyl-prolyl cis/trans-isomerases (PPIases), which are involved in protein folding.
MATERIALS AND METHODS
Bacterial and bacteriophage strains, plasmids, and growth conditions.
F. johnsoniae UW101 (derived from the F. johnsoniae type strain ATCC 17061) was the wild-type strain used in this study, and all mutants were derived from this strain. The 50 nonmotile mutants of F. johnsoniae (obtained from J. Pate) were previously described (5, 20, 39). The bacteriophages active against F. johnsoniae that were used in this study were φCj1, φCj7, φCj13, φCj23, φCj29, φCj42, φCj48, and φCj54 (5, 28, 39). The Escherichia coli strains used were DH5αMCR (GibcoBRL Life Technologies) and S17-1 (33). E. coli strains were grown in Luria-Bertani medium at 37°C, and F. johnsoniae strains were grown in Casitone-yeast extract (CYE) medium at 30°C as previously described (25). To observe colony spreading, F. johnsoniae was grown on PY2 agar medium (1) at 25°C. Chitin utilization was observed essentially as previously described (24), except that MYA medium (0.5 mM MgSO4, 0.05 mM FeSO4, 0.04 mM EDTA, 20 mM potassium phosphate [pH 7.25], 0.1 g of yeast extract per liter, 15 g of agar per liter) was used instead of PY2 agar medium. Chitin powder (practical grade from crab shells; Sigma Chemical Co., St. Louis, Mo.) was prepared as a 2% slurry essentially as described previously (30), and 3 ml of the chitin slurry was allowed to dry on top of solid MYA medium in 9-cm-diameter petri dishes. For radiolabeling experiments, cells were grown in SDY medium (0.5 mM MgSO4, 0.05 mM FeSO4, 0.04 mM EDTA, 0.2 mM CaCl2, 18.7 mM NH4Cl, 22.2 mM glucose, 0.1 g of yeast extract per liter, 20 mM potassium phosphate [pH 7.25]) as previously described (24). Antibiotics were used at the following concentrations when needed: ampicillin, 100 μg/ml; erythromycin, 100 μg/ml; kanamycin, 30 μg/ml; tetracycline, 20 μg/ml. Plasmids and primers used in this study are listed in Table 1.
TABLE 1.
Plasmids and primers used in this studya
| Plasmid or primer | Descriptionb | Reference |
|---|---|---|
| Plasmids | ||
| pT7Blue | ColE1 ori; Apr | Novagen |
| pHP45Ωkan | Plasmid carrying Kmr cassette with transcriptional terminators at each end; Apr Kmr | 8 |
| pCP11 | E. coli-F. johnsoniae shuttle plasmid; Apr (Emr) | 25 |
| pCP23 | E. coli-F. johnsoniae shuttle plasmid; Apr (Tcr) | 1 |
| pCP26 | E. coli-F. johnsoniae shuttle cosmid; Kmr Tcr (Emr) | 20 |
| pCP500 | Cosmid clone carrying gldI; Tcr (Emr) | This study |
| pCP507 | Cosmid clone carrying gldI; Tcr (Emr) | This study |
| pMM258 | 6.3-kbp SacI fragment of pCP500 (spanning ppiB, fjo20, and fjo21) in pCP11; Apr Tcr (Emr) | This study |
| pMM259 | 6.6-kbp SacI fragment of pCP507 (spanning fjo19 and gldI) in pCP11; Apr Tcr (Emr) | This study |
| pMM268 | 2.3-kbp EcoRI-EcoRV fragment of pMM259 (spanning fjo19) in pCP11; Apr (Emr) | This study |
| pMM291 | 1,032-bp fragment spanning gldI in pCP11; Apr (Emr) | This study |
| pMM292 | Identical to pMM291 except that gldI is inserted in the opposite orientation; Apr (Emr) | This study |
| pMM296 | pMM291 with the Kmr cassette from pHP45Ωkan inserted upstream of gldI; Apr Kmr (Emr) | This study |
| pMM297 | Identical to pMM296 except that the Kmr cassette is inserted in the opposite orientation; Apr Kmr (Emr) | This study |
| pTB39 | Recombinant gldB-his in pCP23; expresses GldI with a carboxy-terminal His tag; Apr (Tcr) | 24 |
| pTB45 | Recombinant gldI-his in pCP23; expresses GldI with a carboxy-terminal His tag; Apr (Tcr) | This study |
| Primers | ||
| 459 | 5′ GAATAAAACGAGCTAACGGC 3′; primer used for construction of gldI-containing plasmids pMM291 and pMM292 | |
| 476 | 5′ TCTTACATGACTTTGACTCAGG 3′; primer used for construction of gldI-containing plasmids and pTB45 | |
| 574 | 5′ TTATTAATGATGATGATGATGATGATGATGTGGGTTTAATGTATCTTTTTTAGTTTGAGCTGC 3′; primer used during construction of pTB45 |
Antibiotic resistance phenotypes are indicated as follows: Apr, ampicillin resistance; Emr, erythromycin resistance; Kmr, kanamycin resistance; Tcr, tetracycline resistance. Unless indicated otherwise, antibiotic resistance phenotypes are those expressed in E. coli. Antibiotic resistance phenotypes listed in parentheses are those expressed in F. johnsoniae but not in E. coli.
For primers, both the sequence and description are given.
Cloning of gldI.
GldI was cloned from a cosmid library of wild-type F. johnsoniae DNA in pCP26 essentially as previously described (20). Cosmids were transferred into the nonmotile mutant UW102-41 by conjugation, and complemented (spreading) colonies were isolated. The cosmids pCP500 and pCP507, which share a 7-kbp region of overlap (Fig. 1), each complemented UW102-41. Subclones were generated to determine the minimal region required for complementation. pMM258 was constructed by inserting the 6.3-kbp SacI fragment of pCP500, which has 3.0 kbp of vector DNA (including tetR and tetA) and 3.3 kbp of F. johnsoniae DNA (including ppiB, fjo20, and fjo21), into pCP11. pMM259 was constructed by inserting the 6.6-kbp SacI fragment of pCP507, which has the same 3.0-kbp fragment of vector DNA and 3.6 kbp of F. johnsoniae DNA (including fjo19 and gldI), into pCP11. pMM268 was constructed by cloning the 2.3-kbp EcoRI-EcoRV fragment of pMM259 spanning fjo19 into the SmaI site of pCP11. To construct a clone carrying just gldI, a 1,032-bp fragment spanning gldI was amplified by PCR using primers 476 and 459. This fragment was cloned into the EcoRV site of pT7Blue and transferred as a SacI-XbaI fragment into pCP11 to generate pMM291 and pMM292. pMM292 is identical to pMM291 except that gldI is inserted in the opposite orientation. pMM296 was generated by inserting the 2.1-kbp BamHI fragment of pHP45Ωkan, which carries a kanamycin resistance cassette with transcription termination signals on each end, into the SmaI site of pMM291. pMM297 was identical to pMM296 except that the kanamycin resistance cassette was inserted in the opposite orientation. For complementation analyses, plasmids were introduced into the F. johnsoniae mutants by conjugation or electroporation as previously described (20, 25).
FIG. 1.

Map of the gldI region of F. johnsoniae. Restriction sites are indicated as follows: B, BglII; E, EcoRI; RV, EcoRV; and S, SacI. Kilobase pair positions 2, 4, and 6 are indicated. The presence (+) or absence (−) of complementation of gldI mutants by fragments cloned into shuttle vectors is indicated beneath the map.
Construction of pTB45.
A modified version of gldI which encodes GldI-His (GldI containing eight histidine residues at the carboxy terminus) was constructed to allow isolation and detection of recombinant GldI-His from F. johnsoniae extracts. gldI was amplified using elongase (Life Technologies) and primers 476 and 574. The product was cloned into the SmaI site of pCP23 to generate pTB45.
Nucleic acid sequencing.
Nucleic acid sequencing was performed by the dideoxynucleotide procedure using an automated sequencing system (Applied Biosystems). Sequences were analyzed with MacVector and AssemblyLign software (Oxford Molecular Group Inc., Campbell, Calif.), and comparisons to database sequences were made using the BLAST (2) and FASTA (29) algorithms. Predictions regarding cellular localization were made using PSORT (http://PSORT.NIBB.AC.JP/) (26).
Microscopic observations of cell movement.
Wild-type and mutant cells of F. johnsoniae were examined for movement over glass and agar surfaces and for their ability to propel polystyrene latex spheres by phase-contrast microscopy as previously described (24).
Measurements of bacteriophage sensitivity.
Sensitivity to F. johnsoniae bacteriophages was determined essentially as previously described by spotting 2.5 μl of phage lysates (6 × 107 PFU/ml) onto lawns of cells in CYE overlay agar (20). The plates were incubated for 24 h at 25°C to observe lysis.
Cell fractionation and Western blot analyses.
F. johnsoniae cells were disrupted with a French press and fractionated into soluble and membrane fractions by centrifugation at 223,160 × g for 60 min as described previously (18). Proteins were separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE), and Western blot analyses were performed as previously described (18) except that GldI-His was detected using His-Tag monoclonal antibody (Novagen, Madison, Wis.). Protein concentrations were measured with the bicinchoninic acid reagent (Pierce, Rockford, Ill.).
In vivo radiolabeling with [3H]palmitate and [3H]glutamate.
To identify lipoproteins, cells of wild-type and mutant strains of F. johnsoniae were incubated for 3 h in SDY broth containing 50 μCi of [3H]palmitate or [3H]glutamate per ml. Specifically, [9,10-3H]palmitate (48 Ci/mmol; Perkin-Elmer Life Sciences, Boston, Mass.) or l-[3,4-3H]glutamic acid (51 Ci/mmol; Perkin-Elmer Life Sciences) was used to label the proteins. Radiolabeled proteins were separated by SDS-PAGE and detected by autoradiography as previously described (24). To determine whether GldI is a lipoprotein, cells expressing GldB-His and cells expressing GldI-His were labeled with [3H]palmitate and [3H]glutamate as previously described (24). Cells were lysed, recombinant His-tagged proteins were isolated using Ni-NTA His-Bind resin (Novagen) and separated by SDS-PAGE, and radiolabeled proteins were detected by autoradiography as described previously (24).
Genetic nomenclature.
Genes required for gliding motility were given the name gld followed by a letter. Open reading frames (ORFs) that exhibited strong sequence similarity to genes of known function were named after the corresponding genes. ORFs of unknown function that did not exhibit strong similarity to previously described genes were given the provisional name fjo (for “F. johnsoniae ORF”) followed by a number.
Nucleotide sequence accession number.
The sequence reported in this paper has been deposited in the GenBank database under accession number AF527792.
RESULTS
Identification of gldI.
UW102-41 is a spontaneous nonmotile mutant of F. johnsoniae UW101 (5). Cells of UW102-41 formed nonspreading colonies on PY2 agar (Fig. 2B), whereas wild-type cells formed spreading colonies (Fig. 2A). Wild-type cells also exhibited rapid motility in wet mounts and actively propelled latex spheres along their surfaces, while UW102-41 cells did not exhibit motility in wet mounts and failed to propel latex spheres. A cosmid library containing wild-type F. johnsoniae DNA in pCP26 was transferred by conjugation into UW102-41, and spreading colonies were obtained. pCP500 and pCP507, which share a 7-kbp region of overlap, were isolated from two of the complemented mutants. Introduction of either plasmid into UW102-41 restored the ability of cells to glide over glass surfaces, to propel latex spheres, and to form spreading colonies.
FIG. 2.

Photomicrographs of F. johnsoniae colonies. Colonies were grown for 30 h at 25°C on PY2 agar medium. Photomicrographs were taken with a Kodak DC290 digital camera mounted on an Olympus IMT-2 inverted microscope. (A) Wild-type F. johnsoniae UW101; (B) gldI mutant UW102-41; (C) UW102-41 complemented with pMM291, which carries gldI. Bar, 1 mm.
The nucleotide sequence of the 7-kbp region of overlap between pCP500 and pCP507 was determined, and eight genes were identified (Fig. 1). Plasmids carrying portions of this DNA were used to determine the minimal region required for complementation of UW102-41. Introduction of pMM259 (which contains fjo19 and gldI), pMM291 (which contains gldI), or pMM292 (which contains gldI) into UW102-41 resulted in complementation (Fig. 1 and 2C). The colonies spread over agar, and cells exhibited rapid gliding motility in wet mounts and propelled latex spheres. In contrast, introduction of pMM258 (which contains ppiB, fjo20, and fjo21) and pMM268 (which contains fjo19) into UW102-41 did not restore colony spreading, cell movement, or sphere movement. To determine whether gldI on pMM291 was expressed from its native promoter or from a promoter in the vector, the omega kanamycin resistance cassette (8), which has transcription terminators at each end, was inserted 313 bp upstream of the gldI start codon in pMM291 to generate pMM296 and pMM297. This cassette has previously been shown to disrupt expression of F. johnsoniae genes (24). pMM296 and pMM297 restored colony spreading, motility, and sphere movement to UW102-41, indicating that the 1,032-bp region contained on pMM291 was sufficient for expression of gldI.
Identification of additional gldI mutants.
Strain UW102-41 is one of 50 spontaneous and chemically induced nonmotile mutants isolated by Chang et al. (5) and Wolkin and Pate (39). The genes that are mutated in 18 of these mutants have been identified (1, 18-20, 24). pCP500 was introduced into the remaining 32 mutants to determine whether gldI or nearby genes are defective in any of these mutants. Introduction of pCP500 restored motility to UW102-85 and UW102-92 in addition to UW102-41 but did not restore motility to the other mutants. Introduction of pMM291, which spans just gldI, was sufficient to restore motility to UW102-85, UW102-92, and UW102-41. The exact sites of the mutations in each mutant were determined by amplification and sequencing of the gldI genes. Each mutation was the result of insertion or deletion of a single nucleotide. UW102-41 had a G deleted at position 42 (numbered from the A of the gldI start codon), while UW102-85 had a G inserted at position 342, and UW102-92 had a C deleted at position 91.
Analysis of gldI and surrounding DNA. gldI encodes a predicted primary product of 218 amino acid residues. The amino-terminal sequence (MNYLKISIYSLLFAVLLAGCKHHEE) contains a hydrophobic region terminated by a cysteine (underlined), which is characteristic of lipoproteins. Lipoproteins undergo a series of modifications that result in cleavage of the signal peptide and covalent attachment of multiple lipid moieties to the amino-terminal cysteine (13). These modifications result in localization to either the cytoplasmic membrane or the outer membrane, depending on the particular protein. The predicted molecular mass of the final GldI protein (after cleavage of the signal peptide but without lipid modifications) is 22.3 kDa. The GldI sequence is similar to the sequences of PPIases of the FKBP family (Fig. 3), such as the Legionella pneumophila macrophage infectivity potentiator Mip (27% identity over 139 amino acids) (7) and Neisseria meningitidis MC58 Mip (29% identity over 137 amino acids) (36). Given the moderate level of similarity, we cannot conclude that GldI functions as a PPIase, but critical regions of PPIases, such as the AYG triplet at residues 154 to 156 that is thought to be part of the active site of other FKBPs (11, 31), are present in GldI. GldI is predicted to have a basic isoelectric point (pI 9.49), a trait that it shares with L. pneumophila Mip (predicted pI 8.76) but not with N. meningitidis Mip (predicted pI 5.32).
FIG. 3.

Alignment of F. johnsoniae GldI sequence with L. pneumophila Mip sequence. Identical residues are boxed and shaded. The numbers are amino acid residue positions starting from the amino terminus of each protein. Full-length GldI has 218 amino acids, and Mip has 233 amino acids. Gaps introduced to maximize alignment are indicated by dashes.
The two genes that lie immediately downstream of gldI (ppiA and ppiB) encode putative PPIases that are similar to each other (58% identity over 328 amino acids). The amino-terminal 200-amino-acid sequences of PpiA and PpiB are similar to the sequences of members of the cyclophilin/rotamase family of PPIases, such as Treponema pallidum PpiB (32). F. johnsoniae PpiA and PpiB exhibit 56% identity over 174 amino acids and 53% identity over 163 amino acids to T. pallidum PpiB, respectively. Also, F. johnsoniae PpiA and PpiB each have a carboxy-terminal domain of approximately 125 residues that is similar to those of members of the FKBP family of PPIases (up to 56% amino acid identity), but they exhibit more limited similarity (36 and 32% identity, respectively, over 52 amino acid residues) to F. johnsoniae GldI. The functions of PpiA and PpiB in F. johnsoniae are not known, and there is no evidence linking either gene to gliding motility. The remaining genes in the 7-kbp region encode proteins with functions apparently unrelated to PPIases or to gliding motility. Fjo18 is similar to putative voltage-gated chloride channel proteins such as Pseudomonas syringae PSPT04619 (42% identity over 382 amino acids) (3). Fjo19 is similar to Mycoplasma genitalium MgpA (25% identity over 295 amino acids) whose exact function is not known (9). Fjo20 is similar to members of the drug/metabolite transporter family (National Center for Biotechnology Information [NCBI] conserved domain COG5006), Fjo21 does not display significant similarity to proteins of known function, and Fjo22, which is only partially sequenced, is similar to E. coli aquaporin AqpZ (84% identity over 78 amino acids) (4).
Mutation of gldI does not alter growth rate.
Disruption of a gene encoding a PPIase involved in protein folding could cause growth defects and result in loss of motility as a secondary effect. The growth rates at 30°C of wild-type cells and of cells of the gldI mutant UW102-41 were determined in a medium containing Casitone and yeast extract (CYE) and in a medium containing glucose as the primary carbon and energy source (SDY). The growth rates were similar for the two strains. The doubling times in CYE medium were 45 min (±4 min) for wild-type cells and 42 min (±5 min) for the gldI mutant. In SDY medium, the doubling times were 144 min (±9 min) for wild-type cells and 140 min (±8 min) for the gldI mutant.
Localization of GldI-His and effects of mutations in gldI on other Gld proteins.
To determine the probable localization of GldI, pTB45 expressing GldI-His was introduced into F. johnsoniae UW102-41. GldI-His was functional, since it restored motility to UW102-41 (data not shown). His-Tag monoclonal antisera was used to detect GldI-His in cell extracts. GldI-His migrated with an apparent molecular mass of approximately 27 kDa and was found primarily in the membrane fraction (Fig. 4). The similarity of GldI to PPIases involved in protein folding suggested that it might be required for folding and stability of proteins involved in gliding motility. Western blots developed with antibodies against GldA, GldB, GldG, and GldH were used to determine the effect of a mutation in gldI on the level of each of these proteins. Cells of wild-type F. johnsoniae and of the gldI mutant UW102-41 had approximately equal amounts of GldA, GldB, GldG, and GldH proteins (Fig. 5).
FIG. 4.
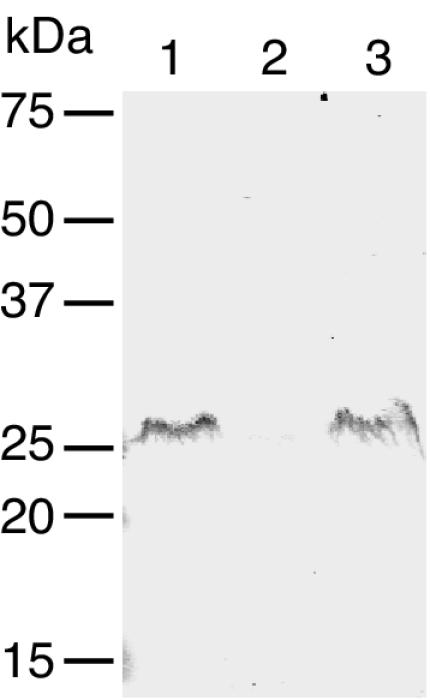
Localization of GldI-His. Cells of F. johnsoniae UW102-41 expressing GldI-His from pTB45 were fractionated, and GldI-His was detected by Western blot analysis. Lane 1, whole cells; lane 2, soluble fraction; lane 3, membrane fraction. Equal amounts of protein were loaded in each lane.
FIG. 5.

Effect of mutation in gldI on other Gld proteins. Cell extracts (20 μg of protein per lane) were examined for GldA (A), GldB (B), GldG (C), and GldH (D) proteins by Western blot analysis. In panels A to D, lane 1 contains wild-type cells and lane 2 contains cells of the gldI mutant UW102-41, while lane 3 contains cells of the gldA mutant F. johnsoniae CJ288 (A), cells of the gldB mutant F. johnsoniae CJ569 (B), cells of the gldFG mutant F. johnsoniae CJ787 (C), and cells of the gldH mutant F. johnsoniae CJ1043 (D).
GldI is a lipoprotein.
Sequence analysis suggested that F. johnsoniae GldI may be a lipoprotein. To detect lipoproteins, cells were labeled with [3H]palmitate as described in Materials and Methods. In a parallel experiment, cells were labeled with [3H]glutamate to label essentially all proteins. A limited subset of wild-type cell proteins were labeled with [3H]palmitate, and the results confirmed that, as previously described (24), label from [3H]palmitate was not rapidly incorporated into amino acids (Fig. 6A, compare lanes 1 and 2). The lipoprotein profiles of cells with mutations in gldI were largely similar to those of wild-type cells, but a band of approximately 70 kDa was not present in the lipoprotein profile of the gldI mutant and the intensities of several other bands were lower than those from the wild-type cells (Fig. 6A, lane 3). The 70-kDa lipoprotein, which is much larger than GldI, was also missing in cells with mutations in gldB, gldD, and gldH (24). The presence of abundant lipoproteins of 22 to 27 kDa complicated detection of GldI (predicted molecular mass of 22.3 kDa after cleavage of signal peptide but without lipid modification).
FIG. 6.

GldI is a lipoprotein. (A) Cells of F. johnsoniae were labeled with either [3H]glutamate (to label nearly all proteins) or [3H]palmitate (to label lipoproteins). Proteins were separated by SDS-PAGE and detected by autoradiography. Lane 1, wild-type cells labeled with [3H]glutamate; lane 2, wild-type cells labeled with [3H]palmitate; lane 3, cells of the gldI mutant UW102-41 labeled with [3H]palmitate; lane 4, cells of UW102-41 complemented with pMM291 (which carries gldI) labeled with [3H]palmitate. (B) Radiolabeling of GldB-His and GldI-His. Cells of F. johnsoniae were labeled with [3H]palmitate or [3H]glutamate. Proteins were isolated by precipitation with Ni-NTA His-Bind resin, separated by SDS-PAGE, and detected by autoradiography. Lane 1, cells expressing GldB-His from pTB39, labeled with [3H]palmitate; lane 2, cells expressing GldB-His from pTB39, labeled with [3H]glutamate; lane 3, cells expressing GldI-His from pTB45, labeled with [3H]palmitate; lane 4, cells expressing GldI-His from pTB45, labeled with [3H]glutamate.
To determine whether GldI was labeled, F. johnsoniae UW102-41 expressing recombinant GldI-His from pTB45 was utilized. Cells were incubated with [3H]palmitate or [3H]glutamate, and proteins were isolated using Ni-NTA His-Bind resin. The eluted proteins were separated by SDS-PAGE, and radiolabeled proteins were detected by autoradiography. Cells expressing the lipoprotein GldB-His from pTB39 (24) were processed similarly as a control. GldI-His, which migrated with an apparent molecular mass of 27 kDa, was labeled by [3H]palmitate, which suggests that it is a lipoprotein (Fig. 6B, lane 3). Cells carrying pTB39 produced radiolabeled GldB-His but, as expected, did not produce a labeled protein with a size similar to that of GldI-His (Fig. 6B, lane 1). This suggests that the 27-kDa band observed in Fig. 6B, lane 3, is GldI-His, rather than some other labeled protein. Incubation with [3H]palmitate resulted in more intense labeling of GldI-His than did incubation with [3H]glutamate (Fig. 6B, lanes 3 and 4), despite the fact that label from [3H]glutamate was more readily incorporated into most proteins (Fig. 6A, lanes 1 and 2). This suggests that labeling was not the result of metabolism of [3H]palmitate into amino acids before incorporation into GldI-His.
The gldI mutant UW102-41 is resistant to bacteriophage infection.
Many nonmotile mutants of F. johnsoniae are resistant to infection by bacteriophages (38). The sensitivity of F. johnsoniae strains UW101, UW102-41, and CJ1172 (UW102-41 carrying pMM291) to the F. johnsoniae bacteriophages φCj1, φCj13, φCj23, φCj28, φCj29, φCj42, φCj48, and φCj54 was tested. F. johnsoniae UW101 was lysed by these phages (Fig. 7A), whereas the gldI mutant UW102-41 was resistant to each of the phages (Fig. 7B). Introduction of wild-type gldI on pMM291 into UW102-41 restored bacteriophage sensitivity in addition to restoring gliding motility (Fig. 7C).
FIG. 7.

Effect of mutation in gldI on bacteriophage resistance. Bacteriophages (2.5 μl of lysates containing approximately 6 × 107 phage/ml) were spotted onto lawns of cells in CYE overlay agar. Bacteriophages were spotted onto three strains of F. johnsoniae, the wild-type strain (A), the gldI mutant strain UW102-41 (B), and strain UW102-41 complemented with pMM291 (which carries gldI) (C). The plates were incubated at 25°C for 24 h to observe lysis. Bacteriophages were applied to the bacteria in the following order from left to right: for the top row, φCj1, φCj13, and φCj23; for the middle row, φCj28, φCj29, and φCj42; for the bottom row, φCj48 and φCj54. The diameter of the petri dish is 9 cm.
The gldI mutant UW102-41 is defective in chitin utilization.
Wild-type cells of F. johnsoniae digest chitin (34), while many nonmotile mutants fail to utilize this insoluble polysaccharide (5, 24). The effect of a mutation in gldI on chitin utilization was determined in MYA medium supplemented with chitin as the primary carbon, energy, and nitrogen source. Wild-type cells readily digested chitin (Fig. 8A), while cells of the gldI mutant UW102-41 failed to digest chitin (Fig. 8B). Complementation with pMM291 restored the ability to digest chitin (Fig. 8C) in addition to restoring gliding motility. Similar results were obtained when utilization of chitin was tested on PY2 medium with chitin (data not shown). PY2-chitin medium contains peptone and yeast extract and allows growth of strains regardless of whether they are able to utilize chitin.
FIG. 8.

Effect of mutation in gldI on the ability to utilize chitin. Approximately 4 × 107 cells of wild-type F. johnsoniae (A), the gldI mutant strain UW102-41 (B), and strain UW102-41 complemented with pMM291 (which carries gldI) (C) were spotted on MYA-chitin medium and incubated for 6 days at 25°C.
DISCUSSION
The mechanism responsible for F. johnsoniae gliding motility is not known. Seven genes (gldA, gldB, gldD, gldF, gldG, gldH, and ftsX) that are required for gliding have previously been described (1, 18-21). GldA, GldF, and GldG are thought to constitute an ABC transporter that is required for cell movement. The functions of the other proteins in gliding are not known. The results presented in this paper identify another gene, gldI, that is required for gliding. Mutations in gldI eliminated gliding motility, and introduction of a wild-type copy of gldI on a plasmid restored motility to the mutants.
GldI exhibits sequence similarity to PPIases involved in protein folding. PPIases catalyze the isomerization of peptide bonds that are N terminal to proline residues (10) and are found in bacteria, archaea, and eukaryotes. Bacterial PPIases may be cytoplasmic, periplasmic, or membrane associated. PPIases can generally be assigned to one of three families that appear to have evolved independently: the cyclophilins, parvulins, and FKBPs. GldI is similar to bacterial members of the FKBP family, such as L. pneumophila Mip (7). L. pneumophila Mip is a cell surface protein that is involved in virulence and contributes to the ability of cells to survive within phagocytic cells (6, 14, 15). GldI may be involved in protein folding in the cell envelope. Cells with mutations in gldI exhibited normal growth rates, indicating that gldI is not essential for growth. GldI may be required for proper assembly of the motility apparatus or may play a role in the function of the apparatus during cell movement. One model to explain gliding proposes that export and import of macromolecules result in cell movement. If these transported macromolecules are proteins, GldI may be required to facilitate their folding and transport.
GldI appears to be a lipoprotein. F. johnsoniae produces many additional lipoproteins at least three of which, GldB, GldD, and GldH, are required for gliding (24). It is not known why such a large fraction of the identified Gld proteins are lipoproteins. The Gld proteins may assemble to form a multiprotein complex in the cell envelope, and membrane anchoring of the Gld lipoproteins may facilitate formation of this complex.
The properties of gldI mutants were similar to those of previously studied gld mutants (1, 18-20, 24). In addition to complete loss of cell movement and colony spreading, cells of gldI mutants, like those of other gld mutants, failed to propel latex spheres, were resistant to a variety of bacteriophages that infect wild-type cells, and failed to digest chitin. It has previously been suggested that gliding motility, chitin utilization, and bacteriophage infection may each rely on one or more transporters that are defective in the gld mutants, but the exact roles of the transporters in cell movement are not known (24).
During the course of this analysis, we found that F. johnsoniae UW101 digested chitin 5 to 10 times more rapidly than we have previously reported (24). Apparently the “wild-type” strain that we had used in previous work was a spontaneous mutant with decreased ability to utilize chitin. This strain, which was formerly referred to as F. johnsoniae UW101 and which we now refer to as F. johnsoniae MM101, was used to isolate all of the Tn4351-induced mutants that we have previously described (1, 18-21, 24). F. johnsoniae MM101 was obtained from J. Pate in 1990 and has been stored at −80°C since then. F. johnsoniae MM101 had probably been maintained without selection for the ability to digest chitin and had lost some but not all ability to attack this insoluble polysaccharide. F. johnsoniae MM101 forms spreading colonies and exhibits wild-type gliding motility.
The wild-type strain used in the present study (UW101) was recently revived from a lyophil prepared by J. Pate in 1982 and was the parent strain for the gldI point mutants described in this study (5). UW101 was also the parent for the strains with point mutations in gldA, gldB, gldD, gldF, gldG, and gldH that have been previously described (1, 5, 18-20, 24, 39). UW101 was originally derived from the F. johnsoniae type strain, ATCC 17061. The type strain is composed of two stable colony types (yellow and nonpigmented) that appear identical except for pigmentation. UW101 (and MM101) cells are descendants of a yellow colony of F. johnsoniae ATCC 17061. MM101 appears to be identical to UW101 except for the partial defect in ability to use chitin. Apparent defects in chitin utilization for strains derived from F. johnsoniae ATCC 17061 have previously been reported (12).
To determine whether UW101 or MM101 is more similar to the parent strain, F. johnsoniae ATCC 17061 was obtained from the American Type Culture Collection, and a single yellow colony was isolated. We refer to this strain as F. johnsoniae FJ1. FJ1 was indistinguishable from UW101 in all properties tested, including motility and the ability to digest chitin. Analysis of 4 kbp of DNA from F. johnsoniae strains UW101, MM101, and FJ1 revealed identical sequences, confirming that they are all derived from the same parent. The decreased chitin utilization ability of MM101 does not alter any of our conclusions from previous studies (1, 18-21, 24), but it is important to be aware of the history of each nonmotile mutant so that comparison of chitin degradative ability is made to the appropriate wild-type parent strain.
Gliding motility is not confined to the Cytophaga-Flavobacterium-Bacteroides group (23). Myxococcus xanthus, a gliding member of the delta subgroup of Proteobacteria, has two motility systems, the S (social) and A (adventurous) gliding motility systems (16, 17). S-motility relies on pilus extension and retraction (35, 40). The mechanism of A-motility is not known but may involve polysaccharide secretion (37). There are many differences between the gliding behavior of F. johnsoniae and M. xanthus (23). Cells of F. johnsoniae move much faster than cells of M. xanthus and exhibit behaviors such as rotary movements and sphere movements that are not characteristic of M. xanthus. F. johnsoniae does not appear to have multiple motility systems, since single mutations in gld genes result in the complete loss of motility. Finally, although many genes involved in F. johnsoniae gliding and M. xanthus motility systems have been identified, there is little sequence similarity between the known motility genes of these bacteria. This may indicate that gliding evolved independently in the Cytophaga-Flavobacterium-Bacteroides group and in the myxobacteria. The motility systems of F. johnsoniae and M. xanthus exhibit some common features despite the limited sequence similarity of known motility genes. Transporters have been linked to gliding of both organisms, and it is possible that the motility systems share additional features (1, 18, 37, 41, 42).
A number of proteins required for F. johnsoniae gliding have been identified, but the mechanism of movement remains uncertain. Further analysis of gldI and other gld genes and proteins will help to determine the mechanisms of F. johnsoniae gliding motility and chitin utilization.
Acknowledgments
This research was supported in part by grants from the National Science Foundation (MCB-9727825 and MCB-0130967) and by a Milwaukee Foundation Shaw Scientist Award to M.J.M.
DNA sequencing was performed by the Automated DNA Sequencing Facility of the University of Wisconsin—Milwaukee Department of Biological Sciences. Preliminary sequence data for Cytophaga hutchinsonii were obtained from The DOE Joint Genome Institute (JGI) (at http://jgi.doe.gov/). We thank D. Saffarini for careful reading of the manuscript.
REFERENCES
- 1.Agarwal, S., D. W. Hunnicutt, and M. J. McBride. 1997. Cloning and characterization of the Flavobacterium johnsoniae (Cytophaga johnsonae) gliding motility gene, gldA. Proc. Natl. Acad. Sci. USA 94:12139-12144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Altschul, S. F., W. Gish, W. Miller, E. W. Myers, and D. J. Lipman. 1990. Basic local alignment search tool. J. Mol. Biol. 215:403-410. [DOI] [PubMed] [Google Scholar]
- 3.Buell, C., V. Joardar, M. Lindeberg, J. Selengut, I. Paulsen, M. Gwinn, R. Dodson, R. Deboy, A. Durkin, J. Kolonay, R. Madupu, S. Daugherty, L. Brinkac, M. Beanan, D. Haft, W. Nelson, T. Davidsen, N. Zafar, L. Zhou, J. Liu, Q. Yuan, H. Khouri, N. Fedorova, B. Tran, D. Russell, K. Berry, T. Utterback, S. Van Aken, T. Feldblyum, M. D'Ascenzo, W. Deng, A. Ramos, J. Alfano, S. Cartinhour, A. Chatterjee, T. Delaney, S. Lazarowitz, G. Martin, D. Schneider, X. Tang, C. Bender, O. White, C. Fraser, and A. Collmer. 2003. The complete genome sequence of the Arabidopsis and tomato pathogen Pseudomonas syringae pv. tomato DC3000. Proc. Natl. Acad. Sci. USA 100:10181-10186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Calamita, G., W. Bishai, G. Preston, W. Guggino, and P. Agre. 1995. Molecular cloning and characterization of AqpZ, a water channel from Escherichia coli. J. Biol. Chem. 270:29063-29066. [DOI] [PubMed] [Google Scholar]
- 5.Chang, L. Y. E., J. L. Pate, and R. J. Betzig. 1984. Isolation and characterization of nonspreading mutants of the gliding bacterium Cytophaga johnsonae. J. Bacteriol. 159:26-35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cianciotto, N., and B. Fields. 1992. Legionella pneumophila mip gene potentiates intracellular infection of protozoa and human macrophages. Proc. Natl. Acad. Sci. USA 89:5188-5191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Engleberg, N., C. Carter, D. Weber, N. Cianciotto, and B. Eisenstein. 1989. DNA sequence of mip, a Legionella pneumophila gene associated with macrophage infectivity. Infect. Immun. 57:1263-1270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Fellay, R., J. Frey, and H. Krisch. 1987. Interposon mutagenesis of soil and water bacteria: a family of DNA fragments designed for in vitro insertional mutagenesis of Gram-negative bacteria. Gene 52:147-154. [DOI] [PubMed] [Google Scholar]
- 9.Fraser, C. M., J. D. Gocayne, O. White, M. D. Adams, R. A. Clayton, R. D. Fleischmann, C. J. Bult, A. R. Kerlavage, G. Sutton, J. M. Kelley, et al. 1995. The minimal gene complement of Mycoplasma genitalium. Science 270:397-403. [DOI] [PubMed] [Google Scholar]
- 10.Galat, A. 2003. Peptidylprolyl cis/trans isomerases (immunophilins): biological diversity-targets-functions. Curr. Top. Med. Chem. 3:1315-1347. [DOI] [PubMed] [Google Scholar]
- 11.Galat, A. 2000. Sequence diversification of the FK506-binding proteins in different genomes. Eur. J. Biochem. 267:4945-4959. [DOI] [PubMed] [Google Scholar]
- 12.Gorski, L., W. Godchaux III, E. R. Leadbetter, and R. R. Wagner. 1992. Diversity in surface features of Cytophaga johnsonae motility mutants. J. Gen. Microbiol. 138:1767-1772. [Google Scholar]
- 13.Hayashi, S., and H. C. Wu. 1990. Lipoproteins in bacteria. J. Bioenerg. Biomembr. 22:451-471. [DOI] [PubMed] [Google Scholar]
- 14.Helbig, J., B. Konig, H. Knospe, B. Bubert, C. Yu, C. Luck, A. Riboldi-Tunnicliffe, R. Hilgenfeld, E. Jacobs, J. Hacker, and G. Fischer. 2003. The PPIase active site of Legionella pneumophila Mip protein is involved in the infection of eukaryotic host cells. Biol. Chem. 384:125-137. [DOI] [PubMed] [Google Scholar]
- 15.Helbig, J., P. Luck, M. Steinert, E. Jacobs, and M. Witt. 2001. Immunolocalization of the Mip protein of intracellularly and extracellularly grown Legionella pneumophila. Lett. Appl. Microbiol. 32:83-88. [DOI] [PubMed] [Google Scholar]
- 16.Hodgkin, J., and D. Kaiser. 1979. Genetics of gliding motility in Myxococcus xanthus (Myxobacteriales): genes controlling movement of single cells. Mol. Gen. Genet. 171:167-176. [Google Scholar]
- 17.Hodgkin, J., and D. Kaiser. 1979. Genetics of gliding motility in Myxococcus xanthus (Myxobacteriales): two gene systems control movement. Mol. Gen. Genet. 171:177-191. [Google Scholar]
- 18.Hunnicutt, D. W., M. J. Kempf, and M. J. McBride. 2002. Mutations in Flavobacterium johnsoniae gldF and gldG disrupt gliding motility and interfere with membrane localization of GldA. J. Bacteriol. 184:2370-2378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hunnicutt, D. W., and M. J. McBride. 2001. Cloning and characterization of the Flavobacterium johnsoniae gliding motility genes gldD and gldE. J. Bacteriol. 183:4167-4175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hunnicutt, D. W., and M. J. McBride. 2000. Cloning and characterization of the Flavobacterium johnsoniae gliding motility genes gldB and gldC. J. Bacteriol. 182:911-918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kempf, M. J., and M. J. McBride. 2000. Transposon insertions in the Flavobacterium johnsoniae ftsX gene disrupt gliding motility and cell division. J. Bacteriol. 182:1671-1679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lapidus, I. R., and H. C. Berg. 1982. Gliding motility of Cytophaga sp. strain U67. J. Bacteriol. 151:384-398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.McBride, M. J. 2001. Bacterial gliding motility: multiple mechanisms for cell movement over surfaces. Annu. Rev. Microbiol. 55:49-75. [DOI] [PubMed] [Google Scholar]
- 24.McBride, M. J., T. F. Braun, and J. L. Brust. 2003. Flavobacterium johnsoniae GldH is a lipoprotein that is required for gliding motility and chitin utilization. J. Bacteriol. 185:6648-6657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.McBride, M. J., and M. J. Kempf. 1996. Development of techniques for the genetic manipulation of the gliding bacterium Cytophaga johnsonae. J. Bacteriol. 178:583-590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nakai, K., and M. Kanehisa. 1991. Expert system for predicting protein localization sites in gram-negative bacteria. Proteins Struct. Funct. Genet. 11:95-110. [DOI] [PubMed] [Google Scholar]
- 27.Pate, J. L., and L.-Y. E. Chang. 1979. Evidence that gliding motility in prokaryotic cells is driven by rotary assemblies in the cell envelopes. Curr. Microbiol. 2:59-64. [Google Scholar]
- 28.Pate, J. L., S. J. Petzold, and L.-Y. E. Chang. 1979. Phages for the gliding bacterium Cytophaga johnsonae that infect only motile cells. Curr. Microbiol. 2:257-262. [Google Scholar]
- 29.Pearson, W. R. 1990. Rapid and sensitive sequence comparison with FASTP and FASTA. Methods Enzymol. 183:63-98. [DOI] [PubMed] [Google Scholar]
- 30.Reichenbach, H. 1992. The genus Lysobacter, p. 3256-3275. In A. Balows, H. Trüper, M. Dworkin, W. Harder, and K. Schleifer (ed.), The prokaryotes, 2nd ed. Springer-Verlag, New York, N.Y.
- 31.Riboldi-Tunnicliffe, A., B. Konig, S. Jessen, M. Weiss, J. Rahfeld, J. Hacker, G. Fischer, and R. Hilgenfeld. 2001. Crystal structure of Mip, a prolylisomerase from Legionella pneumophila. Nat. Struct. Biol. 8:779-783. [DOI] [PubMed] [Google Scholar]
- 32.Shevchenko, D., D. Akins, E. Robinson, M. Li, O. Shevchenko, and J. Radolf. 1997. Identification of homologs for thioredoxin, peptidyl prolyl cis-trans isomerase, and glycerophosphodiester phosphodiesterase in outer membrane fractions from Treponema pallidum, the syphilis spirochete. Infect. Immun. 65:4179-4189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Simon, R., U. Priefer, and A. Puhler. 1983. A broad host range mobilization system for in vivo genetic engineering: transposon mutagenesis in Gram negative bacteria. Bio/Technology 2:784-791. [Google Scholar]
- 34.Stanier, R. Y. 1947. Studies on nonfruiting myxobacteria. I. Cytophaga johnsonae, n. sp., a chitin-decomposing myxobacterium. J. Bacteriol. 53:297-315. [PMC free article] [PubMed] [Google Scholar]
- 35.Sun, H., D. R. Zusman, and W. Shi. 2000. Type IV pilus of Myxococcus xanthus is a motility apparatus controlled by the frz chemosensory system. Curr. Biol. 10:1143-1146. [DOI] [PubMed] [Google Scholar]
- 36.Tettelin, H., N. J. Saunders, J. Heidelberg, A. C. Jeffries, K. E. Nelson, J. A. Eisen, K. A. Ketchum, D. W. Hood, J. F. Peden, R. J. Dodson, W. C. Nelson, M. L. Gwinn, R. DeBoy, J. D. Peterson, E. K. Hickey, D. H. Haft, S. L. Salzberg, O. White, R. D. Fleischmann, B. A. Dougherty, T. Mason, A. Ciecko, D. S. Parksey, E. Blair, H. Cittone, E. B. Clark, M. D. Cotton, T. R. Utterback, H. Khouri, H. Qin, J. Vamathevan, J. Gill, V. Scarlato, V. Masignani, M. Pizza, G. Grandi, L. Sun, H. O. Smith, C. M. Fraser, E. R. Moxon, R. Rappuoli, and J. C. Venter. 2000. Complete genome sequence of Neisseria meningitidis serogroup B strain MC58. Science 287:1809-1815. [DOI] [PubMed] [Google Scholar]
- 37.Wolgemuth, C., E. Hoiczyk, D. Kaiser, and G. Oster. 2002. How myxobacteria glide. Curr. Biol. 12:369-377. [DOI] [PubMed] [Google Scholar]
- 38.Wolkin, R. H., and J. L. Pate. 1986. Phage adsorption and cell adherence are motility-dependent characteristics of the gliding bacterium Cytophaga johnsonae. J. Gen. Microbiol. 132:355-367. [Google Scholar]
- 39.Wolkin, R. H., and J. L. Pate. 1985. Selection for nonadherent or nonhydrophobic mutants co-selects for nonspreading mutants of Cytophaga johnsonae and other gliding bacteria. J. Gen. Microbiol. 131:737-750. [Google Scholar]
- 40.Wu, S. S., and D. Kaiser. 1995. Genetic and functional evidence that type IV pili are required for social gliding motility in Myxococcus xanthus. Mol. Microbiol. 18:547-558. [DOI] [PubMed] [Google Scholar]
- 41.Wu, S. S., J. Wu, Y. L. Cheng, and D. Kaiser. 1998. The pilH gene encodes an ABC transporter homologue required for type IV pilus biogenesis and social gliding motility in Myxococcus xanthus. Mol. Microbiol. 29:1249-1261. [DOI] [PubMed] [Google Scholar]
- 42.Youderian, P., N. Burke, D. J. White, and P. L. Hartzell. 2003. Identification of genes required for adventurous gliding motility in Myxococcus xanthus with the transposable element mariner. Mol. Microbiol. 49:555-570. [DOI] [PubMed] [Google Scholar]