

Significance
Prostate cancer is a major health burden with significant overtreatment because of difficulty segregating high- and low-risk disease. Discovery of biomarkers that stratify risk could have a broad public health impact but requires cohorts with comprehensive molecular and clinical follow-up. We characterize two independent prostate cancer cohorts with genomic and clinical data that include biochemical recurrence and metastasis. We demonstrate that copy number alteration (CNA) burden, a measure of the fraction of a tumor genome that is copy number altered, is prognostic for recurrence and metastasis. CNA burden is also associated with cancer recurrence in an intermediate risk population, and can be identified in biopsies. This work provides a clinicogenomic resource and highlights oncogenomics' potential to identify needed prognostic factors.
Keywords: genomics, prognosis, oncology
Abstract
Primary prostate cancer is the most common malignancy in men but has highly variable outcomes, highlighting the need for biomarkers to determine which patients can be managed conservatively. Few large prostate oncogenome resources currently exist that combine the molecular and clinical outcome data necessary to discover prognostic biomarkers. Previously, we found an association between relapse and the pattern of DNA copy number alteration (CNA) in 168 primary tumors, raising the possibility of CNA as a prognostic biomarker. Here we examine this question by profiling an additional 104 primary prostate cancers and updating the initial 168 patient cohort with long-term clinical outcome. We find that CNA burden across the genome, defined as the percentage of the tumor genome affected by CNA, was associated with biochemical recurrence and metastasis after surgery in these two cohorts, independent of the prostate-specific antigen biomarker or Gleason grade, a major existing histopathological prognostic variable in prostate cancer. Moreover, CNA burden was associated with biochemical recurrence in intermediate-risk Gleason 7 prostate cancers, independent of prostate-specific antigen or nomogram score. We further demonstrate that CNA burden can be measured in diagnostic needle biopsies using low-input whole-genome sequencing, setting the stage for studies of prognostic impact in conservatively treated cohorts.
Prostate cancer is the second leading cause of cancer death and the most common malignancy in men. Given the slow growth rate and low metastatic potential of many primary prostate cancers (1), it is critical to identify those men who can be managed conservatively through active surveillance versus those who need aggressive therapy at time of first diagnosis (2, 3). Today, these treatment decisions are primarily made on the basis of tumor stage, prostate-specific antigen (PSA) level, and the histopathological measure of tumor cell differentiation, the Gleason score. These three factors, together with additional pathological variables assessed in the prostatectomy sample, such as lymph node involvement, are often used to estimate risk of relapse with accuracies in the 70–80% range (3). Postoperative nomograms that incorporate these variables have been developed using large cohorts of typically >1,000 patients and consistently show greater accuracy than preoperative nomograms, where pathological variables are limited to those than can be gleaned from biopsies (4). With increasing interest in active surveillance, however, it is critical to improve risk prediction in the preoperative setting (5, 6).
Toward this end, many groups have investigated molecular and genetic alterations present in tumors that correlate with outcome (3). Tumor-derived RNA signatures have been developed that are associated with the risk of death from prostate cancer, some of which are now in clinical use (7–9). These molecular signatures were generally developed from tumor samples obtained at surgery by examining their association with outcome after surgery, then subsequently evaluated in the preoperative setting using biopsies as the source of tumor RNA. Whereas the clinical application of DNA signatures has lagged behind that of RNA-based signatures, DNA alterations have the potential to serve as orthogonal prognostic markers that are robust to degradation and other technical limitations of RNA-based assays (10, 11).
The discovery of prognostic genomic markers relies heavily on prostate cancer cohorts that have both complete genomic and clinical outcome annotation, but few such cohorts exist (12, 13). We previously reported a cohort of 168 primary cases that combined full clinical annotation with extensive gene expression and copy number profiling of the tumor at surgery (14). This cohort serves as a broadly used resource for biological and biomarker discovery (7, 8, 15–18). In this initial cohort, we found that the pattern of DNA copy number alteration (CNA) in prostate tumors at prostatectomy was associated with biochemical recurrence (BCR) (2). CNAs have also been found at high prevalence in cell-free serum DNA of prostate cancer patients (10). Here we update the clinical outcome data from that initial cohort, which now includes a sufficient number of cases with metastasis as a definitive clinical endpoint. We have also developed a second, independent cohort of 104 primary prostate cancers with long-term clinical follow-up and profiled these for CNA, providing an independent cohort for prognostic discovery and validation.
We now report that CNA burden, defined as the percentage of the tumor genome affected by CNA, is prognostic for prostate cancer relapse, including the definitive clinical endpoint of metastasis, in two independent cohorts. Moreover, CNA burden is prognostic in the intermediate-risk subpopulation defined by Gleason score 7 and can be assessed in formalin-fixed, paraffin-embedded (FFPE) prostate biopsies used in standard clinical practice. This work demonstrates that CNA burden is associated with cancer recurrence and lays the groundwork for further investigation of CNA burden's utility in risk prediction in the pretreatment setting.
Results
Copy Number Landscape of Two Prostate Cancer Cohorts with Long-Term Clinical Follow-Up.
To generate an independent cohort of prostate cancer patients with molecular and clinical follow-up data, we analyzed genome-wide CNAs in 104 prostatectomy cases from Memorial Sloan–Kettering Cancer Center. This cohort, hereafter referred to as the contemporary cohort, had a median follow-up time of 6 y (Table S1 and Dataset S1). We also updated the full clinical and outcome information from our initial cohort (14) of 181 prostatectomy cases (Datasets S2–S4), including 168 cases with high-resolution CNA data (Dataset S2) and a median follow-up of 8 y (Table S1). This initial cohort has served as a widely used prostate cancer resource with copy number, gene expression, and clinical and outcome annotation (7, 15–20); it now includes metastasis as a definitive clinical endpoint in a significant number of cases. The initial and contemporary cohorts have BCR incidence rates of 43 and 53 per 1,000 person-years and metastasis incidence rates of 15 and 5 per 1,000 person-years, respectively. These cohorts have BCR rates of 27% (n = 46) and 23% (n = 24), respectively, and metastasis rates of 11 (n = 19) and 3% (n = 3), respectively, representing one of few large prostate oncogenomic resources with substantial outcome data.
The clinical characteristics of the two independent prostate cancer cohorts are similar (Fig. S1 and Table S1). Pretreatment PSA, age, and lymph node and seminal vesicle invasion did not significantly differ between cohorts (Table S1). The rate of extracapsular extension was higher in the contemporary cohort (Table S1) (P value = 0.05) whereas positive surgical margins were more prevalent in the initial cohort (Table S1) (P value = 0.04). The Gleason score distribution differed between the two cohorts (P value = 0.022), with the contemporary cohort having a higher percentage of Gleason 7 cases and the initial cohort having a higher percentage of Gleason 6 and 8 cases. Nonetheless, these clinical differences did not translate into outcome risk differences. The two cohorts were not significantly different in their risk of BCR, determined by the Stephenson postoperative nomogram (21), which combines clinical and pathological variables such as age, PSA, Gleason grade, and pathological stage (Fig. S1 and Table S1) (P value = 0.8).
The landscape of CNAs observed in the contemporary cohort is highly representative of the gross pattern of aberrations in prostate cancer genomes, including in our initial cohort, that of Barbieri et al. (22), and the interim Cancer Genome Atlas (TCGA) prostate cancer cohort (Fig. 1A); these include frequent genomic losses on chromosomes 6p, 8p, 13q, and 16p, genomic gains of 7 and 8q, and established focal alterations spanning phosphatase and tensin homolog (PTEN), RB1, and tumor protein p53 (TP53) among others (Fig. 1B). The level of CNAs in the new cohort was lower on average than earlier cohorts (Fig. 1), possibly reflecting differences in cohort selection criteria. Nonetheless, the contemporary cohort possesses canonical alterations common to prostate cancer.
Fig. 1.

Copy number landscape of primary prostate cancer. (A) Heat map of CNAs in primary prostate cancer, as seen in the contemporary and initial Memorial Sloan–Kettering Cancer Center primary prostate cancer cohorts, as well as cohorts from Barbieri et al. (22) and TCGA. (B) Significant focal CNAs in the initial and contemporary cohorts. (C) Range of CNA burden in prostate cancer (combined cohorts from this study and those in A) and other urogenital malignancies.
Genome-Wide CNA Is Prognostic for Prostate Cancer Recurrence and Metastasis.
In our initial prostate cancer cohort, some CNA patterns identified by hierarchical clustering were previously found to be correlated with the risk of BCR (14). Clinical application of this finding, however, is complicated by the analytical challenge of assigning individual patients to a risk group based on a clustering analysis derived from a different population.
We therefore asked whether CNA burden, a simple metric of CNA level defined as the percent of the autosomal tumor genome bearing CNAs, could be used as an informative measure of CNA. CNA burden represents the level of copy number gains and losses across the genome in a tumor. To calculate CNA burden for a sample, segments of copy number gains and losses were determined (23), and their total genomic length was summed and calculated as a percentage of the size of the autosomal genome. In primary prostate cancer genomes, CNA burden ranged from less than 0.1% to greater than 50%, with an average of 5% and 4% for the initial and contemporary cohorts, respectively. This finding differs significantly from the higher CNA burden of metastatic prostate cancers (∼32%), as well as of other urogenital neoplasms, such as bladder and renal cell carcinomas (Fig. 1C).
CNA burden, when examined as a continuous variable, was significantly associated with BCR in both the initial and contemporary cohorts [hazard ratios (HR) = 1.09 and 1.05, respectively, for each percent change in CNA burden; P values ≤ 0.0001 and 0.008, respectively] (Table 1). We additionally sought to determine whether CNA burden was associated with BCR when patients were separated into groups based on discrete levels of CNA in their tumor genomes. As a starting point for this analysis, we examined the CNA burden from our earlier hierarchical clustering analysis, which segregated tumors into groups with low, intermediate, and high CNA burdens, independent of clinical outcome (14). We reasoned that the mean CNA burden in the low and intermediate alteration clusters (1.34% and 5.41%, respectively) would serve as logical thresholds for further stratified analysis. These thresholds are not based on risk categories from the prior group, but rather copy number clusters indicative of the identity and level of CNA. Stratification at the intermediate CNA burden threshold (5.41% CNA) was associated with BCR in both the initial and contemporary cohorts, with HRs of 1.99 and 3.85, respectively (P values = 0.021 and 0.001) (Fig. 2 A and B and Table 1). Similarly, this finding holds true for patients stratified by low CNA burden threshold (1.34% CNA) with HRs of 2.03 and 2.42 for the initial and contemporary cohorts, respectively (P values = 0.048 and 0.037, respectively) (Fig. S2). Patients in the initial cohort, with a low CNA burden (<1.34% CNA), had a 13% risk of recurrence within 5 y, compared with a 29% risk of BCR for those with a higher CNA burden (≥1.34% CNA) estimated by Kaplan–Meier. Similarly, patients in the contemporary cohort with a low CNA burden had a 15% risk of BCR, compared with a 38% risk for those with a higher CNA burden (≥1.34% CNA). If cases were dichotomized on the basis of their cohort’s CNA burden quartiles rather than the independently defined CNA cluster medians, CNA burden is still significantly associated with BCR in both cohorts and with metastasis in the initial cohort (insufficient metastatic events in the contemporary cohort) by Cox proportional hazards regression (Table S2). For groups with CNA burden above and below the 75th percentile of their cohort, for example, CNA burden is associated with BCR with HRs of 2.65 and 4.43, respectively, for the initial and validation cohorts (P values = 0.001 and < 0.001) (Fig. S3 and Table S2).
Table 1.
The association of CNA burden with the risk of BCR and metastasis
| Event | Variable | Initial cohort* | Contemporary cohort† | ||||||
| P | HR | 95% CI | P | HR | 95% CI | ||||
| BCR | CNA burden‡ | <0.001 | 1.09 | 1.06 | 1.13 | 0.008 | 1.05 | 1.01 | 1.09 |
| BCR | Stratified CNA burden§ | 0.021 | 1.99 | 1.11 | 3.55 | 0.001 | 3.86 | 1.73 | 8.64 |
| Metastasis | CNA burden‡ | <0.001 | 1.10 | 1.04 | 1.15 | insufficient events | |||
| Metastasis | Stratified CNA burden§ | 0.052 | 2.47 | 0.99 | 6.15 | insufficient events | |||
Initial cohort total n = 168, BCR n = 46 (43 per 1,000 person-years), metastases n = 19 (15 per 1,000 person-years).
Contemporary cohort n = 104, BCR n = 24 (53 per 1,000 person-years), metastases n = 3 (5 per 1,000 person-years).
Per 1% change in CNA burden (continuous).
The reference category is CNA burden ≤5.41% (mean of intermediate CNA clusters).
Fig. 2.
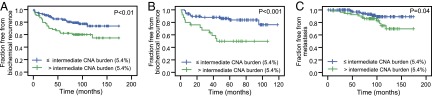
CNA burden in prostate cancer genomes is associated with recurrence and metastasis. Kaplan–Meier plots for BCR in the (A) initial cohort and (B) contemporary cohort are shown. Cases are stratified at intermediate CNA burden (5.41% CNA, the mean of intermediate CNA clusters in ref. 14). Strata with CNA burden greater than (green) or less than or equal to (blue) intermediate CNA burden are shown. (C) Kaplan–Meier plot for metastasis in the initial cohort is shown, stratified by intermediate CNA burden (5.41% CNA as in A and B). The Mantel–Cox log-rank significance value is shown for each.
The longer clinical follow-up now available for the initial cohort allowed us to analyze—to our knowledge for the first time—the association between CNA burden and metastasis as a definitive endpoint (Fig. 2C and Table 1). CNA burden was significantly associated with metastasis in the initial cohort as a continuous variable in a univariate Cox proportional hazards regression model (HR = 1.10; P value = 0.0004) (Table 1) and neared significance as a stratified variable (Cox proportional hazards regression P value = 0.0524, Mantel–Cox log-rank P value = 0.044) (Table 1 and Fig. 2C). Because of the shorter available follow-up time, the contemporary cohort possessed too few metastatic events (n = 3) for this analysis.
CNA Burden Is Associated with Recurrence in Multivariate Clinical Models.
We next assessed whether CNA burden was prognostic in multivariate models when combined with established clinical variables, such as Gleason score and nomogram score. Gleason score, together with PSA level and clinical stage, is used in the preoperative setting to guide whether to treat or conservatively monitor an individual. CNA burden was significantly associated with BCR in both cohorts when corrected for pretreatment PSA (HR = 1.09 and 1.04 for each percent change in CNA burden, respectively, for initial and contemporary cohorts; P values < 0.001 and 0.025) (Table 2). When adjusted for biopsy Gleason score in multivariate models, CNA burden also remained associated with BCR in both cohorts (HR = 1.08 and 1.05, respectively, for initial and contemporary cohorts; P values < 0.0001 and 0.026) (Table 2). Moreover, when biopsy Gleason score categories included separate Gleason 3+4 and 4+3 groups, CNA burden was still associated with BCR in multivariate models with both cohorts (HR = 1.09, P value <0.001, for the initial cohort; HR = 1.05, P value = 0.041, for the contemporary cohort). This result was also true for metastasis in the initial cohort (HR = 1.07, P value = 0.036) where there are sufficient events for analysis.
Table 2.
Multivariate Cox proportional hazards models of the association of CNA burden and clinical variables with BCR
| Variable | Initial cohort (n = 168) | Contemporary cohort (n = 104) | ||||||
| P | HR | 95% CI | P | HR | 95% CI | |||
| CNA burden (continuous)* | <0.001 | 1.09 | 1.05 | 1.13 | 0.025 | 1.04 | 1.01 | 1.09 |
| Pretreatment PSA | 0.001 | 1.01 | 1.00 | 1.01 | 0.046 | 1.03 | 1.00 | 1.07 |
| CNA burden (continuous)* | 0.043 | 1.04 | 1.00 | 1.08 | 0.026 | 1.05 | 1.01 | 1.10 |
| Pathology Gleason score | ||||||||
| 6 | 0.047 | 0.37 | 0.14 | 0.99 | 0.384 | 1.68 | 0.52 | 5.43 |
| 7 | Ref. | — | — | — | Ref. | — | — | — |
| 8 | 0.025 | 2.97 | 1.14 | 7.71 | <0.001 | 23.54 | 5.61 | 73.54 |
| 9 | <0.001 | 8.09 | 3.59 | 18.25 | <0.001 | 15.11 | 4.97 | 38.33 |
| CNA burden (continuous)* | <0.001 | 1.08 | 1.04 | 1.12 | 0.03 | 1.05 | 1.00 | 1.09 |
| Biopsy Gleason score | ||||||||
| ≤6 | 0.008 | 0.40 | 0.21 | 0.79 | 0.228 | 0.56 | 0.22 | 1.44 |
| 7 | Ref. | — | — | — | Ref. | — | — | — |
| 8 | 0.127 | 1.97 | 0.83 | 4.70 | 0.307 | 1.82 | 0.58 | 98.84 |
| 9 | 0.739 | 1.23 | 0.36 | 4.18 | 0.071 | 4.13 | 0.88 | 45.99 |
| CNA burden (continuous)* | 0.318 | 1.02 | 0.98 | 1.06 | 0.011 | 1.05 | 1.01 | 1.10 |
| Stephenson nomogram | <0.001 | 0.02 | <0.01 | 0.06 | <0.001 | 0.02 | 0.01 | 0.09 |
Ref, reference category.
HR is per each 1% change in CNA.
We next examined the prognostic significance of CNA burden when corrected for postoperative variables, such as pathology Gleason score and the Stephenson nomogram. CNA burden was associated with BCR in both cohorts after correcting for pathology Gleason score (HR = 1.04 and 1.05 per 1% change in CNA burden, respectively, for initial and contemporary cohorts; P values = 0.043 and 0.026) (Table 2). In contrast, the postoperative Stephenson nomogram relies on additional postoperative variables, such as extracapsular extension, lymph node involvement, surgical margins, and treatment year to assess postoperative recurrence risk (21). Although the addition of CNA burden did not result in an increase in the concordance index from 0.812 and 0.817 in the initial or contemporary cohorts, respectively, CNA burden was still associated with BCR in the contemporary cohort when combined with the Stephenson 5-y nomogram (HR = 1.05, P value = 0.011) (Table 2), though not in the initial cohort.
We then investigated whether the association of CNA burden with cancer relapse can be attributed to specific focal CNAs that are known to be prognostic. In prior analysis of the initial cohort, we tested all focal CNAs and found few to be individually associated with BCR (14). PTEN loss and MYC gain are the principal focal CNAs that have consistently been found to be associated with prostate cancer recurrence, especially when combined with other clinical variables (24). We therefore examined the prognostic significance of CNA burden when combined with PTEN loss, MYC gain, and TP53 loss in the initial cohort, which had sufficient recurrence and metastatic events for analysis. PTEN copy number loss was positively associated with disease relapse in our cohort, whereas TP53 loss and MYC gain were not. CNA burden remained significantly associated with BCR [P value = 0.001, HR = 1.09, 95% confidence interval (CI) 1.05–1.13, for each percent CNA burden as a continuous variable] when adjusted for PTEN copy number loss, which is independently associated with BCR (P value = 0.001, HR = 3.04, 95% CI 1.55–6.00 in multivariate analysis with CNA burden as a continuous variable). CNA burden remained significantly associated with BCR (P value = 0.001, HR = 1.07, 95% CI 1.03–1.11, for each percent CNA burden as a continuous variable) after additional correction for biopsy Gleason score in addition to PTEN loss, but not after correction for pathology Gleason score. PTEN copy number loss was not significantly associated with metastasis. CNA burden therefore captures information conveyed by CNAs beyond that of these major focal CNAs.
CNA Burden Is Prognostic in Intermediate Risk Gleason 7 Populations.
Gleason 7 tumors fall in an intermediate-risk category, and there is a broad need for new approaches to discriminate indolent from aggressive cancers in this group. We found a wide range of CNA burden across Gleason 7 cancers in both cohorts with ranges of 0.05–25% in the initial cohort and 0.003–50% in the contemporary cohort (Fig. S4). In Gleason 7 tumors, we found that CNA burden was significantly associated with BCR in both the initial and contemporary cohorts (HR = 1.08 and 1.06, respectively; P values = 0.011 and 0.017, respectively) (Fig. 3 and Table 3). CNA burden remained significantly associated with BCR in Gleason 7 disease after correcting for pretreatment PSA in the initial and contemporary cohorts (P values = 0.006 and 0.019, respectively) (Table 3). Moreover, CNA burden was associated with BCR after adjusting for the Stephenson 5-y nomogram in the contemporary cohort (P value = 0.014) (Table 3) and approached significance in the initial cohort (P value = 0.062) (Table 3), although it did not improve the concordance index above 0.7 achieved with the nomogram alone. Too few metastases were available in the Gleason 7 subpopulation for analysis.
Fig. 3.

CNA burden is associated with recurrence in Gleason 7 prostate cancers. Kaplan–Meier plot of BCR in Gleason 7 cases of the combined initial and contemporary cohorts, stratified at intermediate CNA burden (14). The Mantel–Cox log-rank significance value is shown.
Table 3.
CNA burden is associated with prostate cancer recurrence in Gleason 7 cases
| Event | Variable | Initial cohort (n = 168) | Contemporary cohort (n = 104) | ||||||
| P | HR | 95% CI | P | HR | 95% CI | ||||
| Univariate Cox regression | |||||||||
| BCR | CNA burden | 0.011 | 1.08 | 1.02 | 1.14 | 0.017 | 1.06 | 1.01 | 1.11 |
| Multivariable Cox regression | |||||||||
| BCR | CNA burden | 0.006 | 1.08 | 1.02 | 1.15 | 0.019 | 1.06 | 1.01 | 1.11 |
| Pretreatment PSA | 0.001 | 1.00 | 1.00 | 1.01 | 0.912 | 1.00 | 0.94 | 1.07 | |
| BCR | CNA burden | 0.062 | 1.06 | 1.00 | 1.12 | 0.014 | 1.06 | 1.01 | 1.12 |
| Stephenson nomogram | 0.002 | 0.03 | <0.01 | 0.29 | 0.001 | 0.01 | <0.01 | 0.17 | |
CNA Burden Provides Prognostic Information Beyond RNA-Based Signature and Can Be Established in Tumor Biopsies.
Recent work has established the prognostic significance of gene expression-based signatures in prostate cancers, including a cell cycle progression (CCP) signature recently developed for clinical use (8). We compared a nonproprietary version of the RNA-based CCP signature to the DNA-based CNA burden within our initial cohort, for which both DNA copy number and RNA expression data had been generated. Both the CCP signature and CNA burden were independently associated with BCR, with HRs of 1.124 and 1.097, respectively (P value = 0.05 and <0.001, initial cohort cases with both expression and CNA data, n = 113) (Table S3). CNA burden therefore contributes independent information beyond that represented by the CCP expression signature with a comparable HR for each percent CNA burden or percent CCP signature score. CNA burden outperformed the CCP signature for association with BCR in Gleason 7 cancers (Table S3).
To have utility in clinical risk prediction, CNA burden must be measurable at biopsy. We therefore tested the feasibility of assessing CNA burden in FFPE biopsy samples by whole-genome sequencing. We performed proof-of-principle low-pass whole-genome sequencing (between one- to threefold coverage) with varying low-input quantities of FFPE biopsy DNA from four tumors and their adjacent benign controls (Fig. 4 and Dataset S5). Concurrently, we also analyzed these samples by high-resolution array comparative genomic hybridization (aCGH) for comparison (Fig. 4). The pattern and extent of CNAs identified were consistent between aCGH and whole-genome sequencing in even the challenging low-input FFPE DNA setting, spanning from concordant focal deletions (3p13 spanning FOXP1, RYBP, and SHQ1) to arm-level gains and losses of significance to prostate cancer biology (7, 8p, 8q, and 13q). In addition to concordance among discrete CNAs, the amplitudes—and therefore their clonal representation—was also highly consistent (Fig. 4). These results illustrate that whole-genome sequencing can achieve copy number profiling comparable to aCGH and enables characterization of lower tumor burden FFPE biopsies.
Fig. 4.

CNA burden can be established in the diagnostic setting using FFPE needle biopsies. Shown is a heatmap of CNAs in four prostate cancer genomes inferred from either aCGH or low-pass whole-genome sequencing from FFPE needle biopsy libraries generated from either 100-ng or 250-ng input DNA (red and blue are genomic gains and losses, respectively; white is copy-neutral regions). Individual loci of significance in prostate cancers are annotated.
Discussion
Prostate cancer has seen an increase in intermediate and early-stage disease at diagnosis, with widespread use of PSA to screen for prostate cancer and an increase in the number of needle cores taken during biopsy (3). Although Gleason score and other clinicopathologic variables are used to guide treatment decisions, current approaches often do not sufficiently discriminate between those patients who would likely benefit from immediate treatment and those whose treatment can be delayed or avoided completely while their cancer is monitored (25). Recent clinical trials have suggested that 15 patients must be treated to prevent one prostate cancer death overall, and patients under 65 experience a more substantial treatment benefit (26). The challenge faced by physicians and patients with this common cancer is to find better ways to characterize the risk posed by each cancer so that more appropriate treatment decisions can be made. Additional prognostic factors are, therefore, urgently needed to identify cancers unlikely to progress, especially among the intermediate-risk population.
The discovery of molecular biomarkers for prostate cancer has been hindered by the paucity of molecular subtypes with distinct outcomes. Part of the difficulty stems from the long natural history of prostate cancer and its low rate of progression. As a result, whereas many comprehensive genomic studies have defined alterations in prostate cancer genomes, none have had sufficient clinical and outcome annotation to generate prognostic advances. We have collected the clinically annotated cohorts presented here to help address these challenges. The initial cohort has been used extensively by others for prognostic discovery and validation (7, 8, 27) and is updated here by providing a definitive clinical endpoint, metastasis, in significant numbers. The additional independent contemporary cohort now provides a resource for validation of CNA as a prognostic factor and other associations. Because many patients with BCR after surgery will not develop metastases (28), it is important to test prognostic biomarkers with unequivocal clinical endpoints, such a metastases or death. Few associations between genetic alterations and metastasis have been studied in large patient cohorts, and this work may enable their discovery.
We have shown that CNA burden, as a global measure of the level of CNA across tumor genomes, is associated with BCR over a broad spectrum of clinical presentations and adds additional information to currently available clinicopathological variables. CNA burden is also associated with the definitive endpoint of metastasis. Moreover, CNA burden is significantly associated with BCR in intermediate risk Gleason 7 patients, raising the possibility of better stratifying this intermediate risk population. CNA burden varies significantly in other cancers, with many showing large ranges in CNA burden across their populations. It may therefore be fruitful to also explore the prognostic significance of CNA burden in other cancers as well.
As we have shown that CNA burden can be successfully determined in formalin-fixed needle biopsies, future work may explore CNA burden within large biopsy cohorts to determine its prognostic power (29, 30). The fact that CNA burden adds to the impact of existing cell cycle RNA markers provides further motivation for a biopsy cohort study (7, 9). It will be important to know if CNA burden in biopsy samples of low- and intermediate-risk patients will be useful in making the decision for immediate therapy or active surveillance. Active surveillance cohorts may be a particularly useful resource to reduce the effect of treatment on the ability to determine the prognostic association between CNA burden and outcome.
Beyond the prognostic significance of CNA, the work here is a prelude to further studies of CNA in cancer biology. Because structural rearrangements are naturally associated with the breakpoints of discrete significant CNAs, exploring their association with outcomes is an intriguing related approach. Finally, it will be interesting to ask what mechanisms underlie the association between CNA and clinical outcome. Taken together, the data in this work promise to open avenues that improve prognostic markers and our understanding of their underlying mechanisms in cancer.
Materials and Methods
Contemporary Cohort Collection and Annotation.
A total of 104 tumor and matched normal samples were obtained from patients treated by prostatectomy at the Memorial Sloan–Kettering Cancer Center with Institutional Review Board approval. BCR was defined as an increase in PSA of ≥0.2 ng/mL on two occasions. The 2005 Stephenson nomogram for postoperative risk of BCR at 5 y was reported (21). Patient follow-up data were updated through March 2013.
aCGH.
Snap-frozen samples containing greater than 70% tumor cell content were dissected from their frozen blocks. Tumor and pooled reference (Promega) DNA was extracted, labeled, and analyzed by aCGH. Agilent 1M feature arrays were used for the contemporary cohort and the 244K arrays for biopsy cases.
Whole-Genome Sequencing.
Whole-genome sequencing libraries were prepared using 100-ng or 250-ng input FFPE biopsy dsDNA (picogreen) from four patients. Paired-end 100-bp libraries were generated (KAPA LTP kit, Kappa Biosystems) and sequenced by HiSEq. 2000.
DNA CNA.
For aCGH data, all copy-number array data from patients in the contemporary cohort were quantified, normalized, segmented, and analyzed with RAE, as previously described (14). To assess the percent of autosomal genome affected by CNA in both the initial and contemporary cohorts, the per-tumor parameterization by RAE was used (23). For the analyses described here, CNAs are those segments whose value of A0 or D0 (gains and losses or more, respectively) were greater than or equal to 0.75. The total genomic territory spanned by these contiguous segments was summed and a percent was generated using the size of the autosomal human genome.
For sequencing data, reads were aligned to hg19 of the human genome with BWA. The aligned first read-in pair was retained and coverage calculated in 100-kb nonoverlapping windows of the autosomal genome. In each genome, a locally weighted polynomial regression was fit between read coverage and G+C% content in alignable regions not overlapping known gaps in the reference assembly. G+C%-content normalization using the loess fit to normalize read counts in each independent tumor and matched normal genome before segmentation. A pseudocount was added to read counts genome-wide in both samples, the ratio was normalized by library size, and segmented with circular binary segmentation. Individual samples were then analyzed with RAE to determine CAN, as described above.
Statistical Analyses.
For cohort characteristics, P values were determined by Wilcoxon rank sum for continuous variables and by Fisher’s exact test for categorical variables. We tested for a univariate association between CNA and BCR or metastasis in the initial and contemporary cohorts separately. CNA was then added to a multivariable Cox proportional hazards-regression model for the risk of BCR. The proportional hazards model was not checked, as formal testing of the proportional hazards assumption is of limited utility; it is known that for outcomes, such as BCR or metastases after treatment, that the proportional hazards assumption is not met with long follow-up time. For Kaplan–Meier analyses, follow-up time was censored to 14.5 y and the Mantel–Cox log-rank significance reported. For outcome analyses performed using a stratification of cases on the basis of their CNA burden, we grouped case cut-points of 5.41% and 1.34%, which represent the mean CNA burden from patient genomes that defined the intermediate and low alteration clusters reported previously (14) (clusters 1, 3, and 4 vs. 2, respectively). To assess the discriminative accuracy of the models developed, we calculated Harrell’s C concordance statistic after 10-fold cross-validation.
The nonproprietary CCP signature score was defined as the average of the expression level (Affymetrix) of the 30 CCP genes with available expression data in our cohort.
Data Access.
Study array data were deposited in the National Center for Biotechnology Information Gene Expression Omnibus under accession no. GSE54691. The data and annotation also available via the Prostate Cancer Genomics Data Portal: http://cbio.mskcc.org/prostate-portal.
Supplementary Material
Acknowledgments
We thank the members of the Prostate Cancer Oncogenome Group for critical contributions. This work was supported by the Howard Hughes Medical Institute (C.L.S.), the Starr Consortium (C.L.S.), National Institutes of Health Grants P50-CA092629 and R01-CA155169 (to C.L.S.), the Robertson Foundation (N.S.); Stand Up to Cancer (C.L.S.), and the Prostate Cancer Foundation (B.S.C., B.S.T., C.L.S., and N.S.).
Footnotes
The authors declare no conflict of interest.
Data deposition: The data reported in this paper have been deposited in the Gene Expression Omnibus (GEO) database, www.ncbi.nlm.nih.gov/geo (accession no. GSE54691).
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1411446111/-/DCSupplemental.
References
- 1.Draisma G, et al. Lead time and overdiagnosis in prostate-specific antigen screening: Importance of methods and context. J Natl Cancer Inst. 2009;101(6):374–383. doi: 10.1093/jnci/djp001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wilt TJ, et al. Prostate Cancer Intervention versus Observation Trial (PIVOT) Study Group Radical prostatectomy versus observation for localized prostate cancer. N Engl J Med. 2012;367(3):203–213. doi: 10.1056/NEJMoa1113162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Prensner JR, Rubin MA, Wei JT, Chinnaiyan AM. Beyond PSA: The next generation of prostate cancer biomarkers. Sci Trans Med. 2012;4(127):127rv123. doi: 10.1126/scitranslmed.3003180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lughezzani G, et al. Predictive and prognostic models in radical prostatectomy candidates: A critical analysis of the literature. Eur Urol. 2010;58(5):687–700. doi: 10.1016/j.eururo.2010.07.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wong LM, et al. International multicentre study examining selection criteria for active surveillance in men undergoing radical prostatectomy. Br J Cancer. 2012;107(9):1467–1473. doi: 10.1038/bjc.2012.400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cary KC, et al. Predictors of pathologic progression on biopsy among men on active surveillance for localized prostate cancer: The value of the pattern of surveillance biopsies. Eur Urol. 2013 doi: 10.1016/j.eururo.2013.08.060. [DOI] [PubMed] [Google Scholar]
- 7.Irshad S, et al. A molecular signature predictive of indolent prostate cancer. Sci Trans Med. 2013;5(202):202ra122. doi: 10.1126/scitranslmed.3006408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Erho N, et al. Discovery and validation of a prostate cancer genomic classifier that predicts early metastasis following radical prostatectomy. PLoS ONE. 2013;8(6):e66855. doi: 10.1371/journal.pone.0066855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cuzick J, et al. Transatlantic Prostate Group Prognostic value of a cell cycle progression signature for prostate cancer death in a conservatively managed needle biopsy cohort. Br J Cancer. 2012;106(6):1095–1099. doi: 10.1038/bjc.2012.39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Leary RJ, et al. Detection of chromosomal alterations in the circulation of cancer patients with whole-genome sequencing. Sci Trans Med. 2012;4(162):162ra154. doi: 10.1126/scitranslmed.3004742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cheng I, et al. Copy number alterations in prostate tumors and disease aggressiveness. Genes Chromosomes Cancer. 2012;51(1):66–76. doi: 10.1002/gcc.20932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sboner A, et al. Molecular sampling of prostate cancer: A dilemma for predicting disease progression. BMC Med Genomics. 2010;3:8. doi: 10.1186/1755-8794-3-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yu YP, et al. Gene expression alterations in prostate cancer predicting tumor aggression and preceding development of malignancy. J Clin Oncol. 2004;22(14):2790–2799. doi: 10.1200/JCO.2004.05.158. [DOI] [PubMed] [Google Scholar]
- 14.Taylor BS, et al. Integrative genomic profiling of human prostate cancer. Cancer Cell. 2010;18(1):11–22. doi: 10.1016/j.ccr.2010.05.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wang ZA, et al. Lineage analysis of basal epithelial cells reveals their unexpected plasticity and supports a cell-of-origin model for prostate cancer heterogeneity. Nat Cell Biol. 2013;15(3):274–283. doi: 10.1038/ncb2697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lunardi A, et al. A co-clinical approach identifies mechanisms and potential therapies for androgen deprivation resistance in prostate cancer. Nat Genet. 2013;45(7):747–755. doi: 10.1038/ng.2650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ding Z, et al. SMAD4-dependent barrier constrains prostate cancer growth and metastatic progression. Nature. 2011;470(7333):269–273. doi: 10.1038/nature09677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ding Z, et al. Telomerase reactivation following telomere dysfunction yields murine prostate tumors with bone metastases. Cell. 2012;148(5):896–907. doi: 10.1016/j.cell.2012.01.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Markert EK, Mizuno H, Vazquez A, Levine AJ. Molecular classification of prostate cancer using curated expression signatures. Proc Natl Acad Sci USA. 2011;108(52):21276–21281. doi: 10.1073/pnas.1117029108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Qi J, et al. The E3 ubiquitin ligase Siah2 contributes to castration-resistant prostate cancer by regulation of androgen receptor transcriptional activity. Cancer Cell. 2013;23(3):332–346. doi: 10.1016/j.ccr.2013.02.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Stephenson AJ, et al. Postoperative nomogram predicting the 10-year probability of prostate cancer recurrence after radical prostatectomy. J Clin Oncol. 2005;23(28):7005–7012. doi: 10.1200/JCO.2005.01.867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Barbieri CE, et al. Exome sequencing identifies recurrent SPOP, FOXA1 and MED12 mutations in prostate cancer. Nat Genet. 2012;44(6):685–689. doi: 10.1038/ng.2279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Taylor BS, et al. Functional copy-number alterations in cancer. PLoS ONE. 2008;3(9):e3179. doi: 10.1371/journal.pone.0003179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Liu W, et al. Genetic markers associated with early cancer-specific mortality following prostatectomy. Cancer. 2013;119(13):2405–2412. doi: 10.1002/cncr.27954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Daskivich TJ, et al. Overtreatment of men with low-risk prostate cancer and significant comorbidity. Cancer. 2011;117(10):2058–2066. doi: 10.1002/cncr.25751. [DOI] [PubMed] [Google Scholar]
- 26.Bill-Axelson A, et al. SPCG-4 Investigators Radical prostatectomy versus watchful waiting in early prostate cancer. N Engl J Med. 2011;364(18):1708–1717. doi: 10.1056/NEJMoa1011967. [DOI] [PubMed] [Google Scholar]
- 27.Erho N, Buerki C, Triche TJ, Davicioni E, Vergara IA. Transcriptome-wide detection of differentially expressed coding and non-coding transcripts and their clinical significance in prostate cancer. J Oncol. 2012;2012:541353. doi: 10.1155/2012/541353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Antonarakis ES, et al. The natural history of metastatic progression in men with prostate-specific antigen recurrence after radical prostatectomy: Long-term follow-up. BJU Int. 2012;109(1):32–39. doi: 10.1111/j.1464-410X.2011.10422.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Frampton GM, et al. Development and validation of a clinical cancer genomic profiling test based on massively parallel DNA sequencing. Nat Biotechnol. 2013;31(11):1023–1031. doi: 10.1038/nbt.2696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Schweiger MR, et al. Genome-wide massively parallel sequencing of formaldehyde fixed-paraffin embedded (FFPE) tumor tissues for copy-number- and mutation-analysis. PLoS ONE. 2009;4(5):e5548. doi: 10.1371/journal.pone.0005548. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.