

Significance
DNA polymerases are known to select against L-nucleotides, the enantiomers of natural D-nucleotides. However, the structural basis for D-stereoselectivity of a DNA polymerase has not been established, although two L-nucleoside analogs, lamivudine and emtricitabine, have been widely used as anti-HIV and anti-hepatitis B drugs. Here, we report ternary crystal structures of human DNA polymerase λ in complex with DNA and L-deoxycytidine 5′-triphosphate, or its analogs (the triphosphates of lamivudine and emtricitabine). These structures reveal that unlike a polymerase-bound D-nucleotide, an L-nucleotide initially interacts with an active site arginine residue through hydrogen bonds and then pairs with the templating nucleotide. Our work provides a structural basis for the D-stereoselectivity of a polymerase and valuable insight toward design of less toxic antiviral nucleoside analogs.
Keywords: DNA polymerase lambda, antiviral nucleoside analogs, DNA polymerase stereoselectivity, L-nucleotide:arginine paring, pre–steady-state kinetics
Abstract
Although lamivudine and emtricitabine, two L-deoxycytidine analogs, have been widely used as antiviral drugs for years, a structural basis for D-stereoselectivity against L-dNTPs, enantiomers of natural nucleotides (D-dNTPs), by any DNA polymerase or reverse transcriptase has not been established due to lack of a ternary structure of a polymerase, DNA, and an incoming L-dNTP. Here, we report 2.10–2.25 Å ternary crystal structures of human DNA polymerase λ, DNA, and L-deoxycytidine 5′-triphosphate (L-dCTP), or the triphosphates of lamivudine ((−)3TC-TP) and emtricitabine ((−)FTC-TP) with four ternary complexes per asymmetric unit. The structures of these 12 ternary complexes reveal that relative to D-deoxycytidine 5′-triphosphate (D-dCTP) in the canonical ternary structure of Polλ-DNA-D-dCTP, L-dCTP, (−)3TC-TP, and (−)FTC-TP all have their ribose rotated by 180°. Among the four ternary complexes with a specific L-nucleotide, two are similar and show that the L-nucleotide forms three Watson–Crick hydrogen bonds with the templating nucleotide dG and adopts a chair-like triphosphate conformation. In the remaining two similar ternary complexes, the L-nucleotide surprisingly interacts with the side chain of a conserved active site residue R517 through one or two hydrogen bonds, whereas the templating dG is anchored by a hydrogen bond with the side chain of a semiconserved residue Y505. Furthermore, the triphosphate of the L-nucleotide adopts an unprecedented N-shaped conformation. Our mutagenic and kinetic studies further demonstrate that the side chain of R517 is critical for the formation of the abovementioned four complexes along proposed catalytic pathways for L-nucleotide incorporation and provide the structural basis for the D-stereoselectivity of a DNA polymerase.
Nucleoside analog reverse-transcriptase inhibitors (NRTIs), a class of antiviral drugs, are activated to their triphosphate forms by host kinases and then incorporated into the growing viral DNA chain catalyzed by viral reverse transcriptases (RTs), leading to the termination of viral DNA synthesis due to the absence of a 3′-hydroxyl group on the deoxyribose moiety of each NRTI. Among the NRTIs approved against human immunodeficiency virus (HIV) infection, two deoxycytidine analogs, lamivudine [(−)3TC, (−)-β-L-2′,3′-dideoxy-3′-thiacytidine] and its 5-fluorinated derivative, emtricitabine [(−)FTC, (−)-β-L-2′,3′-dideoxy-5-fluoro-3′-thiacytidine] (Fig. 1), possess L-stereochemistry. Both lamivudine and emtricitabine have been shown to be more effective in inhibiting HIV-1 RT and less toxic than their enantiomeric D-isomers (1–4). In addition, both lamivudine, a potent inhibitor of hepatitis B virus (HBV) (5), and telbivudine, the L-analog of thymidine, are approved drugs for the treatment of HBV infection, whereas emtricitabine is currently in clinical trials for this purpose (6). These L-nucleoside analogs demonstrate less clinical toxicity than their corresponding D-isomers, likely because human DNA polymerases possess strong D-stereoselectivity by preferentially binding and incorporating D-dNTPs over unnatural nucleotides with L-stereochemistry (L-dNTPs) during DNA synthesis. Surprisingly, a structural basis for the discrimination against L-dNTPs by any DNA polymerase or RT has not been established, although D-stereoselectivity has been successfully explored in antiviral drug development.
Fig. 1.

Chemical structures of D-dCTP, L-dCTP, (–)3TC-TP, (–)FTC-TP, lamivudine, and emtricitabine. (–)3TC-TP and (–)FTC-TP are the triphosphates of lamivudine [(–)3TC, (–)-β-L-2′,3′-dideoxy-3′-thiacytidine] and emtricitabine [(–)FTC, (–)-β-L-2′,3′-dideoxy-5-fluoro-3′-thiacytidine], respectively. The mirror emphasizes the mirror image relationship between enantiomers D-dCTP and L-dCTP.
Despite high clinical efficacy, NRTIs are often associated with various drug toxicities resulting from the inhibition of host DNA polymerases that share a catalytic mechanism akin to HIV-1 RT (7). There are 16 identified human DNA polymerases that belong to A-, B-, X-, and Y-families. NRTI-associated mitochondrial toxicity has been linked with the inhibition of human DNA polymerase γ (Polγ), an A-family enzyme (8), whereas NRTI-induced genomic instability can be correlated with the inhibition of human replicative B-family polymerases α, δ, and ε (9–11). Recently, our systematic kinetic analysis exploring the relative involvement of various host DNA polymerases in NRTI-associated drug toxicity highlights that DNA damage repair X-family polymerases β and λ (Polλ) and DNA lesion bypass Y-family polymerases η, κ, ι, and Rev1 are more prone to inhibition by the triphosphates of NRTIs than replicative DNA polymerases and can incorporate these analogs as efficiently as HIV-1 RT in vitro (12). Both the absence of a proofreading 3′→5′ exonuclease activity and a flexible active site are attributed to the increased inhibition of human X- and Y-family DNA polymerases over human replicative enzymes.
Although there are hundreds of published ternary crystal structures (polymerase–DNA–D-dNTP) to show how a DNA polymerase or RT binds and incorporates a natural or unnatural incoming nucleotide with D-stereochemistry into DNA, a structural basis for the D-stereoselectivity of a DNA polymerase or RT is still unclear due to the lack of any ternary crystal structure with L-dNTP (polymerase–DNA–L-dNTP). To establish this structural basis, we cocrystallized and solved the ternary structures of human Polλ, a single-nucleotide gapped DNA substrate, and L-dCTP or its analogs (−)3TC-TP and (−)FTC-TP (Fig. 1). Polλ (13–16) fills DNA gaps and plays putative roles in base excision repair (13–17), nonhomologous end joining (18), and V(D)J recombination (19). Our structures illustrate how L-nucleotides, relative to D-dNTPs, are bound within the active site of a polymerase and then proceed through catalysis, and also facilitate the development of less toxic and more potent antiviral L-nucleoside analogs.
Results and Discussion
Difference in the Kinetics of the Binding and Incorporation of L-dCTP and Its Analogs Catalyzed by Human Polλ at 37 °C.
At the active site of a DNA polymerase or RT, the primer 3′-OH makes an in-line nucleophilic attack on the α-phosphate of an incoming D-dNTP, forming a new phosphodiester bond during DNA synthesis. Changing the stereochemistry of the incoming nucleotide from D to L is expected to alter its interactions with the templating nucleotide and active site residues and influence nucleotide binding and incorporation. Our pre–steady-state kinetic analysis confirms that L-dCTP (SI Appendix, Fig. S1), (−)3TC-TP, and (−)FTC-TP were all incorporated with both maximum rate constants (kp) and efficiencies (kp/Kd) several orders of magnitude lower than D-deoxycytidine 5′-triphosphate (D-dCTP) (Table 1). Although the L-stereochemistry did not significantly affect the nucleotide binding affinity (1/Kd) based on similar equilibrium dissociation constants (Kd) of L-dCTP and D-dCTP, the chemical modification in the sugar rings of (−)3TC-TP and (−)FTC-TP contributed to their two- to sixfold higher affinity relative to L-dCTP (Table 1). Our kinetic data further indicate that Polλ preferentially incorporated D-dCTP over L-dCTP with the D-stereoselectivity, defined as (kp/Kd)D-dCTP/(kp/Kd)L-dCTP, of 1.2 × 104, whereas the D-stereoselectivity was reduced to only 100 and 192 for the incorporation of (−)3TC-TP and (−)FTC-TP, respectively (Table 1). Thus, the chemical changes in the ribose of (−)3TC-TP and (−)FTC-TP relaxed the D-stereoselectivity of Polλ by 100-fold and made these L-nucleotide analogs better substrates than L-dCTP. To establish a structural basis for the kinetic differences in the binding and incorporation of D-dCTP, L-dCTP, (−)3TC-TP, and (−)FTC-TP, we performed crystallographic studies with Polλ.
Table 1.
Pre–steady-state kinetic parameters for single nucleotide incorporation
| Nucleotide | kp, s−1 | Kd, μM | kp/Kd, μM−1⋅s−1 | D-stereoselectivity* |
| Catalyzed by wild-type Polλ | ||||
| D-dCTP | 2.02 ± 0.06 | 0.81 ± 0.08 | 2.5 | |
| L-dCTP | (1.4 ± 0.1) × 10−4 | 0.67 ± 0.07 | 2.1 × 10−4 | 1.2 × 104 |
| (–)3TC-TP | (3.0 ± 0.2) × 10−3 | 0.12 ± 0.02 | 2.5 × 10−2 | 100 |
| (–)FTC-TP | (4.7 ± 0.1) × 10−3 | 0.35 ± 0.02 | 1.3 × 10−2 | 192 |
| Catalyzed by the mutant R517A of Polλ | ||||
| D-dCTP | (9.6 ± 0.2) × 10−4 | 0.22 ± 0.02 | 4.4 × 10−3 | |
| L-dCTP | Not determined† | 36 ± 3 | ||
An incoming nucleotide was incorporated opposite the templating nucleotide dG in the single-nucleotide gapped DNA substrate 21-mer⋅19-mer/41-mer (SI Appendix, Fig. S1A) catalyzed by either wild-type human DNA Polλ or its mutant R517A at 37 °C.
D-stereoselectivity = (kp/Kd)D-dCTP/(kp/Kd)L-nucleotide.
No product formation was observed after 7 h.
Similarities and Differences in Overall Ternary Crystal Structures with L-Nucleotides.
Previously, both the ternary complex of a 38.2 kDa human Polλ construct, a single-nucleotide gapped DNA substrate, and D-dCTP (Polλ–DNA–D-dCTP) (SI Appendix, Fig. S2A) (20) and the binary complex of the same Polλ construct and a nearly identical single-nucleotide gapped DNA substrate (Polλ–DNA) (SI Appendix, Fig. S3A) (21) have been crystallized in a single binding conformation, and their structures have been solved at 2.1 and 2.3 Å resolution, respectively. Here, a nearly identical Polλ construct (22), the same single-nucleotide gapped DNA substrate (Materials and Methods), and an L-nucleotide [L-dCTP, (−)3TC-TP, or (−)FTC-TP] were crystallized in space group P21212 (SI Appendix, Table S1) with four different ternary complex molecules (denoted as complexes A, E, I, and M) per asymmetric unit (SI Appendix, Table S2). These crystal structures were refined to a resolution of 2.10–2.25 Å and are referred to as Polλ–DNA–L-dCTP, Polλ–DNA–(−)3TC-TP, and Polλ–DNA–(−)FTC-TP (SI Appendix, Table S1). Notably, the Polλ protein structure in complexes A of Polλ–DNA–L-dCTP, Polλ–DNA–(−)3TC-TP, and Polλ–DNA–(−)FTC-TP and in Polλ–DNA–D-dCTP are almost superimposable with a root-mean-square deviation (rmsd) of 0.70–0.77 Å, whereas modestly larger Polλ protein structural changes are displayed in complexes E, I, and M relative to Polλ–DNA–D-dCTP with rmsds of 0.89–1.37 Å (SI Appendix, Table S2). Interestingly, superposition of the four complexes with the same incoming L-nucleotide—for example, the complexes of Polλ–DNA–L-dCTP in Fig. 2—reveals that complexes A and E are closely related, whereas complexes I and M resemble each other based on the overall similarity of their ternary structures, including the binding conformations of their active site residues, the nascent base pair, and the DNA substrate. Furthermore, all complexes A and E of the three L-nucleotides [L-dCTP, (−)3TC-TP, and (−)FTC-TP] are similar and closely resemble the canonical ternary structure of Polλ–DNA–D-dCTP (SI Appendix, Fig. S2A), with complexes A bearing greater likeness based on their smaller rmsd values and shorter distances between the α-phosphorus atom and the primer 3′-OH than those of complexes E (SI Appendix, Tables S2 and S3). For instance, complex A of Polλ–DNA–L-dCTP and the lone conformation of Polλ–DNA–D-dCTP possess nearly superimposable protein and DNA structures, similar positioning of several active site residues, and analogous binding conformations of the base and triphosphate of the incoming nucleotides (SI Appendix, Fig. S2). In comparison, all complexes I and M with L-dCTP, (−)3TC-TP, and (−)FTC-TP are alike and overlay well with the binary structure of Polλ–DNA, rather than with the ternary structure of Polλ–DNA–D-dCTP. For example, the structure of complex M of Polλ–DNA–L-dCTP closely resembles the Polλ–DNA structure except that the latter lacks an incoming nucleotide (SI Appendix, Fig. S3).
Fig. 2.

Superposition of four different ternary complexes (Polλ–DNA–L-dCTP) within an asymmetric unit. (A) Zoomed superposition of incoming L-dCTP, the templating nucleotide dG, and two divalent metal ions at the active site of human Polλ. The shift for the C6 atom of the templating nucleotide dG from complex I to E is 4.6 Å. (B) Zoomed superposition of incoming L-dCTP and several active site residues. The guanidinium moiety of R517 shifts its position by 4.4 Å from complex M to A, whereas the base of L-dCTP moves closer to the template strand by 3.2 Å from complex M to E. (C) Zoomed superposition of incoming L-dCTP, the templating nucleotide dG, the junction base pair between the template and the upstream primer, and the backbones of the template and upstream primer. The shift for the C6 atom of the “–1” template nucleotide dG is 2.4 Å from complex I to A, whereas the backbone of the template adjusts its position horizontally by 4.6 Å from complex I to E. (D) Superposition of the incoming L-dCTP and the entire single-nucleotide gapped DNA substrate. The nucleotides are shown as lines, DNA backbones as cartoons, the active site residues as sticks, and metal ions as spheres.
Binding Conformations of an Incoming L-Nucleotide Within a Polymerase Active Site.
The structures of complexes A (Fig. 3, Left) and E (SI Appendix, Fig. S4, Left) show that L-dCTP, (−)3TC-TP, and (−)FTC-TP are present in an anti-conformation and form three Watson–Crick hydrogen bonds (2.8–3.0 Å) with the templating nucleotide dG, similar to those in the canonical base pair D-dCTP:dG (2.8–2.9 Å) in Polλ–DNA–D-dCTP (SI Appendix, Fig. S5A). Strikingly, constraints from both the L-stereochemistry and the Watson–Crick base pairing lead to a 180° rotation of the sugar ring of each L-nucleotide (Fig. 3 and SI Appendix, Fig. S4, Left) relative to the ribose of either any nucleotide in DNA or Polλ-bound D-dCTP (SI Appendix, Fig. S5A). Consistently, a modeling study has predicted that the sugar ring of (−)3TC-TP, relative to D-dTTP, undergoes the 180° rotation at the active site of HIV-1 RT (23). Like D-dCTP in Polλ–DNA–D-dCTP (Fig. 4A), the sugar puckers of the L-nucleotides in complexes A and E are in a C3′-endo conformation, whereas their triphosphate moieties adopt a chair-like ( ) conformation observed in canonical polymerase-undamaged DNA-correct D-dNTP ternary structures (24), and interact with both divalent metal ions in the active site (Fig. 4B and SI Appendix, Fig. S5 and Table S3).
) conformation observed in canonical polymerase-undamaged DNA-correct D-dNTP ternary structures (24), and interact with both divalent metal ions in the active site (Fig. 4B and SI Appendix, Fig. S5 and Table S3).
Fig. 3.

Interactions of an incoming L-nucleotide with either the templating nucleotide dG or R517 in Polλ–DNA–L-nucleotide. (A and B) L-dCTP, (C and D) (–)3TC-TP, and (E and F) (–)FTC-TP. A, C, and E are in complexes A, whereas B, D, and F are in complexes M. Only two template nucleotides, the primer 3′-terminal nucleotide, and active site residues R517 and Y505 are displayed as sticks. Hydrogen bonds and the distance between the primer 3′-OH group and the α-phosphorus atom of an incoming L-nucleotide are presented as black dashed lines, with the numbers depicting their lengths in Å. The interactions between the triphosphate of an incoming L-nucleotide and the divalent metal ions at sites A and B, shown as green spheres, are also presented as black dashed lines. The Fo-Fc omit maps (3 σ level) for the incoming L-nucleotides are illustrated in light blue.
Fig. 4.

Conformations adopted by the triphosphate of an incoming nucleotide within Polλ in different ternary complexes. (A) Chair-like conformation () shown by D-dCTP (2PFP). Positions of metal ions at site A (Na+, blue color) and site B (Mg2+, pink color) are displayed as spheres. (B) Chair-like conformation () shown by L-dCTP (complexes A and E), (–)3TC-TP (complexes A, E, and I), and (–)FTC-TP (complexes A and E). (C) N-shaped conformation as shown by L-dCTP (complexes I and M), (–)3TC-TP (complex M), and (–)FTC-TP (complexes I and M). Active site residue R517, forming one or two hydrogen bonds with an incoming L-nucleotide, is also presented. In B and C, Ca2+ ions at sites A and B are displayed as green spheres. The interactions between the triphosphate of an incoming nucleotide and the metal ions at sites A and B are presented as black dashed lines.
Surprisingly, L-dCTP, (−)3TC-TP, and (−)FTC-TP form one (in complexes I) or two (in complexes M) hydrogen bonds with an active site residue R517 in an anti-conformation and do not pair with the templating nucleotide dG, which instead interacts with Y505 through a short hydrogen bond (Fig. 3 and SI Appendix, Fig. S4, Right). To form such unusual L-nucleotide:R517 pairs as exemplified in Polλ–DNA–L-dCTP, the following structural changes occur relative to their positions in complexes A and E (Fig. 2): (i) The base of L-dCTP is closer to the template strand by 2.2–3.2 Å; (ii) the template strand backbone adjusts its position horizontally by 4.2–4.6 Å; (iii) the templating base changes its position downward by 3.5–4.6 Å; (iv) all base pairs in the single-nucleotide gapped DNA substrate adjust their positions, especially the junction base pair between the upstream primer and the template where the −1 template base shifts its position downward by 1.9–2.4 Å; (v) the guanidinium moiety of R517 shifts upward by 3.5–4.4 Å; (vi) the side chains of D427, D429, D490, Y505, and F506 all significantly reposition; and (vii) the two divalent metal ions shift their positions by 1.4–2.4 Å, with a larger movement at site B than at site A. Interestingly, the L-nucleotide:R517 hydrogen bonding pair is reminiscent of the noncanonical pair formed between an incoming D-dCTP and an active site arginine residue in the ternary structures of yeast (25) and human (26) Y-family DNA polymerase Rev1–DNA–D-dCTP, although the nitrogen atoms of the arginine side chain used in Rev1 and Polλ for such unique pairing are different (SI Appendix, Fig. S6). Strikingly, of the four natural D-dNTPs, only D-dCTP can form two hydrogen bonds with the arginine residue in Rev1 (SI Appendix, Fig. S6A), which is why Rev1 is a protein template-dependent, dCTP-specialized polymerase (27, 28). Notably, R517 of Polλ is conserved in three other human X-family DNA polymerases (SI Appendix, Fig. S6C). Thus, these DNA polymerases could form ternary complexes with L-nucleotides similar to complexes M and I. This possibility is currently being investigated in our laboratory.
In addition, unlike the triphosphates in complexes A and E (Fig. 4B) as well as in Polλ–DNA–D-dCTP (Fig. 4A), all triphosphate moieties in complexes I and M are in a novel N-shaped conformation (Fig. 4C), except one in a chair-like conformation (SI Appendix, Fig. S4D), and interact with only the divalent metal ion at site B (Fig. 3 and SI Appendix, Fig. S4, Right). The distance between the α-phosphorus atom and the primer 3′-OH (7.1–9.3 Å) is much longer than those in Polλ–DNA–D-dCTP (4.8 Å) and in complexes A (3.7–4.3 Å) and E (3.9–6.5 Å) (SI Appendix, Table S3), suggesting that complexes I and M are not the ternary structure created immediately before phosphodiester bond formation. As in complexes A and E, constraints from both the L-stereochemistry and the L-nucleotide:R517 hydrogen bonding interactions flip the sugar rings of the three L-nucleotides in complexes I and M by 180° (Fig. 3 and SI Appendix, Fig. S4, Right).
Structural Basis for Potential Catalytic Pathways of L-Nucleotide Incorporation.
Superposition of the binary structure of Polλ–DNA and the ternary structure of Polλ–DNA–D-dNTP has revealed that D-dNTP binding induces DNA and protein conformational changes including an average of 5 Å shift of the template strand relative to the primer strand; repositioning of a loop between β-strands 3 and 4 in the palm domain and β-strand 8 in the thumb domain; motions of the side chains of I492, Y505, F506, R514, and R517, which form part of the nucleotide binding pocket at the active site; and movements of the metal ion ligands D427, D429, and D490 (SI Appendix, Fig. S7) (21). As discussed above, among the four complexes of Polλ–DNA–L-dCTP, complex A and Polλ–DNA–D-dCTP (SI Appendix, Fig. S2) are structurally most similar and so are complex M and Polλ–DNA (SI Appendix, Fig. S3). Furthermore, the distance between the α-phosphorus atom and the primer 3′-OH in the four complexes of Polλ–DNA–L-dCTP follows the order of complex A < E << I < M, and the distances in complexes A and E are close to that in Polλ–DNA–D-dCTP (SI Appendix, Table S3). Together, these results suggest that the binding of L-dCTP to Polλ–DNA likely yields complex M first and ends with complex A before catalysis, and the four complexes of Polλ–DNA–L-dCTP reflect different binding conformations formed along proposed pathway I (Fig. 5). However, it is also possible that L-dCTP binds to Polλ–DNA and forms complex A and/or E (Step 1′) without going through complexes M/I (pathway II in Fig. 5).
Fig. 5.

Proposed pathways for L-dCTP incorporation catalyzed by human Polλ. Only L-dCTP, the templating nucleotide dG, and the surrounding active site residues are presented. Pathways I and II follow black and green arrows, respectively. The active site structures and electron density maps of the binary Polλ–DNA complex (B, 1XSL) and ternary Polλ–DNA–L-dCTP complexes (M, I, E, and A) are overlaid in pairs and indicated by their colors. For each pair, the Fo-Fc omit map (green color) at the 3 σ level (4 σ for complex B) is shown for only one of two overlaid structures (clockwise from complex B → M → I → E → A). The dashed blue arrow indicates the movement of the side chain of R517. The electron density map of the binary complex B is downloaded from Electron Density Server at Uppsala University.
During the first step in pathway I to form complex M, the binding of L-dCTP causes little movement of the side chains of I492, Y505, F506, R514, and R517 but moderately shifts the templating nucleotide dG downward, which is anchored by a 2.8 Å hydrogen bond with the hydroxyl group of Y505 (Fig. 3B). These structural changes allow R517 to form two hydrogen bonds and pair with the incoming L-dCTP. Because complexes I and M are structurally similar (see above), these two binding conformations are assumed to be in an equilibrium. Notably, one of the two hydrogen bonds in L-dCTP:R517 is lengthened from 3.2 Å in complex M (Fig. 3B) to 3.8 Å in complex I (SI Appendix, Fig. S4B) and thereby is abolished, whereas the distance between the α-phosphorus atom and the primer 3′-OH is shortened slightly (SI Appendix, Table S3), suggesting the catalysis direction is going forward from complex M to I. Then, the dramatic conversion of complex I to E (step 2) occurs when the loop between β-strands 3 and 4, the side chains of several active site residues including R517, the divalent metal ions, and the template strand including the templating nucleotide dG all significantly reposition (SI Appendix, Fig. S7 D–F) as observed from Polλ–DNA to Polλ–DNA–D-dNTP (SI Appendix, Fig. S7 A–C). During this conversion step, L-dCTP also shifts its position, forms a Watson–Crick base pair with dG, changes its triphosphate conformation from N-shaped to chair-like (Fig. 5), and considerably shortens the distance from its α-phosphorus atom to the primer 3′-OH (9.2 → 3.9 Å; SI Appendix, Table S3). Step 2 is followed by another hypothetical equilibrium between two closely related conformations in complexes A and E (see above). Finally, L-dCTP is incorporated into DNA, whereas pyrophosphate is released, leading to the reformation of the binary structure of Polλ–DNA (step 3 in Fig. 5).
Like D-dCTP in Polλ–DNA–D-dCTP, the L-nucleotide also forms multiple interactions with the active site residues and the DNA substrate (SI Appendix, Fig. S5) within each of the four distinctive complexes in pathway I. For the interactions in SI Appendix, Fig. S5A, we have previously used mutagenic and kinetic approaches to show that Polλ employs a network of active site residues to tightly bind both correct and incorrect D-dNTPs (29). When comparing SI Appendix, Fig. S5 B versus A, the interaction pattern for L-dCTP in complex A of Polλ–DNA–L-dCTP is very similar to that of D-dCTP in Polλ–DNA–D-dCTP, and thus, L-dCTP is stably anchored in complex A (and similarly in complex E). Consistently, the average B factor of L-dCTP in complexes A (19.0 Å2) and E (31.5 Å2) are either smaller or close to that of D-dCTP (28.4 Å2) in Polλ–DNA–D-dCTP (SI Appendix, Table S3), indicating that the binding of L-dCTP is well ordered in complexes A and E. Relative to D-dCTP (SI Appendix, Fig. S5A), the binding of L-dCTP in complexes I and M (SI Appendix, Fig. S5E) is through a different interaction pattern within the active site of Polλ and is relatively more dynamic, as suggested by its higher average B factors (SI Appendix, Table S3). Specifically, L-dCTP in complexes I and M (SI Appendix, Fig. S5E) is bound by the hydrogen bonds in the L-dCTP:R517 pair, the salt bridge between the γ-phosphate of L-dCTP and R386, the stacking interactions between the ribose of L-dCTP and the aromatic side chains of Y505 and F506 (Fig. 6F), the interaction between the β-phosphate of L-dCTP and the metal ion at site B, and the hydrogen bond between the 3′-OH of L-dCTP and the backbone carbonyl group of F506. Furthermore, relative to the chair-like triphosphate conformation in L-dCTP in complexes A and E as well as in D-dCTP, the N-shaped triphosphate conformation in L-dCTP in complexes I and M is stabilized by an extra 2.8 Å hydrogen bond between the α-phosphate and the backbone amide bond of A510 (SI Appendix, Fig. S5E).
Fig. 6.

Interaction pattern of an incoming nucleotide within the active site of Polλ. (A) Superposition of the active site structure of Polλ–DNA–D-dCTP (2PFP) and complexes A of Polλ–DNA–L-dCTP, Polλ–DNA–(–)3TC-TP, and Polλ–DNA–(–)FTC-TP. The 3′ sulfur atoms in the sugar rings of (–)3TC-TP and (–)FTC-TP are shown as large solid spheres. (B) Superposition of the active site structure of complexes M of Polλ–DNA–L-dCTP, Polλ–DNA–(–)3TC-TP, and Polλ–DNA–(–)FTC-TP. Zoomed stacking interactions are between the active site residues (Y505 and F506) of Polλ and the ribose of D-dCTP (C), L-dCTP (D and F), and (–)3TC-TP (E and G). D and E are of complexes A, whereas F and G are of complexes M. The metal ions at site A and site B are displayed as spheres. The nucleotides and active site residues are presented as lines and sticks, respectively. The distances between the atoms of an incoming nucleotide and the atoms of Y505 and F506 are presented as black dashed lines, with the numbers depicting their lengths in Å.
To provide solution evidence for the existence of complexes I and M in pathway I, R517, which pairs with L-dCTP in these complexes (Fig. 3 and SI Appendix, Fig. S4), was mutated to alanine. This R517A mutant incorporated D-dCTP with a 2,000-fold lower kp (9.6 × 10−4⋅s−1) and a fourfold lower Kd than wild-type Polλ (SI Appendix, Fig. S8A and Table 1). Consistently, we have previously shown that the R517A mutation in Polλ decreases the kp of correct D-dTTP incorporation by 250-fold, although it enhances dTTP binding affinity by twofold (29). The drastic decrease in kp is not surprising, as R517 in Polλ–DNA–D-dCTP interacts with the templating nucleotide, the –1 template nucleotide, and E529 (SI Appendix, Fig. S9A) and thereby stabilizes the template for catalysis (30). Surprisingly, R517A could not incorporate L-dCTP after 7 h at 37 °C (SI Appendix, Fig. S8B). The failure of L-dCTP incorporation suggests that either L-dCTP was unable to bind to the binary complex of R517A–DNA and form a ternary complex R517A–DNA–L-dCTP, or the ternary complex was formed but was catalytically inactive. To distinguish between these possibilities, we estimated the binding affinity of L-dCTP through a competition assay (31, 32), yielding a Kd of 36 µM (SI Appendix, Fig. S8C and Table 1). In contrast to the fourfold higher affinity of D-dCTP, L-dCTP binds to R517A with a 54-fold lower affinity than to wild-type Polλ (Table 1). Furthermore, we tested the inhibitory effect of L-dCTP on D-dCTP incorporation catalyzed by R517A. Our results show that the incorporation of 5 μM D-dCTP was not affected by the presence of 5 μM L-dCTP (SI Appendix, Fig. S8D). This is not surprising, as at the same concentration of 5 μM, L-dCTP was unable to compete against D-dCTP to bind to the R517A⋅DNA complex based on their 164-fold Kd difference (Table 1). Taken together, these kinetic results demonstrate that R517 plays a key role in the binding of L-dCTP, not D-dCTP, in pathway I. It is reasonable to assume that complexes I and M were not formed because the R517A mutation eliminated the key hydrogen bonding interactions between R517 and L-dCTP in these complexes (Fig. 5). Without complexes I/M, complexes A/E could not form due to lack of the conversion (step 2) in pathway I (Fig. 5). However, the weak binding affinity of L-dCTP with the R517A mutant (Kd = 36 µM; Table 1) suggests that a small amount of complexes A/E was formed via pathway II under the conditions in SI Appendix, Fig. S8B. The R517A mutation likely did not impact the L-dCTP binding through pathway II, considering that residue R517 does not directly interact with L-dCTP within complexes A and E (SI Appendix, Fig. S9B). Furthermore, the 54-fold lower affinity of L-dCTP with the R517A mutant than with wild-type Polλ (Table 1) caused by the absence of pathway I suggests the dominance of pathway I over II with wild-type Polλ. The lack of L-dCTP incorporation in SI Appendix, Fig. S8B indicates that the R517A mutation significantly perturbed the interactions within the active site of Polλ in complexes A and E (SI Appendix, Fig. S9B) and rendered these complexes catalytically inactive. Based on the two pathways in Fig. 5 and the above kinetic results, we proposed a simplified kinetic scheme for L-dCTP binding and incorporation by Polλ (Scheme 1). In this scheme, the nonproductive complex (E⋅DNAn⋅dNTP)N likely represents complexes M/I, whereas the productive complex (E⋅DNAn⋅dNTP)P corresponds to complexes A/E.
Scheme 1.
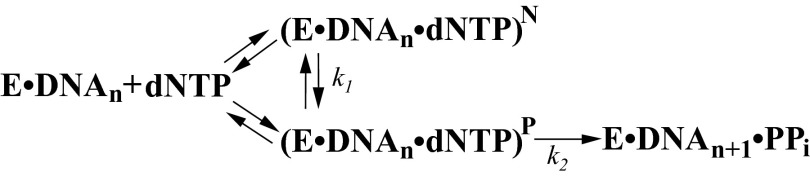
Kinetic pathways for nucleotide incorporation catalyzed by human DNA polymerase λ. L-nucleotide incorporation mainly follows the pathway of E⋅DNAn ↔ (E⋅DNAn⋅dNTP)N ↔ (E⋅DNAn⋅dNTP)P ↔ E⋅DNAn+1⋅PPi, whereas a D-nucleotide is predominantly incorporated via the pathway of En⋅DNA ↔ (E⋅DNAn⋅dNTP)P ↔ E⋅DNAn+1⋅PPi. (E⋅DNAn⋅dNTP)N and (E⋅DNAn⋅dNTP)P represent nonproductive and productive ternary complexes, respectively. PPi denotes pyrophosphate.
Notably, the structures of complexes A, E, I, and M of Polλ–DNA–L-dCTP are very similar to those of the four corresponding complexes of either Polλ–DNA–(–)3TC-TP or Polλ–DNA–(–)FTC-TP (see above). For example, the active site structures of complexes A with the three L-nucleotides are almost superimposable (Fig. 6A), and the same phenomenon can be found with complexes M (Fig. 6B). Thus, it is very likely that Polλ incorporates (–)3TC-TP and (–)FTC-TP, the chemical analogs of L-dCTP, into a single-nucleotide gapped DNA substrate by following the same catalytic pathways (Fig. 5) and kinetic scheme (Scheme 1) as it does L-dCTP. Although not measured here, the rate for each of the steps in Fig. 5 or Scheme 1 is expected to be different among the three L-nucleotides.
Structural Basis for Potential Catalytic Pathways of D-Nucleotide Incorporation.
Unlike the L-nucleotides, D-dNTP has been crystallized with Polλ and DNA in only one binding conformation—for example, the structure of Polλ–DNA–D-dCTP in SI Appendix, Fig. S2A (20, 21). This structural result and the 104-fold higher incorporation efficiency (kp/Kd) of D-dCTP over L-dNTP (Table 1) suggest that Polλ bound and incorporated D-dCTP by predominantly following pathway II, not pathway I (Fig. 5). Consistently, the R517A mutation did not weaken the binding affinity of D-dCTP but actually enhanced it by fourfold (Table 1). Interestingly, the ternary structure of the R517K mutant of Polλ (R517K–DNA–D-dNTP) shows that the template strand is in a similar position as in the canonical ternary structure (SI Appendix, Fig. S2A), but the active site residues are intermediates between those observed in the binary (SI Appendix, Fig. S3A) and ternary (SI Appendix, Fig. S2A) structures with wild-type Polλ (33). Molecular dynamic simulation studies demonstrate that the binding of D-dNTP to Polλ–DNA induces significant DNA motion and the reposition of the side chains of I492, Y505, F506, R514, and R517 before chemistry (30). These results indicate that the conversion from the binary to ternary structures (SI Appendix, Fig. S7 A–C) during D-dCTP incorporation catalyzed by Polλ may go through similar structural intermediate(s) as those observed during L-dCTP incorporation (pathway II, Fig. 5). Consistently, our modeling results suggest that D-dCTP, like L-dCTP, can form hydrogen bonds with R517 at the active site of Polλ and yield the L-nucleotide’s complex I or M-like conformations as long as its triphosphate conformation is flexible and chair-like (SI Appendix, Fig. S10). In conclusion, D-dCTP, as L-dCTP, is bound and incorporated by Polλ via the same two catalytic pathways in Fig. 5 and the kinetic scheme in Scheme 1.
Structural Basis for the D-Stereoselectivity of Polλ and Differing Kinetic Parameters Among the L-Nucleotides.
The D-stereoselectivity [(kp/Kd)D-dNTP/(kp/Kd)L-dNTP] of a DNA polymerase or RT is a product of the nucleotide binding affinity ratio (Kd)L-dNTP/(Kd)D-dNTP and the ratio of incorporation rate constants (kp)D-dNTP/(kp)L-dNTP. Table 1 shows that the D-stereoselectivity of Polλ (1.2 × 104) is contributed by the ratio of incorporation rate constants (1.44 × 104) but decreased slightly by the binding affinity ratio (0.83). These ratios and the D-stereoselectivity as well as kinetic parameters are different among L-dCTP, (−)3TC-TP, and (−)FTC-TP (Table 1), and these differences can be rationalized structurally.
Current kinetic and structural results suggest that pathways I and II govern the binding and incorporation of L-dCTP and D-dCTP, respectively (see above). Consequently, the binding of D-dCTP is determined by the stability of Polλ–DNA–D-dCTP (SI Appendix, Figs. S2A and S5A), whereas all four complexes of each L-nucleotide (Fig. 5) contribute to the binding of the L-nucleotide. Interestingly, the active site interaction patterns for L-dCTP, (–)3TC-TP, and (–)FTC-TP in the same complex group—for example, complexes A (SI Appendix, Fig. S5 B–D)—are analogous. However, in each complex group, the stacking interactions between the side chains of Y505 and F506 and the ribose are stronger in (–)3TC-TP and (–)FTC-TP than in L-dCTP because of the substitution of the C3′ atom of L-dCTP with a more electron-rich sulfur atom in (–)3TC-TP and (–)FTC-TP. The difference in the strength of the stacking interactions is also contributed by the slightly shorter interaction distance with (–)3TC-TP and (–)FTC-TP than with L-dCTP (Fig. 6). The strong stacking interactions outweigh the favorable impact of the hydrogen bond formed between the 3′-OH of L-dCTP and the backbone carbonyl group of F506 (Fig. 6 D and F) and contribute to the two- to sixfold binding affinity difference between the L-dCTP analogs and L-dCTP. In addition, the reason (–)3TC-TP possesses a threefold higher affinity than its 5-fluorinated derivative (–)FTC-TP (Table 1) is because the strong electron-withdrawing 5-fluorine atom in the base of (–)FTC-TP alters the π electron distribution within the cytosine and thereby affects its stacking interactions with the primer 3′-base. In comparison, the stacking interactions in L-dCTP and D-dCTP have similar strength as a result of their chemically identical ribose and comparable distance between the ribose and the side chains of Y505 and F506 (Fig. 6 C, D, and F). Because the binding of L-dCTP (complexes A/E) and D-dCTP are alike in pattern and intensity and L-dCTP in complexes I and M is also bound tightly despite adopting different binding patterns (see above), the L-stereochemistry does not significantly alter nucleotide binding affinity. In contrast, because of an opposite ribose binding orientation caused by the L-stereochemistry, the 3′-OH of L-dCTP, not D-dCTP, forms a hydrogen bond with the backbone carbonyl group of F506 (Fig. 6 C and D), which may lead to a slightly higher binding affinity for L-dCTP than for D-dCTP (Table 1) with a ratio [(Kd)L-dCTP/(Kd)D-dCTP] of 0.83.
Although both D-dCTP and L-dCTP are incorporated by following the same kinetic mechanism in Scheme 1, the incorporation rate of D-dCTP is controlled by k2, whereas the kp of L-dCTP is a function of both k1 and k2 (see above). The conversion (k1) of the nonproductive complexes (complexes M/I) to the productive complexes (complexes A/E) likely slowed down the overall incorporation rate of L-dCTP relative to the kp of D-dCTP. Moreover, the structures of complex A with L-dCTP and the canonical ternary complex of Polλ–DNA–D-dCTP, especially their active site structures, are similar but not identical, leading to very different k2 values for the incorporation of the enantiomers. Owing to the 180° rotation of the ribose, the 3′-OH of L-dCTP and the primer 3′-OH in complex A face each other and may sterically clash (Fig. 3A). To eliminate this problem, both the ribose and triphosphate of L-dCTP significantly adjust their binding conformations from those of D-dCTP (Fig. 6A) and allow water molecule-bridged hydrogen bonding interactions between these 3′-OH groups (SI Appendix, Fig. S5B). These interactions likely weaken the primer 3′-OH group as a nucleophile during phosphodiester bond formation. In addition, relative to the active site structure with D-dCTP (Fig. 6A), the binding of L-dCTP moderately alters the side chain conformations of active site residues D427, D429, D490, Y505, F506, and R514; repositions the divalent metal ions at sites A and B; and forms an extra hydrogen bond between the 3′-OH group of L-dCTP and the backbone carbonyl group of F506 (Fig. 6D). For example, the side chain of F506 in Polλ–DNA–L-dCTP was rotated by 30–50° relative to its conformation in Polλ–DNA–D-dCTP (Fig. 6A). Together, these active site rearrangements, the nucleotide binding conformational changes, and the aforementioned water-mediated hydrogen bonding interactions lead to a very different k2 with L-dCTP than with D-dCTP and eventually a high ratio [(kp)D-dCTP/(kp)L-dCTP] of 1.44 × 104 (Table 1).
Notably, relative to L-dCTP, (−)3TC-TP and (−)FTC-TP lack the 3′-OH group and their C3′ atom is substituted with an electron-rich sulfur atom. These chemical changes in the ribose eliminate the water-mediated hydrogen bonding interactions, somewhat lessen the changes in the active site structure (Fig. 6A and SI Appendix, Fig. S5 C and D), and stabilize nucleotide binding in complex A through stronger stacking interactions between the ribose and the side chains of Y505 and F506 (Fig. 6E). As a result of these improved factors, (−)3TC-TP and (−)FTC-TP possess 21- to 33-fold higher incorporation rates, two- to sixfold tighter binding affinities (see above), and 67- to 119-fold greater incorporation efficiencies than L-dCTP, which collectively lower their D-stereoselectivity values relative to L-dCTP by 100-fold (Table 1).
Structural Insight into Design of Improved Antiviral L-Nucleotides.
If viral RTs bind and incorporate the L-nucleotides similar to Polλ, (−)3TC-TP and (−)FTC-TP will be more potent inhibitors of viral RTs than L-dCTP. This is probably why L-cytidine is not a potent antiviral nucleoside analog. Interestingly, Y115 and F160 of HIV-1 RT occupy the analogous positions in its active site as Y505 and F506 in Polλ. Ternary crystal structures of these L-analogs with HIV-1 RT are necessary to verify the roles of Y115 and F160 in the stacking and binding of an L-nucleotide and provide structural guidance in designing more potent RT inhibitors. For example, to make (−)3TC-TP and (−)FTC-TP stronger anti-HIV inhibitors, one could substitute atoms or groups in their ribose and base to improve the aforementioned stacking interactions with Y115, F160, and the primer 3′-base and enhance their binding and incorporation by HIV-1 RT. Subsequently, one should also consider if these improved RT inhibitors will have a stronger adverse effect on human DNA polymerases than (−)3TC-TP and (−)FTC-TP, resulting in higher in vivo toxicity. To minimize the inhibitory effect of (−)3TC-TP and (−)FTC-TP on human DNA polymerases, especially Polλ, one could substitute cytosine in these L-nucleotides for another base that cannot form hydrogen bonds with R517 and thereby eliminate pathway I (Fig. 5). Regardless, these predications need to be verified by comprehensive crystallographic, kinetic, and toxicological investigation of any rationally designed L-nucleotide inhibitors.
Conclusion
In summary, the 12 ternary structures of human Polλ, DNA, L-dCTP, (−)3TC-TP, or (−)FTC-TP provide structural insight into how an L-nucleotide is bound and incorporated within the active site of Polλ. It would be interesting to see if other DNA polymerases and RTs form similar ternary structures with an incoming L-nucleotide as Polλ. Relative to the ribose of D-dCTP in the canonical ternary structure of Polλ–DNA–D-dCTP, the riboses of all of the L-nucleotides are flipped by 180°. The four ternary structures in a crystal asymmetric unit with each of the L-nucleotides reflect four different binding conformations formed along two proposed catalytic pathways for L-nucleotide incorporation. The two early binding ternary structures in pathway I contain unprecedented L-nucleotide:R517 pairs, whereas the evicted templating nucleotide dG is anchored by a hydrogen bond with the hydroxyl group of Y505. In each L-nucleotide:R517 pair, the triphosphate moiety of the L-nucleotide mostly displays a novel N-shaped conformation. In the two latter ternary structures of pathway I, the incoming L-nucleotide forms a normal Watson–Crick base pair with the templating nucleotide dG. Our site-directed mutagenesis and kinetic studies demonstrate that the side chain of R517 is critical for the formation of the four ternary complexes with each L-nucleotide. Because R517 is conserved in the X-family DNA polymerases, it will be interesting to see if other X-family enzymes bind to an incoming L-nucleotide through the arginine residue. Comparison of the ternary structures with D-dCTP, L-dCTP, (−)3TC-TP, and (−)FTC-TP reveals a structural basis for the difference in their kinetic parameters and associated D-stereoselectivity.
Materials and Methods
Preparation of Protein and DNA.
Human full-length (34) and truncated Polλ (residues 245–575) (22) as well as the R517A mutant of human full-length Polλ (29) were expressed and purified as previously described. DNA oligomers in the single-nucleotide gapped DNA substrate for crystallization including template T11 (5′-CGGCGGTACTG-3′), an upstream primer P6 (5′-CAGTAC-3′), and a downstream 5′-phosphorylated primer P4 (5′-pGCCG-3′) and in the DNA substrate 21-mer⋅19-mer/41-mer (SI Appendix, Fig. S1A) for kinetic studies were purchased from Integrated DNA Technologies. L-dCTP, (–)3TC-TP, and (–)FTC-TP were obtained from Jena Bioscience.
Crystallization and Structure Determination.
Purified Polλ was concentrated to 16 mg/mL and then mixed with an annealed DNA substrate (P6⋅P4/T11) at a molar ratio of 1:3 (protein/DNA) to form a binary complex. A ternary complex was subsequently formed with the addition of 1 mM L-nucleotide [L-dCTP, (–)3TC-TP, or (–)FTC-TP]. Notably, identical Polλ and DNA concentrations and a similar nucleotide concentration (0.9 mM) were used in previous crystallographic studies (35). Crystals were obtained using the hanging drop vapor diffusion method in which each Polλ ternary complex mixture was equilibrated against a reservoir buffer composed of 0.1 M sodium cacodylate (pH 6.5), 0.2 M calcium acetate, and 4% (wt/vol) PEG8000 (36). Notably, noncatalytic Ca(II), rather than catalytic Mg(II), was used here, as Ca(II) has been used regularly to trap incoming nucleotides in preinsertion ternary complexes with other DNA polymerases (37–41). Crystals were harvested and placed in cryosolutions in four different steps of increasing PEG [4–18% (wt/vol)] and ethylene glycol [12.5% (vol/vol)] concentrations before they were flash frozen in liquid nitrogen (36). X-ray diffraction data were collected using LRL-CAT beamline facilities at Advance Photon Source, Argonne National Laboratory. X-ray diffraction data were processed using MOSFLM (42). The structure was solved using the molecular replacement method by PHASER (43) using Protein Data Bank ID code 2PFP (20) as the initial model in the absence of ligands and solvent molecules. Structural refinement was carried out using REFMAC5 (44). COOT (45) was used for visualization and model building. Quality of the models was assessed using PROCHECK (46). Figures were created using PYMOL (47).
Pre–Steady-State Kinetic Assays.
All fast reactions were performed by using a rapid chemical quench-flow apparatus (KinTek). Our published experimental procedures (12) were followed here. Briefly, a preincubated solution of full-length human Polλ (600 nM) and 30 nM [32P]-labeled–21-mer⋅19-mer/41-mer (SI Appendix, Fig. S1A) was mixed with varying concentrations of a nucleotide in buffer L [50 mM Tris⋅HCl, pH 8.4, 5 mM MgCl2, 100 mM NaCl, 0.1 mM EDTA, 5 mM DTT, 10% (vol/vol) glycerol, and 0.1 mg/mL BSA] at 37 °C. The 21-mer⋅19-mer/41-mer is a model DNA substrate for the short-patch base excision repair pathway (48). After various times, the reaction was terminated with 0.37 M EDTA and analyzed by sequencing gel electrophoresis. Each time course of product formation was fit to a single-exponential equation, [Product] = A[1 – exp(–kobst)], using KaleidaGraph (Synergy Software) to yield a reaction amplitude (A) and an observed rate constant of nucleotide incorporation (kobs). The kobs values were then plotted against nucleotide concentrations, and the plot was fit to a hyperbolic equation, kobs = kp[dNTP]/([dNTP] + Kd), to yield an equilibrium dissociation constant (Kd) and a maximum nucleotide incorporation rate constant (kp).
Supplementary Material
Acknowledgments
The authors thank Dr. Joy Feng of Gilead Sciences, Inc. for providing L-dCTP and Ms. Andrea Moon and Dr. Lars C. Pedersen at the Laboratory of Structural Biology, National Institute of Environmental Health Sciences, National Institutes of Health (NIH), for providing initial crystallization conditions. The authors are grateful for the usage of the Lilly Research Laboratories Collaborative Access Team beamline at Sector 31 of the Advanced Photon Source provided by Eli Lilly Company, which operates the facility. The authors are also grateful for the usage of the Advanced Photon Source, an Office of Science User Facility operated for the U.S. Department of Energy (DOE) Office of Science by Argonne National Laboratory, which was supported by the DOE under Contract DE-AC02-06CH11357. This work was supported by NIH Grant GM079403 and National Science Foundation Grant MCB-0960961 (to Z.S.). W.J.Z. was supported by the NIH Chemistry and Biochemistry Interface Program at The Ohio State University (Grant T32 GM008512).
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission. R.K. is a guest editor invited by the Editorial Board.
Data deposition: Atomic coordinates have been deposited in the Protein Data Bank, www.rcsb.org [PDB ID code 4K4G (Polλ–DNA–L-dCTP), 4K4H (Polλ–DNA–(−)3TC-TP), and 4K4I (Polλ–DNA–(−)FTC-TP)].
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1401286111/-/DCSupplemental.
References
- 1.Feng JY, Anderson KS. Mechanistic studies comparing the incorporation of (+) and (-) isomers of 3TCTP by HIV-1 reverse transcriptase. Biochemistry. 1999;38(1):55–63. doi: 10.1021/bi982340r. [DOI] [PubMed] [Google Scholar]
- 2.Feng JY, Shi J, Schinazi RF, Anderson KS. Mechanistic studies show that (-)-FTC-TP is a better inhibitor of HIV-1 reverse transcriptase than 3TC-TP. FASEB J. 1999;13(12):1511–1517. doi: 10.1096/fasebj.13.12.1511. [DOI] [PubMed] [Google Scholar]
- 3.Gumina G, Chong Y, Choo H, Song GY, Chu CK. L-nucleosides: Antiviral activity and molecular mechanism. Curr Top Med Chem. 2002;2(10):1065–1086. doi: 10.2174/1568026023393138. [DOI] [PubMed] [Google Scholar]
- 4.Schinazi RF, et al. Selective inhibition of human immunodeficiency viruses by racemates and enantiomers of cis-5-fluoro-1-[2-(hydroxymethyl)-1,3-oxathiolan-5-yl]cytosine. Antimicrob Agents Chemother. 1992;36(11):2423–2431. doi: 10.1128/aac.36.11.2423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chang CN, Skalski V, Zhou JH, Cheng YC. Biochemical pharmacology of (+)- and (-)-2′,3′-dideoxy-3′-thiacytidine as anti-hepatitis B virus agents. J Biol Chem. 1992;267(31):22414–22420. [PubMed] [Google Scholar]
- 6.Brown JA, Pack LR, Fowler JD, Suo Z. Presteady state kinetic investigation of the incorporation of anti-hepatitis B nucleotide analogues catalyzed by noncanonical human DNA polymerases. Chem Res Toxicol. 2012;25(1):225–233. doi: 10.1021/tx200458s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Joyce CM, Benkovic SJ. DNA polymerase fidelity: Kinetics, structure, and checkpoints. Biochemistry. 2004;43(45):14317–14324. doi: 10.1021/bi048422z. [DOI] [PubMed] [Google Scholar]
- 8.Johnson AA, et al. Toxicity of antiviral nucleoside analogs and the human mitochondrial DNA polymerase. J Biol Chem. 2001;276(44):40847–40857. doi: 10.1074/jbc.M106743200. [DOI] [PubMed] [Google Scholar]
- 9.Moyle G. Toxicity of antiretroviral nucleoside and nucleotide analogues: Is mitochondrial toxicity the only mechanism? Drug Saf. 2000;23(6):467–481. doi: 10.2165/00002018-200023060-00001. [DOI] [PubMed] [Google Scholar]
- 10.Olivero OA, et al. Incorporation of zidovudine into leukocyte DNA from HIV-1-positive adults and pregnant women, and cord blood from infants exposed in utero. AIDS. 1999;13(8):919–925. doi: 10.1097/00002030-199905280-00007. [DOI] [PubMed] [Google Scholar]
- 11.Wutzler P, Thust R. Genetic risks of antiviral nucleoside analogues—A survey. Antiviral Res. 2001;49(2):55–74. doi: 10.1016/s0166-3542(00)00139-x. [DOI] [PubMed] [Google Scholar]
- 12.Brown JA, Pack LR, Fowler JD, Suo Z. Pre-steady-state kinetic analysis of the incorporation of anti-HIV nucleotide analogs catalyzed by human X- and Y-family DNA polymerases. Antimicrob Agents Chemother. 2011;55(1):276–283. doi: 10.1128/AAC.01229-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Aoufouchi S, et al. Two novel human and mouse DNA polymerases of the polX family. Nucleic Acids Res. 2000;28(18):3684–3693. doi: 10.1093/nar/28.18.3684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.García-Díaz M, et al. DNA polymerase lambda (Pol lambda), a novel eukaryotic DNA polymerase with a potential role in meiosis. J Mol Biol. 2000;301(4):851–867. doi: 10.1006/jmbi.2000.4005. [DOI] [PubMed] [Google Scholar]
- 15.Nagasawa K, et al. Identification and characterization of human DNA polymerase beta 2, a DNA polymerase beta-related enzyme. J Biol Chem. 2000;275(40):31233–31238. doi: 10.1074/jbc.M004263200. [DOI] [PubMed] [Google Scholar]
- 16.García-Díaz M, et al. DNA polymerase lambda, a novel DNA repair enzyme in human cells. J Biol Chem. 2002;277(15):13184–13191. doi: 10.1074/jbc.M111601200. [DOI] [PubMed] [Google Scholar]
- 17.Brown JA, Duym WW, Fowler JD, Suo Z. Single-turnover kinetic analysis of the mutagenic potential of 8-oxo-7,8-dihydro-2′-deoxyguanosine during gap-filling synthesis catalyzed by human DNA polymerases lambda and beta. J Mol Biol. 2007;367(5):1258–1269. doi: 10.1016/j.jmb.2007.01.069. [DOI] [PubMed] [Google Scholar]
- 18.Lee JW, et al. Implication of DNA polymerase lambda in alignment-based gap filling for nonhomologous DNA end joining in human nuclear extracts. J Biol Chem. 2004;279(1):805–811. doi: 10.1074/jbc.M307913200. [DOI] [PubMed] [Google Scholar]
- 19.Bertocci B, De Smet A, Weill JC, Reynaud CA. Nonoverlapping functions of DNA polymerases mu, lambda, and terminal deoxynucleotidyltransferase during immunoglobulin V(D)J recombination in vivo. Immunity. 2006;25(1):31–41. doi: 10.1016/j.immuni.2006.04.013. [DOI] [PubMed] [Google Scholar]
- 20.Garcia-Diaz M, Bebenek K, Krahn JM, Pedersen LC, Kunkel TA. Role of the catalytic metal during polymerization by DNA polymerase lambda. DNA Repair (Amst) 2007;6(9):1333–1340. doi: 10.1016/j.dnarep.2007.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Garcia-Diaz M, Bebenek K, Krahn JM, Kunkel TA, Pedersen LC. A closed conformation for the Pol lambda catalytic cycle. Nat Struct Mol Biol. 2005;12(1):97–98. doi: 10.1038/nsmb876. [DOI] [PubMed] [Google Scholar]
- 22.Fiala KA, Abdel-Gawad W, Suo Z. Pre-steady-state kinetic studies of the fidelity and mechanism of polymerization catalyzed by truncated human DNA polymerase lambda. Biochemistry. 2004;43(21):6751–6762. doi: 10.1021/bi049975c. [DOI] [PubMed] [Google Scholar]
- 23.Sarafianos SG, et al. Lamivudine (3TC) resistance in HIV-1 reverse transcriptase involves steric hindrance with beta-branched amino acids. Proc Natl Acad Sci USA. 1999;96(18):10027–10032. doi: 10.1073/pnas.96.18.10027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Vaisman A, Ling H, Woodgate R, Yang W. Fidelity of Dpo4: Effect of metal ions, nucleotide selection and pyrophosphorolysis. EMBO J. 2005;24(17):2957–2967. doi: 10.1038/sj.emboj.7600786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nair DT, Johnson RE, Prakash L, Prakash S, Aggarwal AK. Rev1 employs a novel mechanism of DNA synthesis using a protein template. Science. 2005;309(5744):2219–2222. doi: 10.1126/science.1116336. [DOI] [PubMed] [Google Scholar]
- 26.Swan MK, Johnson RE, Prakash L, Prakash S, Aggarwal AK. Structure of the human Rev1-DNA-dNTP ternary complex. J Mol Biol. 2009;390(4):699–709. doi: 10.1016/j.jmb.2009.05.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Howell CA, Prakash S, Washington MT. Pre-steady-state kinetic studies of protein-template-directed nucleotide incorporation by the yeast Rev1 protein. Biochemistry. 2007;46(46):13451–13459. doi: 10.1021/bi701429v. [DOI] [PubMed] [Google Scholar]
- 28.Brown JA, Fowler JD, Suo Z. Kinetic basis of nucleotide selection employed by a protein template-dependent DNA polymerase. Biochemistry. 2010;49(26):5504–5510. doi: 10.1021/bi100433x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Brown JA, et al. Identification of critical residues for the tight binding of both correct and incorrect nucleotides to human DNA polymerase λ. J Mol Biol. 2010;403(4):505–515. doi: 10.1016/j.jmb.2010.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Foley MC, Arora K, Schlick T. Sequential side-chain residue motions transform the binary into the ternary state of DNA polymerase lambda. Biophys J. 2006;91(9):3182–3195. doi: 10.1529/biophysj.106.092080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Capson TL, et al. Kinetic characterization of the polymerase and exonuclease activities of the gene 43 protein of bacteriophage T4. Biochemistry. 1992;31(45):10984–10994. doi: 10.1021/bi00160a007. [DOI] [PubMed] [Google Scholar]
- 32.Wong I, Patel SS, Johnson KA. An induced-fit kinetic mechanism for DNA replication fidelity: Direct measurement by single-turnover kinetics. Biochemistry. 1991;30(2):526–537. doi: 10.1021/bi00216a030. [DOI] [PubMed] [Google Scholar]
- 33.Bebenek K, et al. Substrate-induced DNA strand misalignment during catalytic cycling by DNA polymerase lambda. EMBO Rep. 2008;9(5):459–464. doi: 10.1038/embor.2008.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Fiala KA, Duym WW, Zhang J, Suo Z. Up-regulation of the fidelity of human DNA polymerase lambda by its non-enzymatic proline-rich domain. J Biol Chem. 2006;281(28):19038–19044. doi: 10.1074/jbc.M601178200. [DOI] [PubMed] [Google Scholar]
- 35.Gosavi RA, Moon AF, Kunkel TA, Pedersen LC, Bebenek K. The catalytic cycle for ribonucleotide incorporation by human DNA Pol λ. Nucleic Acids Res. 2012;40(15):7518–7527. doi: 10.1093/nar/gks413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Garcia-Diaz M, et al. A structural solution for the DNA polymerase lambda-dependent repair of DNA gaps with minimal homology. Mol Cell. 2004;13(4):561–572. doi: 10.1016/s1097-2765(04)00061-9. [DOI] [PubMed] [Google Scholar]
- 37.Swan MK, Johnson RE, Prakash L, Prakash S, Aggarwal AK. Structural basis of high-fidelity DNA synthesis by yeast DNA polymerase delta. Nat Struct Mol Biol. 2009;16(9):979–986. doi: 10.1038/nsmb.1663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Franklin MC, Wang J, Steitz TA. Structure of the replicating complex of a pol alpha family DNA polymerase. Cell. 2001;105(5):657–667. doi: 10.1016/s0092-8674(01)00367-1. [DOI] [PubMed] [Google Scholar]
- 39.Wing RA, Bailey S, Steitz TA. Insights into the replisome from the structure of a ternary complex of the DNA polymerase III alpha-subunit. J Mol Biol. 2008;382(4):859–869. doi: 10.1016/j.jmb.2008.07.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Irimia A, Eoff RL, Guengerich FP, Egli M. Structural and functional elucidation of the mechanism promoting error-prone synthesis by human DNA polymerase kappa opposite the 7,8-dihydro-8-oxo-2′-deoxyguanosine adduct. J Biol Chem. 2009;284(33):22467–22480. doi: 10.1074/jbc.M109.003905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ling H, Boudsocq F, Woodgate R, Yang W. Crystal structure of a Y-family DNA polymerase in action: A mechanism for error-prone and lesion-bypass replication. Cell. 2001;107(1):91–102. doi: 10.1016/s0092-8674(01)00515-3. [DOI] [PubMed] [Google Scholar]
- 42.Battye TG, Kontogiannis L, Johnson O, Powell HR, Leslie AG. iMOSFLM: A new graphical interface for diffraction-image processing with MOSFLM. Acta Crystallogr D Biol Crystallogr. 2011;67(Pt 4):271–281. doi: 10.1107/S0907444910048675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.McCoy AJ, et al. Phaser crystallographic software. J Appl Cryst. 2007;40(Pt 4):658–674. doi: 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Murshudov GN, et al. REFMAC5 for the refinement of macromolecular crystal structures. Acta Crystallogr D Biol Crystallogr. 2011;67(Pt 4):355–367. doi: 10.1107/S0907444911001314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Emsley P, Cowtan K. Coot: Model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr. 2004;60(Pt 12 Pt 1):2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 46.Laskowski RA, Rullmannn JA, MacArthur MW, Kaptein R, Thornton JM. AQUA and PROCHECK-NMR: Programs for checking the quality of protein structures solved by NMR. J Biomol NMR. 1996;8(4):477–486. doi: 10.1007/BF00228148. [DOI] [PubMed] [Google Scholar]
- 47.DeLano WL. The PyMOL Molecular Graphics System. San Carlos, CA: DeLano Scientific; 2002. [Google Scholar]
- 48.Duym WW, Fiala KA, Bhatt N, Suo Z. Kinetic effect of a downstream strand and its 5′-terminal moieties on single nucleotide gap-filling synthesis catalyzed by human DNA polymerase lambda. J Biol Chem. 2006;281(47):35649–35655. doi: 10.1074/jbc.M607479200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.