

Significance
The understanding of how membrane proteins fold pales in comparison with the understanding of globular protein folding. This discrepancy is partly due to the fact that membrane proteins are difficult to work with experimentally. In turn, the lack of high-quality experimental data has caused modeling of membrane proteins to lag behind. Also, the extent to which the translocon assists transmembrane domains in folding is unclear. The number of experimentally determined membrane protein structures has recently increased, and we may now be at the stage where it has become possible to derive transferable simulation models for studying transmembrane protein folding. We describe the optimization of one such model and its application to predicting helical packings within the native topology.
Keywords: energy landscape theory, molecular dynamics
Abstract
We explore the hypothesis that the folding landscapes of membrane proteins are funneled once the proteins’ topology within the membrane is established. We extend a protein folding model, the associative memory, water-mediated, structure, and energy model (AWSEM) by adding an implicit membrane potential and reoptimizing the force field to account for the differing nature of the interactions that stabilize proteins within lipid membranes, yielding a model that we call AWSEM-membrane. Once the protein topology is set in the membrane, hydrophobic attractions play a lesser role in finding the native structure, whereas polar–polar attractions are more important than for globular proteins. We examine both the quality of predictions made with AWSEM-membrane when accurate knowledge of the topology and secondary structure is available and the quality of predictions made without such knowledge, instead using bioinformatically inferred topology and secondary structure based on sequence alone. When no major errors are made by the bioinformatic methods used to assign the topology of the transmembrane helices, these two types of structure predictions yield roughly equivalent quality structures. Although the predictive energy landscape is transferable and not structure based, within the correct topological sector we find the landscape is indeed very funneled: Thermodynamic landscape analysis indicates that both the total potential energy and the contact energy decrease as native contacts are formed. Nevertheless the near symmetry of different helical packings with respect to native contact formation can result in multiple packings with nearly equal thermodynamic occupancy, especially at temperatures just below collapse.
The folding of globular proteins has come to be well understood starting from Anfinsen’s thermodynamic hypothesis (1), by means of statistical energy landscape theory (2–5) and its principle of minimal frustration. Evolution selects the sequences of most globular proteins so that folding is, by and large, thermodynamically controlled and the landscape is dominated by the interactions between residues that are close together in the folded state, i.e., the native contacts. In vivo folding of α-helical transmembrane proteins differs from the usually autonomous folding of globular proteins in that, during translation, another actor, the translocon, generally assists the nascent chain in either translocating across or integrating peptides into the lipid membrane. Topology, by which we mean the “specification of the number of transmembrane helices and their in and/or out orientations across the membrane” (ref. 6, p. 909), in vivo is thus initially established cotranslationally with few exceptions. Large barriers between alternate topologies once the protein is folded, along with the involvement of the translocon catalyst, suggest a role for kinetic control in folding of α-helical transmembrane proteins. In light of these differences, what aspects of energy landscape theory, based as it is on near-equilibrium statistical physics, can be applied for understanding and predicting membrane protein structures and folding mechanisms?
Despite the known differences between globular and membrane protein folding, there is evidence that some of the ideas from energy landscape theory also apply to membrane protein folding. Starting with Khorana’s work (7), numerous α-helical transmembrane proteins have been refolded from a chemically denatured state in vitro (8). This indicates that at least some transmembrane domains may not require the translocon to fold properly. In addition, recent experiments on a few α-helical transmembrane proteins have succeeded in characterizing the structure of transition state ensembles, in a manner like that used for globular proteins. These studies suggest that native contacts are important in the folding nucleus but may not represent the whole story (9, 10). Whether membrane proteins possess energy landscapes as funneled as globular proteins remains an open question. Experimentally, resolving this question is complicated by the fact that the many ways membrane proteins are unfolded also disrupts their membrane environment. One way to circumvent this problem is to use detergent to solubilize the denatured state, but as of yet, it is unknown how closely the resulting folding mechanisms resemble the folding mechanisms in more realistic membrane environments.
We address the role of thermodynamic control and funneled landscapes in α-helical transmembrane protein folding once the protein’s overall topology is set by using coarse-grained molecular dynamics simulations to examine the consequences of the principle of minimal frustration for the second stage of membrane protein folding in which the helices arrange into a specific structure. If the landscapes of α-helical transmembrane membrane proteins indeed are funneled, by using a sufficiently large database of α-helical transmembrane protein structures (11), the principle of minimal frustration provides a strategy to learn an energy function potentially capable of folding α-helical transmembrane proteins via molecular dynamics. We explore this strategy in this paper by extending to membrane proteins an associative memory, water-mediated, structure, and energy model (AWSEM) (12), a coarse-grained molecular dynamics Hamiltonian that has been shown to predict globular protein structures to low resolution. Through an energy landscape optimization we learn the parameters of an intramembrane transferable interaction potential and include an implicit membrane potential. In this work we describe these additions to AWSEM, motivate and detail the optimization and learning algorithm, and evaluate the predictive abilities of the newly optimized AWSEM-membrane force field. We then use thermodynamic landscape analysis to quantify how funnel-like the resulting energy landscapes for folding membrane proteins are when conformations are restricted to have a proper topology within the membrane.
Ingredients of AWSEM-Membrane
Model Overview and Extensions.
We learn the parameters in the energy function by first assuming that membrane proteins do actually have largely funneled landscapes once their topology is set. The quality of structure prediction by annealing the resulting energy function then provides a consistency check on the underlying funnel hypothesis. The prediction of transmembrane domain structure is a two-step process. First, the overall topology must be predicted, and second, the relative arrangement of the helices within the membrane must be determined. Predictions of topology, starting with the idea of the positive-inside rule (13), have been very successful, but dynamical topological rearrangements sometimes occur during the folding process. These exceptions aside, modern methods for predicting topology are quite reliable. Starting from this point, we thus consider only the second stage of membrane protein folding, wherein helices rearrange within a single topological sector, which is assumed to be like that of the final native configuration.
Our force field extends AWSEM—a predictive, coarse-grained molecular dynamics Hamiltonian with transferable tertiary interactions and local-in-sequence interactions determined via bioinformatic pattern matching (12). This model does quite well for globular proteins. In models such as AWSEM, where many atomistic degrees of freedom are integrated over, the interactions necessarily become context dependent. Given the very different environments in which soluble and transmembrane proteins fold, it is expected that retraining some parts of the potential is necessary in order to properly capture the interactions between residues in the membrane. As such, a density-dependent residue–residue interaction potential has been optimized by using the minimal frustration principle. The optimization recipe detailed below is similar to that used to previously to optimize AWSEM for soluble protein structures (14–16). In this case a training set of α-helical transmembrane protein structures is used. The potential retains the three interaction classes used in the potential for globular proteins: direct additive interactions and two sorts of mediated interactions that depend on the density of surrounding protein. Two residues directly interact if their Cβ atoms are close together (<6.5Å). If the Cβ atoms of two residues are separated by 6.5 Å < r < 9.5 Å, they participate in a mediated interaction. If the density of residues around both interacting residues is low, the interaction is said to be membrane mediated, whereas if the density is high, the interaction is termed protein mediated. In the case of globular proteins, the low-density mediated interactions correspond to water-mediated interactions, which have been shown to be important in being simultaneously able to funnel both dry and wet protein–protein interfaces (14). Later we will provide a physicochemical interpretation of the membrane protein parameters found by learning via optimization based on the membrane protein database.
The dominant effects of the lipid membrane are to sequester hydrophobic amino acids into the membrane and mostly, but not entirely, to exclude other amino acid types. This exclusion of polar and charged amino acids from the lipid layer imposes large barriers for topological rearrangements. These effects are captured by Vmembrane, a three-layer implicit membrane potential. The intramembrane region is 30 Å thick, which corresponds to the region that is spanned by the hydrophobic tails of the membrane lipids. Because in this study we focus on transmembrane domains, most of the protein that is outside of the intramembrane region (the loops) actually resides in the interfacial layer occupied by the polar head groups of the lipid molecules, either on the cytoplasmic or periplasmic side. Vmembrane takes an initial assignment of the residue position relative to the membrane as its input. Residues are deemed to be periplasmic, transmembrane, or cytoplasmic. In most of our calculations the assignment is determined from a 3D experimentally determined structure using the TMDET web server (17), thus confining the landscape to the proper topological sector, but we also present results using assignments of location predicted solely from sequence on a purely bioinformatic basis using the Membrane Protein Structure and Topology using Support Vector Machines program, MEMSAT-SVM (18). During the simulation, Vmembrane energetically stabilizes a residue when it is in its initially assigned layer but penalizes the residue when it enters one of the other layers. Another important effect of the membrane is to promote the alignment of transmembrane helices with the oriented lipid tails, leading to a liquid crystalline packing. To model this effect, we applied a weak, nonspecific cylindrical radius of gyration bias that ultimately penalizes configurations having helices aligned parallel to the membrane plane. Full details of the potential are available in SI Text.
There are several advantages for the molecular dynamics implementation of AWSEM-membrane. No explicit representation of the solvent is necessary. This feature along with the coarse-grained representation of the protein chain, at three atoms per residue (Cα, Cβ, O), provides a very significant speedup over fully atomistic models. This intrinsic algorithmic speedup allows not only efficient structure prediction via simulated annealing but also allows the analysis of equilibrium thermodynamics of the landscapes via umbrella sampling. We can thus test whether a transferable energy landscape capable of predicting membrane protein structures is indeed funneled, as was intended by the learning algorithm that gave rise to the landscape, and quantify the extent to which the landscape is funneled.
Energy Landscape Optimization Using an α-Helical Transmembrane Protein Training Set.
Studies on soluble globular proteins indicate that evolution has selected sequences with funneled energy landscapes. The funneledness of a landscape is characterized by the ratio of the folding temperature to the glass transition temperature, TF/TG, within a simple mean field approximation. This ratio is a monotonically increasing function of the ratio of the energy gap between the folded and compact unfolded states, δE, to the energetic ruggedness of the unfolded states characterized by the square root of the variance, ΔE2. The ratio δE/ΔE also determines how confidently a structure can be assigned to being folded. Coarse-grained Hamiltonians that optimize δE/ΔE for a training set of proteins yield energy landscapes with transferable parameters have been shown to successfully predict structures of monomers with quite different sequences and also to predict dimer interfaces of globular proteins de novo (12, 19, 20). AWSEM’s transferable tertiary interactions also equip the model to investigate problems such as the complex energy landscapes of designed proteins and multidomain protein misfolding as well as the mechanism of the initiation of aggregation (21–23).
For a Hamiltonian that depends linearly on a set of parameters {γi}, the energy function may be written as . The γi are the strengths of the interactions that encode the basic forces and the ϕi are the functional forms of the interactions. For such a model, the ratio of the gap to the ruggedness is given by a simple expression , where the vector A depends on the differences in structure between the native protein and molten globules Ai = 〈ϕi〉decoy − ϕnative, whereas B is a matrix that is determined by the fluctuations of the interactions in the molten globule ensemble Bi,j = 〈ϕiϕj〉decoy − 〈ϕi〉decoy〈ϕj〉decoy.
While performing the optimization, it is also useful to constrain the collapse temperature Tc so that folding and collapse occur at nearly the same temperature (24): . When collapse and folding occur at similar temperatures, during simulated annealing the system will not typically get trapped in a nonspecific collapsed state at high temperature before it has a chance to explore other, possibly lower-energy, compact configurations. We thus optimize a more complex target function:
The solution of this geometric problem amounts to solving a system of linear equations γ = B−1[A − λ2A′] up to a scalar multiple. We enforce the folding temperature to be approximately equal to the collapse temperature by determining the Lagrange multiplier λ2 such that Aγ = λ2A′γ. A simple first approximation to the statistics of the molten globule state can be obtained by shuffling a protein’s sequence on top of the native structure. In the case of membrane proteins, we constrain this shuffling to occur only within each respective membrane layer to avoid overemphasizing the apparent mutual attraction of charged and polar residues. This arises mostly from the fact that charged and polar residues are already excluded from the membrane, and this exclusion is taken into account by the implicit membrane potential. Sequence shuffling gives statistics about contacts that would occur in a highly mixed set of molten globules that undergoes no separate ordering transitions.
We average A, A′, and B over a training set of 75 α-helical transmembrane proteins. The training set is a distilled version of the database from Schramm et al. (11). In developing an optimized intramembrane contact potential we decided to exclude information from structures with extended regions outside the membrane. We also excluded chains that did not contain at least two transmembrane helices. Many complex multimers (for example, channels) were also discarded as were chains missing more than 14 nonterminal residues. Nonterminal missing loops were modeled using the ModLoop server (25).
Physicochemical Interpretation of Parameters
The optimization procedure described above yields 630 γ-interaction parameters that can be compared with potentials previously optimized for globular proteins. These parameters can be found in Table S1. We can attempt to interpret the learned parameters on a physicochemical basis. In Fig. 1 the optimized membrane γ-values are shown next to the γ-values for globular proteins. The globular protein gammas show a clear and strong hydrophobic attraction in both the direct and high-density interaction classes. The corresponding signal is weaker for the learned membrane interactions. This damping of the importance of hydrophobicity for funneling within a topological sector is not unreasonable considering that the bilayer is hydrophobic and therefore, to fold completely, membrane proteins can no longer rely too heavily on simple hydrophobic attraction for finding their relative positions within the bilayer. Pairing of oppositely charged residues can be seen in all three interaction classes for membrane proteins. These interactions are strong in the low-density globular γ but absent for the high-density-mediated interactions for globular proteins. In addition to pairing between the highly hydrophobic amino acids valine and isoleucine, pairing between two polar asparagine residues is also found to be highly favorable. The latter pattern has already been noted before and attributed to inter side-chain hydrogen bonding (26, 27).
Fig. 1.

A comparison of the interaction parameters determined for the optimized globular and α-helical membrane protein potentials. The globular interaction parameters (Upper) and membrane interaction parameters (Lower) are shown. For each potential, all three interaction classes are shown: direct, high-density mediated, and low-density mediated. Residue types are ordered by their hydrophobicity, and the strength of interaction is indicated by color with blue being the most destabilizing and red the most stabilizing.
Structure Prediction Results
To test the hypothesis that α-helical transmembrane proteins have funneled energy landscapes within their native topological sector, we used AWSEM-membrane, whose parameters already have been learned based on this hypothesis to predict the tertiary structure of 14 α-helical transmembrane proteins, most of which were also studied in ref. 28 (Table S2).
Many of the targets are subdomains of larger chains and/or multimers. Some of the targets have cofactors, but the cofactors were omitted during the simulations. We first assigned the residues to layers using the actual Protein Data Bank (PDB) structure and the TMDET server (17). For the first round of predictions, we also used the native secondary structure assignment from the structural identification program STRIDE (29) to bias dihedral angles of α-helical residues via Vrama (12). AWSEM-membrane’s local-in-sequence interactions, which use a fragment-based approach (see SI Text for details), do not make use of fragment structures that come from proteins that are significantly globally homologous to the target sequence. We used simulated annealing starting from an unfolded but topologically correct conformation to predict the structures. Initial configurations were generated by first positioning the chain in its native topology (meaning assignment of each residue to one of the three layers) within the implicit membrane. The structures were then unfolded using high temperature and a harmonic biasing potential to low Qw (SI Text). Finally, the temperature was slowly cooled to a quenching temperature. We assess the quality of predictions using both Qc, the fraction of native contacts, and combinatorial extension (CE) alignments (30). AWSEM-membrane, using only information about the topology and secondary structure from experimental structures, gives good predictions for most of the 14 targets according Qc, the fraction of native contacts (Fig. 2). We performed CE alignments and found that for the best-predicted eight targets, the resulting aligned backbone structures had an rmsd of <5 Å when at least 75% of the residues were aligned (see Fig. S1). Two examples of predicted structures are shown in Fig. 3.
Fig. 2.

Qc as a function of protein length of high-quality structures sampled during simulated annealing simulations. Proteins are ordered according to the number of amino acids in the chain, ranging from 91 to 285. Higher Qc values correspond to higher similarity to the experimentally determined structure. The structures that were generated using topology and secondary structure information from the experimentally determined structure are shown as gray circles connected by a dashed line. De novo prediction results using topology and secondary structure information from MEMSAT-SVM are shown as yellow squares for those proteins for which MEMSAT-SVM did not significantly err the topology assignment of the transmembrane helices.
Fig. 3.

Example structure prediction results from simulations using topology and secondary structure information derived from the experimentally determined structure. (Upper) PDB ID code 1J4N, aquaporin water channel AQP1 subdomain. (Lower) PDB ID code 1IWG, multidrug efux transporter subdomain. The experimental structures are shown on the left, and the simulated structures are shown in the middle. For these two structures, color is used to indicate the amino acid index along the chain. At right, a CE structural alignment of the experimental and simulated structures is shown, with the experimental structure in tan and the simulated structure in blue.
Although the AWSEM-membrane intramembrane interaction potential is transferable and depends solely on sequence, the predictions described above do make use of some information from native structure. Both native topology and α-helical secondary structure for each target were derived from the corresponding PDB structure in these first predictions. To test the influence of this native information on the quality of the predictions, we also carried out completely de novo predictions, using MEMSAT-SVM, which depends solely on sequence, to assign the input topology and secondary structure. Simulated structures from two example de novo predictions are shown in Fig. 4. Incorrect secondary structure prediction by MEMSAT-SVM, which biases AWSEM-membrane’s Vrama, is approximately compensated for by both the fragment memory and α-helical hydrogen-bonding potential. Nevertheless rare, major errors in topology prediction do lead to poorer prediction results. We expect de novo predictions will be nearly impossible when the input topology is completely wrong, for example, e.g., if an incorrect number of transmembrane helices is predicted by the bioinformatic input algorithm. More modest differences in topology assignment, such as the shifting by one or two residues at the end of the transmembrane part of a helix, have much less severe consequences on prediction quality. The de novo predictions of many targets without any structural input results in high-quality structures comparable to the results obtained by obtaining topology and secondary structure assignments from the experimentally determined structure. In particular, the de novo prediction quality for the two largest targets that have no significant topological mispredictions, Bacteriorhodopsin (PDB ID code 1PY6) and Rhodopsin (PDB ID code 1U19), was nearly identical to the quality of predictions obtained using the experimentally assigned topology and secondary structure (Fig. 4). The targets for which the native topology was significantly incorrectly predicted by MEMSAT-SVM are not included in Fig. 2, but an example can be found in Fig. S2 and Table S3.
Fig. 4.
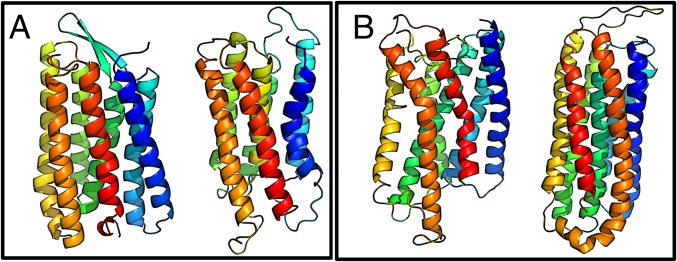
Example structure prediction results from simulations using input topology and secondary structure information derived from bioinformatic sequence-based predictions. (A) 1PY6, Bacteriorhodopsin. (B) 1U19, Rhodopsin. In A and B, The experimental structures are shown on the left, and the simulated structures are shown on the right. Color is used to indicate the amino acid index along the chain.
Thermodynamic Landscape Analysis
AWSEM-membrane is able to predict the helical packing of many membrane proteins via simulated annealing, but what do the energy landscapes of these proteins look like? Landscape analysis based on order parameters is the first step toward understanding folding mechanisms. Because of the high-dimensional nature of the problem, one must first choose the appropriate order parameters. We use Qc, which is expected to correlate with the energy. We also use Cα rmsd, which can separate structures with similar contact maps such as pseudomirror images. The free-energy surface gives information about which states are significantly populated and some idea of the barriers that exist between distinct ensembles. Projecting the potential energy (PE) onto the same order parameters tells us how the average energy depends on the degree of nativeness, i.e., how funneled the landscape is. The lowest PE states dominate at lower temperatures and correspond to the ground states of the Hamiltonian. This gives us an extra check on the simulated annealing results: Sufficiently long simulated annealing should be able to find the ground states that are sampled during the umbrella sampling, but in practice this is not always the case owing to overly rapid annealing. The expectation value of the contact energy allows us to assess the role of native contacts in folding.
We constructed 2D free-energy profiles, F(Qc,rmsd), for the nicotinic acetylcholine receptor subdomain (PDB ID code 2BG9) and for the V-type Na+-ATPase subdomain (PDB ID code 2BL2) from umbrella sampled data along Qw using the multistate Bennett acceptance ratio (31) along with computing expectation values of the PE and the contact energy (Econtact). Fig. 5 shows the results of the landscape analysis on the nicotinic acetylcholine receptor subdomain (2BG9) along with its native structure. Just below the collapse temperature, the landscape of the nicotinic acetylcholine receptor subdomain (2BG9) has two basins that correspond to different helical packings. Both structures have a significant fraction of native contacts formed (Qc ≈ 0.4), but one packing is significantly nonnative (rmsd ≈ 10 Å), whereas the other is quite native in appearance (rmsd ≈ 6 Å). These basins are separated by a low barrier, indicating that interconversion between the states may be rapid. The expectation value of the PE indicates that at lower temperatures the lower rmsd packing is favored, so a larger fraction of the native contacts would form if annealing were taken to this lower temperature. This bias toward the better structure does not apparently come from the interaction potential alone: The expectation value of Vcontact would indicate that the two packings would be roughly equally stable. Nevertheless the landscape for the PE of the nicotinic acetylcholine receptor subdomain (2BG9) is funneled to high Qc and low rmsd. At intermediate temperatures, multiple ensembles corresponding to native and nonnative helical packings are stable. Many of the same contacts form in both packings. This symmetry of contacts with respect to different helical packings is a feature that may be common to the smaller transmembrane domains, as happens for the globular problem, too (32, 33). The degeneracy may also be resolved in the complete multimeric assembly.
Fig. 5.

The experimentally determined structure (Upper Left) and the free-energy profile (Upper Right) for nicotinic acetylcholine receptor subdomain (2BG9) below the collapse temperature. The free energy is plotted versus Qc, the fraction of native contacts (x axis) and the rmsd (y axis). Representative structures are shown from the two free-energy basins. Expectation value of the total PE (Lower Left) and Econtact (Lower Right) are plotted versus the same order parameters.
In Fig. 6 we show the landscape of the V-type Na+-ATPase subdomain (2BL2) below the collapse temperature. As for the nicotinic acetylcholine receptor (2BG9), the landscape for the V-type Na+-ATPase subdomain (2BL2) has two basins that correspond to alternate helical packings with rmsd ≈ 8 and 5 Å. The structures at the higher rmsd resemble the native but two helices have swapped locations. The lower rmsd structures are completely native-like in packing. The expectation value of the PE shows that the decrease in energy is well correlated with the native contact formation. In both packings a high fraction of native contacts has formed (Qc ≈ 0.5). In this case, at low temperatures not only the interaction potential but also the total PE would favor both packings approximately equally. We note that the proline-induced kinks of V-type Na+-ATPase (2BL2) are not captured well by AWSEM-membrane, as shown in the representative structures in Fig. 6. Further refinement of the local-in-sequence signals and special treatment of transmembrane–protein-specific motifs will be needed to improve the performance of the model for this system.
Fig. 6.

The experimentally determined structure (Upper Left) and the free-energy profile (Upper Right) for V-type Na+-ATPase subdomain (2BL2) below the collapse temperature. The free energy is plotted versus Qc, the fraction of native contacts (x axis) and the rmsd (y axis). Representative structures are shown from the two free-energy basins. Expectation value of the total PE (Lower Left) and Econtact (Lower Right) are plotted versus the same order parameters.
Discussion
AWSEM has been extended to model α-helical transmembrane proteins, resulting in the transferable AWSEM-membrane potential that can successfully fold membrane proteins at modest resolution. The AWSEM-membrane force field includes an implicit membrane potential and an optimized intramembrane contact potential that was trained using a set of experimentally determined α-helical transmembrane domain structures. The AWSEM-membrane potential learned by distinguishing native from shuffled structures indicates that although some of the features of interactions between residues are the same both for transmembrane domains and for globular proteins, there are certainly significant differences in the interactions for the two environments. The contact parameters show that the mutual attraction of hydrophobic residues does not contribute as greatly to distinguishing between folded and misfolded states in transmembrane domains as it does for globular proteins. This is quite reasonable given that membrane proteins once inserted fold in a largely hydrophobic environment. Oppositely charged pairs of residues and mutual attraction of polar residues contribute significantly to the selection of the folded structures.
Using only information about the gross topology and secondary structure from experimental structures, AWSEM-membrane is able to fold many α-helical transmembrane proteins. Transmembrane domains with four transmembrane helices or less are especially well predicted, with three of the four smallest proteins predicted to an accuracy of rmsd better than 4 Å. Larger proteins show more significant defects in their predicted structures, but nonetheless possess long regions of native-like structure as indicated by the CE alignment score. Refinement of the AWSEM-membrane force field via iterative self-consistent optimization in which decoys are explicitly generated from molecular dynamics with the same potential should yield even better results. Using a larger training set made possible by the continued growth of available transmembrane protein structural databases will also improve the model. The fact that, for the most part, structure prediction starting from either the exact topology and secondary structure information or with such input inferred by bioinformatic methods yield prediction results of roughly equal quality indicates that the methods for predicting these coarse-grained input data are reasonably well developed. In some cases where significant errors are made in topology prediction, however, the large barrier imposed by the implicit membrane potential would make successful prediction by AWSEM-membrane molecular dynamics very difficult.
Thermodynamic landscape analysis shows that the energy is highly correlated with formation of native contacts. In other words, the energy landscapes of transmembrane domains are indeed funneled, at least within their native topological sector. Nevertheless, the highly symmetric nature of small transmembrane domains with respect to contacts results in distinct helical packings with a similar fraction of native contacts being favored below the collapse temperature. In one case that we examined, this frustration would be resolved by going to lower temperature in the annealing, but in another case it would not. Oligomerization of the domains may also single out one of the basins from the near-degenerate ground states of the monomer. Whether or not this multiplicity of basins for the monomer leads to the kinetic effects of frustration in vitro or whether this frustration is overcome in vivo through the action of the translocon kinetically is an interesting question.
Supplementary Material
Acknowledgments
We thank Garegin Papoian and Davit Potoyan for helpful discussions. B.L.K and N.P.S. were supported by Grant R01 GM44557 and Grant P01 GM071862 from the National Institute of General Medical Sciences. This work was also supported by the Center for Theoretical Biological Physics sponsored by the National Science Foundation (NSF) PHY-0822283. Computational resources were supported in part by the Data Analysis and Visualization Cyberinfrastructure funded by the NSF under Grant OCI-0959097. Additional support was also provided by the D. R. Bullard-Welch Chair at Rice University.
Footnotes
The authors declare no conflict of interest.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1410529111/-/DCSupplemental.
References
- 1.Anfinsen CB. Principles that govern the folding of protein chains. Science. 1973;181(4096):223–230. doi: 10.1126/science.181.4096.223. [DOI] [PubMed] [Google Scholar]
- 2.Bryngelson JD, Wolynes PG. Spin glasses and the statistical mechanics of protein folding. Proc Natl Acad Sci USA. 1987;84(21):7524–7528. doi: 10.1073/pnas.84.21.7524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Leopold PE, Montal M, Onuchic JN. Protein folding funnels: A kinetic approach to the sequence-structure relationship. Proc Natl Acad Sci USA. 1992;89(18):8721–8725. doi: 10.1073/pnas.89.18.8721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bryngelson JD, Onuchic JN, Socci ND, Wolynes PG. Funnels, pathways, and the energy landscape of protein folding: A synthesis. Proteins. 1995;21(3):167–195. doi: 10.1002/prot.340210302. [DOI] [PubMed] [Google Scholar]
- 5.Wolynes PG. Energy landscapes and solved protein-folding problems. Philos Trans A Math Phys Eng Sci. 2005;363(1827):453–464, discussion 464–467. doi: 10.1098/rsta.2004.1502. [DOI] [PubMed] [Google Scholar]
- 6.von Heijne G. Membrane-protein topology. Nat Rev Mol Cell Biol. 2006;7(12):909–918. doi: 10.1038/nrm2063. [DOI] [PubMed] [Google Scholar]
- 7.Huang KS, Bayley H, Liao MJ, London E, Khorana HG. Refolding of an integral membrane protein: Denaturation, renaturation, and reconstitution of intact bacteriorhodopsin and two proteolytic fragments. Journal of Biological Chemistry. 1981;256(8):3802–3809. [PubMed] [Google Scholar]
- 8.Booth PJ. A successful change of circumstance: A transition state for membrane protein folding. Curr Opin Struct Biol. 2012;22(4):469–475. doi: 10.1016/j.sbi.2012.03.008. [DOI] [PubMed] [Google Scholar]
- 9.Curnow P, Booth PJ. The transition state for integral membrane protein folding. Proc Natl Acad Sci USA. 2009;106(3):773–778. doi: 10.1073/pnas.0806953106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Otzen DE. Mapping the folding pathway of the transmembrane protein DsbB by protein engineering. Protein Eng Des Sel. 2011;24(1-2):139–149. doi: 10.1093/protein/gzq079. [DOI] [PubMed] [Google Scholar]
- 11.Schramm CA, et al. Knowledge-based potential for positioning membrane-associated structures and assessing residue-specific energetic contributions. Structure. 2012;20(5):924–935. doi: 10.1016/j.str.2012.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Davtyan A, et al. AWSEM-MD: Protein structure prediction using coarse-grained physical potentials and bioinformatically based local structure biasing. J Phys Chem B. 2012;116(29):8494–8503. doi: 10.1021/jp212541y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.von Heijne G. Membrane protein structure prediction. Hydrophobicity analysis and the positive-inside rule. J Mol Biol. 1992;225(2):487–494. doi: 10.1016/0022-2836(92)90934-c. [DOI] [PubMed] [Google Scholar]
- 14.Papoian GA, Ulander J, Wolynes PG. Role of water mediated interactions in protein-protein recognition landscapes. J Am Chem Soc. 2003;125(30):9170–9178. doi: 10.1021/ja034729u. [DOI] [PubMed] [Google Scholar]
- 15.Papoian GA, Ulander J, Eastwood MP, Luthey-Schulten Z, Wolynes PG. Water in protein structure prediction. Proc Natl Acad Sci USA. 2004;101(10):3352–3357. doi: 10.1073/pnas.0307851100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Schafer N, Kim B, Zheng W, Wolynes P. 2013. Learning to fold proteins using energy landscape theory. arXiv:1312.7283.
- 17.Tusnády GE, Dosztányi Z, Simon I. PDB_TM: Selection and membrane localization of transmembrane proteins in the protein data bank. Nucleic Acids Res. 2005;33(Database issue):D275–D278. doi: 10.1093/nar/gki002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nugent T, Jones DT. Transmembrane protein topology prediction using support vector machines. BMC Bioinformatics. 2009;10:159. doi: 10.1186/1471-2105-10-159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fujitsuka Y, Takada S, Luthey-Schulten ZA, Wolynes PG. Optimizing physical energy functions for protein folding. Proteins. 2004;54(1):88–103. doi: 10.1002/prot.10429. [DOI] [PubMed] [Google Scholar]
- 20.Zheng W, Schafer NP, Davtyan A, Papoian GA, Wolynes PG. Predictive energy landscapes for protein-protein association. Proc Natl Acad Sci USA. 2012;109(47):19244–19249. doi: 10.1073/pnas.1216215109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Truong HH, Kim BL, Schafer NP, Wolynes PG. Funneling and frustration in the energy landscapes of some designed and simplified proteins. J Chem Phys. 2013;139(12):121908. doi: 10.1063/1.4813504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zheng W, Schafer NP, Wolynes PG. Frustration in the energy landscapes of multidomain protein misfolding. Proc Natl Acad Sci USA. 2013;110(5):1680–1685. doi: 10.1073/pnas.1222130110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zheng W, Schafer NP, Wolynes PG. Free energy landscapes for initiation and branching of protein aggregation. Proc Natl Acad Sci USA. 2013;110(51):20515–20520. doi: 10.1073/pnas.1320483110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bryngelson JD, Wolynes PG. A simple statistical field theory of heteropolymer collapse with application to protein folding. Biopolymers. 1990;30(1-2):177–188. [Google Scholar]
- 25.Fiser A, Sali A. ModLoop: Automated modeling of loops in protein structures. Bioinformatics. 2003;19(18):2500–2501. doi: 10.1093/bioinformatics/btg362. [DOI] [PubMed] [Google Scholar]
- 26.Choma C, Gratkowski H, Lear JD, DeGrado WF. Asparagine-mediated self-association of a model transmembrane helix. Nat Struct Biol. 2000;7(2):161–166. doi: 10.1038/72440. [DOI] [PubMed] [Google Scholar]
- 27.Gratkowski H, Lear JD, DeGrado WF. Polar side chains drive the association of model transmembrane peptides. Proc Natl Acad Sci USA. 2001;98(3):880–885. doi: 10.1073/pnas.98.3.880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yarov-Yarovoy V, Schonbrun J, Baker D. Multipass membrane protein structure prediction using Rosetta. Proteins. 2006;62(4):1010–1025. doi: 10.1002/prot.20817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Frishman D, Argos P. Knowledge-based protein secondary structure assignment. Proteins. 1995;23(4):566–579. doi: 10.1002/prot.340230412. [DOI] [PubMed] [Google Scholar]
- 30.Shindyalov IN, Bourne PE. Protein structure alignment by incremental combinatorial extension (CE) of the optimal path. Protein Eng. 1998;11(9):739–747. doi: 10.1093/protein/11.9.739. [DOI] [PubMed] [Google Scholar]
- 31.Shirts MR, Chodera JD. Statistically optimal analysis of samples from multiple equilibrium states. J Chem Phys. 2008;129(12):124105. doi: 10.1063/1.2978177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Levy Y, Cho SS, Shen T, Onuchic JN, Wolynes PG. Symmetry and frustration in protein energy landscapes: A near degeneracy resolves the Rop dimer-folding mystery. Proc Natl Acad Sci USA. 2005;102(7):2373–2378. doi: 10.1073/pnas.0409572102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Noel JK, et al. Mirror images as naturally competing conformations in protein folding. J Phys Chem B. 2012;116(23):6880–6888. doi: 10.1021/jp212623d. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.