

Significance
Many complex diseases are caused by aberrant protein–protein interactions or protein misfolding, which present immense challenges for drug discovery. Here, we present the design, synthesis, and usage of a diverse library of potential pharmacological chaperones that could interfere with undesirable protein–protein interactions and prevent aggregation of misfolded proteins.
Keywords: chemical biology, drug discovery, helical mimetic
Abstract
Pharmacological chaperones are small molecules that bind to proteins and stabilize them against thermal denaturation or proteolytic degradation, as well as assist or prevent certain protein–protein assemblies. These activities are being exploited for the development of treatments for diseases caused by protein instability and/or aberrant protein–protein interactions, such as those found in certain forms of cancers and neurodegenerative diseases. However, designing or discovering pharmacological chaperones for specific targets is challenging because of the relatively featureless protein target surfaces, the lack of suitable chemical libraries, and the shortage of efficient high-throughput screening methods. In this study, we attempted to address all these challenges by synthesizing a diverse library of small molecules that mimic protein α-helical secondary structures commonly found in protein–protein interaction surfaces. This was accompanied by establishing a facile “on-bead” high-throughput screening method that allows for rapid and efficient discovery of potential pharmacological chaperones and for identifying novel chaperones/inhibitors against a cancer-associated protein, myeloid cell leukemia 1 (MCL-1), and a Parkinson disease-associated protein, α-synuclein. Our data suggest that the compounds and methods described here will be useful tools for the development of pharmaceuticals for complex-disease targets that are traditionally deemed “undruggable.”
The majority of drugs on the market today target proteins with defined small-molecule binding sites, including enzymes (47%), transmembrane receptors (34%), and transmembrane channels and transporters (11%) (1). These drugs are usually small molecules that compete with the target protein’s natural substrates, which are typically also small molecules, for binding in the active site or substrate-binding site, for example. However, some of the most pressing and devastating diseases involve proteins that do not possess these natural binding sites, such as those involved in protein–protein interactions (PPIs), associated with many cancers, and protein-misfolding, associated with most neurodegenerative diseases (2–5). For these reasons, there is considerable interest in the design and synthesis of molecules that can modulate PPIs and/or protein stability.
In this article, we show a class of ligands that are intended to stabilize the protein in vitro and in vivo and, as such, are potential pharmacological chaperones. Pharmacological chaperones are small molecules that bind to proteins and stabilize them against thermal denaturation or proteolytic degradation, as well as assisting or preventing certain protein–protein assembly (6–8). Designing pharmacological chaperones against traditionally “undruggable” protein surfaces relies heavily on the availability of three-dimensional structures of the proteins of interests, which are used for the identification of “hotspots” and for the design of ligand mimetic structures (9–11). A variety of classes of compounds have been shown to be effective pharmacological chaperones, including cofactor mimetics (12), catalytic inhibitors (13), ligand mimetics (14), stabilized helical peptides (15–17), and small-molecule secondary structure-mimetics (18, 19). However, discovering pharmacological chaperones for specific targets is challenging for several reasons, including the characteristics of the protein surfaces, the lack of suitable chemical libraries, and the shortage of efficient high-throughput screening (HTS) methods (20, 21). Here, we describe the design and synthesis of a diverse library of small molecules that mimic α-helical secondary structures. The synthetic method is highly accessible, and the resulting compounds showed good aqueous solubility and cell permeability. We then describe the establishment of a facile “on-bead” HTS method that allows for rapid and efficient discovery of potential pharmacological chaperones. Finally, we successfully demonstrate the feasibility of our strategy by identifying myeloid cell leukemia 1 (MCL-1) inhibitors targeting α-helix-groove interaction as well as α-synuclein stabilizers targeting helix–helix interfaces.
Results and Discussion
Design of a Triazine-Piperazine-Triazine Scaffold as α-Helix Mimetics.
α-Helices are involved in mediating many PPIs in which short α-helical peptide segments spanning two to three helical turns play a key role as a recognition motif, and side chains at i, i + 3 or i + 4, and i + 7 are often crucial for PPIs (Fig. 1A). Because α-helix-mediated PPIs are involved in various cellular signaling pathways, there have been tremendous efforts to develop molecules that can mimic helical peptide structures to modulate such interactions (18, 22). These include stabilized helical peptides (15–17, 23) and peptidomimetic foldamers (24–29). Alternatively, nonpeptidic, small-molecule α-helix mimetics have attracted significant interest. In general, small molecules are more preferable over peptides for their cell permeability, proteolytic resistance, and bioavailability. Hamilton and colleagues pioneered the development of nonpeptidic α-helix mimetics. They demonstrated the proof of concept that appropriately functionalized terphenyl 1 (Fig. 1B) can physically mimic α-helical structures and functionally active as inhibitors of α-helix-mediated PPIs (19). This pioneering work inspired considerable efforts devoted to developing small-molecule α-helix mimetics (18, 19). However, the majority of these efforts were stymied by the relatively poor water solubility and synthetic difficulty of α-helix mimetic molecules. To overcome this critical barrier, we set out to develop α-helix mimetic scaffolds with greater synthetic accessibility that yield products with favorable physicochemical properties.
Fig. 1.

(A) α-Helix with i, i + 4, and i + 7 side-chain positions. (B) Terphenyl-based α-helix mimetic scaffold 1. (C) Triazine-piperazine-triazine-based α-helix mimetic scaffold 2. (D) An energy-minimized structure of 2a (R1, R2, R3 = CH3; R4, R5 = H) and overlay with α-helix. (E) Comparison of Clog P values of 1a and 2a.
To obtain such molecules, we designed a triazine-piperazine-triazine scaffold 2 (Fig. 1C). This scaffold is composed of a piperazine ring between two triazine rings. Piperazines are found in many biologically active molecules and can be a good replacement of a phenyl ring in terphenyl structures (30–32). Triazine is a widely used privileged scaffold because of its broad range of biological activities and ease of synthetic manipulation (33, 34). We used this scaffold as the backbone and functionalized its R1, R2, and R3 groups to form α-helix mimetics. To determine the physical resemblance of peptide α-helices, we used in silico molecular dynamics simulation to generate and energy-minimize potential rotamers of the scaffold with arbitrary functional groups. This computational study predicted that spatial arrangements of the three functional groups (R1, R2, and R3) precisely matched the locations of side chains (i, i + 3 or i + 4, and i + 7) on peptidic α-helices (Fig. 1D), suggesting our scaffold could mimic α-helical structures with appropriate placement of side chains.
To gain insight into the properties of our scaffold in solution, we determined its aqueous partition coefficient. The calculated log Poctanol/water (Clog P) value for triazine-piperazine-triazine scaffold 2a is 1.36, which is within the desirable range of hydrophilicity. This is a significant improvement over the current terphenyl scaffold 1a, the Clog P of which is 6.21 (Fig. 1E), suggesting the improved aqueous solubility of the triazine-piperazine-triazine scaffold has more “drug-like” properties.
Moreover, in addition to three variable locations for side chain mimetic functional groups (R1, R2, and R3), scaffold 2 also contains two diversification sites (R4 and R5) for modulating its overall hydrophilicity (Fig. 1C). Most biologically active helical peptides are amphipathic, with one side containing hydrophobic residues (usually at protein-contacting areas) and with hydrophilic residues on the other side, which presumably improves solubility. Such helical peptides bearing solvent-exposed hydrophilic surfaces are energetically favorable when binding to a target protein. In addition, charged residues on amphipathic helical peptides often participate in hydrophilic interactions with charged residues on the protein surface, resulting in an additional contribution to binding affinity and specificity (35). Surprisingly, among many nonpeptidic small-molecule α-helix mimetics, there are only a few amphiphilic α-helix mimetic scaffolds (31, 36–38). Given the two additional sites (R4 and R5) on the opposite side, our scaffold 2 can effectively mimic amphiphilic α-helical structures by introducing various hydrophilic functional groups at R4 and R5, thereby potentially leading to more potent and selective ligands.
Solid-Phase Synthesis.
We have developed a concise, divergent solid-phase synthetic route to prepare the triazine-piperazine-triazine scaffold (SI Appendix, Scheme S1). First, monosubstituted dichlorotriazines 3 were loaded onto Rink-Amide resin. 2-Nitrobenzenesulfonyl-protected piperazine derivatives 5 were coupled to the resin-bound triazine derivatives 4. After removing the N-nitrobenzenesulfonyl protecting group on 6, 2-ethylamino-4,6-dichloro-[1,3,5]triazine 7 was introduced to afford 8. The chloride was displaced with various amines (R3R3′NH). Finally, cleavage reaction with trifluoroacetic acid furnished trifunctionalized compounds 9.
To assess the feasibility of the solid-phase synthesis, a series of compounds was prepared, using various building blocks. The purity and identity of the released crude products were analyzed by liquid chromatography/MS (SI Appendix, Figs. S1 and S2). The average purity of the final products was >92%, demonstrating the efficiency of our solid-phase synthesis. Compared with convergent, lengthy solution-phase synthesis of most terphenyl-based structures, our solid-phase synthetic route is simple and divergent, allowing for the rapid creation of large numbers of diversely functionalized compounds by using many readily available building blocks.
Peptoid-Encoded One-Bead-One-Compound Combinatorial Library.
Although many different HTS methods are available for traditional drug targets such as enzymes, there are limited numbers of screening methods for detecting PPI regulators (20, 21). Indeed, there are only a few reports on the use of HTS to identify α-helix mimetic small molecules (39–41). The most frequently used method to identify α-helix mimetics is a competitive binding assay, using a fluorescence polarization (FP). This assay is designed to find compounds that disrupt a pairwise interaction between a target protein and a fluorescently labeled helical peptide. However, such binding peptides do not always exist, and identifying them is another challenge (especially when high-resolution structural information for protein complexes is not available). More importantly, this type of competitive assay may not be suitable for detecting PPI stabilizers. Hence, development of convenient HTS methods is of considerable interest for facilitating the identification of pharmacological chaperones.
Using our divergent solid-phase synthesis protocol, a large, combinatorial library is readily accessible. For the rapid and efficient selection of best compounds among a library of α-helix mimetics, we developed a convenient HTS method by taking advantage of a bead-based screening platform, which was originally developed by Lam and colleagues for screening peptides (42) and has been successfully applied to cyclic peptides and peptidomimetics such as β-peptides and peptoids (29, 43–47). Relative to conventional HTS assays, on-bead screening has several advantages (48). This method enables a large number of library molecules (literally more than millions) to be screened simultaneously by incubating with a target protein in a single test tube. In addition, bead-based screening does not require any prior knowledge on structures of target proteins and binding partners (either proteins or peptides). Importantly, many protein surfaces often alter their conformation on binding with partner proteins or ligands, creating new binding pockets (49). Such dynamic pockets, induced by the adaptive nature of protein surfaces, are difficult to predict. In this context, unbiased approaches through on-bead screening of a large one-bead one-compound (OBOC) library could be a powerful tool for finding compounds that induce and bind to such unpredictable binding pockets on protein interfaces.
Inspired by Lam’s peptide-based encoding method (50), we constructed a peptoid-encoded OBOC combinatorial library of α-helix mimetics on topologically segregated bilayer beads (Fig. 2). Peptoids would offer several attractive features as coding materials (44, 51): Peptoids can be efficiently synthesized under very mild conditions (52–54), which would be compatible with various reaction conditions used for synthesis of library molecules, and in addition, a vast number of structurally diverse primary amines, as the diversity-generating element in peptoid synthesis, are readily available from commercial sources.
Fig. 2.

General strategy for the identification of α-helix mimetics that bind to a target protein through on-bead screening of peptoid-encoded OBOC library of α-helix mimetics.
A peptoid-encoded combinatorial library was synthesized on bifunctional beads, in which triazine-piperazine-triazines are displayed on the outer layer of the beads, and peptoids as coding tags are separately incorporated in the interior of the beads (SI Appendix, Fig. S3). Therefore, only α-helix mimetics on the bead surface can be exposed to a target protein during on-bead screening, and the structure of hit compounds will be revealed by sequencing the corresponding peptoids (Fig. 2). The diversity of this OBOC combinatorial library was 18 × 3 × 27 = 1,458 (SI Appendix, Figs. S3 and S4).
High-Throughput Screen Against MCL-1.
To demonstrate the efficacy of our peptoid-encoded OBOC library of α-helix mimetics, MCL-1 was chosen as a target protein. MCL-1 is a member of the antiapoptotic B-cell lymphoma 2 (BCL-2) family of proteins that includes BCL-2, BCL-XL, Al, and BCL-w. BCL-2 proteins interact with BCL-2 homology domain 3 (BH3) protein family members such as BAK and block their apoptosis-inducing activity, thereby promoting cell survival. Given the critical roles of BCL-2 in cancer progression, inhibition of the PPIs between BCL-2 prosurvival proteins and BH3 proapoptotic proteins is emerging as an attractive strategy for cancer treatment (55). Many efforts have been made to develop BH3 mimetics to disrupt the BCL-2/BH3 interactions, which are mediated by binding an α-helical peptide segment in BH3 into the hydrophobic cleft of BCL-2 (55). One of the most potent BCL-2 inhibitors is ABT-737 (56). Interestingly, ABT-737 binds to BCL-2 and BCL-XL with high affinity, but not to MCL-1. Not surprisingly, ABT-737 is not effective in cancer cells overexpressing MCL-1 (57). Given that overexpression of MCL-1 is observed in many cancers and is associated with cancer progression and drug resistance, it is generally accepted that inhibition of MCL-1, as well as BCL-2, is a promising strategy for effective cancer treatment (58). Hence, developing MCL-1 inhibitors (either MCL-1-specific inhibitors to use in combination with current BCL-2 inhibitors or pan BCL-2 family inhibitors) is of considerable interest.
For selection of the best MCL-1 ligands from the library molecules, we executed two rounds of on-bead screens, using magnetic separation followed by alkaline phosphatase-based detection (SI Appendix) (44, 47). Briefly, 25 mg (∼10,200 beads, about seven copies of diversity) of the library beads were screened for their binding ability to MCL-1ΔNΔC (amino acids 172–320), a BH3-binding domain of MCL-1, by incubating the library beads with 200 nM biotinylated MCL-1ΔNΔC overnight at 4 °C. After washing unbound protein, followed by exposure to streptavidin-coupled Dynabeads (iron oxide particles), several hundred positive beads were selected by magnetic separation. These initial hit beads were again screened using 100 nM biotinylated MCL-1ΔNΔC for 4 h at 4 °C, followed by probing with streptavidin-conjugated alkaline phosphatase. After treatment with 5-bromo-4-chloro-3-indolyl phosphate, a substrate for colorimetric detection of alkaline phosphatase, 10 blue-colored beads were isolated as putative hits. Peptoids released from individual beads were sequenced by MS/MS (SI Appendix, Fig. S5). Notably, the sequence analysis revealed that the 10 beads contained identical peptoid sequences, falling into 3 kinds of peptoids; five of peptoid-1, three of peptoid-2, and two of peptoid-3 (SI Appendix, Fig. S6). Note that about seven copies of library molecules were used in this screen. This result shows the robustness of our screening platform. The chemical structures of the corresponding triazine-piperazine-triazines (9a–9c) were unambiguously decoded by the coding peptoid sequences (Fig. 3A). For further studies, the hit compounds 9a–9c and their fluorescently labeled derivatives 9a–9c-FL (Fig. 3A) were synthesized (SI Appendix, Fig. S7) and purified by HPLC (SI Appendix, Figs. S8 and S9). Note that the compounds 9a–9c were shown to be reasonably soluble in aqueous solution (∼100 µg/mL in phosphate buffer saline at pH 7.4, which is within a range of water solubility for the favorable oral absorption of drugs) (59).
Fig. 3.

(A) Structures of hit compounds 9a–9c and a negative control 9-nc. (B) Binding curves for 9a–9c binding to MCL-1. (C) Inhibition curves of 9a–9c for fluorescein-labeled BH-3 peptide binding to MCL-1. Error bars represent SD from three independent experiments.
MCL-1 Binding Assays.
To validate the binding ability of the hit compounds to MCL-1ΔNΔC, FP experiments were executed. 9a-FL, 9b-FL, and 9c-FL bound MCL-1ΔNΔC with KD values of 2.8, 3.0, and 1.1 µM, respectively. The binding affinity of a known BH3 peptide (58) used as a positive control was 0.3 µM, which is similar to the reported binding affinity (KD = 245 nM) (Fig. 3B). To verify whether the fluorescein and linker conjugated to 9a–9c have any effects on the binding between the compounds and MCL-1ΔNΔC, we synthesized a fluorescein-linker moiety, Fluor-linker (SI Appendix, Figs. S7 and S9), and used it as a negative control. As shown in Fig. 3B, Fluor-linker did not exhibit any binding to MCL-1ΔNΔC, showing that the fluorophore and the linker have no contribution to the binding affinity of 9a–9c to MCL-1. We then performed a competitive FP assay using nonlabeled compounds 9a–9c to determine whether they are able to antagonize the interaction between MCL-1 and BH3 (Fig. 3C). As expected, 9b and 9c dissociated the MCL-1ΔNΔC/BH3 interaction with Ki values of 9.3 and 17.5 µM, respectively, whereas 9a had weak activity. 9-nc, a negative control compound, did not bind to MCL-1ΔNΔC (Fig. 3C). We chose 9c for more detailed investigation because it has the highest binding affinity among the three hits. To determine whether 9c has selectivity for MCL-1 over other structurally similar proteins, we used competitive FP assays to measure the binding affinity of 9c against BCL-XLΔC (amino acids 1–212), which is a deletion construct of human BCL-XL, another BCL-2 protein member, with the BH3 binding pocket similar to MCL-1. We found that 9c did not significantly inhibit the interaction between BCL-XLΔC and a BH3 peptide (SI Appendix, Fig. S10), indicating that 9c is selective for MCL-1. To understand how 9c might bind to MCL-1ΔNΔC, we computationally docked 9c into the binding site for BH3. The three functional groups at R1, R2, and R3 on the triazine-piperazine-triazine 9c make interactions with the hydrophobic groove on the surface of MCL-1, mimicking the binding mode of the BH3 helical peptide (SI Appendix, Fig. S11).
MCL-1 Cellular Assays.
To examine whether 9c is cell-permeable and can disrupt native interactions between MCL-1 and proapoptotic proteins inside cells, we performed coimmunoprecipitation experiments. Jurkat T cells were treated with DMSO as a carrier control or increasing concentrations of 9c, and cell lysates were immunoprecipitated with anti-MCL-1 antibody and subjected to Western blot, using anti-BAK and anti-MCL-1 antibodies (Fig. 4A). The BAK Western blot analysis showed that immunoprecipitated BAK was dose-dependently decreased in 9c-treated cells. In contrast, 9-nc, a negative control, did not inhibit the MCL-1/BAK interaction (SI Appendix, Fig. S12). This result ensures that 9c is cell permeable and competitively binds to MCL-1, liberating BAK protein. Cellular uptake of 9c was confirmed using confocal microscopy (Fig. 4B).
Fig. 4.

Cellular activity of 9c. (A) Coimmunoprecipitation assay. Jurkat T cells were treated with DMSO or varying concentrations of 9c for 2.5 h. The effect of 9c on the MCL-1/BH3 interaction was analyzed by MCL-1 immunoprecipitation and BAK western analysis. The results are representative of three independent experiments. (B) Confocal microscopic images of A549 cells treated with 1 μM 9c-FL, or 5 (6)-carboxyfluorescein (Flu), for 4 h. (C) Effect of 9c and 9-nc (a negative control) on cell viability of Jurkat T leukemia cells (Left) and WS-1 normal fibroblast cells (Right).
Blocking the MCL-1/BH3 interaction by 9c would liberate proapoptotic proteins such as BAK from MCL-1, resulting in apoptosis induction and cell-growth inhibition. To test this, Jurkat human leukemia T cells and human multiple myeloma U266 cells were treated with DMSO or varying concentrations of 9c and 9-nc, and cell viability was evaluated by MTS assays. Indeed, 9c killed Jurkat T cells and U266 cells in a dose-dependent manner, but not normal cells such as WS-1 human fibroblast cells and human embryonic kidney (HEK) 293 cells (Fig. 4C and SI Appendix, Fig. S13A), indicating it has selective toxicity to malignant cells with MCL-1 overexpression over normal cells. 9-nc, a negative control, had no significant effect on both cancerous cells and normal cells. To validate whether the selective cytotoxicity of 9c is a result of caspase-dependent apoptosis, we conducted apoptosis assays. U266 cells and WS-1 cells were exposed to DMSO or various concentrations of 9c, and caspase activity was monitored by Caspase-Glo 3/7 assay kit. As expected, 9c induced caspase activation in U266 cells, but not in WS-1 cells (SI Appendix, Fig. S13C).
As a consequence, we have successfully demonstrated that a cell-permeable PPI inhibitor can be identified through on-bead screening of a combinatorial library of α-helix mimetics. 9c can be a starting point for development of potent inhibitors targeting MCL-1. Further biological evaluation and structure–activity relationship studies are currently in progress to obtain more potent and selective MCL-1 inhibitors.
α-Synuclein Screening.
To determine whether our compounds are effective pharmacological chaperones in stabilizing protein folding and/or preventing protein aggregation, we used α-synuclein as a representative of protein-folding-related diseases. α-Synuclein aggregation leading to the formation of Lewy bodies is the pathological hallmark of Parkinson disease (60). The mechanism of Lewy body formation is still unclear. However, various lines of evidence indicate that misfolding of α-synuclein and subsequent inappropriate self-association are key steps in Lewy body formation (61). α-Synuclein was traditionally thought to be a natively unfolded protein (62); however, recent evidence by our group and others suggest it also exists as folded tetramers (63, 64), which implies that destabilization of the tetramer might lead to more aggregation-prone species. These findings opened venues for testing new therapeutic approaches for this devastating disease for which there is no cure and for which there are no effective treatments. Here, we use our α-helix-mimetic compounds to test the approach of preventing α-synuclein aggregation by stabilizing its folded structure and/or preventing aberrant self-association with pharmacological chaperones. This is an ideal test case for our chemical library because α-synuclein consists mostly of α-helices (∼65%), and its aggregation could be monitored in real-time (63, 64).
For the screening assay, we engineered a construct of α-synuclein consisting of an N-terminal extension MKCK quad-peptide to introduce a reactive cysteine residue for chemical linking with biotin (wild-type α-synuclein contains no cysteine). The cysteine-containing α-synuclein (cys-Syn) was biotinylated (SI Appendix, Fig. S14) and used for screening, as described earlier for MCL-1. On-bead screening identified two hit compounds, Q1 and Q2, as potential α-synuclein binders (Fig. 5A). We then synthesized and purified these compounds for further experimentation (SI Appendix, Fig. S9).
Fig. 5.

(A) Structure of Q1 and Q2, hit compounds to α-synuclein. (B) FP assays. Binding affinity of hit compounds Q1, Q2, negative controls 9a–9c, and Fluro-linker against wild-type α-synuclein were determined using FP assay. (C) Circular dichroism thermal denaturation assay. Unfolding of α-synuclein in the presence or the absence of hit compounds Q1 and Q2 were monitored using ellipticity at 222 nm, with respect to temperature.
α-Synuclein Binding Assay.
To confirm the binding of the hit compounds, we used FP assay as described earlier for MCL-1. Q1 and Q2 were found to bind to α-synuclein with KD values of 68 and 148 nM, respectively (Fig. 5B). Q1 and Q2 share two identical side chains at R1 and R2 and similar features at R3, with both consisting of 1,2-dialkoxybenzene (Fig. 5A). To investigate the contributions of compound backbone and rophore linker to the observed binding of Q1 and Q2 to α-synuclein, we measured the binding of MCL-1 hit compounds 9a–9c, which have the same backbone structure as Q1–Q2, and Fluor-linker. As shown in Fig. 5B, 9a and 9b showed very weak binding, and Fluor-linker did not show detectable binding. Compound 9c, which contains two identical side chains as the Q compounds, showed moderate affinity to α-synuclein, as anticipated. Conversely, we also examined whether α-synuclein-interacting compounds bind to MCL-1. Consistent with the structural features, Q1, which has similar structure to 9c, moderately inhibited MCL-1/BH3 interactions, whereas Q2 showed little effect (SI Appendix, Fig. S15). In the subsequent cell viability assay, both Q1 and Q2 had no significant effect on the viability of both cancerous leukemia cells and normal cells (SI Appendix, Fig. S15). Taken together, these results indicate that the binding and selectivity of the compounds for their target proteins are conferred by the compounds’ side chains. To further confirm the binding of Q compounds and their effects on α-synuclein thermal stability, we used circular dichroism thermal stability assay. As shown in Fig. 5C and SI Appendix, Fig. S16, Q1 and Q2 shifted the melting temperature of α-synuclein from 37 °C to 40 °C and 41 °C, respectively. The α-helices on α-synuclein are amphiphilic, with long stretches of hydrophobic surfaces (64, 65); the relatively hydrophobic Q1 and Q2 probably interact with the hydrophobic surfaces on α-synuclein. How the binding of these compounds affects the structure of α-synuclein warrants further investigation; however, our objectives here are to test whether our library consists of molecules that could bind and inhibit α-synuclein aggregation, which is detailed here.
α-Synuclein Aggregation Assay.
On the basis of the confirmed binding affinity of Q1 and Q2 to α-synuclein, we set out to determine whether the binding of Q1 and Q2 affects the aggregation propensity of α-synuclein. As mentioned earlier, we initially screened for Q-compounds using wild-type α-synuclein; however, the wild-type does not aggregate readily, so we used an aggregation-prone, disease-associated mutant (A53T) to perform aggregation assays. To ascertain that Q1 and Q2 also bind to A53T, we repeated the FP binding assay using A53T protein and we found that Q1 and Q2 also bind strongly to the A53T mutant (KD values = 268 and 372 nM, respectively), albeit at slightly lower affinity compared with wild-type α-synuclein (Fig. 6A). Compounds 9a–9c also showed similar binding patterns as those observed with wild-type α-synuclein. We selected 9a as a negative control for aggregation assays because it showed no binding to α-synuclein.
Fig. 6.
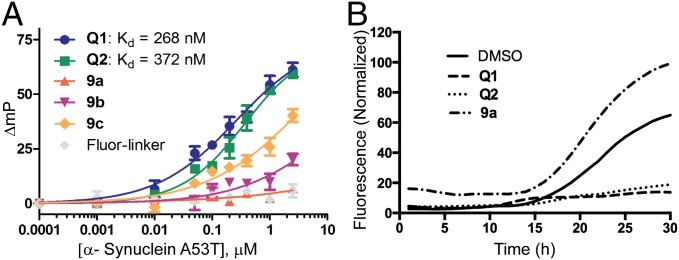
(A) FP assays. Binding affinity of Q1, Q2, 9a–9c, and Fluor-linker against the disease-associated mutant of α-synuclein A53T were measured using FP assay. (B) Thioflavin-T aggregation assays. Effects of Q1, Q2, and negative-control 9a on aggregation of α-synuclein A53T were determined by using Thioflavin-T fluorescence aggregation assay.
We preincubated Q1, Q2, 9a, and DMSO with A53T mutant α-synuclein and monitored its aggregation using Thioflavin-T fluorescence aggregation assay, as previously described (64). We found that both Q1 and Q2 significantly delayed the onset of aggregation and dramatically decreased the total amounts of aggregated fibrils within the 30-h duration of our experiment (Fig. 6B), whereas DMSO and 9a did not inhibit aggregation of A53T mutant α-synuclein.
Although the mechanisms by which the Q-compounds prevent α-synuclein aggregation are still unclear, it is consistent with the idea that the binding of Q1 and Q2 to α-synuclein prevented it from aggregating, either by masking the surfaces that directly involve in aggregation or by preventing its conversion/misfolding into aggregation prone species. In any case, our data demonstrate that our chemical library consists of molecules that effectively inhibited α-synuclein fibrillation and suggests these compounds might be useful also for other proteins associated with protein-misfolding and aggregation-prone related diseases.
Conclusions
Despite their great potential as therapeutic candidates for the treatment of many diseases caused by protein instability and/or aberrant protein–protein interactions, discovering pharmacological chaperones for specific target proteins is among the most difficult challenges in drug discovery and chemical biology. Here, we have presented an approach that allows for large-scale synthesis of a peptoid-encoded OBOC combinatorial library of α-helix mimetic small molecules and rapid identification of potential pharmacological chaperones through a facile “on-bead” HTS. The proof-of-concept screens against a cancer-associated protein MCL-1 and a Parkinson disease-associated protein α-synuclein demonstrated that that triazine-piperazine-triazine-based α-helix mimetics are effective at binding and modulating the activity and behavior of distinctly different helical proteins. MCL-1 is an established canonical helical protein that interacts with other helical structures, suggesting that the hit compounds that we identified likely mimicked certain features of MCL-1’s natural binding partners. α-Synuclein, in contrast, has no known structures; its interacting partners are unknown (aside from self-associating), and aggregation mechanism is still a mystery. What we do know is that when partially folded, it consists of dynamic helical structures, thus demonstrating that our method is capable of obtaining hit compounds even without prior knowledge of the structure of the target protein or its binding partners. Given the convenience of the screening method and structural diversity of the library, we believe our strategy could be a useful approach to developing pharmacological chaperones against a wide range of different helical proteins.
Methods
Synthesis and characterization of 9a–9c and Q1–Q2 can be found in SI Appendix. Protocols for OBOC library synthesis and on-bead screening are detailed in SI Appendix. Methods of molecular docking, protein purification, binding assays, cell viability assays, caspase assays, confocal microscopy experiment, coimmunoprecipitation assays, circular dichroism thermal stability assay, and thioflavin-T aggregation assay are described in SI Appendix.
Supplementary Material
Acknowledgments
We thank Dr. Loren Walensky for the plasmids expressing MCL-1. This research was supported by the National Science Foundation (CHE-1212720 to H.-S.L.); the Basic Science Research Program through the National Research Foundation of Korea, funded by the Ministry of Education, Science and Technology (2012R1A2007768); and POSCO. This research was also supported in part by XSEDE MCB070009 (to W.I.). Q.Q.H. and W.W. are supported by the National Institutes of Health (1R21NS079881-01), Indiana University’s Research Support Funds Grant, and Indiana University School of Medicine’s Biomedical Research Grant.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1320556111/-/DCSupplemental.
References
- 1.Hopkins AL, Groom CR. The druggable genome. Nat Rev Drug Discov. 2002;1(9):727–730. doi: 10.1038/nrd892. [DOI] [PubMed] [Google Scholar]
- 2.Arkin MR, Wells JA. Small-molecule inhibitors of protein-protein interactions: Progressing towards the dream. Nat Rev Drug Discov. 2004;3(4):301–317. doi: 10.1038/nrd1343. [DOI] [PubMed] [Google Scholar]
- 3.Wells JA, McClendon CL. Reaching for high-hanging fruit in drug discovery at protein-protein interfaces. Nature. 2007;450(7172):1001–1009. doi: 10.1038/nature06526. [DOI] [PubMed] [Google Scholar]
- 4.Cohen FE, Kelly JW. Therapeutic approaches to protein-misfolding diseases. Nature. 2003;426(6968):905–909. doi: 10.1038/nature02265. [DOI] [PubMed] [Google Scholar]
- 5.Winklhofer KF, Tatzelt J, Haass C. The two faces of protein misfolding: Gain- and loss-of-function in neurodegenerative diseases. EMBO J. 2008;27(2):336–349. doi: 10.1038/sj.emboj.7601930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Arakawa T, Ejima D, Kita Y, Tsumoto K. Small molecule pharmacological chaperones: From thermodynamic stabilization to pharmaceutical drugs. Biochim Biophys Acta. 2006;1764(11):1677–1687. doi: 10.1016/j.bbapap.2006.08.012. [DOI] [PubMed] [Google Scholar]
- 7.Leandro P, Gomes CM. Protein misfolding in conformational disorders: Rescue of folding defects and chemical chaperoning. Mini Rev Med Chem. 2008;8(9):901–911. doi: 10.2174/138955708785132783. [DOI] [PubMed] [Google Scholar]
- 8.Ringe D, Petsko GA. What are pharmacological chaperones and why are they interesting? J Biol. 2009;8(9):80. doi: 10.1186/jbiol186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Thanos CD, DeLano WL, Wells JA. Hot-spot mimicry of a cytokine receptor by a small molecule. Proc Natl Acad Sci USA. 2006;103(42):15422–15427. doi: 10.1073/pnas.0607058103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Brenke R, et al. Fragment-based identification of druggable ‘hot spots’ of proteins using Fourier domain correlation techniques. Bioinformatics. 2009;25(5):621–627. doi: 10.1093/bioinformatics/btp036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Landon MR, et al. Detection of ligand binding hot spots on protein surfaces via fragment-based methods: Application to DJ-1 and glucocerebrosidase. J Comput Aided Mol Des. 2009;23(8):491–500. doi: 10.1007/s10822-009-9283-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pey AL, et al. Mechanisms underlying responsiveness to tetrahydrobiopterin in mild phenylketonuria mutations. Hum Mutat. 2004;24(5):388–399. doi: 10.1002/humu.20097. [DOI] [PubMed] [Google Scholar]
- 13.Lieberman RL, et al. Structure of acid beta-glucosidase with pharmacological chaperone provides insight into Gaucher disease. Nat Chem Biol. 2007;3(2):101–107. doi: 10.1038/nchembio850. [DOI] [PubMed] [Google Scholar]
- 14.Connelly S, Choi S, Johnson SM, Kelly JW, Wilson IA. Structure-based design of kinetic stabilizers that ameliorate the transthyretin amyloidoses. Curr Opin Struct Biol. 2010;20(1):54–62. doi: 10.1016/j.sbi.2009.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bernal F, Tyler AF, Korsmeyer SJ, Walensky LD, Verdine GL. Reactivation of the p53 tumor suppressor pathway by a stapled p53 peptide. J Am Chem Soc. 2007;129(9):2456–2457. doi: 10.1021/ja0693587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Henchey LK, et al. Inhibition of hypoxia inducible factor 1-transcription coactivator interaction by a hydrogen bond surrogate alpha-helix. J Am Chem Soc. 2010;132(3):941–943. doi: 10.1021/ja9082864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mills NL, Daugherty MD, Frankel AD, Guy RK. An alpha-helical peptidomimetic inhibitor of the HIV-1 Rev-RRE interaction. J Am Chem Soc. 2006;128(11):3496–3497. doi: 10.1021/ja0582051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Azzarito V, Long K, Murphy NS, Wilson AJ. Inhibition of α-helix-mediated protein-protein interactions using designed molecules. Nat Chem. 2013;5(3):161–173. doi: 10.1038/nchem.1568. [DOI] [PubMed] [Google Scholar]
- 19.Cummings CG, Hamilton AD. Disrupting protein-protein interactions with non-peptidic, small molecule alpha-helix mimetics. Curr Opin Chem Biol. 2010;14(3):341–346. doi: 10.1016/j.cbpa.2010.04.001. [DOI] [PubMed] [Google Scholar]
- 20.Heeres JT, Hergenrother PJ. High-throughput screening for modulators of protein-protein interactions: Use of photonic crystal biosensors and complementary technologies. Chem Soc Rev. 2011;40(8):4398–4410. doi: 10.1039/b923660k. [DOI] [PubMed] [Google Scholar]
- 21.Horswill AR, Savinov SN, Benkovic SJ. A systematic method for identifying small-molecule modulators of protein-protein interactions. Proc Natl Acad Sci USA. 2004;101(44):15591–15596. doi: 10.1073/pnas.0406999101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Henchey LK, Jochim AL, Arora PS. Contemporary strategies for the stabilization of peptides in the alpha-helical conformation. Curr Opin Chem Biol. 2008;12(6):692–697. doi: 10.1016/j.cbpa.2008.08.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Phillips C, et al. Design and structure of stapled peptides binding to estrogen receptors. J Am Chem Soc. 2011;133(25):9696–9699. doi: 10.1021/ja202946k. [DOI] [PubMed] [Google Scholar]
- 24.Goodman CM, Choi S, Shandler S, DeGrado WF. Foldamers as versatile frameworks for the design and evolution of function. Nat Chem Biol. 2007;3(5):252–262. doi: 10.1038/nchembio876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Horne WS, Gellman SH. Foldamers with heterogeneous backbones. Acc Chem Res. 2008;41(10):1399–1408. doi: 10.1021/ar800009n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Seebach D, Gardiner J. Beta-peptidic peptidomimetics. Acc Chem Res. 2008;41(10):1366–1375. doi: 10.1021/ar700263g. [DOI] [PubMed] [Google Scholar]
- 27.Hara T, Durell SR, Myers MC, Appella DH. Probing the structural requirements of peptoids that inhibit HDM2-p53 interactions. J Am Chem Soc. 2006;128(6):1995–2004. doi: 10.1021/ja056344c. [DOI] [PubMed] [Google Scholar]
- 28.Chongsiriwatana NP, et al. Peptoids that mimic the structure, function, and mechanism of helical antimicrobial peptides. Proc Natl Acad Sci USA. 2008;105(8):2794–2799. doi: 10.1073/pnas.0708254105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kritzer JA, Luedtke NW, Harker EA, Schepartz A. A rapid library screen for tailoring beta-peptide structure and function. J Am Chem Soc. 2005;127(42):14584–14585. doi: 10.1021/ja055050o. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tosovská P, Arora PS. Oligooxopiperazines as nonpeptidic alpha-helix mimetics. Org Lett. 2010;12(7):1588–1591. doi: 10.1021/ol1003143. [DOI] [PubMed] [Google Scholar]
- 31.Volonterio A, Moisan L, Rebek J., Jr Synthesis of pyridazine-based scaffolds as alpha-helix mimetics. Org Lett. 2007;9(19):3733–3736. doi: 10.1021/ol701487g. [DOI] [PubMed] [Google Scholar]
- 32.Maity P, König B. Synthesis and structure of 1,4-dipiperazino benzenes: Chiral terphenyl-type peptide helix mimetics. Org Lett. 2008;10(7):1473–1476. doi: 10.1021/ol8002749. [DOI] [PubMed] [Google Scholar]
- 33.Min J, et al. Forward chemical genetic approach identifies new role for GAPDH in insulin signaling. Nat Chem Biol. 2007;3(1):55–59. doi: 10.1038/nchembio833. [DOI] [PubMed] [Google Scholar]
- 34.Angell Y, Chen D, Brahimi F, Saragovi HU, Burgess K. A combinatorial method for solution-phase synthesis of labeled bivalent beta-turn mimics. J Am Chem Soc. 2008;130(2):556–565. doi: 10.1021/ja074717z. [DOI] [PubMed] [Google Scholar]
- 35.Popowicz GM, Dömling A, Holak TA. The structure-based design of Mdm2/Mdmx-p53 inhibitors gets serious. Angew Chem Int Ed Engl. 2011;50(12):2680–2688. doi: 10.1002/anie.201003863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Marimganti S, Cheemala MN, Ahn JM. Novel amphiphilic alpha-helix mimetics based on a bis-benzamide scaffold. Org Lett. 2009;11(19):4418–4421. doi: 10.1021/ol901785v. [DOI] [PubMed] [Google Scholar]
- 37.Thompson S, Hamilton AD. Amphiphilic α-helix mimetics based on a benzoylurea scaffold. Org Biomol Chem. 2012;10(30):5780–5782. doi: 10.1039/c2ob25273b. [DOI] [PubMed] [Google Scholar]
- 38.Jung KY, et al. Amphipathic α-helix mimetics based on a 1,2-diphenylacetylene scaffold. Org Lett. 2013;15(13):3234–3237. doi: 10.1021/ol401197n. [DOI] [PubMed] [Google Scholar]
- 39.Shahian T, et al. Inhibition of a viral enzyme by a small-molecule dimer disruptor. Nat Chem Biol. 2009;5(9):640–646. doi: 10.1038/nchembio.192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Shaginian A, et al. Design, synthesis, and evaluation of an alpha-helix mimetic library targeting protein-protein interactions. J Am Chem Soc. 2009;131(15):5564–5572. doi: 10.1021/ja810025g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lee JH, et al. Novel pyrrolopyrimidine-based α-helix mimetics: Cell-permeable inhibitors of protein−protein interactions. J Am Chem Soc. 2011;133(4):676–679. doi: 10.1021/ja108230s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lam KS, et al. A new type of synthetic peptide library for identifying ligand-binding activity. Nature. 1991;354(6348):82–84. doi: 10.1038/354082a0. [DOI] [PubMed] [Google Scholar]
- 43.Joo SH, Xiao Q, Ling Y, Gopishetty B, Pei D. High-throughput sequence determination of cyclic peptide library members by partial Edman degradation/mass spectrometry. J Am Chem Soc. 2006;128(39):13000–13009. doi: 10.1021/ja063722k. [DOI] [PubMed] [Google Scholar]
- 44.Lee JH, Meyer AM, Lim HS. A simple strategy for the construction of combinatorial cyclic peptoid libraries. Chem Commun (Camb) 2010;46(45):8615–8617. doi: 10.1039/c0cc03272g. [DOI] [PubMed] [Google Scholar]
- 45.Lee JH, Kim HS, Lim HS. Design and facile solid-phase synthesis of conformationally constrained bicyclic peptoids. Org Lett. 2011;13(19):5012–5015. doi: 10.1021/ol201773f. [DOI] [PubMed] [Google Scholar]
- 46.Aquino C, et al. A biomimetic polyketide-inspired approach to small-molecule ligand discovery. Nat Chem. 2012;4(2):99–104. doi: 10.1038/nchem.1200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Astle JM, et al. Seamless bead to microarray screening: Rapid identification of the highest affinity protein ligands from large combinatorial libraries. Chem Biol. 2010;17(1):38–45. doi: 10.1016/j.chembiol.2009.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kodadek T. Rethinking screening. Nat Chem Biol. 2010;6(3):162–165. doi: 10.1038/nchembio.303. [DOI] [PubMed] [Google Scholar]
- 49.Arkin MR, et al. Binding of small molecules to an adaptive protein-protein interface. Proc Natl Acad Sci USA. 2003;100(4):1603–1608. doi: 10.1073/pnas.252756299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Liu R, Marik J, Lam KS. A novel peptide-based encoding system for “one-bead one-compound” peptidomimetic and small molecule combinatorial libraries. J Am Chem Soc. 2002;124(26):7678–7680. doi: 10.1021/ja026421t. [DOI] [PubMed] [Google Scholar]
- 51.Kwon YU, Kodadek T. Encoded combinatorial libraries for the construction of cyclic peptoid microarrays. Chem Commun (Camb) 2008;(44):5704–5706. doi: 10.1039/b812735b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Simon RJ, et al. Peptoids: A modular approach to drug discovery. Proc Natl Acad Sci USA. 1992;89(20):9367–9371. doi: 10.1073/pnas.89.20.9367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Gorske BC, Jewell SA, Guerard EJ, Blackwell HE. Expedient synthesis and design strategies for new peptoid construction. Org Lett. 2005;7(8):1521–1524. doi: 10.1021/ol0502984. [DOI] [PubMed] [Google Scholar]
- 54.Olivos HJ, Alluri PG, Reddy MM, Salony D, Kodadek T. Microwave-assisted solid-phase synthesis of peptoids. Org Lett. 2002;4(23):4057–4059. doi: 10.1021/ol0267578. [DOI] [PubMed] [Google Scholar]
- 55.Lessene G, Czabotar PE, Colman PM. BCL-2 family antagonists for cancer therapy. Nat Rev Drug Discov. 2008;7(12):989–1000. doi: 10.1038/nrd2658. [DOI] [PubMed] [Google Scholar]
- 56.Oltersdorf T, et al. An inhibitor of Bcl-2 family proteins induces regression of solid tumours. Nature. 2005;435(7042):677–681. doi: 10.1038/nature03579. [DOI] [PubMed] [Google Scholar]
- 57.van Delft MF, et al. The BH3 mimetic ABT-737 targets selective Bcl-2 proteins and efficiently induces apoptosis via Bak/Bax if Mcl-1 is neutralized. Cancer Cell. 2006;10(5):389–399. doi: 10.1016/j.ccr.2006.08.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Stewart ML, Fire E, Keating AE, Walensky LD. The MCL-1 BH3 helix is an exclusive MCL-1 inhibitor and apoptosis sensitizer. Nat Chem Biol. 2010;6(8):595–601. doi: 10.1038/nchembio.391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Müller CE. Prodrug approaches for enhancing the bioavailability of drugs with low solubility. Chem Biodivers. 2009;6(11):2071–2083. doi: 10.1002/cbdv.200900114. [DOI] [PubMed] [Google Scholar]
- 60.Mezey E, et al. Alpha synuclein in neurodegenerative disorders: Murderer or accomplice? Nat Med. 1998;4(7):755–757. doi: 10.1038/nm0798-755. [DOI] [PubMed] [Google Scholar]
- 61.Fink AL. The aggregation and fibrillation of alpha-synuclein. Acc Chem Res. 2006;39(9):628–634. doi: 10.1021/ar050073t. [DOI] [PubMed] [Google Scholar]
- 62.Weinreb PH, Zhen W, Poon AW, Conway KA, Lansbury PT., Jr NACP, a protein implicated in Alzheimer’s disease and learning, is natively unfolded. Biochemistry. 1996;35(43):13709–13715. doi: 10.1021/bi961799n. [DOI] [PubMed] [Google Scholar]
- 63.Bartels T, Choi JG, Selkoe DJ. α-Synuclein occurs physiologically as a helically folded tetramer that resists aggregation. Nature. 2011;477(7362):107–110. doi: 10.1038/nature10324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Wang W, et al. A soluble α-synuclein construct forms a dynamic tetramer. Proc Natl Acad Sci USA. 2011;108(43):17797–17802. doi: 10.1073/pnas.1113260108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Chandra S, Chen X, Rizo J, Jahn R, Südhof TC. A broken alpha -helix in folded alpha -Synuclein. J Biol Chem. 2003;278(17):15313–15318. doi: 10.1074/jbc.M213128200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.