

Significance
The Hedgehog (Hh)/Gli signaling pathway is a key regulator of embryonic patterning and tissue homeostasis, and its inappropriate activation can lead to several human cancers, including basal cell carcinoma, medulloblastoma, and meningioma. To better understand the mechanisms that control Hh pathway state, we have conducted a genome-scale cDNA overexpression screen for signaling proteins that promote Gli-dependent transcription. Our studies reveal Arhgap36 to be a potent Gli activator, yielding the first functional insights, to our knowledge, for this Rho GAP family member. We also find that ARHGAP36 is overexpressed in a subset of medulloblastomas, suggesting that this Gli-activating protein plays an important role in tumorigenesis.
Abstract
Hedgehog (Hh) pathway activation and Gli-dependent transcription play critical roles in embryonic patterning, tissue homeostasis, and tumorigenesis. By conducting a genome-scale cDNA overexpression screen, we have identified the Rho GAP family member Arhgap36 as a positive regulator of the Hh pathway in vitro and in vivo. Arhgap36 acts in a Smoothened (Smo)-independent manner to inhibit Gli repressor formation and to promote the activation of full-length Gli proteins. Arhgap36 concurrently induces the accumulation of Gli proteins in the primary cilium, and its ability to induce Gli-dependent transcription requires kinesin family member 3a and intraflagellar transport protein 88, proteins that are essential for ciliogenesis. Arhgap36 also functionally and biochemically interacts with Suppressor of Fused. Transcriptional profiling further reveals that Arhgap36 is overexpressed in murine medulloblastomas that acquire resistance to chemical Smo inhibitors and that ARHGAP36 isoforms capable of Gli activation are up-regulated in a subset of human medulloblastomas. Our findings reveal a new mechanism of Gli transcription factor activation and implicate ARHGAP36 dysregulation in the onset and/or progression of GLI-dependent cancers.
Hedgehog (Hh) signaling is an evolutionarily conserved regulator of embryonic patterning and tissue homeostasis (1, 2), and its uncontrolled activation can cause basal cell carcinoma, medulloblastoma, and other human cancers (3). In vertebrates, Hh target gene expression is primarily regulated by the transcription factors Gli2 and Gli3, which bind to the protein scaffold Suppressor of Fused (Sufu) and traffic constitutively through the primary cilium (4, 5). Sufu promotes the cilium-dependent phosphorylation and proteolytic processing of Gli2 and Gli3 into N-terminal repressors (Gli2/3R), thereby suppressing Hh pathway activity in quiescent cells (6–9). These events are inhibited in cells responding to Hh ligands (Sonic, Indian, or Desert; Shh, Ihh, or Dhh), which bind to the Patched family of 12-transmembrane receptors (Ptch1 and Ptch2) and alleviate its repression of Smoothened (Smo), a G protein-coupled receptor-like protein. Activated Smo accumulates within the primary cilium (10, 11) and induces the dissociation of Sufu–Gli complexes (4, 5), abrogating Gli2/3R formation and promoting the cilium-dependent conversion of full-length Gli proteins (Gli2/3FL) into transcriptionally active forms (Gli2/3A) (12, 13). Hh signaling coincides with the steady-state accrual of Gli2/3 at the distal tip of the primary cilium (8, 14), and Gli2/3A translocate to the nucleus to drive the expression of Ptch1, the constitutively active factor Gli1, and other Hh target genes (1, 15).
Within this general framework, the canonical and noncanonical processes that lead to Gli2/3A formation remain enigmatic. To identify signaling proteins that can promote Gli activation, we surveyed a genome-scale collection of ORFs for Hh pathway agonists. Using a flow cytometry-based cDNA overexpression screen that permits quantitative analyses with single-cell resolution, we have discovered the putative Rho GTPase-activating protein Arhgap36 as a positive regulator of the Hh pathway. We find that Arhgap36 overexpression recapitulates multiple aspects of Hh signal transduction, including reduced Gli3R levels, ciliary accumulation of Gli2, and Gli1 expression. These effects are independent of Smo and require kinesin family member 3a (Kif3a) and intraflagellar transport protein 88 (Ift88). Arhgap36 also functionally interacts with Sufu and can biochemically associate with this Gli antagonist. We further observe that ARHGAP36 isoforms are up-regulated in a subset of medulloblastomas and that these signaling proteins are potent activators of Gli function. Based on these results, we propose a model in which Arhgap36 promotes noncanonical, oncogenic Gli activation through interactions with Sufu.
Results
Arhgap36 Is a Positive Regulator of the Hh Pathway.
To identify novel Hh pathway agonists, we surveyed the human ORFeome collection (v5.1), which includes 15,483 ORFs encoding 12,794 nonredundant genes (16). Our screening strategy used an NIH-3T3 cell-derived line stably transfected with a Gli-dependent enhanced green fluorescent protein reporter (Shh-EGFP cells) (17, 18), retroviral pools encoding sublibraries of the ORF collection, and a fluorescence-activated cell sorting (FACS)-based assay for ascertaining Hh pathway activity in single cells (Fig. 1A and SI Appendix, Fig. S1). We segregated the human ORFs into 169 nonoverlapping pools and used recombination cloning to shuttle each sublibrary en masse into a Moloney murine leukemia virus (MMLV)-derived vector. Each ORF was cloned in frame with three C-terminal FLAG tags, followed by an internal ribosomal entry site (IRES)-driven mCherry fluorescent reporter, and the Shh-EGFP cells were infected with the corresponding retroviral pools. The number of infected cells (red fluorescence) with increased Gli reporter expression (green fluorescence) for each sublibrary was then assessed by flow cytometry.
Fig. 1.
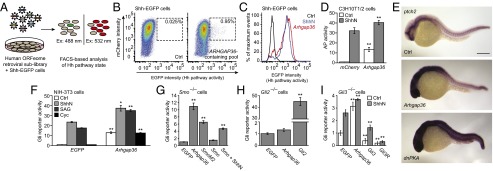
Identification of Arhgap36 as a Hh pathway activator. (A) Schematic representation of the cDNA overexpression screen. (B) FACS scatter plots revealing a retroviral pool that induces Hh ligand-independent pathway activation. Retroviral transduction of mCherry alone served as a negative control, and the percentage of total cells within the sorting gate (dashed box) for each condition is indicated. Deconvolution of this sublibrary identified ARHGAP36 as the active component. (C) FACS histogram plots demonstrating the response of Shh-EGFP cells to ShhN or exogenous Arhgap36. (D) Alkaline phosphatase (AP) levels in C3H10T1/2 cells transduced with mCherry or Arhgap36 and then cultured in the absence or presence of ShhN for 48 h. Data are the average ± SEM, n = 8. (E) Expression levels of ptch2 in uninjected zebrafish embryos (Top) or those injected with Arhgap36 (Middle) or dnPKA (Bottom) mRNA. Lateral views of 24-hpf embryos are shown. (Scale bar: 200 μm.) (F) Hh pathway activities in NIH-3T3 cells cotransfected with Gli-dependent firefly luciferase and SV40-driven Renilla luciferase reporters and either EGFP or Arhgap36. The cells were then treated with ShhN, 200 nM SAG, or 5 µM cyclopamine for 30 h. Data are the average ± SEM, n = 3. (G) Hh pathway activities in Smo−/− MEFs cotransfected with the luciferase reporters and designated cDNAs. (H) Hh pathway activities in Gli2−/− MEFs cotransfected with the luciferase reporters and designated cDNAs for 48 h. (I) Hh pathway activities in Gli3−/− MEFs cotransfected with the luciferase reporters and designated cDNAs and then cultured in the absence of presence of ShhN for 30 h. Data for G–I are the average ± SEM, n = 4. Statistical analyses: Single and double asterisks indicate P < 0.05 and P < 0.01, respectively (D, Arhgap36 vs. mCherry for each cell culture condition; F, Arhgap36 vs. EGFP for each cell culture condition; G–I, each designated gene versus EGFP).
Using this screening regimen, we discovered two retroviral sublibraries that increased Gli reporter expression. One pool contained GLI1 and was not examined further. By testing overlapping subsets of the second pool, we identified the putative Rho GAP gene ARHGAP36 as a positive Hh pathway regulator (Fig. 1B and SI Appendix, Fig. S2). The ability of ARHGAP36 to activate Gli-dependent transcription in the absence of Hh ligand appears to be unique among the 33 other ARHGAP genes in the human ORFeome collection (SI Appendix, Table S1). Accordingly, we transduced Shh-EGFP cells with six representative ARHGAP homologs, and none up-regulated Hh pathway activity (SI Appendix, Fig. S3).
To corroborate this result, we confirmed that retrovirally overexpressed mouse Arhgap36 induced EGFP reporter levels comparable to those achieved by ShhN stimulation (Fig. 1C), whereas the closest homolog, Arhgap6, had no effect on the reporter under these conditions (SI Appendix, Fig. S3). NIH-3T3 cells transiently transfected with Arhgap36 and a Gli-dependent firefly luciferase reporter similarly exhibited near-maximum levels of Hh pathway activity (SI Appendix, Fig. S4). We also retrovirally transduced Arhgap36 into C3H10T1/2 cells, which differentiate into alkaline phosphatase-expressing osteoblasts upon Hh pathway activation (19). Arhgap36 promoted alkaline phosphatase expression in these cells, and its activity could be further enhanced by ShhN (Fig. 1D).
We next investigated whether Arhgap36 expression can drive Hh target gene expression in zebrafish embryos by injecting zygotes with Arhgap36 mRNA and assessing ptch2 expression at 24 h postfertilization (hpf). In comparison with uninjected wild-type embryos, those with exogenous Arhgap36 had increased ptch2 transcription in the developing brain and other anterior structures, but not in somitic cells, which also respond to Hh ligands (Fig. 1E). The differential activity of Arhgap36 in neural and muscle progenitors contrasted the effects of dominant-negative protein kinase A (dnPKA) mRNA, which up-regulated ptch2 expression in both cell types (Fig. 1E). We also observed differential Arhgap36 expression in a murine tissue array, with the highest transcript levels in embryonic tissues and the adult central nervous system (SI Appendix, Fig. S5). Arhgap36-induced Gli activation may therefore have developmental stage- and/or tissue-specific requirements.
Arhgap36 Action Is Smo-Independent and Gli2-Dependent.
To better understand how Arhgap36 activates the Hh pathway, we assessed its functional interactions with canonical Hh signaling proteins. We first transiently transfected NIH-3T3 cells with Arhgap36 and the Gli-dependent luciferase reporter described above and then treated them with ShhN, the Smo agonist SAG (20), the Smo antagonist cyclopamine (21), or control medium. Hh pathway activity driven by exogenous Arhgap36 was resistant to cyclopamine and additive to that induced by ShhN- and/or SAG (Fig. 1F). Smo−/− murine embryonic fibroblasts (MEFs) transiently transfected with Arhgap36 and the luciferase reporter exhibited an analogous increase in Hh pathway activity (Fig. 1G). We similarly transfected MEFs lacking either Gli2 or Gli3 with Arhgap36 and the luciferase reporter. Gli2−/− MEFs were unresponsive to exogenous Arhgap36 (Fig. 1H), but Arhgap36 overexpression could up-regulate reporter activity to some extent in Gli3−/− MEFs (Fig. 1I). Thus, Arhgap36 acts between Smo and the Gli transcription factors to drive Hh target gene expression, primarily through the activation of Gli2.
Arhgap36 Modulates Gli2 Trafficking and Gli3 Processing.
We next investigated the effects of Arhgap36 on specific aspects of Gli function. To determine whether this Rho GAP family member can influence Gli2/3 localization, we retrovirally transduced Shh-EGFP cells with Arhgap36 and briefly cultured the cells in the absence or presence of ShhN. The cells were then fixed, and Gli2 localization was quantitatively assessed by immunofluorescence. Exogenous Arhgap36 caused Gli2 to accrue within the primary cilium (Fig. 2 A and B), achieving steady-state levels comparable to that induced by ShhN. The combined actions of Arhgap36 and ShhN led to even greater ciliary accumulation of Gli2.
Fig. 2.

Effects of Arhgap36 on Gli trafficking and processing. (A) Ciliary Gli2 levels in Shh-EGFP cells retrovirally transduced with either mCherry or Arhgap36 and then cultured in the absence or presence of ShhN for 1 h. Representative immunofluorescence micrographs are shown with staining for Gli2, acetylated tubulin (primary cilia), and DAPI (nuclei). (Scale bar: 2 µm.) (B) Quantification of Gli2 immunofluorescence within primary cilia. Data are the average ± SEM, n ≥ 50 cells. (C) Gli3 forms in Shh-EGFP cells transduced with either mCherry or Arhgap36 and concurrently treated with ShhN-free medium, ShhN, or ShhN plus 5 µM cyclopamine for 24 h. A representative blot is shown with protein bands corresponding to Gli3R, Gli3FL, and the phosphorylated form of Gli3FL labeled. Gli1 levels indicate Hh pathway state, and importin β levels were used as gel-loading controls. (D and E) Quantification of Gli3FL/Gli3R ratios and Gli1 levels for each experimental condition. Data are the average ± SEM, n = 3 independent blots. (F) Hh pathway activities in Kif3a−/− MEFs cotransfected with Gli-dependent firefly luciferase and SV40-driven Renilla luciferase reporters, either EGFP or Arhgap36, and varying amounts of exogenous Kif3a. The cells were then cultured in absence or presence of ShhN for 30 h. Data are the average ± SEM, n = 4. Statistical analyses: Single and double asterisks indicate P < 0.05 and P < 0.01, respectively (A–E, Arhgap36 versus mCherry for each cell culture condition; F, Arhgap36 versus EGFP for each Kif3a transfection and cell culture condition).
We also evaluated the effects of Arhgap36 on Gli3 processing. Shh-EGFP cells transduced with Arhgap36 were cultured with or without ShhN, and the ShhN-stimulated cells were concurrently treated with cyclopamine or a vehicle control. The resulting Gli3 forms were then analyzed by quantitative immunoblotting, and Gli1 levels were similarly measured to ascertain Hh pathway state. Arhgap36 overexpression emulated the ability of ShhN to increase the Gli3FL/Gli3R ratio and Gli1 levels in these cells, to decrease total Gli3 levels, and to generate a Gli3 form with reduced electrophoretic mobility (Fig. 2 C–E). This latter effect is due to Gli3 phosphorylation (5), which likely coincides with Gli3 activation and destabilization. Consistent with our epistatic analyses, these Arhgap36-dependent changes were not reversed by cyclopamine.
Arhgap36-Induced Hh Pathway Activation Requires Kif3a and Ift88.
Because the primary cilium is required for Gli activation (6, 7), we next examined whether this signaling center participates in Arhgap36 function. Anterograde ciliary transport is driven by kinesin-2 (22), and cells lacking the Kif3a subunit cannot form primary cilia or respond to ShhN (23). We therefore transfected Kif3a−/− MEFs with Arhgap36, variable amounts of exogenous Kif3a, and the Gli-dependent luciferase reporter. The cells were then cultured in the absence or presence of ShhN. Arhgap36 overexpression did not significantly increase Gli reporter activity in Kif3a−/− MEFs, but coexpression of Kif3a restored Hh signaling and Arhgap36 function (Fig. 2F and SI Appendix, Fig. S6). Arhgap36 similarly could not induce Gli reporter activity in cells lacking Ift88, a component of the IFT-B complex that is required for ciliary retrograde transport and ciliogenesis (SI Appendix, Fig. S7) (24). These results suggest an integral role for primary cilia in Arhgap36-induced Gli activation.
Functional Analyses and Localization of Arhgap36 Domains.
Although Arhgap36 is predicted to be a Rho GAP family member, it lacks the “arginine finger” motif that participates in Rho GTPase activity (25). The replacement of this structural element with a threonine (T246) suggests that catalytic GAP domain function may not be required for Arhgap36-induced Gli activation. To explore this possibility further, we mutated two other Arhgap36 residues, K283 and R287, which are structurally equivalent to those necessary for Rho GAP function in Graf (26). NIH-3T3 cells transiently transfected with these mutants, and a Gli-dependent luciferase reporter achieved Hh pathway activities comparable to those observed with wild-type Arhgap36 (SI Appendix, Fig. S8).
Because mutations within the predicted Rho GAP domain did not have any discernible effects, we investigated the functions of other regions within this signaling protein (Fig. 3A). In addition to the region with Rho GAP homology (amino acids 215–414), Arhgap36 contains unique N-terminal and C-terminal sequences (amino acids 1–214 and 415–490, respectively). Transiently expressing either the Rho GAP or C-terminal domains alone in NIH-3T3 fibroblasts did not perturb basal or ShhN-induced pathway activity to a significant extent, as determined by the luciferase reporter (Fig. 3B). In contrast, overexpressing the N-terminal portion of Arhgap36 (Arhgap36-N) inhibited Hh signaling in these cells. Arhgap36-N also reduced cellular levels of Gli1 and Gli2 (SI Appendix, Fig. S9), both of which are expressed in a Hh pathway-dependent manner (27, 28). However, Arhgap36-N overexpression could not overcome the ability of full-length Arhgap36 to activate the Gli reporter, suggesting that Arhgap36-induced Gli activation primarily involves other domains within this Rho GAP family member (SI Appendix, Fig. S10).
Fig. 3.

Arhgap36 domain analysis and interactions with Sufu. (A) Schematic representation of Arhgap36 with the N-terminal, Rho GAP, and C-terminal domains labeled. (B) Hh pathway activities in NIH-3T3 cells cotransfected with Gli-dependent firefly luciferase and SV40-driven Renilla luciferase reporters and EGFP, wild-type Arhgap36, or the indicated Arhgap36 domains. Data are the average ± SEM, n = 3. (C) Hh pathway activities in Sufu−/− MEFs cotransfected with the luciferase reporters and either EGFP or the designated cDNAs. Sufu and Sufu/SAG were used to verify Sufu-dependent Arhgap36 function and the Hh pathway-competence of the cells. Data are the average ± SEM, n = 4. (D) Immunoprecipitation studies of HEK-293T cells cotransfected with EGFP-Sufu and either ARHGAP26-3xFLAG or Arhgap36-3xFLAG. Statistical analyses: Single and double asterisks indicate P < 0.05 and P < 0.01, respectively (Arhgap36 or its individual domains versus EGFP for each cell culture condition).
Intrigued by these findings, we compared the subcellular localizations of full-length Arhgap36 and its N-terminal domain in NIH-3T3 cells, using retroviral constructs encoding each polypeptide with C-terminal FLAG tags. The full-length protein was localized to the plasma membrane and intracellular vesicles, whereas Arhgap36-N was enriched in primary cilia, as determined by colocalization with Arl13b (SI Appendix, Fig. S11A). Arhgap36 function and localization therefore may be regulated by motifs within its N-terminal domain.
Arhgap36 Can Functionally and Biochemically Interact with Sufu.
The inhibitory activity of Arhgap36-N also allowed us to investigate its epistatic relationship with Sufu. We cotransfected Sufu−/− MEFs with the Gli-dependent firefly luciferase reporter and cDNAs encoding either full-length Arhgap36 or its N-terminal domain. Neither form of Arhgap36 altered the constitutive Hh pathway activity in these cells, but exogenous Sufu rescued the activities of both polypeptides (Fig. 3C), indicating that Sufu is required for at least some aspects of Arhgap36 function. Full-length Arhgap36 and Arhgap36-N also had opposing effects on Sufu levels upon their overexpression in NIH-3T3 cells, although these changes were of moderate statistical significance (P = 0.09–0.12) (SI Appendix, Fig. S9).
To determine whether Arhgap36 can biochemically interact with Sufu, we then expressed in HEK-293T cells an EGFP-Sufu fusion protein and either Arhgap36 or the non-Hh pathway-regulating homolog ARHGAP26 bearing C-terminal FLAG epitopes. We observed that the exogenous Arhgap36 can coimmunoprecipitate with EGFP-Sufu and, therefore, may directly modulate Sufu function, whereas the overexpressed ARHGAP26 did not interact with the Sufu fusion protein (Fig. 3D).
ARHGAP36 Is Overexpressed in a Subset of Medulloblastomas.
A lack of endogenous Arhgap36 transcript levels in NIH-3T3–derived cells (SI Appendix, Fig. S12A) and the inability of Arhgap36 siRNAs to abrogate Hh signaling in C3H10T1/2 cells (SI Appendix, Fig. S12 B and C) suggest that this signaling protein may not be an obligate component of the pathway. Moreover, morpholino antisense oligonucleotides directed against the two closest Arhgap36 homologs in zebrafish did not disrupt embryonic ptch2 expression, either individually or in combination (SI Appendix, Figs. S13–S15). These observations argue against a conserved role for Arhgap36 in developmental Hh signaling.
Because Arhgap36 overexpression in zebrafish embryos preferentially activated the Hh pathway in neural tissues, we examined whether ARHGAP36 might contribute to the etiology and/or progression of medulloblastoma. We first analyzed previously reported microarray datasets for medulloblastoma allografts derived from Ptch1+/−;p53−/− mice and propagated in the absence or presence of the Smo antagonist NVP-LDE225 (29). We found Arhgap36 to be the most up-regulated gene in NVP-LDE225–resistant medulloblastomas induced by long-term drug treatment in comparison with allografts treated briefly with the Smo inhibitor or vehicle alone (Fig. 4A).
Fig. 4.

ARHGAP36 up-regulation in medulloblastoma and a model for Arhgap36-dependent Hh pathway regulation. (A) Arhgap36 transcript levels in NVP-LDE225–sensitive and resistant medulloblastoma allografts derived from Ptch+/−;p53−/− mice (data from ref. 29). (B) ARHGAP36 transcript levels in human medulloblastomas (groups 1–4) and normal cerebellar tissue, as determined by microarray analysis. (C) Schematic representation of the five predicted ARHGAP36 isoforms with the N-terminal, Rho GAP, and C-terminal domains labeled. (D) Alignments of the RNA-seq read coverage for three medulloblastoma samples, the ARHGAP36 genomic locus (Genome Reference Consortium GRCh37/hg19), and exon-intron structures for the predicted ARHGAP36 isoforms (UniprotKB/Swiss-Prot Q6ZRI8KB designations). Reads for all predicted ARHGAP36 exons except for the first were observed, indicating that the tumors were only capable of generating isoforms 2, 3, and 5. (E) Hh pathway activities in NIH-3T3 cells cotransfected with Gli-dependent firefly luciferase and SV40-driven Renilla luciferase reporters and either EGFP or individual ARHGAP36 isoforms with C-terminal V5 tags. The cells were then cultured in the absence or presence of ShhN for 30 h. Data are the average ± SEM, n = 3, and the double asterisks indicate P < 0.01 (ARHGAP36 isoforms versus EGFP for each cell culture condition). (F) Hh pathway activities in murine cerebellar GNPs retrovirally transduced with mCherry, Arhgap36, or ARHGAP36 (isoform 2) and treated with 3 µM cyclopamine for 48 h, as measured by endogenous Gli1 expression. Data are the average ± SEM, n = 6, and the double asterisks indicate P < 0.01 (ARHGAP36 isoforms versus EGFP for each cell culture condition). (G) Schematic representation of noncanonical Hh pathway activation by Arhgap36 and the inhibition of canonical Hh signaling by Arhgap36-N. Statistical analyses: Single and double asterisks indicate P < 0.05 and P < 0.01, respectively (E, ARHGAP36 isoforms versus EGFP for each cell culture condition; F, Arhgap36 or ARHGAP36 isoform 2 versus mCherry).
We next compiled transcriptional profiles of 436 medulloblastoma and 18 normal cerebellum samples, using a microarray series generated through the PedBrain Tumor Project of the International Cancer Genome Consortium and published microarray datasets (30–33). The medulloblastoma samples were segregated according to molecular subtypes as described (34, 35), which includes tumors associated with WNT pathway activation (group 1), SHH pathway activation (group 2), or more complex etiologies (groups 3 and 4). Similar to the murine medulloblastoma allografts that are NVP-LDE225–sensitive, ARHGAP36 was not highly transcribed in the group 2 tumors, which frequently carry loss-of-function PTCH1 mutations and can respond to SMO antagonists (36). However, ARHGAP36 expression was found to be up-regulated several hundredfold in a subset of group 3 and 4 medulloblastomas (Fig. 4B).
ARHGAP36 transcript levels in group 3 and group 4 medulloblastomas did not correlate with those of PTCH1 and GLI1 (SI Appendix, Fig. S16), suggesting that ARHGAP36 does not drive SHH target gene expression in the tumor bulk by the time of diagnosis and resection. However, ARHGAP36 forms expressed in these tumors (isoforms 2, 3, or 5) are potent inducers of Gli-dependent transcription in NIH-3T3 cells (Fig. 4 C–E and SI Appendix, Figs. S17–S19). As with murine Arhgap36, the active ARHGAP36 isoforms localized to the plasma membrane and intracellular vesicles in these cells, with the most N-terminally truncated form (isoform 3) exhibiting detectable levels in the primary cilium (SI Appendix, Fig. S11B). Both murine Arhgap36 and its human ortholog were also able to activate Gli1 expression in murine granule neuron precursors (GNPs) (Fig. 4F), which contribute to cerebellar development in Hh pathway-dependent manner and are believed to be cells of origin for medulloblastoma (37–39).
Discussion
By implementing a genome-scale, FACS-based screen of mammalian ORFs, we have identified Arhgap36 as a potent Hh pathway agonist. Arhgap36 overexpression induces changes in Gli function that have been associated with Hh signaling, such as reduced Gli3R formation, Gli3FL phosphorylation, increased ciliary Gli2 levels, and Gli1 up-regulation. Arhgap36 activity is independent of Smo, and this Rho GAP family member can functionally and biochemically interact with Sufu. Taken together, our findings uncover a new mechanism of Gli regulation and support a model in which Arhgap36 promotes noncanonical Gli activation by inhibiting Sufu function (Fig. 4G). These signaling events may occur within the primary cilium, because the subcellular distributions of Arhgap36, Sufu, and Gli converge at this subcellular compartment and Arhgap36-induced Gli activation requires Kif3a and Ift88 function. Arhgap36 function can also act in cooperation with ShhN or SAG to drive Hh target gene expression.
Rho GAP domain-containing proteins typically stimulate guanine nucleotide hydrolysis to generate inactive, GDP-bound forms of Rho GTPase (40), and Arhgap36 could similarly inhibit a Rho signaling protein that promotes Sufu activity. However, Arhgap36 lacks the “arginine finger” motif that stimulates GTPase hydrolysis (25), and GAP domain mutants can retain Gli-inducing activity. Arhgap36 may therefore act through a noncatalytic mechanism, as has been observed with the p85 subunit of phosphatidylinositol 3-kinase, the phosphatidylinositol-4,5-bisphosphate 5-phosphatase OCRL, and n-chimaerin (41–43). In addition, the neural-specific function of Arhgap36 in zebrafish embryos likely reflects a requirement for other signaling proteins. Arhgap36 might inhibit Sufu by recruiting accessory proteins, or Arhgap36 itself may be regulated by tissue-specific factors.
Finally, our results implicate ARHGAP36 in the onset and/or progression of medulloblastoma. ARHGAP36-induced GLI activation may be a mechanism by which group 2 medulloblastomas acquire resistance to Smo antagonists, in addition to SMO mutations and chromosomal GLI2 amplicons (29, 36, 44). Furthermore, the up-regulation of ARHGAP36 in group 3 and 4 tumors alludes to a more complex relationship between GLI activity and medulloblastoma biology than has been discerned through the bulk transcriptional profiling of advanced-stage tumors. Our studies confirm that the ARHGAP36 isoforms expressed in these cancers are potent Gli activators in fibroblasts and cerebellar GNPs, and our zebrafish studies demonstrate the ability of Arhgap36 to drive Hh target gene expression in neural progenitors. These observations raise the possibility that GLI activation in GNPs or other neural progenitors is sufficient for medulloblastoma initiation, with other signaling mechanisms subsequently maintaining the tumor in certain cases. Accordingly, previous studies have shown that late-stage group 3 and 4 medulloblastomas lack primary cilia (45), which could account for an eventual loss of ARHGAP36-induced Hh pathway activation in these cancers. The segregation of ARHGAP36-expressing medulloblastomas from the group 2 subtype further suggests that this Rho GAP family member could actuate other signaling pathways that influence disease progression.
Tumor cell lineage studies will be necessary to test these ideas and elaborate the mechanistic links between ARHGAP36 up-regulation, GLI activation, and oncogenesis. Studies of ARHGAP36 function and its binding proteins may also reveal new SHH pathway regulators, including potential targets for future anticancer therapies.
Materials and Methods
Reagents and procedures used in this report are detailed in SI Appendix, SI Materials and Methods.
Cell Lines.
Shh-EGFP (17, 18) and Shh-LIGHT2 cells (46) have been described. Smo−/− MEFs were provided by P. Beachy (Stanford University, Stanford, CA), and Sufu−/− MEFs were provided by R. Toftgård (Karolinska Institute, Stockholm). Gli2−/− and Gli3−/− MEFs were provided by W. Bushman (University of Wisconsin, Madison, WI). Kif3a−/− and Ift88−/− MEFs were provided by J. Reiter (University of California, San Francisco). NIH-3T3, C3H10T1/2, and HEK-293T cells were purchased from the American Type Culture Collection.
Constructs and Reagents.
All PCR products were generated with Phusion polymerase (New England Biolabs) by using the primers and templates listed in SI Appendix, Table S2, and all plasmids were sequence-verified. siRNA reagents and sources are listed in SI Appendix, Table S3. Antibody sources and working dilutions are listed in SI Appendix, Table S4. Morpholino oligonucleotide sequences are listed in SI Appendix, Table S5. SAG was synthesized as described (20), and cyclopamine was provided by Infinity Pharmaceuticals.
Retroviral ORFeome Sublibraries.
The human ORFeome collection (v5.1) in the pDONR233 backbone was transferred into pBMN-3xFLAG-IRES-mCherry-DEST by using LR Clonase II (Invitrogen). To produce pDNR223 input pools for the LR recombination reactions, 96-well plates containing sublibraries of the human ORFeome collection were grown in 96-well blocks containing Superior Broth (SB; Athena) with 25 µg/mL spectinomycin at 37 °C overnight, and the contents of each block were pooled. DNA was isolated from 10 mL of each pool by using Qiagen Miniprep kits. Retroviral medium was then generated by cotransfecting HEK-293T cells with the pBMN-3xFLAG-IRES-mCherry-DEST vector containing a human ORFeome cDNA sublibrary and pCL-ECO. Retroviral supernatant were collected from transfected cells and either used immediately or stored at −80 °C.
Medulloblastoma Microarray and RNA-seq Analyses.
Transcriptional profiling data from human medulloblastomas was compiled from previously published series (30–33) and a new series generated at the German Cancer Research Center (Gene Expression Omnibus accession nos. GSE37418, GSE49243, and GSE10327), for a total of 436 medulloblastoma and 18 normal cerebellum samples. This merged dataset was restricted to gene expression data generated on Affymetrix U133 Plus 2.0 arrays. RNA sequencing data for ARHGAP36 was provided by the PedBrain Tumor Project/International Cancer Genome Consortium (P. Lichter and S. Pfister; European Genome-Phenome Archive accession no. EGAS00001000771). Fresh frozen medulloblastoma and normal cerebellum samples used in these transcriptional profiling and RNA-seq studies were obtained after institutional review board approval and appropriate patient/family consent through the PedBrain Tumor Project/International Cancer Genome Consortium (www.pedbraintumor.org).
Transcriptional profiling data for murine tumors was obtained from a published series (29).
Supplementary Material
Acknowledgments
We thank P. Beachy, W. Bushman, T. Caspary, D. Esposito, J. Hartley, A. Liu, G. Nolan, V. Pasque, J. Reiter, R. Rohatgi, M. Scott, R. Toftgård, and R. Tsien for reagents; P. Lichter for early access to data from the International Cancer Genome Consortium PedBrain Tumor Project; D. Vaka for assistance with bioinformatics work; M. Clutter, M. Galic, T. Howes, and K. Schulz for helpful discussions; and M. Clutter and the Stanford Shared FACS Facility staff (M. Bigos, C. Crumpton, O. Herman, and J. Van Dyke) for technical assistance. This work was supported by National Institutes of Health (NIH)/National Cancer Institute Grants R01 CA136574 (to J.K.C.) and U01 CA172687 (to Y.-J.C.), NIH Director’s Pioneer Award DP1 HD075622 (to J.K.C.), the St. Baldrick’s Foundation (Y.-J.C.), the Stanford Center for Children’s Brain Tumors (Y.-J.C.), an Alex’s Lemonade Stand Foundation Young Investigator Award (to J.N.), a Beirne Faculty Scholar Endowment (to Y.-J.C.), German Cancer Aid Grants 109252 and 108456 (to S.M.P.), and the Federal Ministry of Education and Research through International Cancer Gene Consortium PedBrain and National Genome Research Network-Plus Grant 01GS0883 (to S.M.P.).
Footnotes
The authors declare no conflict of interest.
*This Direct Submission article had a prearranged editor.
Data deposition: The data reported in this paper have been deposited in the Gene Expression Omnibus (GEO) database, www.ncbi.nlm.nih.gov/geo (accession nos. GSE37418, GSE49243, and GSE10327). Short-read sequencing data have been deposited at the European Genome-Phenome Archive (EGA), www.ebi.ac.uk/ega/ (accession no. EGAS00001000771).
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1322362111/-/DCSupplemental.
References
- 1.Hui CC, Angers S. Gli proteins in development and disease. Annu Rev Cell Dev Biol. 2011;27:513–537. doi: 10.1146/annurev-cellbio-092910-154048. [DOI] [PubMed] [Google Scholar]
- 2.Zacharias WJ, et al. Hedgehog signaling controls homeostasis of adult intestinal smooth muscle. Dev Biol. 2011;355(1):152–162. doi: 10.1016/j.ydbio.2011.04.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Teglund S, Toftgård R. Hedgehog beyond medulloblastoma and basal cell carcinoma. Biochim Biophys Acta. 2010;1805(2):181–208. doi: 10.1016/j.bbcan.2010.01.003. [DOI] [PubMed] [Google Scholar]
- 4.Tukachinsky H, Lopez LV, Salic A. A mechanism for vertebrate Hedgehog signaling: Recruitment to cilia and dissociation of SuFu-Gli protein complexes. J Cell Biol. 2010;191(2):415–428. doi: 10.1083/jcb.201004108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Humke EW, Dorn KV, Milenkovic L, Scott MP, Rohatgi R. The output of Hedgehog signaling is controlled by the dynamic association between Suppressor of Fused and the Gli proteins. Genes Dev. 2010;24(7):670–682. doi: 10.1101/gad.1902910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Haycraft CJ, et al. Gli2 and Gli3 localize to cilia and require the intraflagellar transport protein polaris for processing and function. PLoS Genet. 2005;1(4):e53. doi: 10.1371/journal.pgen.0010053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Liu A, Wang B, Niswander LA. Mouse intraflagellar transport proteins regulate both the activator and repressor functions of Gli transcription factors. Development. 2005;132(13):3103–3111. doi: 10.1242/dev.01894. [DOI] [PubMed] [Google Scholar]
- 8.Chen MH, et al. Cilium-independent regulation of Gli protein function by Sufu in Hedgehog signaling is evolutionarily conserved. Genes Dev. 2009;23(16):1910–1928. doi: 10.1101/gad.1794109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wang B, Fallon JF, Beachy PA. Hedgehog-regulated processing of Gli3 produces an anterior/posterior repressor gradient in the developing vertebrate limb. Cell. 2000;100(4):423–434. doi: 10.1016/s0092-8674(00)80678-9. [DOI] [PubMed] [Google Scholar]
- 10.Corbit KC, et al. Vertebrate Smoothened functions at the primary cilium. Nature. 2005;437(7061):1018–1021. doi: 10.1038/nature04117. [DOI] [PubMed] [Google Scholar]
- 11.Rohatgi R, Milenkovic L, Scott MP. Patched1 regulates hedgehog signaling at the primary cilium. Science. 2007;317(5836):372–376. doi: 10.1126/science.1139740. [DOI] [PubMed] [Google Scholar]
- 12.Wang C, Pan Y, Wang B. Suppressor of fused and Spop regulate the stability, processing and function of Gli2 and Gli3 full-length activators but not their repressors. Development. 2010;137(12):2001–2009. doi: 10.1242/dev.052126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pan Y, Bai CB, Joyner AL, Wang B. Sonic hedgehog signaling regulates Gli2 transcriptional activity by suppressing its processing and degradation. Mol Cell Biol. 2006;26(9):3365–3377. doi: 10.1128/MCB.26.9.3365-3377.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kim J, Kato M, Beachy PA. Gli2 trafficking links Hedgehog-dependent activation of Smoothened in the primary cilium to transcriptional activation in the nucleus. Proc Natl Acad Sci USA. 2009;106(51):21666–21671. doi: 10.1073/pnas.0912180106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wilson CW, Chuang PT. Mechanism and evolution of cytosolic Hedgehog signal transduction. Development. 2010;137(13):2079–2094. doi: 10.1242/dev.045021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Brasch MA, Hartley JL, Vidal M. ORFeome cloning and systems biology: Standardized mass production of the parts from the parts-list. Genome Res. 2004;14(10B):2001–2009. doi: 10.1101/gr.2769804. [DOI] [PubMed] [Google Scholar]
- 17.Cupido T, et al. The imidazopyridine derivative JK184 reveals dual roles for microtubules in Hedgehog signaling. Angew Chem Int Ed Engl. 2009;48(13):2321–2324. doi: 10.1002/anie.200805666. [DOI] [PubMed] [Google Scholar]
- 18.Hyman JM, et al. Small-molecule inhibitors reveal multiple strategies for Hedgehog pathway blockade. Proc Natl Acad Sci USA. 2009;106(33):14132–14137. doi: 10.1073/pnas.0907134106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kinto N, et al. Fibroblasts expressing Sonic hedgehog induce osteoblast differentiation and ectopic bone formation. FEBS Lett. 1997;404(2-3):319–323. doi: 10.1016/s0014-5793(97)00014-8. [DOI] [PubMed] [Google Scholar]
- 20.Chen JK, Taipale J, Young KE, Maiti T, Beachy PA. Small molecule modulation of Smoothened activity. Proc Natl Acad Sci USA. 2002;99(22):14071–14076. doi: 10.1073/pnas.182542899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chen JK, Taipale J, Cooper MK, Beachy PA. Inhibition of Hedgehog signaling by direct binding of cyclopamine to Smoothened. Genes Dev. 2002;16(21):2743–2748. doi: 10.1101/gad.1025302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Blacque OE, Cevik S, Kaplan OI. Intraflagellar transport: From molecular characterisation to mechanism. Front Biosci. 2008;13:2633–2652. doi: 10.2741/2871. [DOI] [PubMed] [Google Scholar]
- 23.Marszalek JR, Ruiz-Lozano P, Roberts E, Chien KR, Goldstein LS. Situs inversus and embryonic ciliary morphogenesis defects in mouse mutants lacking the KIF3A subunit of kinesin-II. Proc Natl Acad Sci USA. 1999;96(9):5043–5048. doi: 10.1073/pnas.96.9.5043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pazour GJ, et al. Chlamydomonas IFT88 and its mouse homologue, polycystic kidney disease gene tg737, are required for assembly of cilia and flagella. J Cell Biol. 2000;151(3):709–718. doi: 10.1083/jcb.151.3.709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rittinger K, et al. Crystal structure of a small G protein in complex with the GTPase-activating protein rhoGAP. Nature. 1997;388(6643):693–697. doi: 10.1038/41805. [DOI] [PubMed] [Google Scholar]
- 26.Jelen F, et al. Dissecting the thermodynamics of GAP-RhoA interactions. J Struct Biol. 2009;165(1):10–18. doi: 10.1016/j.jsb.2008.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dahmane N, Lee J, Robins P, Heller P, Ruiz i Altaba A. Activation of the transcription factor Gli1 and the Sonic hedgehog signalling pathway in skin tumours. Nature. 1997;389(6653):876–881. doi: 10.1038/39918. [DOI] [PubMed] [Google Scholar]
- 28.Regl G, et al. Human GLI2 and GLI1 are part of a positive feedback mechanism in Basal Cell Carcinoma. Oncogene. 2002;21(36):5529–5539. doi: 10.1038/sj.onc.1205748. [DOI] [PubMed] [Google Scholar]
- 29.Buonamici S, et al. Interfering with resistance to smoothened antagonists by inhibition of the PI3K pathway in medulloblastoma. Sci Transl Med. 2010;2(51):51ra70. doi: 10.1126/scitranslmed.3001599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Roth RB, et al. Gene expression analyses reveal molecular relationships among 20 regions of the human CNS. Neurogenetics. 2006;7(2):67–80. doi: 10.1007/s10048-006-0032-6. [DOI] [PubMed] [Google Scholar]
- 31.Kool M, et al. Integrated genomics identifies five medulloblastoma subtypes with distinct genetic profiles, pathway signatures and clinicopathological features. PLoS ONE. 2008;3(8):e3088. doi: 10.1371/journal.pone.0003088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Fattet S, et al. Beta-catenin status in paediatric medulloblastomas: Correlation of immunohistochemical expression with mutational status, genetic profiles, and clinical characteristics. J Pathol. 2009;218(1):86–94. doi: 10.1002/path.2514. [DOI] [PubMed] [Google Scholar]
- 33.Robinson G, et al. Novel mutations target distinct subgroups of medulloblastoma. Nature. 2012;488(7409):43–48. doi: 10.1038/nature11213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cho YJ, et al. Integrative genomic analysis of medulloblastoma identifies a molecular subgroup that drives poor clinical outcome. J Clin Oncol. 2011;29(11):1424–1430. doi: 10.1200/JCO.2010.28.5148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Northcott PA, et al. Medulloblastoma comprises four distinct molecular variants. J Clin Oncol. 2011;29(11):1408–1414. doi: 10.1200/JCO.2009.27.4324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rudin CM, et al. Treatment of medulloblastoma with hedgehog pathway inhibitor GDC-0449. N Engl J Med. 2009;361(12):1173–1178. doi: 10.1056/NEJMoa0902903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wechsler-Reya RJ, Scott MP. Control of neuronal precursor proliferation in the cerebellum by Sonic Hedgehog. Neuron. 1999;22(1):103–114. doi: 10.1016/s0896-6273(00)80682-0. [DOI] [PubMed] [Google Scholar]
- 38.Wallace VA. Purkinje-cell-derived Sonic hedgehog regulates granule neuron precursor cell proliferation in the developing mouse cerebellum. Curr Biol. 1999;9(8):445–448. doi: 10.1016/s0960-9822(99)80195-x. [DOI] [PubMed] [Google Scholar]
- 39.Dahmane N, Ruiz i Altaba A. Sonic hedgehog regulates the growth and patterning of the cerebellum. Development. 1999;126(14):3089–3100. doi: 10.1242/dev.126.14.3089. [DOI] [PubMed] [Google Scholar]
- 40.Moon SY, Zheng Y. Rho GTPase-activating proteins in cell regulation. Trends Cell Biol. 2003;13(1):13–22. doi: 10.1016/s0962-8924(02)00004-1. [DOI] [PubMed] [Google Scholar]
- 41.Zheng Y, Bagrodia S, Cerione RA. Activation of phosphoinositide 3-kinase activity by Cdc42Hs binding to p85. J Biol Chem. 1994;269(29):18727–18730. [PubMed] [Google Scholar]
- 42.Dambournet D, et al. Rab35 GTPase and OCRL phosphatase remodel lipids and F-actin for successful cytokinesis. Nat Cell Biol. 2011;13(8):981–988. doi: 10.1038/ncb2279. [DOI] [PubMed] [Google Scholar]
- 43.Kozma R, Ahmed S, Best A, Lim L. The GTPase-activating protein n-chimaerin cooperates with Rac1 and Cdc42Hs to induce the formation of lamellipodia and filopodia. Mol Cell Biol. 1996;16(9):5069–5080. doi: 10.1128/mcb.16.9.5069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Dijkgraaf GJ, et al. Small molecule inhibition of GDC-0449 refractory smoothened mutants and downstream mechanisms of drug resistance. Cancer Res. 2011;71(2):435–444. doi: 10.1158/0008-5472.CAN-10-2876. [DOI] [PubMed] [Google Scholar]
- 45.Han YG, et al. Dual and opposing roles of primary cilia in medulloblastoma development. Nat Med. 2009;15(9):1062–1065. doi: 10.1038/nm.2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Taipale J, et al. Effects of oncogenic mutations in Smoothened and Patched can be reversed by cyclopamine. Nature. 2000;406(6799):1005–1009. doi: 10.1038/35023008. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.