

Abstract
The extensive links between proteotoxic stress, protein aggregation and pathologies ranging from aging to neurodegeneration underscore the importance of understanding how cells manage protein misfolding. Using live-cell imaging, we here determine the fate of stress-induced misfolded proteins from their initial appearance until their elimination. Upon denaturation, misfolded proteins are sequestered from the bulk cytoplasm into dynamic ER-associated puncta that move and coalesce into larger structures in an energy-dependent but cytoskeleton-independent manner. These puncta, which we name Q-bodies, concentrate different misfolded and stress-denatured proteins en-route to degradation, but do not contain amyloid aggregates, which localize instead to the IPOD. Q-body formation and clearance depends on an intact cortical ER and a complex chaperone network that is affected by rapamycin and impaired during chronological aging. Importantly, Q-body formation enhances cellular fitness during stress. We conclude that spatial sequestration of misfolded proteins in Q-bodies is an early quality control strategy occurring synchronously with degradation to clear the cytoplasm from potentially toxic species.
Introduction
Misfolded proteins challenge the integrity of the cellular proteome and compromise cell viability1 and compromise cell viability2. Their accumulation in insoluble protein aggregates is linked to neurodegenerative amyloid disorders, including Alzheimer's and Huntington's Disease3. Accordingly, cells evolved elaborating Quality Control (QC) machineries that eliminate misfolded proteins and maintain protein homeostasis4, 5. Molecular chaperones are central to this process, as they recognize non-native conformations and triage polypeptides for either refolding or degradation through the ubiquitin-proteasome pathway (UPS) or autophagy4, 5. In addition, cells can actively sequester misfolded proteins in defined QC compartments6-8. Upon proteasome impairment, misfolded proteins partition into spatially and functionally distinct compartments6, 7: the JUNQ (juxtanuclear quality-control compartment), which sequesters misfolded polypeptides in a detergent soluble state; and the IPOD (insoluble protein deposit), which sequesters terminally aggregated polypeptides9, 10. Amyloid proteins, such as poly Q-expanded Huntingtin, are partitioned primarily to the IPOD, even in the absence of proteasome inhibition5. Protein sequestration into these inclusions is proposed as an alternative cellular defense when QC machineries fail11-13.
QC is currently viewed as a two-tiered system, whereby misfolded proteins are first either refolded or degraded through the action of chaperones and the UPS, and then, when triage fails, transported in a cytoskeleton-dependent manner to cellular QC inclusions14. By following, in real time, QC substrates in the absence of proteasome inhibition, we find instead that misfolded proteins are rapidly concentrated in many dynamic inclusions, which we term Q-bodies, even as they are degraded by the proteasome. Q-body formation and movement is independent of the cytoskeleton, but requires the cortical endoplasmic reticulum (ER) and the concerted action of a chaperone network. We propose that sequestration of misfolded proteins is an integral and early aspect of cellular QC that does not necessarily ensue from proteostasis impairment or cellular dysfunction. Our data have implications for understanding the genesis of terminal protein inclusions characterizing a wide number of human pathologies1.
Results
Sequestration of misfolded proteins into dynamic puncta is an early quality control response
To understand QC in unperturbed yeast cells, we used live-cell microscopy to follow the fate of a thermolabile allele of UBC9 (Ubc9-2 herein Ubc9ts) that is folded at 28°C (Ubc9ts-N; Fig. 1a) but denatures above 33°C (Ubc9ts-Den; Fig. 1a, b and Fig. S1a)6. Ubc9ts-GFP expressed from a galactose-inducible promoter at 28°C generated folded Ubc9ts (Ubc9ts-N), localized diffusely throughout the cell. We shut-off Ubc9ts expression by addition of glucose and shifted the cells to 37°C, causing the pre-existing Ubc9ts to misfold and be degraded with a half-life of approximately 15-20 min (Fig. 1c and d). The UPS is the major degradation pathway for misfolded Ubc9ts since inhibition of autophagy by deletion of Atg815 produced only a minor stabilization of Ubc9ts (Fig. 1e and Fig. S1b). As reported6, proteasome inhibition leads to Ubc9ts accumulation into the JUNQ and IPOD (Fig. 1c).
Figure 1. Misfolded proteins are sequestered in Q-bodies upon heat stress.

(a) Ubc9ts-GFP fate upon heat stress. (b) Ubc9ts-GFP expressed in WT cells at 28°C was shifted for 15 min at 37°C. GFP (upper panel) and DIC (lower panel) are shown. Scale bars equal 1.5 μm. (c) pdr5Δ cells expressing Ubc9ts-GFP were grown at 28°C in galactose medium and shifted to 37°C in glucose medium with (+) or without (-) 100 μM MG132. Images show Ubc9ts-GFP at the shift (t=0) and after 60 min (t=60). Dotted lines highlight cell outlines. Scale bar equals 1μm. (d) As in c, Ubc9ts-GFP was immunoblotted with anti-GFP antibodies and quantified as the relative ratio to the initial amount for each condition. Results represent mean and Standard Deviation (SD) of three independent experiments. (e) pdr5Δ and pdr5Δatg8Δ cells expressing Ubc9ts-GFP were grown as in c. Ubc9ts-GFP was immunoblotted with anti-GFP antibodies (Fig. S1b) and quantified relative to the initial amount for each condition. Results represent mean and SD of three independent experiments. (f) WT cells expressing Ubc9ts-GFP at 28°C in galactose medium were shifted at 37°C in glucose medium. Time series images show GFP signal in a cell 1 min (1′) to 30 min (30′) after the shift (video S1). Scale bar equals1 μm. (g) Inset in f, highlights the coalescence of Ubc9ts-GFP Q-bodies (arrows), from 10 to 13 min after the shift. (h) Number of puncta in the medial focal plane over time in WT (main panel; puncta assessed from a total population of n=63 cells over three independent experiments, 1 field counted per experiment) or in pdr5Δ cells (puncta assessed from a total population of n=36 cells over three independent experiments, 1 field counted per experiment) with or without MG132 (secondary panel). Results represent mean of puncta for n cells and SD. (**) p<0.005, compared to untreated cells for the same indicated time. (i) Trajectories of three Ubc9-GFP Q-bodies in WT cells. Positions of particles are represented from each frame of a movie (1 frame/15 sec) at the medial focal plane. Consecutive positions are connected by lines: black, before coalescence; grey, after coalescence. Green and red dots indicate the initial and final positions, respectively. Initial, final and coalescing times are indicated, and average speed (+/- SD) over the entire trajectory.
The dynamics and fate of misfolded Ubc9ts-GFP during the process of degradation was visualized in live cells by epifluorescence microscopy following expression shut-off (Fig. 1f, video S1). Shortly after shifting to 37°C, an increasing number of dim scattered Ubc9ts-GFP puncta appeared throughout the cytosol. Within 5 minutes, 5 to 10 puncta were clearly observed in the medial focal plane (Fig. 1f). Between 5 and 30 minutes, the diffuse signal disappeared as the dim Ubc9ts-GFP puncta coalesced into fewer and brighter structures that continued to merge even as Ubc9ts was cleared from the cell (Fig. 1f, g and video S1). Wild-type Ubc9-GFP remained diffuse and soluble at 37°C and did not form puncta (Fig. S1c), confirming these inclusions arise from Ubc9ts misfolding. Quantification of the average number of puncta in a medial focal plane at 5 min intervals during a 60 min movie indicated a consistent decrease in puncta number from 5 to 30 min (Fig. 1h). All puncta disappeared within an hour. Similar results were obtained when Ubc9ts was misfolded at 33°C, indicating that this pathway is not specific to higher temperature stress (Fig. S1a). Upon proteasome inhibition, the formation and early coalescence of Ubc9ts-GFP puncta occur similarly to untreated cells, albeit with a slightly higher number of puncta (Fig. 1h), which continue coalescing into 1-3 large inclusions6 (Fig. 1c, d). These results suggest that the JUNQ, the IPOD, and other inclusions observed upon proteasome inhibition7 may result from the accumulation of coalesced puncta over time as their clearance is impaired.
We conclude that misfolded proteins do not remain diffusely distributed in the cytosol, but are also not deposited into a static pre-existing compartment. Instead, they are collected and processed through a dynamic cellular pathway into multiple punctate structures throughout the cell which by coalescence, mature into larger inclusions. By analogy to the dynamic P-body-mediated RNA QC16, we propose to name these protein QC structures Q-bodies.
Q-body dynamics are energy dependent but cytoskeleton independent
Tracking the trajectory of Q-bodies in vivo did not reveal any directional movement suggesting they do not move on defined tracks (Fig. 1i). The average speed of puncta was 5 to 15 nm/s, i.e. about 10 times slower than endocytic particles moving on the cytoskeleton17 and did not change appreciably before (black trace) and after (gray trace) coalescence with another puncta (Fig. 1i). This led us to examine the role of the cytoskeleton on Q-body dynamics. The actin and microtubule cytoskeletons were disrupted with 200 μM of latrunculin A (LatA) and 15 μm/mL nocodazole (Noc), respectively, followed by imaging of Ubc9ts-GFP (Fig. 2a, b and c top panels). A 10 min LatA treatment prior to induction of Ubc9ts misfolding completely disrupted the actin cytoskeleton, as confirmed by imaging the actin binding protein Abp1-GFP (Fig. 2a). Nonetheless, Q-bodies were still formed, processed and degraded similar to WT (Fig. 2a, video S2). Tracking analysis indicated that LatA treatment did not measurably affect the average speed or directionality of Q-bodies over their lifetime (Fig. 2a, lower panel). Of note, prolonged incubation (2 hours) with LatA did perturb Q-body formation and movement (Fig. 2b) suggesting that prolonged disruption of the actin cytoskeleton impairs protein homeostasis. The tubulin cytoskeleton is also dispensable for Q-body formation and dynamics. 10 min treatment with Noc disrupted microtubules, but did not affect Q-body dynamics and clearance (Fig. 2c). We conclude that neither Q-body formation nor dynamics requires a functional actin and tubulin cytoskeleton.
Figure 2. Energy dependence but cytoskeleton independence of Q-body dynamics.

(a) WT cells expressing Ubc9ts-GFP or Abp1-GFP (lower panel) were grown at 28°C in galactose medium and treated with (right panel) or without (left panel) 200 μM Latrunculin A (LatA) for 10 min prior to a shift to 37°C in glucose medium. 5 min series of images show Ubc9ts-GFP or Abp1-GFP signal (video S2). Scale bars equal 1 μm. Trajectories of three Ubc9-GFP puncta in WT cells with or without LatA are represented (lower panel) as described in Fig. 1i. (b) WT cells expressing Ubc9ts-GFP and Abp1-GFP were grown as in a but treated with (right panel) or without (left panel) LatA for 2h prior to the shift. Scale bars equal 1 μm. (c) WT cells expressing Ubc9ts-GFP or Tub1-GFP were grown as in a but treated with (lower panel) or without (upper panel) 15 μg/ml Nocodazole (Noc) for 10 min prior to the shift. 5 min series of images show Ubc9ts-GFP or Tub1-GFP signal. Scale bars equal 1 μm. (d) WT cells expressing Ubc9ts-GFP were grown as in a but treated for 30 min with (right panel) or without (left panel) 10 mM azide and deoxyglucose prior to the shift. 5 min series of images show Ubc9ts-GFP signal (video S3). Scale bars equal 1 μm. Trajectories of three Ubc9-GFP puncta in cells with and without azide and deoxyglucose treatment are represented (lower panel) as described in Fig. 1i.
We next assessed whether Q-body formation and movement require energy. Intracellular ATP depletion with sodium azide and deoxyglucose for 30 min (Fig. 2d) did not abrogate Q-body formation, suggesting that this step is energy independent. However, Q-body dynamics, coalescence and clearance were highly perturbed (Fig. 2d, video S3). We conclude that that the concentration of misfolded protein into Q-bodies is energy independent, whereas their movement, coalescence and clearance require ATP.
Q-body formation and processing requires an intact cortical ER
We next sought to identify cellular structures associated with Q-bodies. Because the JUNQ colocalizes with the perinuclear ER6, Ubc9ts was co-expressed with the nuclear marker Npl3, or with perinuclear ER protein Hmg1 (Fig. 3a and b)18. Z-sections of deconvolved images revealed the distribution of Ubc9ts Q-bodies vis-à-vis the nucleus (Fig. 3a and b). In each cell analyzed, at least one puncta was juxtanuclear, while the others were distributed throughout the cell.
Figure 3. Q-body dynamics relies on an intact cortical ER.

(a) Cells expressing Npl3-RFP and Ubc9ts-GFP were imaged after 10 min at 37°C. Two-color deconvolved images show Npl3-RFP signal (nucleus) in red and Ubc9ts-GFP signal in green. Scale bar equals 1 μm. (b) Cells expressing Hmg1-GFP (perinuclear ER, green) and Ubc9ts-CHFP (red) were imaged as in a. Arrow indicates Q-body in proximity to the ER. Scale bar equals 1 μm. (c) Cells expressing Rtn1-GFP and Ubc9ts-CHFP were imaged after 10 min at 37°C. Two-color images of three Z-focal plans (0.2 μm intervals) show Rtn1-GFP signal (cortical ER) in green and Ubc9ts-CHFP signal in red (video S4). Arrows indicate Q-bodies in proximity to the cortical ER. Scale bar equals1 μm. (d) Inset from c representing a series of 0.2 μm Z-section. Scale bar equals 1 μm. (e) Ubc9ts-GFP was expressed in WT and rnt1Δrtn2Δyop1Δ cells at 28°C in galactose medium and shifted at 37°C in glucose medium. 5 min series of images show Ubc9ts-GFP signal over 30 min. Scale bars equal 1 μm. (f) As in e, cells were harvested at indicated time from the shift and Ubc9ts-GFP was immunoblotted with anti-GFP antibodies. Ubc9ts-GFP was quantified as the relative ratio to the initial amount from one experiment representative of four independent experiments. (g) Schematic of ER-associated Q-body processing of misfolded Ubc9ts (red star). Green represents the ER network throughout the cell.
No obvious colocalization of Q-bodies was observed with early endosomes (GFP-Snc1), late endosomes (GFP-Pep12), vacuole (MDY64), autophagic structures (CHFP-Atg8) and the spindle pole body (Spc42-GFP) (Fig. S2a, b). Given the vicinity of one inclusion with the perinuclear ER, we examined Q-body association with the cortical ER, extending as a tubular network throughout the cell19. Two-color Z-stacks of deconvolved images from cells expressing Ubc9ts-CHFP and the cortical ER marker Rtn1-GFP revealed a close proximity between Q-bodies and cortical ER tubules (Fig 3c, d, and video S4).
The role of the cortical ER in Q-body dynamics was assessed in rtn1Δrtn2Δyop1Δ cells, whose cortical ER morphology is disrupted20-22. A large Ubc9ts inclusion was already observed at the permissive temperature (Fig. 3e). At 37°C, a few additional dim puncta appeared and coalesced with the large inclusion (Fig. 3e, video S5). After 30 minutes at 37°C, when most misfolded Ubc9ts was cleared in WT cells, the large inclusion persisted in over 50% of rtn1Δrtn2Δyop1Δ cells, consistent with a delay in Ubc9ts degradation (Fig. 3f). Thus, the formation, dynamics and clearance of cytoplasmic Q-bodies rely on the integrity of a dynamic ER network (Fig. 3g).
The Hsp70-Hsp90 chaperone network mediates Q-body formation and clearance
The ATP-dependence of Q-body clearance resonates with previous data implicating the ATP-dependent Hsp70 and Hsp90 systems in misfolded protein QC (Table S1)23-27. Indeed, Hsp70, Hsp90 and the Hsp70 cofactor Hsp110/Sse1 are also required for degradation of misfolded Ubc9ts (Fig. 4b). At 37°C, the Hsp70 Ssa1 colocalized with all misfolded Ubc9ts Q-bodies (Fig. 4a). The role of Hsp70 in the Q-body pathway was examined using two Hsp70-deficient strains: ssa1Δssa2Δ, lacking the two major SSA isoforms, and ssa1ts ssa2Δssa3Δssa4Δ, containing a single temperature-sensitive SSA isoform and lacking the others28. Both strains yielded similar results (Fig. 4c and Fig. S3b). Even before the temperature shift, most cells contained a static bright cortical inclusion, indicating that Hsp70 contributes to conformational maintenance of Ubc9ts-GFP at the permissive temperature (Fig. 4c, video S6). After the shift, the number of puncta remained unchanged over 30 min: no coalescence of the faint additional puncta was detected and most of the misfolded protein was found in the large inclusion (Fig. 4c and d). Thus, Hsp70 is required for Q-body formation, maturation and clearance, consistent with biochemical experiments (Fig. 4b). Q-bodies still formed in Hsp90-deficient cells (hsc82Δhsp82ts)25, but their movement and coalescence were dramatically affected, associated with a strong defect in misfolded Ubc9ts-GFP degradation (Fig. 4b, e, f and video S7). Thus, Hsp90 also participates in Q-body dynamics and degradation, but acts after the initial formation of inclusions.
Figure 4. The maturation and degradation of Q-bodies rely on the Hsp70-Hsp90 system.
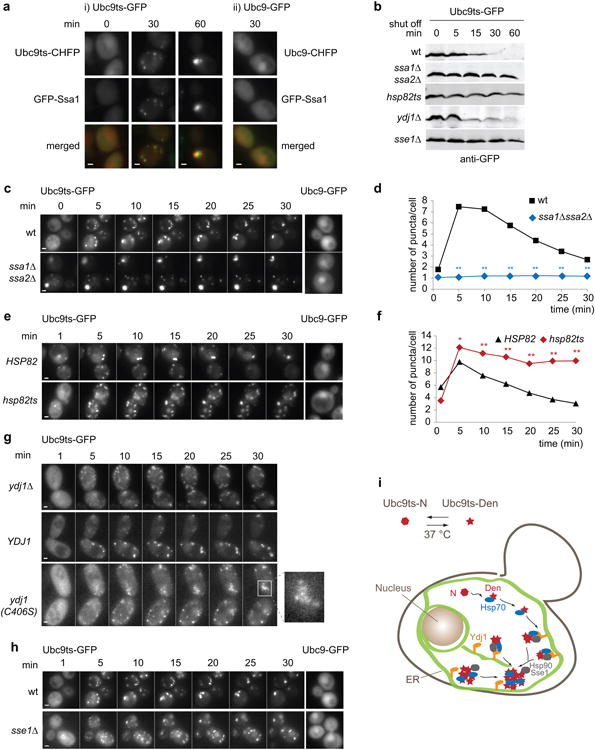
(a) WT cells co-expressing GFP-Ssa1 and Ubc9ts-CHFP, or GFP-Ssa1 and Ubc9-CHFP, were grown at 28°C in galactose medium and shifted to 37°C in glucose medium. Two-color images show GFP-Ssa1 signal in green and Ubc9ts-CHFP signal in red at 0, 30 and 60 min after the shift. Scale bars equal 1.5 μm. (b) Cells expressing Ubc9ts-GFP at 28°C in galactose medium were shifted at 37°C (t=0) in glucose medium. Cells were harvested at indicated times and Ubc9ts-GFP immunoblotted using anti-GFP antibodies. (c) Ubc9ts-GFP was expressed in WT and ssa1Δssa2Δ cells at 28°C in galactose medium and shifted at 37°C in glucose medium. 5 min series of images show Ubc9ts-GFP signal over 30 min (i). Cells expressing Ubc9-GFP were similarly prepared and imaged 15 min after the shift (ii). Scale bars represent 1 μm. (d) Average number of puncta per cell in WT (black squares) and ssa1Δssa2Δ (blue diamonds) cells over time. Puncta assessed from a total population of n=35 cells over three independent experiments (1 field counted per experiment).(**) p<0.005 compared to WT for the same indicated time. (e) WT and hsp82ts cells expressing Ubc9ts-GFP or Ubc9-GFP were prepared and imaged as in c. Scale bars represent 1 μm. (f) Average number of puncta per cell in WT (black triangles) and hsp82ts (red diamonds) cells over time. Puncta assessed from a total population of n=25 cells over two independent experiments (1 field counted per experiment).(*) p<0.05 (**) p<0.005 compared to WT for the same indicated time. (g) ydj1Δ strains expressing Ubc9ts-GFP alone, and in combination with YDJ1, ydj1(C406S) or empty vector was prepared and imaged as in c. Shown an inset of a ydj1(C406S)-expressing cell. Scale bars represent 1 μm. (h) WTorsse1Δ cells expressing Ubc9ts-GFP or Ubc9-GFP were prepared and imaged as in c. Scale bars represent 1 μm. (i) Role of the Hsp70-Hsp90-Hsp110 system in Q-bodies pathway for misfolded Ubc9ts (red star). Green represents the ER network throughout the cell.
The activity of Hsp70 and its cooperation with Hsp90 is regulated by J-domain proteins (Fig. S3a). Since Ydj1 is ER-associated through a farnesyl group29, we examined whether it helps to process ER-associated Q-bodies. ydj1Δ cells contained more diffuse misfolded Ubc9ts than WT cells and numerous dim puncta at the periphery (Fig. 4g). Cells expressing the farnesylation defective mutant ydj1(C406S)30 also displayed clusters of tiny cortical puncta that appeared unable to merge, suggesting a defect in Q-body coalescence (Fig. 4g and video S8). However, Ubc9ts was eventually degraded with similar kinetics as wild-type (Fig. 4g, video S8 and Fig. S3c), perhaps due to the high level of redundancy between J-domain proteins31. Indeed, disruption of both Ydj1 and another ER-anchored J-domain protein, Hlj1, in the hlj1Δ ydj1-151 strain32 strongly affected Q-body formation. Only faint Ubc9ts-GFP puncta were discernible in hlj1Δ ydj1-151 cells at 37°C (Fig. S3d). Thus, ER-anchored J-domain proteins, but perhaps also other homologues, participate in Q-body formation. The Hsp70 nucleotide exchange factor Sse1 also affected Q-body dynamics (Fig. 4h and video S6). sse1Δ cells exhibited a significant delay in Q-body clearance, associated with Ubc9ts degradation impairment (Fig. 4b and Fig. S3e). We conclude that a chaperone network involving Hsp70, Hsp90 and their co-chaperones mediates the active formation and maturation of Q-bodies, as well as their degradation (Fig. 3i and Table S1).
Balance between addition and dissolution activities controls Q-body dynamics
We next examined the role of the disagregase Hsp104 and the small HSPs implicated in prion formation33, 34 and aggregate management in proteasome-inhibited cells6, 7, 14. Live-cell imaging demonstrated that both Hsp104 and Hsp42 associate with Ubc9ts-CHFP Q-bodies formed upon heat stress (Fig. 5a-c; video S9). At 37°C, in absence of proteasome inhibition, Hsp104 and Hsp42 persist in the punctate Q-bodies structures even after Ubc9ts-CHFP degradation (Fig. 5a; video S9), suggesting that endogenous heat-denatured proteins are also concentrated and processed through the Q-body pathway. Accordingly, cells not expressing Ubc9ts, or expressing native Ubc9-GFP, also formed Hsp104-GFP and Hsp42-GFP containing puncta upon heat stress, which move and coalesce with similar dynamics as Ubc9ts Q-bodies (Fig. 5b; c). We conclude that Hsp42 and Hsp104 are components of endogenous Q-bodies, which form upon heat stress as a physiological response to misfolded proteins.
Figure 5. Balance between addition and dissolution activities controls Q-body dynamics.

(a) Cells expressing Hsp104-GFP and Ubc9ts-CHFP or Ubc9-GFP were grown at 28°C in galactose medium and shifted at 37°C in glucose medium. 5 min series of a two-color movie show Hsp104-GFP signal in green and Ubc9ts-CHFP signal in red over 60 min (i). Cells expressing Hsp104-GFP and Ubc9-CHFP were imaged 15 min after the shift (ii). Scale bar equals 1 μm. (b) As in a, but cells were imaged 10 min after the shift. Two-color deconvolved images show Hsp104-GFP signal in green and Ubc9ts-CHFP or Ubc9-CHFP signal in red. Scale bars represent 1 μm. (c) Cells expressing Hsp42-GFP (green) and Ubc9ts-CHFP or Ubc9-CHFP (red) were imaged as in b. Scale bars equal 1 μm. (d) WT, hsp104Δ, hsp42Δ, and hsp26Δ cells expressing Ubc9ts-GFP (i) or Ubc9-GFP (ii) were prepared as in a. 5 min series of images show Ubc9ts-GFP signal over 30 min. Ubc9-GFP signal is shown 15 min after the shift. Scale bars represent 1 μm. (e) Average number of puncta per cell in the WT (black squares), hsp104Δ (orange diamonds), hsp26Δ (gray circles) strains over time. Puncta assessed from a total population of n=35 cells over three independent experiment (1 field counted per experiment). (*) p<0.05 (**) p<0.005 compared to WT for the same indicated time. (f) Indicated strains expressing Ubc9ts-GFP were grown as in a and harvested at indicated times from the shift (t=0). Ubc9ts-GFP was immunoblotted using anti-GFP antibodies. (g) WT and hsp42Δhsp104Δ cells expressing Ubc9ts-GFP and Ubc9-GFP were prepared and imaged as in d. Scale bars represent 1 μm. (h) WT and the hsp42Δsse1Δ cells expressing Ubc9ts-GFP and Ubc9-GFP were prepared and imaged as in d. Scale bars represent 1 μm. (i) Dissolution-addition balance between Hsp104 and Hsp42: Q-body coalescence results from an Hsp104-mediated process of dissolution followed by Hsp42 stimulated re-addition of the released misfolded protein into existing inclusions.
Interestingly, untreated and proteasome-inhibited cells exhibited differences in their Hsp104 and Hsp42 distribution. In untreated cells, Hsp42-GFP colocalized with all the Ubc9ts containing puncta (Fig. 5c), while in proteasome-inhibited cells, Hsp42 was absent from some inclusions7. Hsp104 did not co-localize with perinuclear puncta without proteasome inhibitor (Fig. 5b). The lack of Hsp104 in perinuclear puncta was confirmed by two-color Z-stack deconvolution and analysis of DAPI-labeled fixed cells (Fig. S4). Possibly, proteasome inhibition alters inclusion dynamics and composition, as inclusions continue coalescing and persist much longer in the cell.
We evaluated how Hsp104 and the small HSPs Hsp42 and Hsp26 participate in Q-body processing. In hsp26Δ cells, the inclusion dynamics and Ubc9ts half-life were unaffected (Fig. 5d-f). In hsp104Δ cells, misfolded Ubc9ts-GFP progressed from dim puncta to the formation of medium-intensity puncta, similar to WT cells. These medium-intensity puncta did not coalesce into few brighter inclusions as in WT cells, indicating that the progression of the pathway is blocked by loss of Hsp104 (Fig. 5d and e, video S10). In hsp42Δ, only a handful of dim puncta formed after 10 min indicating a severe defect in Q-body formation (Fig. 5d, and video S10). Remarkably, Ubc9ts degradation was unaffected in both hsp104Δ and hsp42Δ strains (Fig. 5f). Thus, sequestration into larger inclusions is not essential for misfolded protein degradation.
The fact that puncta coalescence, but not degradation, requires a disaggregase such as Hsp104 is counter-intuitive; a simple model would predict that disaggregase deletion leads to formation of larger hard-to-degrade inclusions. We hypothesized that puncta coalescence may involve Hsp104-mediated dissolution of some puncta followed by re-addition of the released misfolded protein into existing inclusions (Fig. 5i). In this view, loss of puncta in hsp42Δ cells would not reflect an absolute requirement of Hsp42 for Q-body formation but rather a shift in the balance between Hsp42-stimulatedaddition and Hsp104-stimulateddissolution. Indeed Ubc9ts inclusions were observed in the double hsp42Δhsp104Δ cells (Fig. 5g, video S10), indicating that Hsp42 is not required for inclusion formation. However, puncta intensity in hsp42Δhsp104Δ was heterogeneous and their coalescence was affected, consistent with Hsp104 promoting Q-body maturation into large inclusions. Notably, Ubc9ts degradation was unaffected in hsp42Δhsp104Δ cells (Fig. 5f). Hsp104 is not the only chaperone promoting Q-body dissolution, since the hsp42Δsse1Δ mutant also forms inclusions (Fig. 5h), albeit with severely affected dynamics. Thus, a chaperone-mediated balance between addition and dissolution regulates Q-body formation and maturation (Fig. 5i).
Soluble misfolded proteins, but not amyloidogenic IPOD substrates, transit through the Q-body pathway
The generality of the Q-body pathway was assessed by coexpressing Ubc9ts with two distinct QC substrates: misfolded variants of the tumor suppressor VHL or the thermolabile firefly luciferase (Luc)25, 35. These misfolded proteins also formed Q-bodies in distinguishable from those observed for Ubc9ts. The puncta formed by VHL or Luc completely colocalized with Ubc9ts Q-bodies, moved and coalesced together throughout the time-course of degradation (Fig. 6a, b; video S11, and Fig. S5a). Importantly, VHL did not colocalize non-specifically with wild-type Ubc9, which remains folded at 37°C (Fig. 6a). VHL puncta were generated regardless of the presence of Ubc9 and proceeded through the Q-body pathway (Fig. S5b). This indicates that different misfolded proteins are recruited to the same cellular structures and are handled by the same QC pathway.
Figure 6. Different types of misfolded proteins, but not amyloids, are processed together through the Q-body pathway.

(a) WT cells expressing Ubc9ts-GFP and CHFP-VHL was grown at 28°C in galactose medium and imaged at 37°C in glucose medium. 5 min series of images show Ubc9ts-GFP signal in green and CHFP-VHL signal in red (i). Cells expressing Ubc9-GFP and CHFP-VHL were similarly prepared and imaged 15 min after the shift (ii). Scale bar equals 1 μm. (b) 15 sec interval series of an inset from cell represented in a (top panel) and from a cell co-expressing Luc-GFP and Ubc9-CHFP (bottom panel, and Fig. S5a). Scale bars equal 1 μm (c). WT cells expressing Htt-Q97-CHFP (red) and Ubc9ts-GFP (green) were prepared and imaged as in a. Scale bar equals 1 μm. (d) 15 sec interval series of an inset from cell represented in c (bottom panel) and from a cell co-expressing GFP-VHL and Rnq1-CHFP (top panel, and Fig. S5c). Scale bars equal 1 μm. (e) Two-color Z-stack projection of cells expressing both Htt-Q97-GFP (in green) and Rnq1-CHFP (in red). Scale bar equals 1 μm. (f) GFP signal of WT, ssa1Δssa2Δ, hsp42Δ, hsp104Δ cells expressing Ubc9ts-GFP or Luc-GFP at 37°C; or Htt-Q97-GFP or Rnq1-GFP at 28°C. Scale bars equal 1 μm.(g-h) Percentages of cells with puncta from a population analyzed in f. Respective total population sizes for WT, ssa1ssa2, hsp42, and hsp104 cells are [ (g): orange n=(2418, 375, 3013, 591) cells; yellow n=(1035, 472, 442, 681) cells; (h): purple, n=(971, 519, 1047, 617) cells; green n=(1165, 410, 1235, 853) cells] over three independent experiments.(6 fields counted per experiment) (*) p<0.05 (**) p<0.005 compared to WT for the same indicated time.
We examined whether the Q-body pathway overlaps with insoluble amyloidogenic IPOD substrates such as the prion Rnq1 and the polyglutamine-expanded Huntingtin exon1 fragment, Htt-Q97 (Fig. 6e)6. Live-cell imaging of misfolded Ubc9ts or VHL in cells coexpressing either Htt-Q97 or Rnq1 indicated that most Ubc9ts or VHL puncta moved and evolved independently of the IPOD. A few dim puncta transiently colocalized with the IPOD, but without merging with it (Fig. 6c, d; video S11 and Fig. S5c). Notably, Q-body dynamics were slightly altered in the presence of Htt-Q97 or Rnq1, suggesting that the presence of amyloids in the IPOD can affect protein homeostasis. Ubc9ts and VHL were still cleared, while Rnq1 and Htt-Q97 persisted for the duration of the time-course (Fig. 6c, d; video S11 and Fig. S5c). These results reveal two major fates for misfolded proteins in the absence of proteasome inhibition.
Distinct role of chaperones in Q-body and IPOD formation
The different mechanisms to cope with misfolded proteins could stem from their distinct interactions with the chaperone machinery. Indeed, chaperones participate differently in Q-body and IPOD formation, both qualitatively (Fig. 6f) and when quantifying the number of cells with inclusions over the population (Fig. 6g, h). Q-body substrates formed one major inclusion in ssa1Δssa2Δ cells; large peripheral inclusions in hsp104Δ, and no inclusions in hsp42Δ cells. In contrast, IPOD substrates did not form any inclusions in hsp104Δ mutant as described36-38, and significantly less inclusions in ssa1Δssa2Δ cells (Fig. 6f, h). Unlike Q-bodies, IPOD inclusions were still formed in hsp42Δ mutant, even though they appeared dimmer (Fig. 6f). Hsp42 did not colocalize with the IPOD substrates in non-stressed cells, and weakly at higher temperatures (Fig. S5d and e), consistent with it being dispensable for IPOD formation. This may reflect the higher tendency of amyloidogenic proteins to self-assemble. Alternatively, IPOD substrates may be less dependent on the action of Hsp42 because they are more refractory to the disaggregase activity of Hsp104 and other chaperones.
Q-bodies arise in response to proteotoxic stress
We next examined whether Q-bodies arise generally upon proteotoxic stress. Cells were exposed to ethanol, which denatures pre-existing proteins39, or treated with the proline analog azetidine 2-carboxylic acid (AZC), which causes misfolding upon incorporation into newly synthetized proteins40. Exposure at 37°C served as a positive control (Fig. 7a). Ubc9ts-GFP and Hsp104-GFP were used to visualize Q-bodies. Ethanol treatment was sufficient to generate Hsp104-GFP and Ubc9ts-GFP positive Q-bodies similar to those observed during heat stress (Fig. 7a). While their kinetics of appearance was faster during heat stress, Q-body dynamics and coalescence appeared similar in all treatments (Fig. 7a).
Figure 7. The Q-body pathway responds to proteotoxic stress, chronologic aging and nutrient signaling.

(a) Ubc9ts-GFP-expressing cells were grown at 28°C in galactose medium and Hsp104-GFP-expressing at 28°C in glucose medium. Cells were imaged at 28°C in glucose medium after a 10 min treatment with 10% ethanol (EtOH), 30 min treatment with 5 mg/mL AZC, or at 37°C with no treatment. GFP signal is shown in 5 min series movies over 30 min. Scale bars equal 1 μm. (b) Cells expressing Ubc9ts-GFP (i) or Hsp104-GFP (ii) were grown at 28°C for 5 hrs (young) or for 3 days (aged) and imaged at 37°C. GFP signal is shown in 5 min series movies. Scale bars equal 1 μm. (c) Ubc9ts-GFP-expressing cells were grown at 28°C in galactose medium and imaged at 37°C in glucose medium in the absence (-) or presence (+) of 0.2 μg/mL rapamycin 10 min prior the shift. GFP signal is shown in 5 min series movies over 30 min. Scale bars equal 1 μm. (d) Quantification of the average number of puncta per cell in untreated (-, circles) or rapamycin-treated (+, triangles) cells over time as presented in c. Puncta assessed from a total population of n=40 cells over three independent experiment (3 fields counted per experiment).(*) p<0.05 (**) p<0.005 compared to untreated cells for the same indicated time.
We determined whether generating newly synthesized misfolded proteins affects the state of pre-existing folded proteins. Synthesis of Ubc9ts was repressed by glucose addition at 28°C yielding a pool of pre-existing folded and diffusely localized Ubc9ts, which served as a sensor of general proteostasis in the cell. Newly translated misfolded proteins were generated by AZC treatment at 28°C. Strikingly, the pre-existing folded Ubc9ts-GFP re-localized to Q-bodies at 28°C after AZC addition, even though no new Ubc9ts protein was being translated (Fig. 7a). These results indicate that misfolding of newly made proteins shifts the proteostasis balance of the cell to induced misfolding of a pre-existing labile protein, likely because its folding depends on the continued action of chaperones.
The Q-body pathway is regulated by cellular status
Protein homeostasis is regulated by cellular status and is impaired during aging, which may account for the late-onset of many amyloid diseases41-43. We examined whether cellular state or aging modulate the formation and processing of Q-bodies. Budding yeast enter a non-diving G0-like state when a culture is grown to stationary phase, a paradigm often referred to as chronological aging44. Aged non-dividing cells expressing diffuse Ubc9ts-GFP were shifted to 37°C and imaged. None of the aged cells was capable of inducing Q-bodies, unlike what is observed in young dividing cells (Fig. 7b and Fig. S6). However, we did observe that aged cells contained pre-existing Hsp104-positive puncta as well as a distinct large inclusion containing Hsp42 (Fig. 7b), previously reported as a hallmark of chronologically aged cells45.
The Q-bodies pathway is also regulated by TOR signaling, known to modulate protein homeostasis in response to nutrient and cellular state42, 46, 47. Treatment with 0.2 μg/ml rapamycin immediately prior to a 37°C shift profoundly affected the Q-body pathway (Fig. 7c, d, and video S12) leading to rapid clearance of Q-bodies from the cytoplasm. Our data imply a regulatory role of TOR signaling in the Q-body pathway, and suggest that aged cells have lost their responsiveness to spatially sequester misfolded proteins.
Fitness advantage of chaperones providing spatial sequestration of misfolded proteins
Despite their role in Q-body formation and dissolution, Hsp42 and Hsp104 are dispensable both for misfolded protein degradation and growth at 37°C (Fig. S7). However, the existence of a general pathway for the active spatial sequestration of misfolded proteins argues for a potential benefit to the cells. We hypothesized that sequestration into Q-bodies may purge the cellular milieu from potentially deleterious misfolded species, thereby providing a fitness advantage to cells during stress. To test this, we carried out competition experiments comparing the relative fitness of WT cells and cells lacking either Hsp42, Hsp26 or Hsp104 under normal and heat stress conditions. Equal amounts of WT and mutant cells were mixed and grown together at 28°C or 37°C, with daily dilutions, for 5 days. At each dilution, the proportion of WT and mutant cells was examined by comparing the number of kanamycin-resistant (kanR) colony-forming units (CFU) [i.e. mutant cells] vs. kanamycin-sensitive CFUs [i.e. WT cells] (Fig. 8a). Deletion of Hsp26 provides neither a fitness cost nor a benefit at either temperature. The equal proportion of WT and hsp26Δ cells was maintained throughout the 5 days of growth at either temperature (Fig. 8b), also indicating that kanR does not affect the fitness of mutant cells. Deletion of Hsp42 provided neither cost nor benefit at 28°C (Fig. 8c, left panel). However, at 37°C, deletion of Hsp42 was remarkably disadvantageous as WT cells out competed hsp42Δ cells after 2 days (Fig. 8c, right panel). Thus, Hsp42 provides a strong fitness advantage for cells subjected to stress. A parallel analysis for cells lacking Hsp104 proved surprising. Deletion of Hsp104 also reduced the fitness advantage of cells grown at 37°C (Fig. 8d, right panel) suggesting that protein disaggregation is advantageous under conditions of stress. However, under normal growth conditions, i.e. 28°C, Hsp104 has a strong fitness cost, as hsp104Δ cells were much fitter than WT cells (Fig. 8d, left panel). Since the major biochemical and cellular function of Hsp104 is to disaggregate misfolded and aggregated proteins, this suggests that a strong disaggregase activity is disadvantageous in the absence of stress-induced protein misfolding.
Figure 8. Fitness advantage of spatial sequestration of misfolded proteins in Q-bodies.

(a) Schematic of competition assay; [KanS], kanamycin-sensitive phenotype; [KanR], kanamycin-resistant phenotype. (b) Percentage of KanR hsp26Δ colony-forming units (CFU) over KanS WT CFU was quantified at 0 and 5 days. (c-d) Percentage of KanR hsp42Δ CFU (c) or hsp104Δ CFU (d) over KanS WT CFU was quantified over time at 28°C (left panels) or 37°C (right panels). Data represent three independent experiments (n = 350 CFU for each day of the experiment).
Discussion
Misfolded protein clearance mechanisms were proposed to be hierarchical, whereby misfolded proteins are first stabilized by chaperones for either refolding or degradation with sequestration into inclusions as a second line of defense when proteostasis fails. Our study calls for a revision of this concept. We find that misfolded protein sequestration is a physiological and early response to the presence of misfolded proteins. Degradation occurs concurrently with Q-body processing, implying that sequestration is not necessarily a consequence of degradation failure. Q-bodies form in response to many stresses but become observable in larger inclusions upon proteostasis collapse, because their clearance is abrogated while their formation and coalescence continues. Compartmentalization into Q-bodies is not essential for degradation but may enhance cell fitness by sequestering potentially toxic misfolded species. Our findings provide a simple explanation to the genesis of large inclusions upon protein homeostasis dysfunction and UPS impairment that does not require invoking activation of special inclusion-formation mechanisms.
Misfolded protein concentration into Q-bodies is an active process that requires molecular chaperones. The Hsp70-Hsp90-Hsp110 system, which also participates in the degradation and refolding of misfolded proteins48, 49, is central to the formation and dynamics of Q-bodies. These chaperones likely constitute the core of the cytoplasmic QC system. Q-bodies are associated with the ER, which may provide a basis for Q-body movement and coalescence through its tubular and dynamic network throughout the cytosol. ER-associated J-domain proteins, Ydj1 and Hlj129, 32, are important for Q-body concentration and maturation, consistent with their role recruiting Ssa1 to the ER membrane32, 50. However, their function is not essential, likely due to functional overlaps between J-domain proteins31.
The small HSP Hsp42 and the disaggregase Hsp104 contribute to Q-body dynamics. Epistasis analyses of mutations in Hsp42, Hsp104 and Hsp110/Sse1 suggest that at least part of Q-body dynamics results from the balance between addition and dissolution activities. A simple model explaining our data is that puncta formation involves the action of different chaperones, notably Hsp42 and J-domain proteins, whereas Q-body growth occurs at the expense of Hsp104 or Hsp110/Sse1-mediated dissolution of others. Interestingly, a similar cycle of addition-dissolution is proposed to govern P-granule dynamics and the asymmetric partition of maternally inherited mRNAs51. However, coalescence may also involve direct merging of Q-bodies with the help of chaperones and additional factors.
Q-body formation, dynamics and clearance are independent of either actin or tubulin cytoskeleton, consistent with previous analysis of the dynamics of Hsp104-associated proteins aggregates52 and in contrast to alternative proposals7, 14. Importantly, our finding that prolonged depolymerization of the actin cytoskeleton has deleterious effects on the Q-body pathway suggests an explanation to reconcile these discrepancies. Unassembled actin may bind to and titrate QC components, generally impairing cellular proteostasis. The cytoskeleton may also be required for asymmetric partitioning of inclusions, a question that is not addressed here. Future studies incorporating our findings should illuminate the link between the cytoskeleton, protein homeostasis, and the inheritance of misfolded protein inclusions.
Without proteasome inhibition, misfolded proteins are not directly targeted to a static pre-existing QC compartment, but concentrated throughout the cell in multiple dynamic Q-bodies. The link between Q-bodies and compartments previously observed in presence of proteasome inhibition6-8, 14 remains to be determined. These structures may be endpoints of the Q-body pathway resulting from the coalescence of Q-bodies when clearance is impaired. The observation that some perinuclear puncta lack Hsp104 can explain the appearance of the JUNQ upon proteasome inhibition; puncta with slower dissolution rates would eventually grow at the expense of those with higher Hsp104 levels and faster disaggregation kinetics. It will be interesting to determine whether proteasome inhibition alters the composition and properties of these structures, for instance by stabilizing labile QC factors as suggested in 8.
Amyloidogenic proteins sequestered in the insoluble IPOD seem to bypass the Q-body pathway. Perhaps IPOD substrates interact aberrantly with chaperones that recruit substrates to the Q-body pathway thereby failing to be cleared. Alternatively, dense amyloid structures may be more refractory to disaggregation, thereby eluding the Q-body cycle. Interestingly, Q-body dynamics are impaired in presence of an IPOD suggesting that the IPOD alters cellular proteostasis. Several studies reported that amyloid inclusions sequester chaperones, causing aggregation of unrelated proteins without their co-aggregation with the amyloid inclusion53, 54. The impairment of the Q-body pathway during aging could render aged cells more sensitive to stress and misfolding, consistent with their enhanced sensitivity to protein misfolding55. Understanding the regulation of the Q-body pathway and its relation to other reported QC structures may provide new therapeutical targets for conformational disorders, including Alzheimer's and Huntington's disease.
What is the function of Q-bodies? Sequestering misfolded proteins is dispensable for degradation but may help clear the cytosol of potentially toxic misfolded species, store labile proteins for later use and/or facilitate QC by concentrating chaperones and substrates on a two-dimensional surface. Loss of chaperones involved in Q-body formation and dynamics leads to a fitness reduction in stressed cells. Surprisingly, the robust disaggregase Hsp104 is deleterious under normal growth conditions. This could explain the long-standing enigma of why Hsp104 is only found in yeast, plants and bacteria, generally subjected to repeated and severe stress56, but not in metazoan cells, which control their stress responses through organism-wide cell non-autonomous circuits57, 58. For these multicellular organisms, the advantage provided by Hsp104 during stress may not compensate for the disadvantage of having a potent cellular disaggregase under normal conditions. These results highlight the delicate tuning of QC pathways during evolution.
Concentration of misfolded proteins in Q-bodies creates a dynamic and flexible management system conferring fitness pending the impact of the immediate environmental changes challenging the cell. The relationship between the biochemical functions of chaperones and their spatial organization within the cell will be instrumental to understand folding impairments during pathological states.
Supplementary Material
Acknowledgments
We thank Christopher Toret and Veronique Albanese for experimental advice and discussions; Christopher Toret and Raul Andino for critical reading of the manuscript. SET was initially supported by a fellowship from Fondation pour la Recherche Medicale (France). WV was supported by the Marie Curie International Outgoing Fellowship Programme. This work was supported by grant from NIH and a Senior Scholar Award from the Ellison Foundation to JF.
Footnotes
Author Contribution: SET and JF conceived the project, SET performed most experiments, WV performed experiments in Fig. 1d, e, Fig 3e, f, and Fig. S1b, all authors interpreted the experiments and contributed to writing.
References
- 1.Balch WE, Morimoto RI, Dillin A, Kelly JW. Adapting proteostasis for disease intervention. Science. 2008;319:916–919. doi: 10.1126/science.1141448. [DOI] [PubMed] [Google Scholar]
- 2.Lindquist SL, Kelly JW. Chemical and biological approaches for adapting proteostasis to ameliorate protein misfolding and aggregation diseases: progress and prognosis. Cold Spring Harb Perspect Biol. 2011;3 doi: 10.1101/cshperspect.a004507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chiti F, Dobson CM. Protein misfolding, functional amyloid, and human disease. Annual Review of Biochemistry. 2006;75:333–366. doi: 10.1146/annurev.biochem.75.101304.123901. [DOI] [PubMed] [Google Scholar]
- 4.Houck SA, Singh S, Cyr DM. Cellular responses to misfolded proteins and protein aggregates. Methods Mol Biol. 2012;832:455–461. doi: 10.1007/978-1-61779-474-2_32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chen B, Retzlaff M, Roos T, Frydman J. Cellular strategies of protein quality control. Cold Spring Harb Perspect Biol. 2011;3:a004374. doi: 10.1101/cshperspect.a004374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kaganovich D, Kopito R, Frydman J. Misfolded proteins partition between two distinct quality control compartments. Nature. 2008;454:1088–1095. doi: 10.1038/nature07195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Specht S, Miller SB, Mogk A, Bukau B. Hsp42 is required for sequestration of protein aggregates into deposition sites in Saccharomyces cerevisiae. J Cell Biol. 2011;195:617–629. doi: 10.1083/jcb.201106037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Malinovska L, Kroschwald S, Munder MC, Richter D, Alberti S. Molecular chaperones and stress-inducible protein-sorting factors coordinate the spatiotemporal distribution of protein aggregates. Mol Biol Cell. 2012;23:3041–3056. doi: 10.1091/mbc.E12-03-0194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Johnston JA, Ward CL, Kopito RR. Aggresomes: a cellular response to misfolded proteins. J Cell Biol. 1998;143:1883–1898. doi: 10.1083/jcb.143.7.1883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhang X, Qian SB. Chaperone-mediated hierarchical control in targeting misfolded proteins to aggresomes. Mol Biol Cell. 2011;22:3277–3288. doi: 10.1091/mbc.E11-05-0388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Douglas PM, Summers DW, Cyr DM. Molecular chaperones antagonize proteotoxicity by differentially modulating protein aggregation pathways. Prion. 2009;3:51–58. doi: 10.4161/pri.3.2.8587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cohen E, Bieschke J, Perciavalle RM, Kelly JW, Dillin A. Opposing activities protect against age-onset proteotoxicity. Science. 2006;313:1604–1610. doi: 10.1126/science.1124646. [DOI] [PubMed] [Google Scholar]
- 13.Arrasate M, Mitra S, Schweitzer ES, Segal MR, Finkbeiner S. Inclusion body formation reduces levels of mutant huntingtin the risk of neuronal death. Nature. 2004;431:805–810. doi: 10.1038/nature02998. [DOI] [PubMed] [Google Scholar]
- 14.Liu B, et al. The polarisome is required for segregation and retrograde transport of protein aggregates. Cell. 2010;140:257–267. doi: 10.1016/j.cell.2009.12.031. [DOI] [PubMed] [Google Scholar]
- 15.Nakatogawa H, Ichimura Y, Ohsumi Y. Atg8, a ubiquitin-like protein required for autophagosome formation, mediates membrane tethering and hemifusion. Cell. 2007;130:165–178. doi: 10.1016/j.cell.2007.05.021. [DOI] [PubMed] [Google Scholar]
- 16.Sheth U, Parker R. Targeting of aberrant mRNAs to cytoplasmic processing bodies. Cell. 2006;125:1095–1109. doi: 10.1016/j.cell.2006.04.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Toshima JY, et al. Spatial dynamics of receptor-mediated endocytic trafficking in budding yeast revealed by using fluorescent alpha-factor derivatives. Proc Natl Acad Sci U S A. 2006;103:5793–5798. doi: 10.1073/pnas.0601042103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wright R, Basson M, D'Ari L, Rine J. Increased amounts of HMG-CoA reductase induce “karmellae”: a proliferation of stacked membrane pairs surrounding the yeast nucleus. J Cell Biol. 1988;107:101–114. doi: 10.1083/jcb.107.1.101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Shibata Y, Voeltz GK, Rapoport TA. Rough sheets and smooth tubules. Cell. 2006;126:435–439. doi: 10.1016/j.cell.2006.07.019. [DOI] [PubMed] [Google Scholar]
- 20.Voeltz GK, Prinz WA, Shibata Y, Rist JM, Rapoport TA. A class of membrane proteins shaping the tubular endoplasmic reticulum. Cell. 2006;124:573–586. doi: 10.1016/j.cell.2005.11.047. [DOI] [PubMed] [Google Scholar]
- 21.Shibata Y, et al. The reticulon and DP1/Yop1p proteins form immobile oligomers in the tubular endoplasmic reticulum. J Biol Chem. 2008;283:18892–18904. doi: 10.1074/jbc.M800986200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.West M, Zurek N, Hoenger A, Voeltz GK. A 3D analysis of yeast ER structure reveals how ER domains are organized by membrane curvature. J Cell Biol. 2011;193:333–346. doi: 10.1083/jcb.201011039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.McDonough H, Patterson C. CHIP: a link between the chaperone and proteasome systems. Cell Stress Chaperones. 2003;8:303–308. doi: 10.1379/1466-1268(2003)008<0303:calbtc>2.0.co;2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mayer MP, Bukau B. Hsp70 chaperones: cellular functions and molecular mechanism. Cell Mol Life Sci. 2005;62:670–684. doi: 10.1007/s00018-004-4464-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.McClellan AJ, Scott MD, Frydman J. Folding and quality control of the VHL tumor suppressor proceed through distinct chaperone pathways. Cell. 2005;121:739–748. doi: 10.1016/j.cell.2005.03.024. [DOI] [PubMed] [Google Scholar]
- 26.Coppinger JA, et al. A chaperone trap contributes to the onset of cystic fibrosis. PLoS One. 2012;7:e37682. doi: 10.1371/journal.pone.0037682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Schneider C, et al. Pharmacologic shifting of a balance between protein refolding and degradation mediated by Hsp90. Proc Natl Acad Sci U S A. 1996;93:14536–14541. doi: 10.1073/pnas.93.25.14536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Becker J, Walter W, Yan W, Craig EA. Functional interaction of cytosolic hsp70 and a DnaJ-related protein, Ydj1p, in protein translocation in vivo. Mol Cell Biol. 1996;16:4378–4386. doi: 10.1128/mcb.16.8.4378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Caplan AJ, Tsai J, Casey PJ, Douglas MG. Farnesylation of YDJ1p is required for function at elevated growth temperatures in Saccharomyces cerevisiae. J Biol Chem. 1992;267:18890–18895. [PubMed] [Google Scholar]
- 30.Flom GA, Lemieszek M, Fortunato EA, Johnson JL. Farnesylation of Ydj1 is required for in vivo interaction with Hsp90 client proteins. Mol Biol Cell. 2008;19:5249–5258. doi: 10.1091/mbc.E08-04-0435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kampinga HH, Craig EA. The HSP70 chaperone machinery: J proteins as drivers of functional specificity. Nat Rev Mol Cell Biol. 2010;11:579–592. doi: 10.1038/nrm2941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Youker RT, Walsh P, Beilharz T, Lithgow T, Brodsky JL. Distinct roles for the Hsp40 and Hsp90 molecular chaperones during cystic fibrosis transmembrane conductance regulator degradation in yeast. Mol Biol Cell. 2004;15:4787–4797. doi: 10.1091/mbc.E04-07-0584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Shorter J, Lindquist S. Hsp104, Hsp70 and Hsp40 interplay regulates formation, growth and elimination of Sup35 prions. EMBO J. 2008;27:2712–2724. doi: 10.1038/emboj.2008.194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tipton KA, Verges KJ, Weissman JS. In vivo monitoring of the prion replication cycle reveals a critical role for Sis1 in delivering substrates to Hsp104. Mol Cell. 2008;32:584–591. doi: 10.1016/j.molcel.2008.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gupta R, et al. Firefly luciferase mutants as sensors of proteome stress. Nat Methods. 2011;8:879–884. doi: 10.1038/nmeth.1697. [DOI] [PubMed] [Google Scholar]
- 36.Chernoff YO, Lindquist SL, Ono B, Inge-Vechtomov SG, Liebman SW. Role of the chaperone protein Hsp104 in propagation of the yeast prion-like factor [psi+] Science. 1995;268:880–884. doi: 10.1126/science.7754373. [DOI] [PubMed] [Google Scholar]
- 37.Sondheimer N, Lindquist S. Rnq1: an epigenetic modifier of protein function in yeast. Mol Cell. 2000;5:163–172. doi: 10.1016/s1097-2765(00)80412-8. [DOI] [PubMed] [Google Scholar]
- 38.Meriin AB, et al. Huntington toxicity in yeast model depends on polyglutamine aggregation mediated by a prion-like protein Rnq1. J Cell Biol. 2002;157:997–1004. doi: 10.1083/jcb.200112104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Piper PW. The heat shock and ethanol stress responses of yeast exhibit extensive similarity and functional overlap. FEMS Microbiol Lett. 1995;134:121–127. doi: 10.1111/j.1574-6968.1995.tb07925.x. [DOI] [PubMed] [Google Scholar]
- 40.Trotter EW, et al. Misfolded proteins are competent to mediate a subset of the responses to heat shock in Saccharomyces cerevisiae. J Biol Chem. 2002;277:44817–44825. doi: 10.1074/jbc.M204686200. [DOI] [PubMed] [Google Scholar]
- 41.Morimoto RI. Proteotoxic stress and inducible chaperone networks in neurodegenerative disease and aging. Genes Dev. 2008;22:1427–1438. doi: 10.1101/gad.1657108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Conn CS, Qian SB. mTOR signaling in protein homeostasis: less is more? Cell Cycle. 2011;10:1940–1947. doi: 10.4161/cc.10.12.15858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Taylor RC, Dillin A. Aging as an event of proteostasis collapse. Cold Spring Harb Perspect Biol. 2011;3 doi: 10.1101/cshperspect.a004440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Fabrizio P, Longo VD. The chronological life span of Saccharomyces cerevisiae. Aging Cell. 2003;2:73–81. doi: 10.1046/j.1474-9728.2003.00033.x. [DOI] [PubMed] [Google Scholar]
- 45.Narayanaswamy R, et al. Widespread reorganization of metabolic enzymes into reversible assemblies upon nutrient starvation. Proc Natl Acad Sci U S A. 2009;106:10147–10152. doi: 10.1073/pnas.0812771106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Peters TW, et al. Tor1 regulates protein solubility in Saccharomyces cerevisiae. Mol Biol Cell. 2012;23:4679–4688. doi: 10.1091/mbc.E12-08-0620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kapahi P, et al. With TOR, less is more: a key role for the conserved nutrient-sensing TOR pathway in aging. Cell Metab. 2010;11:453–465. doi: 10.1016/j.cmet.2010.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Glover JR, Lindquist S. Hsp104, Hsp70, and Hsp40: a novel chaperone system that rescues previously aggregated proteins. Cell. 1998;94:73–82. doi: 10.1016/s0092-8674(00)81223-4. [DOI] [PubMed] [Google Scholar]
- 49.Mandal AK, et al. Hsp110 chaperones control client fate determination in the hsp70-Hsp90 chaperone system. Mol Biol Cell. 2010;21:1439–1448. doi: 10.1091/mbc.E09-09-0779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Huyer G, et al. Distinct machinery is required in Saccharomyces cerevisiae for the endoplasmic reticulum-associated degradation of a multispanning membrane protein and a soluble luminal protein. J Biol Chem. 2004;279:38369–38378. doi: 10.1074/jbc.M402468200. [DOI] [PubMed] [Google Scholar]
- 51.Ouellet J, Barral Y. Organelle segregation during mitosis: Lessons from asymmetrically dividing cells. J Cell Biol. 2012;196:305–313. doi: 10.1083/jcb.201102078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Zhou C, et al. Motility and segregation of Hsp104-associated protein aggregates in budding yeast. Cell. 2011;147:1186–1196. doi: 10.1016/j.cell.2011.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Gidalevitz T, Krupinski T, Garcia S, Morimoto RI. Destabilizing protein polymorphisms in the genetic background direct phenotypic expression of mutant SOD1 toxicity. PLoS Genet. 2009;5:e1000399. doi: 10.1371/journal.pgen.1000399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Olzscha H, et al. Amyloid-like aggregates sequester numerous metastable proteins with essential cellular functions. Cell. 2011;144:67–78. doi: 10.1016/j.cell.2010.11.050. [DOI] [PubMed] [Google Scholar]
- 55.Kikis EA, Gidalevitz T, Morimoto RI. Protein homeostasis in models of aging and age-related conformational disease. Adv Exp Med Biol. 2010;694:138–159. doi: 10.1007/978-1-4419-7002-2_11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Shorter J. Hsp104: a weapon to combat diverse neurodegenerative disorders. Neurosignals. 2008;16:63–74. doi: 10.1159/000109760. [DOI] [PubMed] [Google Scholar]
- 57.Gidalevitz T, Prahlad V, Morimoto RI. The stress of protein misfolding: from single cells to multicellular organisms. Cold Spring Harb Perspect Biol. 2011;3 doi: 10.1101/cshperspect.a009704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Durieux J, Wolff S, Dillin A. The cell-non-autonomous nature of electron transport chain-mediated longevity. Cell. 2011;144:79–91. doi: 10.1016/j.cell.2010.12.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.