

Abstract
The increasing prevalence of Type 2 diabetes has emphasized the need to optimize treatment regimens. Metformin, the most widely used oral agent, is recommended as first-line drug therapy by multiple professional organizations. Response to metformin varies significantly at the individual level; this heterogeneity may be explained in part by genetic factors. Understanding these underlying factors may aid with tailoring treatment for individual patients as well as with designing improved Type 2 diabetes therapies. The past 10 years have seen substantial progress in the understanding of the pharmacogenetics of metformin response. The majority of this work has focused on genes involved in the pharmacokinetics of metformin. Owing to the uncertainty surrounding its mechanism of action, studies of pharmacodynamic genetics have been relatively few; genome-wide approaches have the potential to illuminate the molecular details of metformin response. In this review we summarize current knowledge about metformin pharmacogenetics and suggest directions for future investigation.
Keywords: antidiabetic, biguanide, metformin, pharmacogenetics, Type 2 diabetes mellitus
Diabetes is a widespread problem, affecting 25.8 million people in the US and 347 million worldwide; in adults, Type 2 diabetes (T2D) accounts for 90–95% of cases. Diabetes is the leading cause of kidney failure, and a major cause of heart disease and stroke, in US adults [1,101]. Metformin, a biguanide, is recommended by both the American Diabetes Association and the European Association for the Study of Diabetes as first-line oral therapy for T2D [2] and is the most widely used oral agent worldwide. Additionally, there is interest in the use of metformin for diabetes prevention, treatment of polycystic ovary syndrome (PCOS) and treatment of gestational diabetes. Clinically, there is considerable variation in response to metformin at the individual level. Genetic factors may, at least in part, account for some of this variability, and in recent years there has been great interest in exploring the influence of genetic variants on metformin pharmacokinetics and action. As with all drugs, understanding pharmaco genomics may help clinicians personalize medical care and select the right drug for the right patient. With metformin in particular, the details underlying the molecular mechanism of action are not completely understood, which both complicates the approach to exploring its pharmacogenomics but also gives this field great potential for new discoveries and insights. In this review, we summarize what is known about the association of genetic variants with metformin pharmacokinetics and response, and suggest paths for future exploration.
Background
We begin with a brief overview of metformin pharmacodynamics and pharmacokinetics; for more in-depth treatment of these subjects, we refer the reader to the excellent reviews by Graham et al. [3], Viollet et al. [4], Gong et al. [5], and Rena et al. [6].
Pharmacodynamics
Clinically, metformin lowers both fasting and postprandial glucose, primarily by reducing hepatic glucose production, and possibly also by increasing peripheral glucose utilization [7,8]. Metformin also appears to lower fatty acid and triglyceride levels [7,9].
The molecular mechanisms of metformin action are not fully understood. Metformin activates AMPK in the liver [10], apparently via an upstream kinase regulator known as LBK1 [11]. However, Foretz et al. used two mouse models, lacking either AMPK or LKB1 in hepatocytes, to demonstrate a preserved glucose-lowering effect in both strains [12]. Metformin does not directly act on either AMPK or LKB1, but there is evidence that AMPK activation is secondary to the effect of metformin on mitochondria. Metformin has been shown in several studies to inhibit the mitochondrial respiratory chain complex I, ultimately leading to a reduction in ATP synthesis [13,14]. This effect of metformin does not require AMPK activation; rather, AMPK is activated by metabolic stresses that increase the intracellular ADP:ATP and AMP:ADP ratio, and this may be an alternate explanation of how metformin activates AMPK [4,15,16]. More recently, Miller et al. demonstrated that biguanides antagonize the glucose-raising effects of glucagon in the liver. Metformin and fellow biguanide phenformin blocked glucagon-dependent increases in levels of cyclic AMP, activity of the cAMP-dependent kinase PKA and hepatic glucose output. These findings were replicated in AMPK-deficient hepatocytes, indicating that the effects are independent of AMPK. The authors suggest that biguanides exert their effect via AMP, which can bind an inhibitory `P' site on adenylyl cyclase, the enzyme responsible for cAMP production [17].
Pharmacokinetics
Metformin is a hydrophilic molecule that is actively transported via organic cation transporters in the intestine, liver and kidney (Figure 1); passive distribution across cell membranes is limited by its low lipid solubility [3]. In the intestine, the PMAT (encoded by the gene SLC29A4) may be the primary source of metformin uptake, as expression of this transporter has been shown to be localized to the luminal side of enterocytes and in vitro work shows avid uptake of metformin [18]. OCT3 (encoded by SCL22A3) is also localized on the luminal side of enterocytes and may contribute to gastrointestinal metformin uptake [19]. OCT1 (encoded by SLC22A1), expressed on the basolateral membrane of enterocytes, may be responsible for transport of metformin into the intersitial fluid [19]. Both OCT1 and OCT3 are expressed on the basolateral membrane of hepatocytes [3,20,21]; in Oct1 knockout mice, the hepatic metformin concentration is greatly reduced, indicating that Oct1 is the major hepatic transporter in mice [22]. MATE1 (encoded by SLC47A1) may contribute to hepatic excretion, as it is expressed in the liver and is a carrier of metformin [23,24]; however, in humans, biliary excretion of metformin appears negligible, as there is no fecal excretion of metformin after intravenous dosage [3]. In the kidney, metformin is taken up into renal epithelial cells by OCT2 (encoded by SLC22A2), expressed on the basolateral membrane [25], and excreted into the urine via MATE1 and MATE2 (encoded by SLC47A2) [23,24,26]. OCT1 and PMAT are both expressed on the apical membranes of renal cells, and may contribute to metformin reabsorption [5].
Figure 1.
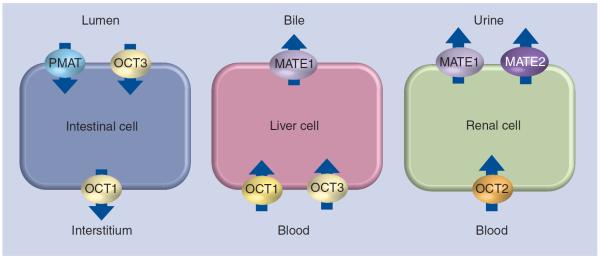
Transporters involved in metformin absorption, hepatic uptake and urinary excretion.
Pharmacogenetics
To date, the exploration of metformin pharmacogenetics has primarily occurred via a candidate gene approach; much of this work has focused on the organic cation transporters as understanding of their role in metformin pharmacokinetics has increased (Table 1). Only a few studies have explored candidates for metformin pharmacodynamics, which likely reflects the ambiguity surrounding the molecular mechanism of response.
Table 1.
Summary of variants in metformin transporter genes.
| SNPID | DNA | AA change | Reported effect of minor allele or variant |
|---|---|---|---|
| SLC22A1 (OCT1) | |||
|
| |||
| -43T>G | Associated with better response to metformin in T2D patients [33] | ||
|
| |||
| rs12208357 | 181C>T | R61C | Increased metformin AUC, higher Cmax, lower volume of distribution [32]; attenuated effect of metformin on OGTT [22]; increased CLrenal [32]; no association with metformin response in T2D patients [35]; lower trough metformin levels and attenuated effect of metformin on HbA1c in T2D subjects [36] |
|
| |||
| rs683369 | 480G>C | L160F | Reduced benefit of metformin on diabetes prevention [40] |
|
| |||
| rs34130495 | 1201G>A | G401S | Increased metformin AUC, higher Cmax, lower volume of distribution [31]; attenuated effect of metformin on OGTT [22]; increased CLrenal [32]; lower trough metformin levels and attenuated effect of metformin on HbA1c in T2D subjects [36] |
|
| |||
| rs72552763 | 1365GAT>del | 420del | Increased metformin AUC, higher Cmax, lower volume of distribution [31]; attenuated effect of metformin on OGTT [22]; increased CLrenal [32]; no association with metformin response in T2D patients [35]; lower trough metformin levels and attenuated effect of metformin on HbA1c in T2D subjects [36] |
|
| |||
| rs34059508 | 1393G>A, 1393G>C | G465R | Increased metformin AUC, higher Cmax, lower volume of distribution [31]; attenuated effect of metformin on OGTT [22]; increased CLrenal [32]; lower trough metformin levels and attenuated effect of metformin on HbA1c in T2D subjects [36] |
|
| |||
| rs628031 | 1222A>G | M408V | Associated with risk of nonresponse to metformin in T2D patients [33]; associated with decreased risk of side effects [41] |
|
| |||
| rs622342 | Intron | Attenuated effect of metformin on HbA1c [34]; no association with effect of metformin on HbA1c [36,38]; or trough metformin levels in T2D subjects [36]; no association with benefit of metformin on diabetes prevention [40] | |
|
| |||
| rs461473 | Intron | Associated with enhanced metformin effect on HbA1c in T2D subjects [36] | |
|
| |||
| rs36056065 | 8 bp insertion | Associated with decreased risk of side effects [41] | |
|
| |||
| SLC22A2 (OCT2) | |||
|
| |||
| 495A>G | M165I | Altered transporter function in vitro; no effect on metformin transport [28] | |
|
| |||
| 596C>T | T199I | Reduced in vitro transporter function [42]; reduced CLrenal and higher Cmax [43] | |
|
| |||
| 602C>T | T201M | Reduced in vitro transporter function [42]; reduced CLrenal and higher Cmax [43]; no association with metformin response in T2D patients [33] | |
|
| |||
| rs316019 | 808G>T | A270S | Altered transporter function in vitro; no effect on metformin transport [28]; reduced in vitro transporter function [42]; reduced CLrenal and higher Cmax [43]; reduced CLrenal but no effect on Cmax [44]; increased CLrenal [44]; no association with metformin response [33,36,38] or trough metformin levels [36] in T2D patients; no association with benefit of metformin on diabetes prevention [40]; increased CLrenal in healthy volunteers homozygous for the reference variant of MATE1 g.-66T>C [46] |
|
| |||
| rs8177516 | 1198C>T | A400C | Altered transporter function in vitro; no effect on metformin transport [28] |
| 1294A>C | K432G | Altered transporter function in vitro; no effect on metformin transport [28] | |
|
| |||
| rs662301 | Intron | Enhanced benefit of metformin on diabetes prevention [40] | |
|
| |||
| SLC22A3 (OCT3) | |||
|
| |||
| rs8187715 | T44M | Increased in vitro metformin uptake [20] | |
|
| |||
| rs8187717 | A116S | Decreased in vitro metformin uptake [20] | |
|
| |||
| rs8187725 | T400I | Decreased in vitro metformin uptake [20] | |
|
| |||
| SLC47A1 (MATE1) | |||
|
| |||
| rs2289669 | Intron | Enhanced effect of metformin on HbA1c in T2D patients [38,53]; no association with trough metformin levels or effect of metformin on HbA1c in T2D subjects [36] | |
|
| |||
| rs8065082 | Intron | Increased benefit of metformin on diabetes prevention [40] | |
|
| |||
| rs2252281 | g.-66T>C | Decreased promoter activity; enhanced effect of metformin on OGTT [51,52]; no association with trough metformin levels or effect of metformin on HbA1c in T2D subjects [36]; interaction with effect of OCT2 A270S on CLrenal [46] | |
|
| |||
| rs111060521 | 28G>T | V10L | Normal in vitro transporter function [47] |
|
| |||
| rs111060524 | 191G>A | G64D | Reduced transporter function in vitro [47,49], no effect on pharmacokinetics in vivo [50] |
| 373C>T | L125F | Reduced transporter function in vitro [49]; no effect on pharmacokinetics in vivo [48] | |
|
| |||
| rs35790011 | 1012G>A | V338I | Reduced transporter function in vitro [49] |
| 1438G>A | V480M | Reduced transporter function in vitro [49] | |
|
| |||
| rs35395280 | 1490G>C | C497S | Reduced transporter function in vitro [49] |
| 1557G>C | Q519H | Normal in vitro transporter function [49] | |
|
| |||
| rs111060527 | 983A>C | D328A | No effect on pharmacokinetics in vivo [48] |
|
| |||
| rs2289669/rs8065082 | Intron | Enhanced effect of metformin on HbA1c in T2D patients [38,53]; no association with effect of metformin on HbA1c in T2D subjects [36]; enhanced benefit of metformin on diabetes prevention [40] | |
|
| |||
| SLC47A2 (MATE2-K) | |||
|
| |||
| rs12943590 | g.-130G>A | n/a | Increased promoter activity [50,52]; weaker response to metformin in T2D patients [50]; attenuated effect of metformin on OGTT [52] |
|
| |||
| PMT5634 | 485C>T | P162L | Reduced metformin uptake and reduced protein expression in vitro [50] |
|
| |||
| rs111060532 | 632_633GC>TT | G211V | Reduced transporter function in vitro [47]; no effect on pharmacokinetics in vivo [48] |
|
| |||
| rs34399035 | 1177G>A | G393R | Reduced metformin uptake and reduced protein expression in vitro [50]; no association with trough metformin levels or effect of metformin on HbA1c in T2D subjects [36] |
AA: Amino acid; AUC: Area under the curve; CLrenal: Renal clearance; HbA1c: Glycated hemoglobin; OGTT: Oral glucose tolerance test; T2D: Type 2 diabetes.
OCT1/SLC22A1
Characterization of variants in vitro & in healthy volunteers
Early studies established that the OCT1 gene SLC22A1 is highly polymorphic, and that non-synonymous variants affect its function as a cation transporter [27–30]. In 2007, Shu and colleagues established that hepatic uptake of metformin via OCT1 is essential for the glucose-lowering effects of the drug, and that in healthy volunteers with at least one of four reduced function variants (R61C/rs12208357, G401S/rs34130495, 420del/rs72552763 and G465R/rs34059508) the effects of a short (two dose) course of metformin on glucose tolerance tests are attenuated [22]. A follow-up study by the same group established that individuals with reduced-function alleles had higher area under the curve (AUC) of metformin plasma concentration over time, higher Cmax and lower oral volume of distribution in the 24 h following the second metformin dose [31]. Tzvetkov et al. studied the pharmacokinetics of a single 500 mg dose of metformin in 103 healthy volunteers and found that G465R was associated with increased renal clearance (CLrenal). When the reduced function variants (R61C, G401S, 420del and G465R) were analyzed as a group, the CLrenal of metformin increased with increasing number of hypofunctioning alleles [32].
Effect of variants on clinical response
Whether OCT1 variants affect treatment response in patients with T2D is less clear. A study by Shikata et al. divided a retro spective cohort of patients with T2D (n = 33) into responders versus nonresponders, based on the ability to achieve and maintain a reduction in glycated hemoglobin (HbA1c) of 0.5% after metformin initiation. Variants were identified by sequence analysis of SLC22A1 exons; of note, the reduced-function variants R61C, G401S, 420del and G465R were not seen in this cohort. Of the variants found, -43T>G and M408V/rs628031 were negative and positive predictors, respectively, of metformin response [33]. As the M408V variant had been previously shown to have no effect on transport function [27,29,30] and -43T>G is in a noncoding region, the authors examined the effect of these variants on SLC22A1 expression; neither had a significant effect, although there was a trend towards reduced expression in the M408V variant. The Rotterdam study, which analyzed OCT1 tagging SNPs in a population of 102 T2D patients with incident metformin use, found an association between the intronic SNP rs622342 and reduction in HbA1c after metformin treatment [34]. A larger study in 1531 patients with T2D and incidental metformin use from the GoDARTS population focused on the two most common poly morphisms, R61C and 420del. The authors found no association of OCT1 variants with metformin response, whether defined as initial HbA1c reduction, odds of achieving a target HbA1c of less than 7%, average HbA1c on monotherapy or hazard of monotherapy failure [35]. All three of these studies were observational, in which the metformin dose, prescription of other antidiabetic medications, and timing of HbA1c measurements were at the discretion of the treating provider. The Japanese study included patients taking other T2D agents; the Rotterdam study excluded those on acarbose, rosiglitazone, pioglitazone, or insulin but not sulfonylureas; and the GoDARTS study excluded those whose nonmetformin T2D treatments changed over the 6-month period of observation, dividing the remainder into drug-naive (n = 1,014) and stably treated (n = 517) cohorts. The length of time on metformin therapy required for study inclusion varied from 2 weeks in the Rotterdam study, to 1 month in the Japanese study, and 6 months in the GoDARTS study. The GoDARTS authors took the additional step of genotyping the excluded subgroups and found no difference in genotype frequencies compared with either the initial cohort or final study groups.
A later study by Christensen et al. examined 159 patients with T2D participating in the South Danish Diabetes Study, a prospective inter vention study in which patients were randomized in a 2 × 2 ×2 factorial design to receive insulin aspart versus NPH insulin, metformin versus placebo, and rosiglitazone versus placebo (thus, metformin was given in four of the eight treatment arms); of note, metformin and rosiglitazone were given from the start of the study, while insulin dose was titrated over a 3-month period [36,37]. The authors found that trough steady-state metformin level was significantly lower in patients heterozygous for 420del; haplotype analysis of the four reduced-function variants (R61C, G401S, 420del and G465R) showed a decrease in trough steady-state metformin levels with increasing number of variant alleles, a trend that was additive and statistically significant. With respect to HbA1c, a significantly attenuated response to metformin over the initial 6 months was seen in subjects with increasing number of reduced function alleles; analysis of specific genotypes showed that this attenuated response was significant in those heterozygous for G401S but not in any of the other reduced-function variants. Subjects homozygous for the intronic SNP rs461473 showed an enhanced drop in HbA1c compared with the reference genotype. There was no association of the variant rs622342 identified by the Rotterdam study with either metformin level or HbA1c response [36]. Tkáč et al., attempting to replicate the Rotterdam study findings, also genotyped rs622342 in a prospective study of 148 drug-naive T2D patients started on metformin therapy; as with the Christensen study, there was no association with rs622342 and change in HbA1c after 6 months of therapy [38].
The influence of OCT1 on metformin response in phenotypes other than diabetes has also been explored. Gambineri et al. studied the influence of the hypofunctioning variants R61C, G401S, 420del and G465R on response to metformin treatment, characterized by multiple clinical and biochemical parameters, in women with PCOS. A total of 150 women, 84 homo zygous for the reference allele at all four positions, and 68 with at least one variant allele (52 with one, 13 with two and one with three polymorphisms in total), were treated prospectively with 1000 to 2700 mg daily of metformin for 6 months. In both the reference and variant groups, metformin treatment resulted in a significant reduction in BMI, improvement of menstrual status, increase in sex hormone binding globulin levels, and decrease in fasting glucose and insulin. However, with regards to total cholesterol and triglycerides, only the reference cohort showed a significant decrease in levels, whereas in the variant carriers there was no significant change. On the oral glucose tolerance test (OGTT), both the reference cohort and the 52 women with one variant allele had a similar reduction in OGTT-stimulated insulin secretion after metformin treatment; however, the 14 subjects with two or more OCT1 variants did not have a significant change in OGTT-stimulated insulin, although low power may have played a role [39]. Jablonski et al. used tagging SNPs in SLC22A1 to evaluate the impact of genetic variants on the protective effect of metformin on diabetes onset in 2994 participants in the Diabetes Prevention Program (DPP) [40]. The missense SNP L160F/rs683369 showed nominal significance, with a protective effect of the major allele on diabetes onset; however, the significance of this finding is uncertain, as this variant was not previously seen to have a significant impact on OCT1 function [22,30] and it is not in high linkage disequilibrium (LD; i.e., correlated via shared haplotypes) with any SNP previously associated with metformin level or response. No other SLC22A1 tagSNP, including rs622342, was associated with the protective effect of metformin [40]. Tarasova et al. examined the association of five variants (R61C, 420del, G465R, M408V and the 8 bp insertion rs36056065) with the risk of metformin side effects in a case–control study of 246 subjects (53 cases and 193 controls); all control subjects were identified via a government-funded biobank, while the majority of cases were recruited from the investigators' university hospital. Cases were defined as those who had experienced diarrhea, nausea, flatulence, abdominal pain, asthenia and vomiting while on metformin; no cases of lactic acidosis were identified during subject selection. After correction for multiple testing, the minor variants of M408V and the 8 bp insertion rs36056065 were significantly associated with decreased risk of side effects; these two SNPs are in strong LD with each other (r2 = 0.929) [41]. As noted above, while M408V had not previously been shown to have an effect on OCT1 transport function [27,29,30], the minor variant was associated with risk of nonresponse to metformin by Shikata et al. [33]. The effects of variation at rs36056065 on OCT1 function have not been characterized by any prior study; however, the 8 bp insertion of the minor variant occurs at the 3′ end of exon 7 and does not alter the amino acid sequence of OCT1, so it is unclear how this variant affects metformin response.
OCT2/SLC22A2
Characterization of variants in vitro & in healthy volunteers
The renal transporter OCT2 appears to have less functional variation compared with OCT1. An initial genetic and functional analysis by Leabman et al. in 2002 identified eight non-synonymous variants in 247 ethnically diverse samples; the four most common (M165I, A270S/rs316019, R400C and K432Q) were shown to have altered transporter function. The authors noted that nonsynonymous variants had significantly more skewed allele frequencies compared with synonymous variation, suggesting selection pressure against functional OCT2 variation; the most common functional variant was A270S [28]. In a Korean population, Kang et al. identified three nonsynonymous variants: A270S was the most common variant, while the novel variants T199I and T201M had low minor allele frequency (<1%); all had reduced transporter function compared with wild-type [42]. Shikata et al. sequenced SLC22A2 in addition to SLC22A1 (as discussed above), identifying A270S and T201M; they too noted that the number of nonsynonymous variants and their allelic frequencies were lower than in OCT1 as well as other known transporter genes [33].
The effect of variation in OCT2 with respect to metformin pharmacokinetics and response has been studied to a lesser extent than OCT1. Song et al. assessed the impact of the OCT2 variants T199I, T201M and A270S on metformin pharmacokinetics in vitro and in 26 healthy volunteers (n = 26; nine with reference genotype and 17 with a variant allele at one of the three loci); the OCT2 variants had reduced metformin uptake in vitro, and were associated with reduced CLrenal and higher peak plasma concentration in vivo [43]. Similarly, Wang et al. found A270S to be associated with reduced CLrenal in healthy volunteers (n = 15) although they did not find a difference in plasma metformin concentration between genotypes [44]. Somewhat surprisingly, Chen et al. found that healthy volunteers hetero zygous for the A270S variant (n = 9) had higher CLrenal compared with the reference group (n = 14); in heterozygotes, metformin plasma concentrations were higher in the first hour after metformin administration, but similar at later time points; there was no difference in peak plasma concentration [45]. In the pharmaco kinetic study described above by Tzvetkov et al., there was no significant association of A270S or tagging SNPs in OCT2 with CLrenal of metformin [32]. A recent study by Christensen et al. found an interaction between A270S and the MATE1 promoter variant g.-66T>C on CLrenal in 50 healthy volunteers: while neither variant alone had a significant effect, in patients homozygous for the reference g.-66T>C allele, presence of the minor variant of A270S predicted increased CLrenal in an additive fashion [46].
Effect of variants on clinical response
Studies of patient populations have yet to find an association with OCT2 variants and metformin response. In addition to sequencing, Shikata et al. assessed the association of A270S and T201M with metformin response in the 33 T2D patients categorized as responders or nonresponders; neither were associated with risk of metformin nonresponse in their small study population (n = 33) [33]. In the Danish T2D intervention trial analyzed by Christensen et al. described above, A270S did not show association with either metformin levels or change in HbA1c [36]. Tkáč et al., in their prospective study of 148 drug-naive T2D patients treated with metformin, similarly did not find an association with A270S and change in HbA1c after 6 months of therapy [38]. In the DPP, Jablonski et al. genotyped A270S along with 43 OCT2 tagging SNPs and did not find association of any with the effectiveness of metformin to delay diabetes onset [40]. Tarasova et al., in their study of metformin side effects, did not find an association with A270S [41].
OCT3/SLC22A3
Only a few studies have examined the influence of variants in OCT3 on metformin pharmacokinetics or effectiveness. Chen et al. identified six nonsynonymous variants through sequence analysis, three of which (rs68187715/T44M, rs8187717/A116S and rs8187725/T400I) had altered uptake of substrates including metformin in vitro [20]. Tzvetkov et al. used tagging SNPs to examine the impact of OCT3 variants on pharmaco kinetics in healthy volunteers and found no significant association of any with CLrenal of metformin [32].
MATE1/SLC47A1 & MATE2-K/SLC47A2
Characterization of variants in vitro & in healthy volunteers
Similar to OCT2/SLC22A2, the renal efflux transporters MATE1 and MATE2-K appear to have a low frequency of nonsynonymous variation in their coding regions. Kajiwara et al. identified five nonsynonymous MATE1 (V10L, G64D, A310V, D328A and N474S) and two nonsynonymous MATE-2K variants (K64N and G211V) in Japanese subjects, all of which (except V10L) showed reduced transporter function in vitro; allele frequencies ranged from 0.6 to 2.2% [47]. A follow-up report by the same group showed no difference in plasma-concentration time profiles after single-dose metformin administration in 48 subjects carrying one of the three most common MATE1/MATE2-K variants (G64D and L125F in SLC47A1, and D328A in SLC47A2) versus those with the reference genotype. Of note, no subject was homozygous for any MATE variant [48]. Chen et al. sequenced SLC47A1 in 272 individuals with ethnically diverse backgrounds and found six non-synonymous variants (G64D, L125F, V338I/rs35790011, V480M, C497S/rs35395280 and Q519H), none of which occurred in individuals of European ancestry. Of these, three were singleton variants (i.e., found in one study subject), two had allele frequencies of approximately 5% in the Mexican– or African–American samples, and one had an allele frequency of approximately 2% in African–Americans. In vitro functional analysis showed that all six nonsynonymous variants exhibited reduced uptake of metformin, although to varying degrees [49]. This group also sequenced SLC47A2 in the same cohort, finding four nonsynonymous variants (P162L/PMT5634, G393R/rs34399035, T505I/rs113679066 and A525T/rs133234335). Of these, one nonsynonymous variant (P162L) had an allele frequency of approximately 5% in African–Americans; the remaining variants had allele frequencies of less than 1% in all ethnic groups. Both this variant and the less common G393R showed significantly reduced transporter activity in vitro [50].
More recent work has focused on the influence of promoter variants in MATE1 and MATE2-K on metformin response. Following up their work on coding variants in MATE1, Ha Choi et al. sequenced the basal promoter region, and found that the most common variant, g.-66T>C/rs2252281 (minor allele frequency: 2.9–4.4%), was associated with decreased luciferase reporter activity in vitro and in vivo, suggesting a negative impact on MATE1 expression [51]. The same group also found four promoter region variants in their sequence analysis of MATE2-K (g.-166C>G/PMT5598, g.-130G>A/rs12943590, g.-46G>A/PMT5596 and g.-45C>T/PMT5595). The variant g.-130G>A was fairly common, with allele frequencies ranging from 26.2–48.5% among the different ethnicities; the other promoter variants were significantly less common at frequencies of 1.5% or less. This more common variant was associated with a significant increase in promoter activity; the others did not show any effect in reporter assays [50]. A follow-up study focusing on these promoter variants (MATE1 g.-66T>C and MATE2-K g.-130G>A) found that both were associated with altered post-metformin glucose tolerance in 57 healthy volunteers, with the MATE1 expression-inhibiting variant associated with increased response to metformin and the MATE2-K expression-enhancing variant associated with reduced metformin response [52].
Effect of variants on clinical response
In addition to the in vitro MATE-2K promoter variant findings described above, Choi et al. examined a retrospective cohort of 253 patients with T2D on metformin monotherapy and found that the variant g.-130G>A was associated with a weaker response to metformin in patients homozygous for the variant, with a markedly diminished reduction in HbA1c after metformin initiation [50]. In the South Danish Diabetes Study, Christensen et al. genotyped the MATE1 promoter variant g.-66T>C, but did not find a significant association with metformin response [36].
Tagging SNPs have also been used to assess the association of variants in MATE1/SLC47A1 with metformin response, both in the Rotterdam study and the DPP. In the Rotterdam study, the intronic SNP rs2289669 was associated with change in HbA1c after metformin initiation, with the minor A allele associated with a greater drop in HbA1c [53]. Similarly, Tkáč et al. observed a greater reduction in HbA1c in homozygous risk allele carriers [38]. In the DPP, the intronic SNP rs8065082, which is in LD with rs2289669 (r2 = 0.8), was associated with the protective effect of metformin, with the minor allele associated with lower diabetes incidence in the metformin arm but not in the placebo cohort. However, the association of rs2289669 with metformin response was unable to be replicated by Christensen et al. [36].
Candidate genes involved in metformin pharmacodynamics
Relatively few studies have explored potential pharmacodynamic pharmacogenomics of metformin. Legro et al. explored the effect of genetic variation in several candidate genes for treatment response in a prospective intervention study of fertility-enhancing treatment options for women with PCOS, where subjects were randomized to clomiphene, metformin or combined therapy. A total of 312 subjects agreed to participate in the genetic substudy (98 and 112 in the metformin and combined arms, respectively); the investigators found an association with STK11, the gene encoding the kinase LKB1, and metformin-induced improvement in ovulation [54]. Dong et al. examined the association of the serine racemase intronic SNP rs391300, which had recently been identified as a novel T2D susceptibility locus in a Taiwanese cohort, with metformin response in a prospective study of newly diagnosed T2D patients (n = 44); they found that carriers of the minor A allele (which was associated with protection from T2D in the Taiwanese) had significantly greater improvements in fasting and postprandial glucose and fasting cholesterol after 12 weeks of metformin therapy [55]. Jablonski et al. took a broad approach to assessing the genetics of the response to preventive interventions in the DPP, examining, in addition to metformin transporter genes, genes associated with monogenic diabetes, T2D, glycemic traits, lipid metabolism, monogenic obesity, putative metformin targets, cellular energy, hormonal regulation and response to exercise. Of these candidate genes for pharmacodynamic interactions, they detected nominal interactions with STK11, the AMPK subunit genes PRKAA1, PRKAA2 and PRKAB2, and two downstream targets of AMPK, PGC1A and HNF4A with metformin response [40].
Genome-wide association study
To date, there has been one hypothesis-free, genome-wide approach to assessing variants involved in metformin response. The GoDARTS and UKPDS metformin pharmacogenetics study groups investigated the genetics of metformin response in a discovery cohort of 1024 Scottish individuals with T2D and incident metformin use. A locus on chromosome 11, tagged by rs11212617, was associated with metformin response; this finding was replicated in two independent cohorts of 1783 additional Scottish subjects and 1113 individuals from the UK, with an overall genome-wide significant p-value of 2.9 × 10−9. This locus is in a large area of LD encompassing seven genes, ATM, CUL5, NPAT, C11orf65, EXPH5, ACAT1 and KDELC2. ATM was suggested as the most likely candidate given previous association with insulin resistance and T2D, and prior laboratory reports suggesting a role in AMPK activation [56]. However, the preliminary work carried out by this group linking ATM with metformin-induced AMPK activation was called into question in a subsequent report showing off-target effects of the ATM inhibitor KU-55933 [57]. More work is needed to clarify the causal variant and mechanism underlying the association tagged by rs11212617.
Conclusion & future perspective
Over the past few years, measurable progress has been made with respect to understanding the genetic contribution to the variability in clinical response to metformin. Much work has focused on variants in transporter genes, which clearly play a role in metformin pharmacokinetics, but to date the impact of these variants on clinical response in T2D patients is uncertain. Part of this gap may be owing to the difference of assessing clinical response compared with studying detailed pharmacokinetic measures in a controlled laboratory setting. It may be that evolutionary conservation of cation transporters, plus redundancy of transporters in vivo, has made variation in these genes of less clinical consequence. In addition, the effect of transporter variants on metformin uptake and excretion may be overcome by empiric titration by the practitioner of metformin dose to clinical response. However, the majority of metformin pharmacogenetic studies have been hindered by small sample size. The two large-scale populations explored to date, GoDARTS and the DPP, give conflicting results as to the significance of transporter genes in clinical practice; this disparity could stem from the different outcomes measured (diabetes treatment vs prevention), the diverse populations under study (patients with diabetes vs those with prediabetes), and the disparate study designs (retrospective observational vs prospective interventional). Additional large studies in the T2D population may help to clarify the significance of variation in these genes clinically.
In contrast to pharmacokinetics, the pharmacodynamic aspect of metformin pharmacogenetics has been less thoroughly explored. The uncertainty of the molecular basis for the clinical effects of metformin may have hindered previous investigation, but this ambiguity also provides an opportunity for pharmacogenetic studies to increase our understanding of basic biology. Moreover, T2D is a heterogeneous disorder; as our knowledge of the genetic architecture of T2D grows, our understanding of the genetic underpinnings of response to T2D treatments may improve. An agnostic genetic evaluation of metformin response may help identify the molecules and networks it targets. To date, there has been one such study, by the GoDARTS–UKPDS group, which produced a robust signal at a locus on chromosome 11. While promising, this locus only explains 2.5% of the variance in metformin response. Continued efforts using genome-wide approaches, at both the common and rare variant level, have the great potential to expand our understanding of metformin action and response.
In conclusion, the past 10 years have seen considerable progress in our comprehension of the pharmacogenetics of metformin response, yet there is still a wide gap to cross before translation to clinical practice can be realized. We expect in the next 5–10 years, improved understanding will be gained by studies that increase sample size, explore genome-wide associations and examine rare variation. Further insight into the genetic influence on the clinical response to metformin holds promise both for personalizing medicine and for serving as a basis for design of improved therapies.
Executive summary.
Genes involved in metformin transport
-
■
OCT1 is highly polymorphic, with four reduced-function variants identified that have demonstrable effect on metformin pharmacokinetics in healthy subjects. Evidence for impact of OCT1 reduced-function variants on clinical response is less clear, with effects seen in some but not all studies; many studies are hindered by small sample size and/or design limitations.
-
■
OCT2 has far less functional variation than OCT1, and conflicting results have been found for the impact of known variants on metformin pharmacokinetics in healthy subjects; no association with clinical outcome has been demonstrated with OCT2 variants.
-
■
The renal efflux transporters MATE1 and MATE2-K have a very low frequency of functional variation; promoter variants in MATE1 and MATE2-K have been associated with altered metformin response.
Genes involved in metformin pharmacodynamics
-
■
Studies of pharmacodynamic genetics have been relatively few owing to the uncertainty surrounding the mechanism of action of metformin.
-
■
STK11 was associated with metformin effectiveness in both polycystic ovary syndrome patients and subjects in the Diabetes Prevention Program.
-
■
The AMPK subunit genes PRKAA1, PRKAA2 and PRKAB2, and two downstream targets of AMPK, PGC1A and HNF4A, were also associated with metformin response in the Diabetes Prevention Program.
Associations discovered by genome-wide association studies
-
■
A locus on chromosome 11 (rs11212617) was associated with metformin response; it is unclear which of the seven genes in linkage disequilibrium with this SNP harbors the causal variant.
Conclusion & future perspective
-
■
While considerable progress has been made in our comprehension of the pharmacogenetics of metformin response, there is still a wide gap to cross before translation to clinical practice can be realized. Studies that increase sample size, explore genome-wide associations and examine rare variation have the potential to give insight into the genetic influence on the clinical response to metformin.
Acknowledgments
JN Todd is supported by NIH Training Grant T32 DK007260. JC Florez is supported by National Institute of Diabetes and Digestive and Kidney Diseases grants R01 DK072041 and R01 DK088214 and a Massachusetts General Hospital Scholars Award.
Footnotes
Financial & competing interests disclosure The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
No writing assistance was utilized in the production of this manuscript.
References
Papers of special note have been highlighted as:
■ of interest
■ of considerable interest
- 1.Danaei G, Finucane MM, Lu Y, et al. National, regional, and global trends in fasting plasma glucose and diabetes prevalence since 1980: systematic analysis of health examination surveys and epidemiological studies with 370 country-years and 2.7 million participants. Lancet. 2011;378(9785):31–40. doi: 10.1016/S0140-6736(11)60679-X. [DOI] [PubMed] [Google Scholar]
- 2.Inzucchi SE, Bergenstal RM, Buse JB, et al. Management of hyperglycemia in Type 2 diabetes: a patient-centered approach: position statement of the American Diabetes Association (ADA) and the European Association for the Study of Diabetes (EASD) Diabetes Care. 2012;35(6):1364–1379. doi: 10.2337/dc12-0413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Graham GG, Punt J, Arora M, et al. Clinical pharmacokinetics of metformin. Clin. Pharmacokinet. 2011;50(2):81–98. doi: 10.2165/11534750-000000000-00000. [DOI] [PubMed] [Google Scholar]
- 4.Viollet B, Guigas B, Sanz Garcia N, Leclerc J, Foretz M, Andreelli F. Cellular and molecular mechanisms of metformin: an overview. Clin. Sci. (Lond.) 2012;122(6):253–270. doi: 10.1042/CS20110386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gong L, Goswami S, Giacomini KM, Altman RB, Klein TE. Metformin pathways: pharmacokinetics and pharmacodynamics. Pharmacogenet. Genomics. 2012;22(11):820–827. doi: 10.1097/FPC.0b013e3283559b22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rena G, Pearson ER, Sakamoto K. Molecular mechanism of action of metformin: old or new insights? Diabetologia. 2013;56(9):1898–1906. doi: 10.1007/s00125-013-2991-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bailey CJ. Biguanides and NIDDM. Diabetes Care. 1992;15(6):755–772. doi: 10.2337/diacare.15.6.755. [DOI] [PubMed] [Google Scholar]
- 8.Stumvoll M, Nurjhan N, Perriello G, Dailey G, Gerich JE. Metabolic effects of metformin in non-insulin-dependent diabetes mellitus. N. Engl. J. Med. 1995;333(9):550–554. doi: 10.1056/NEJM199508313330903. [DOI] [PubMed] [Google Scholar]
- 9.Bailey CJ, Turner RC. Metformin. N. Engl. J. Med. 1996;334(9):574–579. doi: 10.1056/NEJM199602293340906. [DOI] [PubMed] [Google Scholar]
- 10.Zhou G, Myers R, Li Y, et al. Role of AMP-activated protein kinase in mechanism of metformin action. J. Clin. Invest. 2001;108(8):1167–1174. doi: 10.1172/JCI13505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Shaw RJ, Lamia KA, Vasquez D, et al. The kinase LKB1 mediates glucose homeostasis in liver and therapeutic effects of metformin. Science. 2005;310(5754):1642–1646. doi: 10.1126/science.1120781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Foretz M, Hebrard S, Leclerc J, et al. Metformin inhibits hepatic gluconeogenesis in mice independently of the LKB1/AMPK pathway via a decrease in hepatic energy state. J. Clin. Invest. 2010;120(7):2355–2369. doi: 10.1172/JCI40671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Owen MR, Doran E, Halestrap AP. Evidence that metformin exerts its anti-diabetic effects through inhibition of complex 1 of the mitochondrial respiratory chain. Biochem. J. 2000;348(Pt 3):607–614. [PMC free article] [PubMed] [Google Scholar]
- 14.El-Mir MY, Nogueira V, Fontaine E, Averet N, Rigoulet M, Leverve X. Dimethylbiguanide inhibits cell respiration via an indirect effect targeted on the respiratory chain complex I. J. Biol. Chem. 2000;275(1):223–228. doi: 10.1074/jbc.275.1.223. [DOI] [PubMed] [Google Scholar]
- 15.Stephenne X, Foretz M, Taleux N, et al. Metformin activates AMP-activated protein kinase in primary human hepatocytes by decreasing cellular energy status. Diabetologia. 2011;54(12):3101–3110. doi: 10.1007/s00125-011-2311-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hawley SA, Ross FA, Chevtzoff C, et al. Use of cells expressing gamma subunit variants to identify diverse mechanisms of AMPK activation. Cell Metab. 2010;11(6):554–565. doi: 10.1016/j.cmet.2010.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Miller RA, Chu Q, Xie J, Foretz M, Viollet B, Birnbaum MJ. Biguanides suppress hepatic glucagon signalling by decreasing production of cyclic AMP. Nature. 2013;494(7436):256–260. doi: 10.1038/nature11808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhou M, Xia L, Wang J. Metformin transport by a newly cloned proton-stimulated organic cation transporter (plasma membrane monoamine transporter) expressed in human intestine. Drug Metab. Dispos. Biol. Fate Chem. 2007;35(10):1956–1962. doi: 10.1124/dmd.107.015495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Muller J, Lips KS, Metzner L, Neubert RH, Koepsell H, Brandsch M. Drug specificity and intestinal membrane localization of human organic cation transporters (OCT) Biochem. Pharmacol. 2005;70(12):1851–1860. doi: 10.1016/j.bcp.2005.09.011. [DOI] [PubMed] [Google Scholar]
- 20.Chen L, Pawlikowski B, Schlessinger A, et al. Role of organic cation transporter 3 (SLC22A3) and its missense variants in the pharmacologic action of metformin. Pharmacogenet. Genomics. 2010;20(11):687–699. doi: 10.1097/FPC.0b013e32833fe789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nies AT, Koepsell H, Winter S, et al. Expression of organic cation transporters OCT1 (SLC22A1) and OCT3 (SLC22A3) is affected by genetic factors and cholestasis in human liver. Hepatology. 2009;50(4):1227–1240. doi: 10.1002/hep.23103. [DOI] [PubMed] [Google Scholar]
- 22.Shu Y, Sheardown SA, Brown C, et al. Effect of genetic variation in the organic cation transporter 1 (OCT1) on metformin action. J. Clin. Invest. 2007;117(5):1422–1431. doi: 10.1172/JCI30558. [DOI] [PMC free article] [PubMed] [Google Scholar]; ■ Key work establishing that OCT1 is essential for both hepatic uptake of metformin as well as the glucose-lowering effects of the drug.
- 23.Otsuka M, Matsumoto T, Morimoto R, Arioka S, Omote H, Moriyama Y. A human transporter protein that mediates the final excretion step for toxic organic cations. Proc. Natl Acad. Sci. USA. 2005;102(50):17923–17928. doi: 10.1073/pnas.0506483102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tanihara Y, Masuda S, Sato T, Katsura T, Ogawa O, Inui K. Substrate specificity of MATE1 and MATE2-K, human multidrug and toxin extrusions/H(+)-organic cation antiporters. Biochem. Pharmacol. 2007;74(2):359–371. doi: 10.1016/j.bcp.2007.04.010. [DOI] [PubMed] [Google Scholar]
- 25.Takane H, Shikata E, Otsubo K, Higuchi S, Ieiri I. Polymorphism in human organic cation transporters and metformin action. Pharmacogenomics. 2008;9(4):415–422. doi: 10.2217/14622416.9.4.415. [DOI] [PubMed] [Google Scholar]
- 26.Masuda S, Terada T, Yonezawa A, et al. Identification and functional characterization of a new human kidney-specific H+/organic cation antiporter, kidney-specific multidrug and toxin extrusion 2. J. Am. Soc. Nephrol. 2006;17(8):2127–2135. doi: 10.1681/ASN.2006030205. [DOI] [PubMed] [Google Scholar]
- 27.Kerb R, Brinkmann U, Chatskaia N, et al. Identification of genetic variations of the human organic cation transporter hOCT1 and their functional consequences. Pharmacogenetics. 2002;12(8):591–595. doi: 10.1097/00008571-200211000-00002. [DOI] [PubMed] [Google Scholar]
- 28.Leabman MK, Huang CC, Kawamoto M, et al. Polymorphisms in a human kidney xenobiotic transporter, OCT2, exhibit altered function. Pharmacogenetics. 2002;12(5):395–405. doi: 10.1097/00008571-200207000-00007. [DOI] [PubMed] [Google Scholar]
- 29.Sakata T, Anzai N, Shin HJ, et al. Novel single nucleotide polymorphisms of organic cation transporter 1 (SLC22A1) affecting transport functions. Biochem. Biophys. Res. Commun. 2004;313(3):789–793. doi: 10.1016/j.bbrc.2003.11.175. [DOI] [PubMed] [Google Scholar]
- 30.Shu Y, Leabman MK, Feng B, et al. Evolutionary conservation predicts function of variants of the human organic cation transporter, OCT1. Proc. Natl Acad. Sci. USA. 2003;100(10):5902–5907. doi: 10.1073/pnas.0730858100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Shu Y, Brown C, Castro RA, et al. Effect of genetic variation in the organic cation transporter 1, OCT1, on metformin pharmacokinetics. Clin. Pharmacol. Ther. 2008;83(2):273–280. doi: 10.1038/sj.clpt.6100275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tzvetkov MV, Vormfelde SV, Balen D, et al. The effects of genetic polymorphisms in the organic cation transporters OCT1, OCT2, and OCT3 on the renal clearance of metformin. Clin. Pharmacol. Ther. 2009;86(3):299–306. doi: 10.1038/clpt.2009.92. [DOI] [PubMed] [Google Scholar]
- 33.Shikata E, Yamamoto R, Takane H, et al. Human organic cation transporter (OCT1 and OCT2) gene polymorphisms and therapeutic effects of metformin. J. Hum. Genet. 2007;52(2):117–122. doi: 10.1007/s10038-006-0087-0. [DOI] [PubMed] [Google Scholar]
- 34.Becker ML, Visser LE, van Schaik RH, Hofman A, Uitterlinden AG, Stricker BH. Genetic variation in the organic cation transporter 1 is associated with metformin response in patients with diabetes mellitus. Pharmacogenomics J. 2009;9(4):242–247. doi: 10.1038/tpj.2009.15. [DOI] [PubMed] [Google Scholar]
- 35.Zhou K, Donnelly LA, Kimber CH, et al. Reduced function SLC22A1 polymorphisms encoding organic cation transporter 1 (OCT1) and glycaemic response to metformin: a Go-DARTS study. Diabetes. 2009;58(6):1434–1439. doi: 10.2337/db08-0896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Christensen MM, Brasch-Andersen C, Green H, et al. The pharmacogenetics of metformin and its impact on plasma metformin steady-state levels and glycosylated hemoglobin A1c. Pharmacogenet. Genomics. 2011;21(12):837–850. doi: 10.1097/FPC.0b013e32834c0010. [DOI] [PubMed] [Google Scholar]
- 37.Gram J, Henriksen JE, Grodum E, et al. Pharmacological treatment of the pathogenetic defects in Type 2 diabetes: the randomized multicenter South Danish Diabetes Study. Diabetes Care. 2011;34(1):27–33. doi: 10.2337/dc10-0531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tkáč I, Klimcakova L, Javorsky M, et al. Pharmacogenomic association between a variant in SLC47A1 gene and therapeutic response to metformin in Type 2 diabetes. Diabetes Obes. Metab. 2013;15(2):189–191. doi: 10.1111/j.1463-1326.2012.01691.x. [DOI] [PubMed] [Google Scholar]
- 39.Gambineri A, Tomassoni F, Gasparini DI, et al. Organic cation transporter 1 polymorphisms predict the metabolic response to metformin in women with the polycystic ovary syndrome. J. Clin. Endocrinol. Metab. 2010;95(10):E204–E208. doi: 10.1210/jc.2010-0145. [DOI] [PubMed] [Google Scholar]
- 40.Jablonski KA, Mcateer JB, De Bakker PI, et al. Common variants in 40 genes assessed for diabetes incidence and response to metformin and lifestyle interventions in the diabetes prevention program. Diabetes. 2010;59(10):2672–2681. doi: 10.2337/db10-0543. [DOI] [PMC free article] [PubMed] [Google Scholar]; ■■ Broad-based candidate gene approach to the genetics of metformin response in a large multiethnic population studied in the context of a randomized clinical trial.
- 41.Tarasova L, Kalnina I, Geldnere K, et al. Association of genetic variation in the organic cation transporters OCT1, OCT2 and multidrug and toxin extrusion 1 transporter protein genes with the gastrointestinal side effects and lower BMI in metformin-treated Type 2 diabetes patients. Pharmacogenet. Genomics. 2012;22(9):659–666. doi: 10.1097/FPC.0b013e3283561666. [DOI] [PubMed] [Google Scholar]
- 42.Kang HJ, Song IS, Shin HJ, et al. Identification and functional characterization of genetic variants of human organic cation transporters in a Korean population. Drug Metab. Dispos. Biol. Fate Chem. 2007;35(4):667–675. doi: 10.1124/dmd.106.013581. [DOI] [PubMed] [Google Scholar]
- 43.Song IS, Shin HJ, Shim EJ, et al. Genetic variants of the organic cation transporter 2 influence the disposition of metformin. Clin. Pharmacol. Ther. 2008;84(5):559–562. doi: 10.1038/clpt.2008.61. [DOI] [PubMed] [Google Scholar]
- 44.Wang ZJ, Yin OQ, Tomlinson B, Chow MS. OCT2 polymorphisms and in-vivo renal functional consequence: studies with metformin and cimetidine. Pharmacogenet. Genomics. 2008;18(7):637–645. doi: 10.1097/FPC.0b013e328302cd41. [DOI] [PubMed] [Google Scholar]
- 45.Chen Y, Li S, Brown C, et al. Effect of genetic variation in the organic cation transporter 2 on the renal elimination of metformin. Pharmacogenet. Genomics. 2009;19(7):497–504. doi: 10.1097/FPC.0b013e32832cc7e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Christensen MM, Pedersen RS, Stage TB, et al. A gene–gene interaction between polymorphisms in the OCT2 and MATE1 genes influences the renal clearance of metformin. Pharmacogenet. Genomics. 2013;23(10):526–534. doi: 10.1097/FPC.0b013e328364a57d. [DOI] [PubMed] [Google Scholar]
- 47.Kajiwara M, Terada T, Ogasawara K, et al. Identification of multidrug and toxin extrusion (MATE1 and MATE2-K) variants with complete loss of transport activity. J. Hum. Genet. 2009;54(1):40–46. doi: 10.1038/jhg.2008.1. [DOI] [PubMed] [Google Scholar]
- 48.Toyama K, Yonezawa A, Tsuda M, et al. Heterozygous variants of multidrug and toxin extrusions (MATE1 and MATE2-K) have little influence on the disposition of metformin in diabetic patients. Pharmacogenet. Genomics. 2010;20(2):135–138. doi: 10.1097/FPC.0b013e328335639f. [DOI] [PubMed] [Google Scholar]
- 49.Chen Y, Teranishi K, Li S, et al. Genetic variants in multidrug and toxic compound extrusion-1, hMATE1, alter transport function. Pharmacogenomics J. 2009;9(2):127–136. doi: 10.1038/tpj.2008.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Choi JH, Yee SW, Ramirez AH, et al. A common 5′-UTR variant in MATE2-K is associated with poor response to metformin. Clin. Pharmacol. Ther. 2011;90(5):674–684. doi: 10.1038/clpt.2011.165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ha Choi J, Wah Yee S, Kim MJ, et al. Identification and characterization of novel polymorphisms in the basal promoter of the human transporter, MATE1. Pharmacogenet. Genomics. 2009;19(10):770–780. doi: 10.1097/FPC.0b013e328330eeca. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Stocker SL, Morrissey KM, Yee SW, et al. The effect of novel promoter variants in MATE1 and MATE2 on the pharmacokinetics and pharmacodynamics of metformin. Clin. Pharmacol. Ther. 2013;93(2):186–194. doi: 10.1038/clpt.2012.210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Becker ML, Visser LE, van Schaik RHN, Hofman A, Uitterlinden AG, Stricker BHC. Genetic variation in the multidrug and toxin extrusion 1 transporter protein influences the glucose-lowering effect of metformin in patients with diabetes: a preliminary study. Diabetes. 2009;58(3):745–749. doi: 10.2337/db08-1028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Legro RS, Barnhart HX, Schlaff WD, et al. Ovulatory response to treatment of polycystic ovary syndrome is associated with a polymorphism in the STK11 gene. J. Clin. Endocrinol. Metab. 2008;93(3):792–800. doi: 10.1210/jc.2007-1736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Dong M, Gong ZC, Dai XP, et al. Serine racemase rs391300 G/A polymorphism influences the therapeutic efficacy of metformin in Chinese patients with diabetes mellitus Type 2. Clin. Exp. Pharmacol. Physiol. 2011;38(12):824–829. doi: 10.1111/j.1440-1681.2011.05610.x. [DOI] [PubMed] [Google Scholar]
- 56.Zhou K, Bellenguez C, Spencer CC, et al. Common variants near ATM are associated with glycemic response to metformin in Type 2 diabetes. Nat. Genet. 2011;43(2):117–120. doi: 10.1038/ng.735. [DOI] [PMC free article] [PubMed] [Google Scholar]; ■■ The sole genome-wide association study of metformin response to date.
- 57.Yee SW, Chen L, Giacomini KM. The role of ATM in response to metformin treatment and activation of AMPK. Nat. Genet. 2012;44(4):359–360. doi: 10.1038/ng.2236. [DOI] [PMC free article] [PubMed] [Google Scholar]
Website
- 101.Centers for Disease Control and Prevention . National diabetes fact sheet: national estimates and general information on diabetes and prediabetes in the United States, 2011. US Department of Health and Human Services, Centers for Disease Control and Prevention; GA, USA: 2011. www.cdc.gov/diabetes/pubs/factsheet11.htm. [Google Scholar]