

Abstract
This paper reviews the recent advances in the synthesis of catabolically stable sugar mimetics, C-oligosaccharides. These compounds are synthetic analogs of the naturally occurring O-oligosaccharides, in which the interglycosidic oxygen has been replaced by a methylene group. This review is organized in terms of chemistry used to assemble C-oligosaccarides under the sub-headings: anionic approaches, cationic methods, reductive glycosyl samarium chemistry, cyclization methodology, and free radical chemistry.
1. INTRODUCTION
Oligosaccharides (oligo means few in Greek) are composed of 2 to 10 glycosidically-linked mono-saccharides [1]. Oligosaccharides are hydrolyzed readily by aqueous acid to form an oligosaccharides constituent monosaccharides. Oligosaccharides represent a unique class of glycosides, in which the “aglycon” is also a carbohydrate residue instead of a simple alcohol.
Oligosaccharides and glycocojugates are involved in a multitude of biological processes including cell recognition, cell differentiation and cell adhesion. In addition, a number of protein-carbohydrate interactions mediate critical biological process such as cell signaling in growth and differentiation, and fertilization [2]. While these above processes are generally beneficial, carbohydrates also play major roles in a number of detrimental processes, including inflammation, viral and bacterial infections, and tumor metastasis. Major achievements in the development of carbohydrate based therapeutics include the synthesis of a heparin pentasaccharide, which displays selective antithrombic activity, and the design and synthesis of derivatives of sialyl Lewis X antigen, that are interest as potential anti-inflammatory agents [3].
However, the glycosidic bonds connecting individual saccharides units within carbohydrate-based therapeutics are unstable to mild acid and to glycosidase enzymes in vivo, thus agents with enhanced stability need be developed [4]. Considerable research has focused on the synthesis of C-glycosides, stable mimics of the naturally occurring O-saccharides. These molecules, in which the interglycosidic oxygen has been replaced by a methylene group, can be considered as chemically inert isosteres of natural O- and N-glycosides, and are stable to mild acid in vitro and glycosidase enzymes in vivo [5].
C-Oligosaccharides are defined as compounds with two or more sugar units linked by one carbon atom through (1→1), (1→2), (1→3) or (1→4) linkages, or linked by two carbon atoms for (1→1) or (1→6) linkages. Synthetically prepared C-glycosides have so far proved useful for probing stereoelectronic effects, which can control the conformation of oligosaccharides, and can also prevent viral and bacterial attachment to lectins in adhesion assays, to the same extent as O-glycosides [6]. C-glycosides can inhibit some carbohydrate processing enzymes. Oligosaccharides and glycoconjugates are biosynthesized through complicated, highly efficient enzyme mediated routes. The structure of the final glycoconjugate is governed by the action of a series of enzymes. Alteration in the regulation of these enzymes is believed to be responsible for the biosynthesis of carbohydrates associated with disease processes. Inhibitors of glycosidase and glycosyltransferase enzymes are currently receiving attention as new pharmaceutical agents. Moreover, a number of enzyme inhibitors have proven therapeutic efficacy for the treatment of diseases such as influenza and AIDS [7].
Biological studies have shown that the natural inhibitors highlighted in Fig. 1, exert their effects on a range of glycosidases and are therefore of limited use as medicinal agents [8]. Consequently, more specific inhibitors are currently being sought that can be engineered to more accurately resemble the substrates that they are mimicking and thereby provide greater selectivity. Early tests on C-glycosides have provided evidence for their potential to act as oligosaccharide processing inhibitors [7].
Fig. (1).
Natural inhibitors.
The area of C-linked glycoside synthesis has been extensively reviewed over the past 15 years [9]. The organization of this review is based on the type of chemistry used to assemble the target C-oligosaccharides. The chemistry involved includes anionic and cationic chemistry, reductive samariation, cyclization methodology and free radical approaches. Methods that afford the C-oligosaccharides with high stereoselectivity at the anomeric center (α or β) are clearly desirable. This selectivity can be controlled by either starting with a stereochemically defined C-glycoside, which is then coupled with another sugar, or by controlling the stereochemistry of the newly formed center.
2. ANIONIC APPROACHES
2.1. Enolate Anions
Fraser-Reid and Jarosz coupled two monosaccharide units through a directed aldol reaction [10]. Aldehyde 1 was deprotonated with (Me3Si)2NLi (LiHMDS) to give the chelated enolate 2, which was condensed with aldehyde 3 to give 4a as the major product (Scheme 1).
Scheme (1).

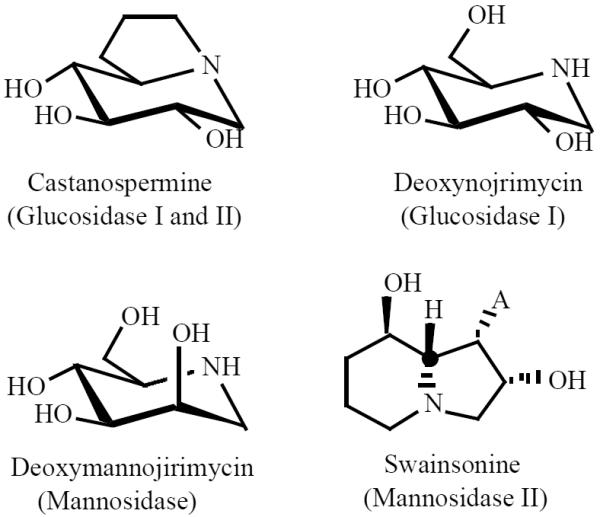
The zinc-copper-mediated reaction between 5 and 6 gives rise to the generation of single product 7, in 78% yield demonstrating a remarkable double stereoselectivity with a strong preference for α-C-glycosylation of the ulosyl bromide together with the elaboration of the (R) configuration for the CHOH group linking the pyranoid moieties (Scheme 2) [11].
Scheme (2).
Vogel utilized a “naked sugar” approach for the synthesis of an aza-C-disaccharide. Enolate derived from 8 was coupled with aldehyde 9 to give the adduct 10 in 61% yield. The ketone was reduced and the resulting diol was acetylated. This was followed by selenoxide elimination to afford olefin 11, which was converted to 12 as shown in Scheme 3 [12].
Scheme (3).

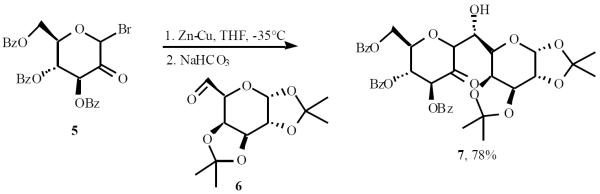
Cross-aldol reaction of racemic aldehyde 14 with enantiomerically pure lithium enolate of (−)-(1R,4R,5R,6R)- or (−)-(1S,4S,5S,6S)-13, results in mixtures composed mostly of enantiomerically pure (+)-15 and (-)-15, respectively, in 37–48% (only one of the isomers is shown in Scheme 4) [13].
Scheme (4).
Vogel and co-workers use a novel approach to C-disaccharide synthesis that involves conjugate addition and enolate trapping [14]. Levoglucosenone 16 was treated with Me2AlSPh to give the aluminium enolate 17. Condensation of 17 with aldehyde 18 gave the aldol product that was reduced with DIBAL to 20. Treatment of 20 with acidic methanol afforded the (1→3)-C-disaccharide 21 (Scheme 5).
Scheme (5).

Enone 22 was synthesized and condensation of trapping enolate with aldehyde 23 led to a mixture of aldols 24 and 25 that were separated by column chromatography on silica gel and isolated in 15% and 29% yield, respectively. Reduction of the major aldol 25 with NaBH4 in MeOH/THF was highly stereoselective and provided the C-disaccharide 26 in 77% yield (Scheme 6) [15].
Scheme (6).

When reacted with PhMe2SiZnMe2Li in THF at −78 °C enone 27 afforded a single adduct 28. Treatment of ketone 28 with dicyclohexylboron chloride and Et3N at −15 °C followed by addition of 29, the resulting aldol-borate was oxidized with 35% H2O2 giving adduct 30 in 24% yield. Stereoselective reduction of aldol 30 with Me4NBH(AcO)3 afforded diol 31 in 59% yield (Scheme 7) [15].
Scheme (7).

2.2. Nitro Stabilized Anions
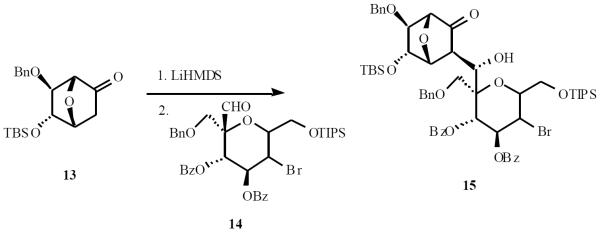
Vasella and Martin pioneered the use of nitro-based anions for C-saccharide synthesis. Anomeric nitro derivative 32 was treated with TBAF and exposed to aldehyde 33 to give the adduct 34 in 78% yield. Acetylation of 34 followed by reductive denitration afforded 35 (Scheme 8) [16].
Scheme (8).
Martin's work has also relied on the Henry reaction. Aldol condensation of nitro sugar 36 with aldehyde 33 gave 37. Dehydration of 37 and conjugate reduction led to 38. Removal of nitro group by radical-based reduction followed by the removal of protecting groups gave rise to the (1→6)-β-C-disaccharide 40 (Scheme 9) [17].
Scheme (9).

An open chain sugar derived aldehyde was used to access a C-trehalose derivative through the Henry reaction. Condensation of 36 with aldehyde 41 in the presence of KF and crown ether 18-crown-6, gave adduct 42, which was immediately converted to 43 with a sequence of reactions in 29% overall yield. The intermediate was finally transformed to the C-linked-(1→1)-disaccharide 44 (C-β,β-trehalose) (Scheme 10) [17].
Scheme (10).

Martin and coworkers applied the same methodology to prepare an O,C-trisaccharide, namely methyl O-β-d-glucopyranosyl-(1→4)-C-β-d-glucopyranosyl-(1→6)-d-glucopyranoside 48 [18]. The condensation of compound 45 with 6-aldehydo-glucopyranoside 46 in the presence of KF in an aprotic medium afforded the desired pseudotrisaccharide 47 in 73% yield. Application of the same chemistry to 47 gave the target compound 48 (Scheme 11).
Scheme (11).

Kobertz and coworkers utilized Martin's methodology to prepare C-allolactose. Nitro sugar 49 was condensed with aldehyde 50 afforded the adduct 51 as a mixture of diastereomers. The hydroxyl group was converted to the corresponding phenyl thiocarbonate ester 52. Treatment with tin hydride and a radical initiator effected elimination reaction, yielding only E-olefin 53. Finally the olefin was reduced with dimide (generated in situ) and benzyl ethers were removed by dissolving metal reduction to afford the target disaccharide 54 (Scheme 12) [19].
Scheme (12).

Gurjar et al. synthesized the first methyl α-C-D-araf-(1→5)-α-D-araf 60, the C-disaccharide segment of motif C of Mycobacterium tuberculosis, through nitro-aldol condensation. The coupling reaction between 55 and 56 gave diasteromeric mixture 57, which was subjected to three subsequent steps, i.e. dehydration, selective reduction of conjugated olefin and denitration, to give the penta-O-benzyl C-disaccharide 59 (Scheme 13) [20].
Scheme (13).

Witczak has used nitro anions for C-disaccharide synthesis. Michael addition of the nitro anion derived from 61 to levoglucosenone 62 gave adduct 63 in 43% yield. The sterics in 62 dictates the facial bias of the addition, which can only occur from the α-face. Reductive denitration followed by reduction of lactol and ketone afforded 64. Ring opening, deprotection and acetylation gave 65 (Scheme 14) [21].
Scheme (14).

2.3. Sulfur Stabilized Anions
The Taylor group employed the Meyers variant of the Ramberg-Bäcklund reaction to synthesize C-disaccharides [22]. The penta-O-benzyl thioglucose 66 was coupled with iodide compound 67 followed by oxidation of the formed sulfide gave sulfone 68. Exposure of the sulfone to the Meyers variant of the Ramberg-Bäcklund reaction afforded exo-glycal 69 as a mixture of isomers in 48% yield. Hydrogenation of this mixture stereoselectively reduced double bond and cleaved the benzyl groups. Peracetylated C-trehalose 70 was furnished by acetylation of free sugar (Scheme 15).
Scheme (15).

This approach is well suited to analog synthesis simply by varying the alkylating agent. Thus, the homologous (1→1')-C-disaccharide, containing a two-carbon bridge (“homoisotrehalose”), was readily prepared (Scheme 16) [23]. Lactol 71 was converted into sulfonate 74 by a straightforward three-step sequence involving Moffat-type C-glycosylation to give ester 72, followed by reduction and mesylation. Next, 74 was coupled with thiol 75 in a similar alkylation-oxidation sequence to that above. The resulting sulfone 77 underwent a halogenative Ramberg-Bäcklund rearrangement (RBR) to give exo-glycal 78 as a 88:12 mixture of (Z)- and (E)-isomers in 57% yield. Subsequent reduction/debenzylation followed by acetylation produced the novel, ethylene-bridged disaccahride 79.
Scheme (16).

This group also reported a rapid synthesis of a range of C-disaccharides involving a tandem Horner-Wadsworth-Emmons (HWE)/conjugate-addition C-glycosylation followed by a tandem halogenation-RBR sequence, utilizing Meyers variant of the RBR [24]. The HWE reagent 81 proceeded the HWE/conjugate-addition sequence with diisopropylidene mannofuranose 80 in 86% yield, to produce the adduct sulfone 82 (as a mixture of diastereomers). The key halogenation-RBR was carried out using the conditions devised by Chan et al. giving E-alkene 83 as the only isolable product. Alkene hydrogenation and concomitant debenzylation, followed by acetylation for characterization purposes, gave the novel disaccharide 84 in 69% yield over the two reactions (Scheme 17).
Scheme (17).

A two-directional variant of this methodology can be used to prepare symmetrical C-disaccharides, as shown in Scheme 18 [24]. The double HWE reaction on diisopropylidene mannofuranose 80 worked extremely well in the HWE/conjugation-addition C-glycosydation sequence giving diastereomeric sulfones 85, in a combined yield of 75%. RBR of the major stereoisomer (β,α-79) using the Meyers-Chan procedure gave alkene 86 in 66% unoptimized yield, and hydrogenation produced the novel C-linked bisfuranose disaccharide 87 in 76% yield.
Scheme (18).

Norris et al. deprotonated 88 with n-BuLi and the resulting anion was coupled with an excess lactone 89 at low temperature to afford the adduct 90 in 77% yield. SPh groups were removed with Raney nickel, and lactol 91 was isolated as a syrup that contained two components in approximately a 4:1 ratio. The two compounds were thought to be α- and β-lactols with the latter being major. It was straightforward to reduce the lactols with a high degree of stereocontrol to afford the C-disaccharide 92 with a 51% isolated yield (Scheme 19) [25].
Scheme (19).

2.4. Phosphorous Stabilized Anions
The most common type of reaction discussed in this section is Wittig approach to C-oligosaccharide synthesis. This methodology is particularly suited for synthesis of (1→6)-C-saccharides, since one sugar can hold the aldehyde while the other the ylid or phosphonate anion.
Dondoni has utilized this approach to prepare a variety of β-d-(1→6)-C- disaccharides. The general approach is shown in Scheme 20. Coupling of β-linked galactopyranosyl or mannofuranosyl derivative (93, 94, 95, or 96) with ylide from phosphonium iodide 97 or 98 gave alkene 99. Alkene was reduced and removal of deprotecting groups gave the corresponding β-d-(1→6)-C- disaccharide 100 [26].
Scheme (20).

α-d-(1→6)-C-disaccharide 103 was also synthesized using the approach (Scheme 21).
Scheme (21).

Dondoni applied this methodology in an iterative fashion to prepare the corresponding β-(1→6)-C-trigalactosides and β-(1→6)-C-tetragalactosides [27]. Starting with coupling of ylide 104, generated in situ with aldehyde 33, gave adduct 105 (E/Z 1:9). Removal of silyl protecting group followed by oxidation led to aldehyde 106, which was coupled with the ylide from 104 to give C-trisaccharide 107 in 36% yield. The same sequence was repeated with 107 afforded the protected C-tetrasaccharide alkene 108. Compounds 108 was desilylated and exposed to hydrogen over Pd(OH)2 on carbon followed to peracetylation gave protected C-tetrasaccharide 109 (Scheme 22).
Scheme (22).

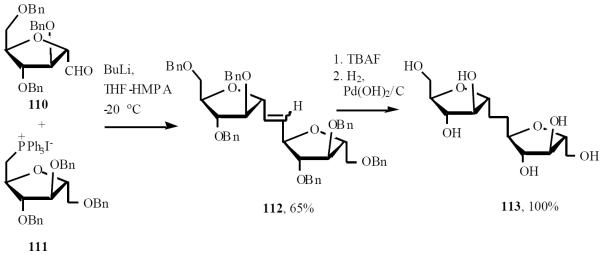
Taking advantage of their experience on the stereo-selective synthesis of C-oligosaccharides by iterative Wittig olefination, this group developed new building blocks and a modified protocol for the straightforward assembly of C-α-(1→5)-d-oligoarabinofuranosides [28]. While the two monofunctionalized sugar moieties, 110 and 111 were suitable precursors to the head and tail of the oligosaccharidic chain, the difunctionalized compound 114 was a repeating unit (Scheme 23). Building block 114 was designed to allow, after the Wittig coupling, for regeneration of the formyl group in a single step under conditions that do not affect the chemical and stereochemical integrity of the growing saccharidic chain (Scheme 24).
Scheme (23).
Scheme (24).

Colinas et al. reported a method for the synthesis of C-disaccharides by a sequence involving the Wittig reaction as key step and further glycosydation of these compounds to afford new C, O-trisaccharides [29]. The enol ether 122a-b were prepared by the Wittig reaction of the α, β mixture of 2-deoxiglycosyl phosphonium salts 120a-b with aldehyde 121. Hydrogenation of the exo-glycals afforded the β-C-disaccharides with good yields. The O-glycosidation employing the exo-glycals and the glycosyl acceptor 124 is illustrated in Scheme 25.
Scheme (25).

2.5. C-1 Vinyl Anions
Schmidt was the first to explore the addition of sulfoxide-stabilized vinyl C-1-organolithiums to carbohydrate-based carbonyl compounds [30]. The vinyl sulfoxide 126 was deprotonated with LDA to give 127. Addition of 127 to aldehyde 128 gave a mixture of diastereoisomers, of which the major 129 was formed in a 10:1 ratio. Desulfurization, hydroboration and removal of benzyl groups gave the (1→5)- β–C-disaccharide 130 (Scheme 26).
Scheme (26).

Schmidt and coworkers used this methodology for a highly efficient synthesis of an analog of C-trehalose 134, Scheme 27 [31]. Anion 131 was converted to aldehyde 132 and condensation of this aldehyde with the same anion gave adduct 133, which was then converted to the target C-disaccharide 134.
Scheme (27).

2.6. Acetylide-Based Anions
Sinaÿ was the first to prepare a C-disaccharide. His approach involved the coupling of a C-6 alkyne with an anomeric lactone followed by reduction of the formed hemiacetal [32] Aldehyde 135 was converted to the dibromoalkene 136, which was treated with n-butyllithium to give an intermediate acetylide 137. The latter, upon nucleophilic addition to lactone 138, gave a mixture of lactol 139 in high yield. Stereoselective reduction of lactol 139 was followed by hydrogenation of benzyl groups and the triple bond, affording the (1→6)-β-C-disaccharide 140 (Scheme 28).
Scheme (28).

The lactone-addition methodology was later applied by Sinaÿ and co-workers in an iterative fashion for the preparation of higher homologues [33]. Hydroxylmethylene in compound 141 was converted to acetylide lithium in compound 142 by a sequence of reactions. Condensation of compound 142 with the galacto lactone 143 gave 144 as mixture of anomers. The lactol were stereoselectively reduced to and triple bond was hydrogenated to give 145. Methyl glycoside 145 was hydrolyzed to the corresponding lactols, which were oxidized to lactone 146. The same sequence was then repeated twice to give C-trisaccharide 147 and C-tetrasaccharide 148 (Scheme 29).
Scheme (29).

The di-, tri- and tetrasaccharide derivatives (145, 147 and 148) were all debenzylated and tested for their binding affinity to three different monoclonal immunoglobulins with specificity for different O-β-(1→6)-d-galactopyranosyl determinants. These C-saccharide analogs had similar binding properties to the corresponding O-saccharides.
Schmidt has also used acetylide-lactone condensation to assemble C-disaccharides of ketoses [34]. Lithium acetylide 149 was condensed with ketone 150 to give a mixture of alcohol 151 after removal of the pivoloyl group. Attempted cyclization under Lewis acid catalysis resulted only in the removal of the acetonide. Accordingly, the diol was tied back as the carbonate and the acetylene complex as the dicobalt species 152. The isomers were then separately cyclized to give a 1:1 mixture of C-disaccharide 153. The cobalt complex on α-C-disaccharide was removed under standard conditions to give 154 and deprotection of the carbonate was followed by hydrogenation and peracetylation to give 155 (Scheme 30). Similar chemistry was also carried out with the corresponding β-isomer.
Scheme (30).

Van Boom and co-workers used lithiated alkynyl derivative 156 in presence of ZnCl2 to selectively open ring of the 1,2-anhydrosugar 157, affording the α-C-(alkynyl)-glycoside 158. Hydrogenolysis of the benzyl groups was followed by peracetylation to give the (1→6)-α-C-disaccharide 159 (Scheme 31) [35].
Scheme (31).

Normally, attack of the nucleophile proceeds from an equatorial direction to give the β-isomer. The unexpected stereochemical outcome is rationalized by the generation of an initial zinc acetylide species, which facilitates intramolecular delivery to the α-face (Scheme 32).
Scheme (32).

Wightman synthesized C-disaccharide analogs of the α-D-arabinofuranosyl-(1→5)-α-D-arabinofuranosyl motif of mycobacterial cell walls through alkynyl intermediates [36]. The lactone 164 was treated with lithio(trimethylsilyl)ethyne at low temperature to give lactol in 66% yield of 165. Treatment of this with triethylsilane and BF3·Et2O gave the separable isomers, 166α (60%) and 166β (13%). Desilylation of the major isomer, 166α gave 167 in 97% yield. Treatment of 167 with n-BuLi, followed by addition of lactone 164 gave 85% yield of the hemiacetal 168, which on reduction with Et3SiH and BF3·Et2O gave the disubstituted alkyne 169 (83%) as the major product. Reduction-hydrogenolysis of 169 then gave the C-disaccharide 170 in near-quantitative yield (Scheme 33).
Scheme (33).

3. REDUCTIVE GLYCOSYL SAMARIUM SPECIES
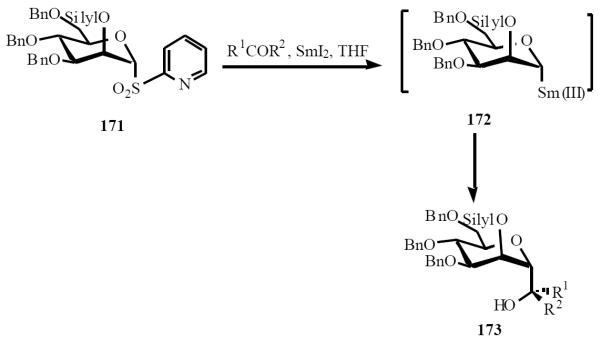
The introduction of SmI2 as reducing agent can be credited to Kagan and has since been utilized in numerous synthetic schemes [37]. Sinaÿ and Beau introduced SmI2 chemistry into C-glycoside synthesis through intermolecular reactions with appropriate electrophiles, and intramolecular reactions of silyl tethered substrates [38]. Anomeric samarium species are highly stable and not likely to undergo β-elimination. Treatment of the aryl sulfone 171 with SmI2 affords the nucleophilic intermediate 172 through two single electron transfer processes. Introduction of a suitable electrophile (carbonyl compounds), affords the α-C-mannopyrannoside 173 in good yield and with excellent stereo-selectivity (Scheme 34) [38a].
Scheme (34).
Preparation of C-glycosides of ulosonic acids using SmI2 was pioneered by Linhardt and coworkers [39]. Coupling of pyridyl sulfone 174 with aldehyde 175 in the presence of SmI2 gave an excellent yield of the N-acetylneuraminic acid C-disaccharide 176 as single isomer. This stereochemical outcome was rationalized on the basis of the Felkin-Anh model for predicting the stereochemical outcome of a kinetically controlled addition of a nucleophile to a chiral adehyde (Scheme 35, A).
Scheme (35).

Reductive samarium method was utilized to synthesize sialo-antigen C-analogs by the Linhardt group. A fully protected C-analogue of the SialylTn antigen, α-D-Neu5Ac-(2→6)α-D-GalNAc-(1→O)-Ser 182, was prepared [40]. C-glycosylation of the neuraminic acid sulfone donor with aldehyde acceptor in presence of excess SmI2 afforded the α-C-glycoside 180 as an R/S mixture (1:1). The critical intermediate, aldehyde acceptor 178, was prepared in 13 steps. Chemoselective resolution to compound 180 was undertaken by oxidation with DMSO/Ac2O to keto-bridged compound 181, followed by stereoselective reduction with Zn(BH4)2 to regenerate the bridge hydroxyl function, gave 182 in >90% de (Scheme 36).
Scheme (36).

A common versatile N-acetylneuraminic acid C-disaccharide precursor of the C-glycoside analogs of gangliosides GM4 and GM3 has been synthesized [41]. Samarium(II) iodide coupling of N-acetylneuraminic acid sulfone 179 with a 3-formyl galactopyranoside derivative 183 afforded the corresponding C-disaccharide 184 as a mixture of (R) and (S) isomers at the newly formed hydroxymethylene bridge. This diastereoisomeric mixture was resolved after debenzylation of the galactoside residue, and the chirality of each isomer assigned after acetylation. Conversion of the p-methoxyphenyl group in the peracetylated (S) isomer 187 into the corresponding thiophenyl glycoside 188 was accomplished, affording the key intermediate for the synthesis of C-glycoside analogs of GM4, GM3, and other related gangliosides (Scheme 37).
Scheme (37).

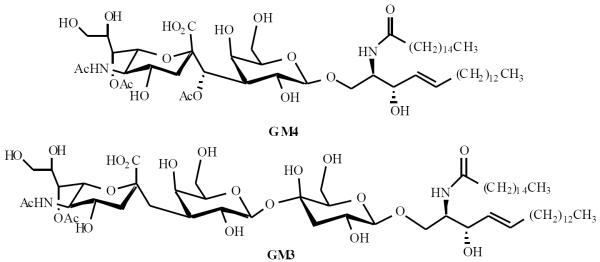
Ganglioside GM4 is a very important cell adhesion molecule. The C-disaccharide of GM4 is currently being synthesized in Linhardt's lab using standard O-glycosylation of C-disaccharide with protected ceramide. Once the ceramide has been attached, it can be oxidized to afford an aldehyde group for coupling to keyhole limpet hemocyanin (KLH) by reductive amination for use as carbohydrate-based vaccine. GM3, a related trisaccharide containg ganglioside, plays an important role in cell growth and differentiation, Fig. 2. The synthesis of C-oligosaccharide GM3 is currently underway following a similar route to that of C-GM4 synthesis [42].
Fig. (2).
Beau also reported a sialyl C-disaccharide synthesis which relied on the unprecedented use of a stable and crystalline 2-pyridyl sulfide of the N-acetylneuraminic acid derivative 189 as the anomeric precursor in a samarium-Reformastky procedure [43]. The coupling procedure with sulfide 189 and aldehyde 190 afforded the carbon-linked dimer 191 in 93% yield as a 1:1 diastereomeric mixture.
Alcohols 191 were converted into thiocarbonates and deoxygenated by employing triphenyltin hydride, catalytic AIBN, and pentafluorophenol, which yielded C-disaccharide 192 as a single compound in 65% yield for the 2 steps. Desilylation, hydrogenolysis, acetylation, and final acetolysis of the methyl glycoside provided the carbon-linked mimic 193 of the disaccharidic component of the sialylTn tumor antigen (Scheme 38).
Scheme (38).

4. CATIONIC METHODS
This section is categorized according to reactions that involve the generation of anomeric ions followed by the capture with sugar-based nucleophiles.
4.1. Exo-glycals as Nucleophiles
Nitotra et al. have found that treatment of exo-glycal 194 with BF3·OEt2 followed by a water quench, afforded the C-disaccharide 195 (Scheme 39). It is noteworthy to noteworthy that the attack of the enol ether on the anomeric oxonium ion occurs with high stereoselectivity on the α-face, only 195 was obtained [44].
Scheme (39).

4.2. Silyl-based Nucleophiles
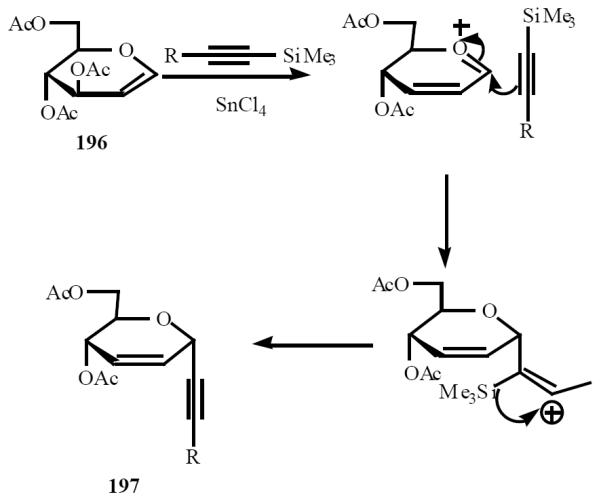
Isobe has designed and developed alkynylation as a key reaction for the introduction of a carbon chain onto sugars. Those alkynylated compounds are called “sugar acetylenes” [45]. The mechanism includes eliminative formation of cation to which silylacetylene can coordinate on the α-face (Scheme 40).
Scheme (40).
Double C-glycosylation between glycal 198 and bis (silylacetylene) gave C-disaccharide “like” compounds 199 and 200 (Scheme 41).
Scheme (41).

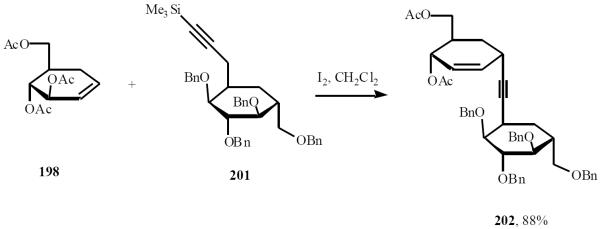
Silylpropargyl-sugar 201 smoothly reacted with D-glucal to furnish exclusively the α-acetylene glycoside product 202 in 88% (Scheme 42) [46].
Scheme (42).
5. CYCLIZATION METHODOLOGY
This approach to C-oligosaccharide synthesis relies on the coupling of an open chain fragment with an intact monosaccharide to give an intermediate that is then cyclized to deliver the C-oligosaccharide target. This section is divided by the type of chemistry used to initially assemble the two pre-cyclization fragments.
5.1. Wittig Reaction-Cyclization
The Kishi group was the first to use Wittig-cyclization approach to synthesize C-disaccharides. Starting with Wittig reaction between 203 and 204, the intermediate 205 was afforded as a single stereoisomer in 82% yield. Osmylation then gave a mixture of diols with the major one formed in a 6:1 ratio. Selective protection of the diols gave 206a and 206b. The mono-para-methoxybenzyl ether 206a was then transformed into the hemiketal 207 in 75% overall yield in three steps, i.e., (1) Swern oxidation, (2) acetonide hydrolysis (4 N HCl/THF/rt), and (3) benzoylation (3 equiv of PhCOCl/py/CH2Cl2/rt). Silane reduction of 207 in an acidic medium should preferentially yield the equatorially substituted β-C-glycoside which was deblocked and converted to the methyl- β-C-disaccharide, 209. Exact parallel experiments, applied to the minor isomer, gave the corresponding C-disaccharide 210 (Scheme 43) [47].
Scheme (43).

Armstrong has employed Wittig-osmylation protocol for the preparation of a small library of α-C-(1→6)-disaccha-rides [48]. Readily available allyl C-glycoside 211 was homologated to diene ester 212 and subjected to osmylation followed by reduction to afford a 5:1 mixture (213/214: 215/216) of lactols that were readily separable by chromatography. Following a deprotection of an equimolar mixture of 213 and 214, 217 and the D/L hybrid C-disaccharide of d-glucose-O-(1→6)-L-galactose 218 were obtained. The D/D and D/L derivatives 219 and 220 of glucose-α-(1→6)-idose were also obtained as a near equimolar mixture after deprotection of 215 and 216 (Scheme 44).
Scheme (44).

5.2. Anion Addition-Cyclization
Kishi developed an approach for the synthesis of the C–trehalo sugars, which relied on nucleophilic addition of one intact sugar fragment to a fundamental aldehyde, i.e., Kishi-Nozaki reaction [49]. Coupling of monosaccharide 221 and aldehyde 222 gave 223. The triple bond was partially reduced and osmylation was followed by selective protection to afford 224. The acetonide was converted to epoxide 225 by standard methods and removal of the PMB group. Acid-catalyzed cyclization followed by aglyconic hydrolysis and protection of hydroxyl group with (p-methoxyphenyl)diphenylmethyl chloride (MMTrCl) gave 226. Inversion of the C-2 hydroxyl and removal of protecting groups delivered α, α-C-trehalose 227 (Scheme 44).
Kishi and co-workers also employed aldol reactions as an efficient access to the C-trisaccharide related to the type II ABO(H) blood group determinant [50]. Aldol condensation of the enolate of methyl ketone 228 with aldehyde 229 gave a 4:1 mixture of hydroxy ketones 230 and 231. The major ketol was cyclized, desulfurized and oxidized to afford the disaccharide ketone 233. The ketone in THF was treated with LiHMDS (3 equiv) and TMEDA, followed by the addition of MgBr2. Addition of the aldehyde 234 led to almost exclusive formation of the equatorial products 235 and 236 in a 1:2 ratio. Mesylation of 235 and 236 followed by treatment with liquid ammonia gave enones 237a and 237b, which were reduced to yield exclusively the C-2'-equatorial ketone 238. Sodium borohydride reduction of the C-3' carbonyl proceeded with the desired stereoselectivity to yield the protected C-trisaccharide 239, which was deblocked to provide the polyol 240 (Scheme 46).
Scheme (46).

Sutherlin and Amstrong cleverly applied a recursive stereochemical deconvolution (RSD) combinatorial approach for the synthesis of a small library of C-trisaccharides as potential inhibitors for the cell surface proteins of bacterium Helicobactor pylori [51]. The workers chose to carry out variations in galactose ring of these trisaccharides. An efficient Nozaki-Kishi coupling of vinyl bromide 241 with aldehyde 242 in a diasteromeric ratio of 1:2, followed by protection with TBSOTf led to 243 and 244. Hydroboration followed by oxidation gave aldehydes 245 and 246, respectively. Addition of allylmagnesium bromide to aldehyde 245 followed by removal of the silyl group afforded 247, to 246 giving 248 and 249. Epoxidation gave a 1:1 mixture of the corresponding epoxides which were cyclized, to give the C-trisaccharide 250–255, after removal of the benzyl groups (Scheme 47).
Scheme (47).

Schmidt reported a stereoselective, 6-exo-trig selective, electrophilic cylization approach for de novo synthesis of a methylene-bridged Neu5Ac-α-(2,3)-Gal C-disaccharide, the C-analog of an important motif in glycosphingolipids [52]. The open chain precusors 259a/b (a 3:2-mixture of diaseteromeric alcohols) were formed by the addition of lithiated iodide 256 to open chain aldehyde 258. Subsequent C1 incorporation using Tebbe-reagent, formation of a cyclic carbonate, and deprotection of the two isopropylidene ketals afforded tetrol 264 which, upon treatment with phenylselenyl trifilate, was steroselectively cyclized in a 6-exo-trig selective manner, affording a diasteromeric mixture of 251 and 252 in a ratio of 7:1. A seleno-Pummerer rearrangement to afford aldehyde 267 in 5 steps in 90% overall yield. Oxidation of the aldehyde and subsequent steps led to the desired C-disaccharide 268 (Scheme 48).
Scheme (48).

5.3. Esterification-Cyclization
Work from the Postema group has focused on a ring closing metathesis (RCM) approach for the synthesis of C-saccharides [53a]. The chemistry is versatile and has been used for the synthesis of a small library of differentially linked β-C-disaccharides through a radical allylation-RCM strategy [53b]. The synthesis of the β-(1→4)-glucopyranosyl glucopyranoside 280 represents an example of this chemistry. The known triacetyl glucopyranoside 269 was converted to iodide 270 in good yield. Radical allylation of 271 proceeded to afford a mixture of equatorial and axial allylation products in a 6:1 ratio. Oxidative cleavage of the double bond gave the corresponding aldehydes 273a and 273b, which were separable. Pinnick oxidation of the major aldehyde 273a then gave the C-4 equatorial acid 274. DCC-mediated coupling of alcohol 275 and acid 274 proceeded to afford ester 276 in good yield. Methylenation of 276 gave 277 and exposure to catalysist 278 gave a 41% yield of the C-disaccharide glycal, which was exposed to an excess of BH3·THF to give 64% yield of the β-C-disaccharide 279. Removal of the benzyl groups and global peracetylation gave the peracetylated (1→4)-β-C-disaccharide 281 in good yield (Scheme 49).
Scheme (49).

5.4. Ring Expansion-Cyclization
Nelson developed a new strategy for asymmetric and stereoselective synthesis of C-linked disaccharide mimetics, the C58-C71 fragment of palytoxin [54]. The diol (R,R)-282 was converted into the di-dihydropyran (DHP) template 283, 284 and 285. The di-DHPs were functionized in a two directional sense using dihydroxylation reactions. The two-directional approach was often very efficient indeed: for example, in the synthesis of 286, six new stereogenic centers were introduced with almost complete stereocontrol using a reduction and a dihydroxylation reaction. The approach is not restricted to the synthesis of C2-symmetrical disaccharide mimetics. Using the unsymmetrical template 285, which has both a pseudo-axial and a pseudo-equatorial hydroxyl groups, the unsymmetrical mimetics 289 and 290 were prepared (Scheme 50).
Scheme (50).

6. RADICAL APPROACHES
The radical approach for C-C bond formation is a popular method within organic chemistry. The use of radical chemistry in carbohydrate synthesis has certain advantages. Firstly, the reaction conditions are very mild and tolerant of a range of functional and protecting groups. Second, anomeric radicals are stable with respect to elimination and epimeration. Most significantly, the chemistry required to incorporate an appropriate substitute at C-1, employed in the initial hemolytic cleavage step, is common within carbohydrate chemistry [55]. The use of such radical technique can be subdivided into two classes, intermolecular and intramolecular approaches.
6.1. Intermolecular Radical Approaches
Giese was the first to employ an intermolecular radical-based approach for synthesis of C-disaccharides [56]. Addition of an alkene to an anomeric radical, under appropriate conditions, allowed exclusive entry to the α-linked C-glycoside, a result which was mirrored by the work of Baldwin [57]. The high α-selectivity of the reaction was in contrast with other reports. Thus radicals generated at the C-2, C-3 and C-4 centers all preferred to occupy equatorial positions [58]. When the anomeric bromide 291 was treated with tin hydride and AIBN, resulting anomeric radical, and reacted with lactone 292 in a Michael addition, it afforded 293. In this reaction hydrogen abstraction at the resulting radical center occurs from axial face to selectively generate the equatorial C-2' substituent. Reduction and acetylation of 293 furnished C-disaccharide 294 (Scheme 51) [59].
Scheme (51).

Witczak used the facial bias of levoglucosene to stereoselectively add a carbohydrate-based radical in the preparation of the 3-deoxy-(1→4)-β-C-disaccharide 299 [60]. Addition of the radical derived from iodide 295 to 296 gave adduct 297 in 26% yield. Removal of the anomeric hydroxyl of 297 was achieved by treatment with triethyl silane, borontrifluoride ether complex followed by stereoselective reduction of the keto function at C-2' with the formation of 298 in 89% yield. Debenzylation followed by acetolysis of 1,6-anhydro ring produced the peracetylated target C-disaccharide 299 in 19% overall yield from 295 (Scheme 52).
Scheme (52).

Vogel has developed general methods to transform enantiomerically pure 7-oxabicyclo[2,2,1]heptyl derivatives (“naked sugars”) into C-disaccharides. Because of these chiral bicyclic skeletons, they undergo highly stereoselective reactions with predictable diasteroselectivities. An arsenal of methods is available that allow to substitute these “naked” carbon centers with a variety of functional groups with all possible relative configurations. The methods allow preparation of both enantiomers of a given target with the same ease. Predictably high stereoselectivity of the reactions of the bicyclic chirons adds to the flexibility of approach, which can lead to high molecular diversity [61].
α-C-(1→3)-mannopyranoside of N-acetyl-galactosamine, potent β-galactosidase inhibitors, was synthesized by this approach [61]. Enone 302 was readily prepared from 300. The addition of the radical derived from bromide 303 to enone 302 afforded the α-isomer 304. The ketone was then reduced and the selenide eliminated via the intermediacy of the selenoxide, to give 305. Acid-promoted (CF3SO3H) 7-oxa ring opening of 305 in MeCN produced the amino-conduritol derivative 306 resulting from the quenching of the allylic cation intermediate by the solvent (Ritter reaction). Ozonolysis of the chloroalkene 306 generated an acyl chloride-aldehyde intermediate that reacted with MeOH to produce a mixture of methyl uronates. The major compound 307 was silylated and reduced. The crude polyol obtained was acetylated to produce 308. Desilylated and ammonolysis afforded a mixture of α-/β-pyranoses 309 and corresponding α-/β-furanoses (Scheme 53).
Scheme (53).

Similar chemistry was carried out to synthesize non-protected α-C(1→2)-L-fucopyranoside of D-galactopyrano-side, corresponding to a blood group mimetic [62]. Giese's radical fucosidation of racemic enone 310 with α-L-bromoacetofucose 311, provided the mixture of diastereomeric α-C-fucoside 312a and 312b in reproducible yield of 78%. Reduction of the mixture gave a 1:1 mixture of endo-alcohols 313 and 314, which were isolated pure in 40% and 35.5% yield, respectively. Oxidative elimination of the benzeneselenyl group of 313 followed by acetylation gave endo acetate 315 in 85% yield. Acid-promoted ring opening of 315 and ozonolysis of the resulted chloroalkene gave methyl uronic ester 316 in modest yield (10%). Treatment of 316 with Cl3CCN and NaH, followed with BF3·OEt2 furnished the totally protected C-disaccharide 317 in 79%. The reduction of 317 and treatment with DOWEX-H+ resin provided the desired C-fucoside 318 in 70% (Scheme 54).
Scheme (54).

6.2. Intramolecular Radical Approaches
One disadvantage with the intermolecular approaches is that during such radical mediated reactions, electron deficient alkenes are required to facilitate C-C bond formation. The intramolecular approach of Sinaÿ overcomes this drawback by bring two substrates together via a temporary covalent silaketal connector. This enables the use of a wider range of alkenes, which can include alkene-functionalized sugars, thus leading to the generation of C-disaccharides [63]. Based on the early work of Stork, Sinaÿ employed the tether reaction in the synthesis of both α- and β-C-disaccharides, with this work subsequently being employed in the synthesis of biologically significant natural products [64]. The availability of different hydroxyl groups on each monosaccharide unit allowed the fine-tuning of stereochemical outcome by selective positioning of the silyl tether [65]. Development of this early stereochemical control led to the tethering of the donor 320 with alcohol 319 through a di-isopropyl silicon tether 321. Subsequent treatment with Bu3SnH and AIBN, led to a 9-endo-trig cyclization to form the C-disaccharide 322 in an 80% yield with exclusive formation of the α-product. The silyl tether was removed with Bu4NF, and deprotection under standard conditions afforded the desired disaccharide 323 in excellent yield (Scheme 55).
Scheme (55).

In parallel, the development of a 3,2' silyl tether allowed access to the β-linked product as demonstrated in Sinaÿ's synthesis of methyl β-C-lactoside 328 [66]. Alcohol 324 and 320 were connected via the dimethylsilyl tether, an 8-endo-trig cyclization followed to exclusively afford the β-disaccharide 326 in 45% yield. Removal of the tether and deprotection of benzyl ether gave β-C-disaccharide 328 (Scheme 56).
Scheme (56).

This group also selected iodosugars bearing a free hydroxyl group as progenitors of the radical donors [67]. In a typical experiment, the radical donor 329 was connected to the radical acceptor 330 through a silaketal tether, following their procedure. Tributyltinhydride mediated 8-endo-trig radical cyclization of compound 331 gave, after detethering of the non isolated intermediate 332, the protected β-C-disaccharide 333 in 37% overall yield (from starting materials 330 and 329). Peracetate 335 was achieved after hydrogenolysis. The C-laminaribioside 334 is of potential biological interest as it mimics the repeating disaccharide unit of fungal β-(1→3)-glucans, which have antitumor and immunomodulating properties (Scheme 57).
Scheme (57).

CONCLUSIONS
In this paper, the biological importance of C-oligosaccharides as subunits of natural products and as a new generation of carbohydrate based on therapeutics has been illustrated. Different methods are available for the synthesis of C-oligosaccharides. As with traditional saccharide synthesis, it has been necessary to develop a number of specific approaches to facilitate the regioselective and stereoselective preparation of the desired targets. Work in this area continues and it is expected that many interesting new C-oligosaccharides will be prepared using the chemistry described in this review. It is also likely that new chemistry will be developed in the future for the preparation of these biologically important targets.
Scheme (45).

ACKNOWLEDGEMENTS
The authors thank Dr. Sultan Baytas for her critical reading of this manuscript.
REFERENCES
- [1].Kennedy JF, White CA. Bioactive Carbohydrates In Chemistry, Biochemistry and Biology. Ellis Horwood Ltd.; Chichester,UK: 1983. [Google Scholar]
- [2].Wassarman PW. Mammalian Fertilization; Molecular Aspects of Gamete Adhesion, xocytosion, Exocytosis, and Fusion. Cell. 1999;96:175–183. doi: 10.1016/s0092-8674(00)80558-9. [DOI] [PubMed] [Google Scholar]
- [3].Wong C-H, editor. Carbohydrate-based Drug Discovery. Wiley-VCH; Weinheim; Cambridge: 2003. [Google Scholar]
- [4].(a) Sharon N, Lis H. Lectins as Cell Recognition Molecules Science. 1987;246:227–234. doi: 10.1126/science.2552581. [DOI] [PubMed] [Google Scholar]; (b) Dwek RA. Chem. Rev. 1996;96:683–720. doi: 10.1021/cr940283b. [DOI] [PubMed] [Google Scholar]
- [5].Postema M. C-Glycoside synthesis, CRC, Boc Ratom. 1995 [Google Scholar]
- [6].(a) Weatherman RV, Mortell KH, Cheruenak M, Kiessling LL, Toone EJ. Specificity of C-Glycoside Complexation by Mannose/Glucose Specific Lectins. Biochem. 1996;35:3619–3624. doi: 10.1021/bi951916z. [DOI] [PubMed] [Google Scholar]; (b) Ravishankar R, Surolia A, Vijayan M, Lim S, Kishi Y. Preferred Conformation of C-Lactose at the Free and Peanut Lectin Bound States. J. Am. Chem. Soc. 1998;120:11297–11303. [Google Scholar]
- [7].(a) Wong C-H, Halcomb RL, Ichikawa Y, Kajimoto T. Enzymes in organic synthesis; Application to the problems of carbohydrate recognition part2. Angew. Chem. Int. Ed. Engl. 1995;34:521–546. [Google Scholar]; (b) Howard S, Withers SG. Bromoketone C-Glycosides, a New Class of β–Glucanase Inactivators. J. Am. Chem. Soc. 1998;120:10326–10331. [Google Scholar]; (c) Wang Q, Wolff M, Polat T, Du Y, Linhardt RJ. C-glycoside of Neuraminic Acids as Neura-minidase Inhibitors. Bioorg. Med. Lett. 2000;10:941–944. doi: 10.1016/s0960-894x(00)00132-3. [DOI] [PubMed] [Google Scholar]; (d) Phili R, Chang J, Partis RA, Mueller RA, Chrest FJ, Passaniti A. The Alpha-glucosidase I Inhibitor Castanospermine Alters Endothelial cell Glycosylation, Prevents Angiogenesis, and Inhibits Tumor Growth. Cancer Res. 1995;55:2927–2935. [PubMed] [Google Scholar]
- [8].Ossor A, Elbein AD. Carbohydrates in Chemistry and Biology. II. Vol. 3. Wiley/VCH; 2000. pp. 513–539. [Google Scholar]
- [9].(a) Sinaÿ P. Synthesis of oligosaccharide Mimetics. Pure Appl. Chem. 1997;69:459–463. [Google Scholar]; (b) Beau J-M, Gallagher T. Nucleophilic C-glycosyl Donors for C-glycoside Synthesis. Top. in Curr. Chem. 1997;187:1–54. [Google Scholar]; (c) Nicotra F. Synthesis of C-glycosides of Biological Interest. Top. in Curr. Chem. 1997;187:55–83. [Google Scholar]; (d) Du Y, Linhardt RY. Recent Adv. in Stereo-Selective C-Glycoside Synthesis. Tetrahedron Lett. 1998;54:9913–9959. [Google Scholar]; (e) Liu L, Mckee M, Postema MHD. Synthesis of C-saccharides and Higher Congeners. Current Org. Chem. 2001;5:1133–1167. [Google Scholar]
- [10].Jarosz S, Fraser-Reid B. Synthesis of A Higher Carbon Sugar Via Directed Aldol Condensation. Tetrahedron Lett. 1989;30:2359–2362. [Google Scholar]
- [11].Lichtenthaler FW, Lergenmiiller M, Schwidetzky S. C-Glycosidations of 2-Ketohexosyl With Electrophilic, Radical, and Nudeophilic Anomeric Carbons. Eur. J. Org. Chem. 2003:3094–3103. [Google Scholar]
- [12].Baudat A, Vogel P. Aza-C-disaccharides; Synthesis of 6-Deoxygalactonojirimycin-β-C(1→3) Linked with D-Altrofuranosides and D-Galactose. J. Org. Chem. 1997;62:6252–6260. [Google Scholar]
- [13].Turner D, Vogel P. Synthesis of 4-Hydroxy-2,6-Bis(hydroxymethyl) tetrahydropyran-2-Carbaldehyde Derivatives. Synlett. 1998:304–306. [Google Scholar]
- [14].Zhu Y-H, Vogel P. Short, Stereoselective Synthesis of C-(1→3)-Linked Disaccharides. Tetrahedron Lett. 1998;39:31–34. [Google Scholar]
- [15].Zhu Y-H, Vogel P. Synthesis of a (2R, 6R)-2-(Hydroxymethyl)-6-Propa-1, 2-Dienyl-2H-Pyran-3(6H)-One Derivative, A New Enone for The Convergent Construction of C-Glycosides of C-Disaccharides. Synlett. 2001:82–86. [Google Scholar]
- [16].Baumberger F, Vasella A. Deoxynitrosugars. The 6th Communication. Stereolectronic Control in the reductive denitration of tertiary nitro ethers. A Synthesis of “C-glycosides”. Helv. Chim. Acta. 1983;66:2210–2221. [Google Scholar]
- [17].Martin OR, Lai W. Synthesis and Conformational Studies of β-(1→6)- and β,β-(1→1)-Linked C-disaccharides. J. Org. Chem. 1993;58:176–185. [Google Scholar]
- [18].Spak SJ, Martin R. The synthesis of an analogue of Methyl O,C-Analogue of Methyl 4'-O-β-D-Glucopyranosylgentiobioside. Tetrahedron. 2000;56:217–224. [Google Scholar]
- [19].Kobertz WR, Bertozzi CR, Bednarski MD. C-Glycosyl Aldehyde: Synthons for C-Linked Disaccharides. J. Org. Chem. 1996;61:1894–1897. doi: 10.1021/jo9517095. [DOI] [PubMed] [Google Scholar]
- [20].Gurjar MK, Nagaprasad R, Ramana CV. First Synthesis of Methyl α-C-D-Arabino-furanosyl-(1→5)-α-D-Arabinofuranoside; the C-Disaccharide Segment of Motif C of Mycobaterium Tuberculosis. Tetrahedron Lett. 2000;43:7577–7579. [Google Scholar]
- [21].Witczak ZJ. Synthesis of C-Glycosyl Compounds and Other Natural Products from Levoglucosenone. Pure Appl. Chem. 1994;66:2189–2192. [Google Scholar]
- [22].Griffin FK, Paterson D, Taylor RJK. Ramberg-Backlund Approaches to the Synthesis of C-Linked Disaccharides. Angew Chem. Int. Engl. 1999;38:2939–2942. doi: 10.1002/(sici)1521-3773(19991004)38:19<2939::aid-anie2939>3.0.co;2-s. [DOI] [PubMed] [Google Scholar]
- [23].Paterson BE, Griffin FK, Alcaraz M-L, Taylor RJK. A Ramberg-Bäcklund Approach to the Synthesis of C-Glycosides, C-Linked Disaccharides, and C-Glycosyl Amino Acids. Eur. J. Org. Chem. 2002:1323–1336. [Google Scholar]
- [24].McAllister GD, Paterson DE, Taylor RJK. A Simplified Ramberg-Bachlund Approach to Novel C-Glycosides and C-Linked Disaccharides. Angew. Chem. Int. Ed. Engl. 2003;42:1387–1391. doi: 10.1002/anie.200390356. [DOI] [PubMed] [Google Scholar]
- [25].McCartney JL, Christopher TM, Cicchillo RM, Bernardina MD, Wagner TR, Norris P. J. Org. Chem. 2003;68:10152–10155. doi: 10.1021/jo0350256. [DOI] [PubMed] [Google Scholar]
- [26].Dondoni A, Zuurmond HM, Bascarate A. Synthesis of α-and β-D-(1→6)-C-Disaccharides by Witting Olefination of Formyl C-Glycosides with Glycopyranose 6-phosphoranes. J. Org. Chem. 1997;62:8114–8124. doi: 10.1021/jo971177n. [DOI] [PubMed] [Google Scholar]
- [27].Dondoni A, Kleban M, Zuurmond H, Marra A. Synthesis of (1→6)-C-Oligogalactosides by Interative Wittig Olefination. Tetrahedron Lett. 1998;39:7991–7994. [Google Scholar]
- [28].Dondoni A, Marra A. A New Synthetic Approach to Mycobacterial Cell Wall α-(1→5)-D-Arabinofuranosyl C-Oligosaccharides. Teterahedron Lett. 2003;44:4067–4071. [Google Scholar]
- [29].Colinas PA, Ponzinibbio A, Lieberknecht A, Bravo RO. Wittig Reaction of Glycosyl Phosphonium Salts; A Stereoselective Route to C-Disaccharides and C, O-Trisaccharides. Tetrahedron Lett. 2003;44:7985–7988. [Google Scholar]
- [30].Schmidt RR, Preuss R. Synthesis of Carbon. Tetrahedron Lett. 1989;30:3409–3412. [Google Scholar]
- [31].Dietrich H, Schmidt RR. Synthesis of Carbon-Bridged C-Lactose and Derivatives. Liebigs. Ann. Chem. 1994:975–981. [Google Scholar]
- [32].Rouzaud D, Sinaÿ P. The First Synthesis of a “C-Disaccharide”. J. Chem. Soc., Chem. Commun. 1983:1353–1354. [Google Scholar]
- [33].Xin Y-C, Zhang Y-M, Mallet J-M, Glaudemans CPJ, Sinaÿ P. Synthesis of C-Oligosaccharides That Mimie Their Natural O-Analogues Immunodeterminants in Binding to Monoclonal Immunoglobulins. Eur. J. Org. Chem. 1999:471–476. [Google Scholar]
- [34].Streicher H, Geyer A, Schmidt RR. C-Disaccharides of Ketoses. Chem. Eur. J. 1996;2:502–510. doi: 10.1002/chem.19960020508. [DOI] [PubMed] [Google Scholar]
- [35].Leeuwenburgh MA, Timmers CM, Vander Marel GA, Van Boom JH, Mallet J-M, Sinaÿ PG. Stereoselective Synthesis of α-C-(Alkynyl)-Glycosides Via Ring-Opening of α-1,2-Anhydrous. Tetrahedron Lett. 1997;38:6251–6254. [Google Scholar]
- [36].Aslam T, Fuchs MGG, Le Formal A, Wightman RH. Synthesis of C-Disaccharide Analogue of the α-DArabinofuranosyl-(1→5)-α-Arabinofuranosyl Motif of Mycobacterial Cells Walls via Alkynyl Intermediates. Tetrahedron Lett. 2005;46:3249–3252. [Google Scholar]
- [37].(a) Girad P, Namy JL, Kagan HB. Divalent Lanthanide Derivative in Organic Synthesis. 1. Mild Preparation of Samarium Iodide and Ytterbium Iodide and Their Use as Reducing or Coupling Agents. J. Am. Chem. Soc. 1980;102:2693–2698. [Google Scholar]; (b) Molander GA, Harris CR. Sequencing Reactions with Samarium(II) Iodide. Chem. Rev. 1996;96:307–338. doi: 10.1021/cr950019y. [DOI] [PubMed] [Google Scholar]
- [38].(a) Mazéas D, Skrydstrup T, Doumeix O, Beau J-M. Samarium Iodide-Induced Intramolecular C-Glycoside Formation; Efficient Radical Formation in the Absence of an Additive. Angew. Chem. Int. Ed. Engl. 1994;33:1383–1386. [Google Scholar]; (b) Mazéas D, Skrydstrup T, Beau J-M. A Highly Stereoselective Synthesis of 1, 2-trans-C-Glycosides Via Glycosyl Samarium(III) Compounds. Angew. Chem. Int. Ed. Engl. 1995;34:909–912. [Google Scholar]; (c) Jarreton O, Skrydstrup T, Beau J-M. The Stereospecific Synthesis of Methyl α-C-Mannobioside A Potential Inhibitor of M. Tuberculosis Binding to Human Macrophages. J. Chem. Soc., Chem. Commun. 1996:1661–1662. [Google Scholar]; (d) Jarreton O, Skrydstrup T, Espionosa J-F, Jiménez-Barbero J, Beau J-M. Samarium Diiode Prompted C-Glycosylation: An Application to The Stereospecific Synthesis of α-(1→2)-C-Mannobioside and Its Derivatives. Chem. Eur. J. 1999;5:430–441. [Google Scholar]; (e) Mipuel N, Doisneau G, Beau J-M. Reductive Samariation of Anomeric 2-Pyridyl Sulfones with Catalytic: An Unexpected Improvement in The Synthesis of 1,2-Trans-Diequatorial C-Glycosyl Compounds. Angew. Chem., Int. Ed. Engl. 2000;39:4111–4114. doi: 10.1002/1521-3773(20001117)39:22<4111::aid-anie4111>3.0.co;2-c. [DOI] [PubMed] [Google Scholar]
- [39].Vlahov IR, Vlahova PI, Linhardt RJ. Diastereocontrolled Synthesis of Carbon Glycosides of N-Acetylneuraminic Acid Via Glycosyl Samarium(III) Intermediates. J. Am. Chem. Soc. 1997;119:1480–1481. doi: 10.1021/ja962703f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Kuberan B, Sikkander SA, Tomiyama H, Linhardt RJ. Synthesis of a C-Glycoside Analogue of sTn; An HIV-and Tumor-Associated Antigen. Angew. Chem., Int. Ed. Engl. 2003;42:2073–2075. doi: 10.1002/anie.200351099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Bazin HG, Du Y, Polat T, Linhardt RJ. Synthesis of a Versatile Neuraminic Acid “C”-Disaccharide Precursor for the Synthesis of C-Glycoside Analogues of Gangliosides. J. Org. Chem. 1999;64:7254–7259. [Google Scholar]
- [42].Chen C-C, Ress DK, Linhardt RJ. Samarium Iodide-Mediated Synthesis of Neuraminic Acid C-Glycosides. In: Roy R, editor. Synthesis of Glycommetics. Vol. 896. ACS Syposium Series; 2004. pp. 53–80. [Google Scholar]
- [43].Abdallah Z, Doisneau G, Beau J-M. Synthesis of a Carbon-Linked Mimic of The Disaccharide Component of The Tumor-Relate SialyTn Antigen. Angew. Chem., Int. Ed. Engl. 2003;42:5209–5212. doi: 10.1002/anie.200351969. [DOI] [PubMed] [Google Scholar]
- [44].Lay L, Nicotra F, Panza L, Russo G, Caneva E. Synthesis of C-disaccharides through dimerization of exo-glycals. J. Org. Chem. 1992;57:1304–1306. [Google Scholar]
- [45].Isobe M, Nishizawa R, Hosokawa S, Nishikawa T. Stereocontrolled Synthesis and Reactivity of Sugar Acetylenes. J. Chem. Soc., Chem. Commun. 1998:2665–2676. [Google Scholar]
- [46].Saeeng R, Sirion U, Sahakitpichan P, Isobe M. Iodine Catalyzes C-Glycosidation of D-Glucal with Silylacetykene. Tetrahedron Lett. 2003;44:6211–6215. [Google Scholar]
- [47].(a) Babirad SA, Wang Y, Kishi Y. Synthesis of C-Disaccharides. J. Org. Chem. 1987;52:1370–1372. [Google Scholar]; (b) Wang Y, Babirad SA, Kishi Y. Preferred Conformation of C-Glycosides. 8 Synthesis of 1,4-Linked Carbon Disaccharides. J. Org. Chem. 1992;57:468–481. [Google Scholar]
- [48].Armstrong RW, Sutherlin DP. Strategies for the Synthesis of C-Disaccharides Containing D and L Sugars. Tetrahedron Lett. 1994;35:7743–7746. [Google Scholar]
- [49].Wei A, Kishi Y. Preferred Conformation of C-Glycosides. 12. Synthesis and Conformational Analysis of α,α-, α, β-, and β, β-Trehaloses. J. Org. Chem. 1994;59:88–96. [Google Scholar]
- [50].Haneda T, Goekjian PG, Kim SH, Kishi Y. Preferred Conformation of C-Glycosides. 10. Synthesis and Conformation Analysis of Carbon Trisaccharides. J. Org. Chem. 1992;57:490–498. [Google Scholar]
- [51].Sutherlin DP, Armstrong RW. Synthesis of 12 Stereochemistry and Structurally Diverse C-Trisaccharides. J. Org. Chem. 1997;62:5267–5283. [Google Scholar]
- [52].Notz W, Hartel C, Waldscheck B, Schmidt RR. De Novo Synthesis of a Methylene-Bridged Neu5Ac-α-(2,3)-Gal C-Disaccharide. J. Org. Chem. 2001;66:4250–4260. doi: 10.1021/jo015543l. [DOI] [PubMed] [Google Scholar]
- [53].(a) Postema MHD, Calimente D, Liu L, Behrmanm TL. An Olefin Metathesis Route for the Preparation of (1→6)-Linked-C-Disaccharide Glycals. A Convergent and Flexible Approach to C-Saccharide Synthesis. J. Org. Chem. 2000;65:6061–6068. doi: 10.1021/jo0005159. [DOI] [PubMed] [Google Scholar]; (b) Postema MH, Piper JL, Liu L, Faust M, Andreana P. Synthesis and Partial Biological Evaluation of a Small Library of Differentially Linked β-C-Disaccharide. J. Org. Chem. 2003;68:4748–4754. doi: 10.1021/jo030039x. [DOI] [PubMed] [Google Scholar]
- [54].Nelson A. The Development of Strategies and Methods for The Synthesis of Biologically Active Compounds. New J. Chem. 2004;28:771–776. [Google Scholar]
- [55].Togo H, He W, Waki Y, Yokoyama M. C-Glycosylation Technology With Free Radiant Reactions. Synlett. 1998:700–717. [Google Scholar]
- [56].Giese B, Dupuis J. Diastereoselective Synthesis of C-Glycopyranosides. Angew. Chem. Int. Ed. Engl. 1983;22:622–623. [Google Scholar]
- [57].Adlington RM, Baldwin JE, Basak A, Kozyrod RP. Application of Radical Addition Reactions to tho Synthesis of a C-Glucoside and A Functionalized Amino Acid. J. Chem. Soc., Chem. Commun. 1983:144–145. [Google Scholar]
- [58].Giese B, González-Gómez JA, Witzel T. The Scope of Radical CC-Coupling by The “Tin Method”. Angew. Chem. Int. Ed. Engl. 1984;23:69–70. [Google Scholar]
- [59].Giese B, Witzel T. Synthesis of “C-Disaccharides” by Radical C-C Bond Formation. Angew. Chem. Int. Ed. Engl. 1986;25:450–451. [Google Scholar]
- [60].Witczak ZJ, Chhabra R, Chojnacki J. C-Disaccharides I. Stereoselective Approach to β-(1→4)-3-Deoxy-C-Disaccharides from Levoglucosenone. Carb. Res. 1997;301:167–175. [Google Scholar]
- [61].Vogel P. Synthesis of Rare Carbohydrates and Analogues Starting from Enantiomerically Pure 7-Oxabicyclo[2.2.1]heptyl Derivatives (“Naked Sugars”) Current Org. Chem. 2000;4:455–480. [Google Scholar]
- [62].Viodé C, Vogel P. Synthesis of The C-Disaccharide α-C(1→3)-L-Fucopyranoside of N-Acetylgalactosamine. J. Carb. Chem. 2001;20:733–746. [Google Scholar]
- [63].Xin YC, Mallet JM, Sinaÿ P. An Expeditious Synthesis of A C-Disaccharide Using A Temporary Silaketal Connection. J. Chem. Soc., Chem. Commun. 1993:864–865. [Google Scholar]
- [64].(a) Petitou M, Herault P, Lormeau J-C, Helmboldt A, Mallet J-M, Sinaÿ P, Herbert J-M. Introducing A C-Interglycosidic Bond in A Biologically Active Pentasaccharide Hardly Affects Its Biological Properties. Bioorg. Med. Chem. 1998;6:1509–1516. doi: 10.1016/s0968-0896(98)00094-7. [DOI] [PubMed] [Google Scholar]; (b) Sinaÿ P. Advances in The Synthesis of the Carbohydrate Mimics. Pure. Appl. Chem. 1998;70:1495–1499. [Google Scholar]
- [65].Sinaÿ P. Chemical Synthesis of Oligosaccharide Mimetics. Pure. Appl. Chem. 1998;70:407–410. [Google Scholar]
- [66].Sinaÿ P. Chemical Synthesis of Oligosaccharide Mimetics. Pure. Appl. Chem. 1998;69:459–463. [Google Scholar]
- [67].Vauzeilles B, Sinaÿ P. Selective Radical Synthesis of β-C-Disaccharides. Tetrahedron Lett. 2001;42:7269–7272. [Google Scholar]