

Abstract
Background
Alterations in glucose metabolism and epithelial-mesenchymal transition (EMT) constitute two important characteristics of carcinoma progression toward invasive cancer. Despite an extensive characterization of each of them separately, the links between EMT and glucose metabolism of tumor cells remain elusive. Here we show that the neuronal glucose transporter GLUT3 contributes to glucose uptake and proliferation of lung tumor cells that have undergone an EMT.
Results
Using a panel of human non-small cell lung cancer (NSCLC) cell lines, we demonstrate that GLUT3 is strongly expressed in mesenchymal, but not epithelial cells, a finding corroborated in hepatoma cells. Furthermore, we identify that ZEB1 binds to the GLUT3 gene to activate transcription. Importantly, inhibiting GLUT3 expression reduces glucose import and the proliferation of mesenchymal lung tumor cells, whereas ectopic expression in epithelial cells sustains proliferation in low glucose. Using a large microarray data collection of human NSCLCs, we determine that GLUT3 expression correlates with EMT markers and is prognostic of poor overall survival.
Conclusions
Altogether, our results reveal that GLUT3 is a transcriptional target of ZEB1 and that this glucose transporter plays an important role in lung cancer, when tumor cells loose their epithelial characteristics to become more invasive. Moreover, these findings emphasize the development of GLUT3 inhibitory drugs as a targeted therapy for the treatment of patients with poorly differentiated tumors.
Keywords: Epithelial-mesenchymal transition, Glucose transporter, GLUT3, Non-small cell lung cancer, SLC2A3, ZEB1
Background
The reprogramming of energy metabolism constitutes one of the hallmarks of cancer [1]. In order to build the necessary biomass required for proliferation, tumor cells increase their glucose consumption [2]. Glucose transporters of the GLUT (SLC2A) family are at the first step of cellular glucose utilization, mediating glucose entry by facilitative diffusion. The GLUT family is composed of 14 members that transport glucose or other substrates in different tissues, with different efficiencies [3]. In various tumor types, an increased expression of GLUT1 has been reported [4]. Furthermore, GLUT1 levels are higher in colorectal cancer cell lines harboring KRAS or BRAF mutations compared with isogenic clones and confer cell survival properties in low-glucose conditions [5]. Interestingly, in an oncogenic Kras(G12D)-dependent mouse model of pancreatic ductal adenocarcinoma (PDAC), oncogene withdrawal led to reduced Glut1 expression and glucose uptake [6]. In contrast to GLUT1, little is known about the regulation and function of another glucose transporter, GLUT3, in cancer. GLUT3 was originally referred to as the neuronal GLUT [7]; with a high affinity for glucose (KM approximately 1.5 mM) and the highest calculated turnover number of all glucose transporters, it ensures efficient glucose uptake by neurons of the central nervous system. With the exception of neurons and a few hematopoietic cell types, GLUT3 is lowly or not expressed in most organs of healthy adults. However, pathological GLUT3 expression has been reported in gastric, testicular, ovarian, and non-small cell lung cancer (NSCLC) [8,9]. Recently, GLUT3 was shown to be highly expressed in glioblastoma and to promote the growth of brain tumor initiating cells [10]. GLUT3 was also reported to be a transcriptional target of NF-κB and HMGA1, in mouse embryonic fibroblasts and human colorectal tumor cells, respectively [11,12]. However, the regulation and contribution of GLUT3 to lung tumor progression remain unknown.
The epithelial-mesenchymal transition (EMT) is a process that occurs early in embryonic development, notably during gastrulation, in which epithelial cells undergo cytoskeletal changes and lose cell-cell contacts to gain mesenchymal traits and become more motile [13]. During carcinoma progression, pathological EMT occurs to promote tumor cell invasion and metastasis. One of the essential steps in the EMT process is the loss of E-cadherin, an adherens junction protein that maintains cell-cell adhesion and epithelial tissue integrity. Mechanistically, the EMT process can be triggered by different transcription factors that include TWIST, ZEB1, ZEB2, SNAIL, and SLUG [14]. In our study, we discovered a strong association between the EMT program and the induction of the glucose transporter GLUT3 in NSCLC and extended this observation to tumor cells from another cancer type, hepatocellular carcinoma (HCC). We demonstrate that GLUT3 is a direct transcriptional target of ZEB1. We further show that GLUT3 expression contributes to proliferation of lung tumor cells and is an independent prognostic factor of poor overall survival in NSCLC.
Methods
Plasmid constructs
Human GLUT3, mouse Snail, and mouse Zeb1 cDNAs, purchased from Thermo Scientific (Waltham, MA, USA; MHS1010-7429646, MMM1013-7510291, and MMM1013-99828709, respectively), were amplified by polymerase chain reaction (PCR) using forward 5′-CTCATCGATGCCACCATGGGGACACAGAAGGT-3′ and reverse 5′-CTCCCCGGGTTAGACATTGGTGGTGG-3′ (GLUT3), forward 5′-CTCATCGATGCCACCATGCCGCGCTCCTTC-3′ and reverse 5′-CTCGAATTCTCAGCGAGGGCCTCC-3′ (Snail), and forward 5′-CTCATCGATGCCACCATGGCGGATGGCCCC-3′ and reverse 5′-CTCGAATTCCTAAGCTTCATTTGT-3′ (Zeb1) oligos. The PCR products were digested with ClaI and XmaI (GLUT3) or ClaI and EcoRI (Snail and Zeb1) and cloned into identically digested pRDI292-CMV lentiviral vector (gift of D. Trono, EPFL, Lausanne). Genomic DNA was used to amplify by PCR (a) an approximately 1,000-bp region ending close to the transcription start site of human SLC2A3 (gene encoding GLUT3), with the oligos forward 5′-CTCGAGCTCGAGACTAGCAGAAAGTG-3′ and reverse 5′-CTCCTCGAGCGACAAGCCCCCAGCCCCACCCCACCCCACCCCACCCCCCTGAAGCAA-3′, or (b) a region containing the SLC2A3 intron 2 sequence, with the oligos forward 5′-CTCGAGCTCACTGGGGTCATCAATGCTCC-3′ and reverse 5′-CTCCTCGAGGGTTGGTGGAAGAACAGAC-3′. After SacI and XhoI digestion, the fragments were cloned into an identically digested luciferase reporter plasmid containing a minimal promoter (kindly provided by J. Huelsken, EPFL, Lausanne) to generate prom.-LUC or int.2-LUC constructs, respectively. Deletion of the E-box-like motif CACCTC from the intron 2 sequence was achieved by site-directed mutagenesis, using oligos forward 5′-CCACTCTTTATAGTGATGCACATCCTG-3′ and reverse 5′-CATCACTATAAAGAGTGGGAGGAAGAAC-3′, combined with the oligos indicated above in (b). shRNAs specific to GLUT3 were either from Thermo Scientific (TRCN0000042880) or designed using the pSICOLIGOMAKER 1.5 program (created by A. Ventura, Memorial Sloan-Kettering Cancer Center, New York). In the latter case, forward 5′-TGCAAGGATGTCACAAGAAATTCAAGAGATTTCTTGTGACATCCTTGCTTTTTTC-3′ and reverse 5′-TCGAGAAAAAAGCAAGGATGTCACAAGAAATCTCTTGAATTTCTTGTGACATCCTTGCA-3′ oligos were annealed and ligated into a pSicoR lentiviral vector. The fidelity of all the PCR amplifications and oligo syntheses was confirmed by sequencing. Control pLKO.1 was from Thermo Scientific.
Immunoprecipitation
Cells (8 × 107 per immunoprecipitation) were lysed in NP-40 buffer (0.2% NP-40, 150 mM NaCl, 20 mM Tris pH 8.0, 10 mM EDTA) containing a protease inhibitor cocktail (complete, Roche, Basel, Switzerland) and 1 mM Na3VO4 for 15 min on ice, followed by three quick steps of freezing in liquid N2 and thawing at 37°C. Pre-clearing was achieved using sepharose-6B (Sigma-Aldrich, St. Louis, MO, USA) for 60 min at 4°C on a rotating wheel. Immunoprecipitation was performed using a 1:1 mixture of sepharose-6B and protein-G sepharose (Sigma-Aldrich), together with 2 μg control or ZEB1 antibody, overnight at 4°C on a rotating wheel. After four steps of washing in lysis buffer, sample buffer was added, and the samples were boiled and loaded on a polyacrylamide gel for electrophoresis followed by Western blot.
Western blotting
Except when used for immunoprecipitation, cells were lysed in RIPA buffer (20 mM Tris pH 8, 50 mM NaCl, 0.5% Na-deoxycholate, 0.1% SDS, 1 mM Na3VO4, protease inhibitor cocktail (complete, Roche)) for 5 min on ice. Proteins were loaded on 8% or 10% polyacrylamide gels for electrophoresis (150 V, 1 h). Transfer was performed on PVDF membranes (100 V, 1 h).
Cell culture conditions
The human embryonic kidney (HEK) 293 T cells and the human hepatoma cell lines, which include HLF, HLE, Huh-1 (all three kindly provided by K. Morikawa, Showa University, Tokyo, Japan), Huh-7, and Hep3B, were grown in DMEM. The human non-small cell lung cancer cell lines include A549, SW1573, NCI-H23, NCI-H2122, NCI-H441, NCI-H460, NCI-H727, NCI-H2009, NCI-H1944, and Calu-6; they were all obtained from ATCC (Manassas, VA, USA) and all grown in RPMI. All cell media were supplemented with 10% FBS. For the experiments with high or low glucose, cells were cultured for 4 days in normal RPMI (containing 11 mM glucose), supplemented by 10% FBS, or with RPMI without glucose, supplemented by 10% FBS, resulting in a final concentration of 11.5 mM and 0.5 mM glucose, respectively. After 2 days of culture, the medium was replaced with fresh medium to maintain the initial concentrations of glucose.
Cell counting
To measure cell number, a trypan blue exclusion assay was performed. Cells were mixed with trypan blue, and the number of live, dead, and total cells was counted on an automated cell counter (Countess, Life Technologies, Carlsbad, CA, USA).
2-Deoxy-d-[3H]glucose uptake
Cells (1 × 106 per well, seeded the day before the experiment) were cultured on six-well plates. 2-Deoxyglucose uptake assays were performed as described previously [15]. Radioactivity was determined by scintillation counting. For sample normalization, protein concentration was measured by BCA protein assay.
Transfection
siRNAs were from Life Technologies. siRNA transfection was performed with RNAiMAX transfection reagent (Life Technologies), according to the manufacturer’s instructions. For each siRNA, 10 nM was used. cDNA transfection of 293 T cells was performed with Lipofectamine 2000 (Life Technologies), according to the manufacturer’s instructions. For each well of a 12-well plate, a mix of 125 ng Renilla-Luciferase (phRL-TK, Promega, Madison, WI, USA) and 1.6 μg GLUT3-Luciferase (promoter, intron 2-WT or intron 2-Δ-CACCTC) was used. Twenty-four hours after transfection, cells were lysed and a luciferase reporter assay was performed (Dual-Luciferase Reporter Assay System, Promega). The reported luciferase activity was the ratio between Firefly (GLUT3 construct)-Luciferase and Renilla-Luciferase, which was then normalized to 1 to get fold activities. For double transfection, 293 T cells were transfected first with siRNAs and re-transfected 72 h later with plasmids, 24 h prior to lysis.
Anchorage-independent growth assays
Cells (9 × 104) were plated in triplicates in six-well plates in 0.4% agar in RPMI on top of a layer of 0.8% agar with RPMI. Cells were allowed to grow at 37°C for 3 weeks. Colonies were counted using a microscope. A colony was defined as an aggregate of 50 or more cells.
RNA purification, reverse transcription, and real-time PCR amplification
RNA was purified using Trizol (Life Technologies), according to the manufacturer’s instructions. RNA (1 μg) was reverse-transcribed using the High-Capacity cDNA Reverse Transcription Kit (Life Technologies). cDNA (5 ng) was used for real-time PCR amplification, using commercially available Taqman probes for human 18S, GAPDH, SLC2A3, SLC2A1, SLC2A4, SLC2A12, VIM, ZEB1, SNAI1, and CDH1 (Life Technologies). Data were normalized to GAPDH or 18S levels. For measurement of mature miR-200b, reverse transcription was performed with the TaqMan MicroRNA Reverse Transcription Kit (Life Technologies), according to the manufacturer’s instructions, separately for miR-200b and the normalization control, RNU24. cDNA (0.9 ng) was used for real-time PCR amplification.
Immunocytochemistry
Cells (4 × 107) were fixed in 4% paraformaldehyde, embedded in paraffin, and mounted on slides. Following antigen retrieval with 10 mM Na-citrate and blocking, cells were stained with anti-GLUT3 or anti-E-cadherin antibodies overnight at 4°C, followed by washing and staining with biotin-conjugated secondary antibodies for 1 h at room temperature. After washing, avidin-biotin horseradish peroxidase complexes were added for 30 min (ABC kit, Vectastain), and the complexes were revealed with a DAB peroxidase substrate kit (Vector Laboratories, Burlingame, CA, USA). Counterstain was performed using Harris hematoxylin.
Reagents
Recombinant human TGF-β2 was from PeproTech (Rocky Hill, NJ, USA). Antibodies used were anti-GLUT3 (#400062, Calbiochem, San Diego, CA, USA), anti-E-cadherin (#3195, Cell Signaling Technology, Danvers, MA, USA), anti-vimentin (#5741, Cell Signaling), anti-SNAIL (#3879, Cell Signaling), anti-ZEB1 (#sc-25388, Santa Cruz Biotechnology, Inc., Dallas, TX, USA), anti-β-tubulin (#sc-9104, Santa Cruz Biotechnology), anti-CtBP (#sc-17759, Santa Cruz Biotechnology), anti-p300 (#sc-585, Santa Cruz Biotechnology), and normal rabbit IgG (#12-370, Millipore, Billerica, MA, USA).
Virus production and infection
293 T cells were transfected using the calcium-phosphate precipitation method, co-transfecting the lentiviral plasmid of interest in conjunction with pMD2G (VSV-G protein) and pCMVR8.74 (lentivirus packaging vector, kind gift of D. Trono, EPFL, Lausanne). Viral supernatants were harvested 24 and 36 h post-transfection, filtered, and used directly for infection of cell lines, as described. Puromycin selection was performed to select cells with stable pRDI292-CMV (delivering control, human GLUT3, mouse Snail, or mouse Zeb1 cDNA) or pLKO.1 (with control or GLUT3 shRNA) genomic integration. Fluorescence-activated cell sorting was used to select cells with stable pSicoR constructs, based on GFP expression.
Statistics
Unless specified differently, P values were determined by Student’s t tests.
ChIP-seq analysis
Publicly available replicate ChIP-seq data for ZEB1 (GSM803411), RNA polymerase II (GSM803485), H3K27ac (GSM733771), as well as naked DNA (Input) (GSM733742) [16] were re-analyzed using Bowtie 2 [17] for mapping to the human GRCh37 genome with the parameters ‘--very-sensitive -M 10 -p 8’ and CCAT 3.0 [18] for peak-calling using the parameters ‘fragmentSize 200 slidingWinSize 100 movingStep 50 isStrandSensitiveMode 1 minCount 13 outputNum 100000 randomSeed 123456 minScore 5 bootstrapPass 80’. Data were visualized using IGV Browser [19].
ChIP-PCR
ChIP was performed as described previously [20], using 6 × 107 SW1573 and 5 μg of each antibody. To amplify regions within the promoter region of SLC2A3, the following primers were used: 5′-ACTGCCCTGATAGTTGGTCTGG-3′ with 5′-TTTGCCAGTGTTCCTTTCTTCG-3′ (−608 and −523 bp upstream of SLC2A3 TSS); 5′-ACTGCCCTGATAGTTGGTCTGG-3′ with 5′-GAGGGAAAGACAGCCTGAGAGA-3′ (−608 and −482 bp upstream of SLC2A3 TSS). To amplify regions from the intron 2, the following primers were used: 5′-CATCACAGTTGCTACAATCGGC-3′ with 5′-ACCATGCCTGGCCTTAAATTCT-3′ (2,396 and 2,559 bp downstream of SLC2A3 TSS); 5′-ACCATGCCTGGCCTTAAATTCT-3′ with 5′-AGCCTCAGGAGTAGCTGGGACT-3′ (2,452 and 2,594 bp downstream of SLC2A3 TSS); 5′-GTGAGTGCCAGGCCACAATAAT-3′ with 5′-TGTGTTGCTCAGGATGGTGTTT-3′ (2,466 and 2,647 bp downstream of SLC2A3 TSS); 5′-AGTCCCAGCTACTCCTGAGGCT-3′ with 5′-TTCCGGGAGTAAGTGAGCTTTG-3′ (2,573 and 2,791 bp downstream of SLC2A3 TSS); 5′-AAACACCATCCTGAGCAACACA-3′ with 5′-TGAGGTGAAAGAGTGGGAGGAA-3′ (2,626 and 2,831 bp downstream of SLC2A3 TSS); 5′-TTCCTCCCACTCTTTCACCTCA-3′ with 5′-GCACCGATGTTCACAGTCTACC-3′ (2,810 and 2,926 bp downstream of SLC2A3 TSS); 5′-AAGCTGGGTTCCCTTAGCAGAG-3′ with 5′-AAAGGGTTGGTGGAAGAACAGA-3′ (2,877 and 3,117 bp downstream of SLC2A3 TSS); 5′-CAGTCTGTTCTTCCACCAACCC-3′ with 5′-AGGACCAGAGAGACGTGAGCAG-3′ (3,093 and 3,212 bp downstream of SLC2A3 TSS). The intergenic region upstream of GAPDH was amplified, using primers 5′-ATGGGTGCCACTGGGGATCT-3′ and 5′-TGCCAAAGCCTAGGGGAAGA-3′, as described previously [21]. As positive control, a DNA sequence of CDH1 known to bind ZEB1 was amplified using primers 5′-GGCCGGCAGGTGAACCCTCA-3′ and 5′-GGGCTGGAGTCTGAACTGA-3′, as described previously [22]. Real-time PCR was performed using SyBR green. Fold enrichment was calculated as follows: 2^ΔΔct (ΔΔct = Δctspecific antibody − ΔctIgG). Data were normalized using the negative control, an untranscribed region upstream of GAPDH, and represented as fold enrichment/fold enrichment of the negative control locus.
Microarray analysis
We collected publicly available datasets from journal articles and Gene Expression Omnibus (GEO) repository, selecting those with medium to large sample size [23,24]. The .CEL files were imported into R/Bioconductor (http://www.bioconductor.org/) and RMA normalized [25]. A total of 462 early-stage untreated lung NSCLC samples were available, with associated patients’ overall survival. The probe IDs with higher variation for each gene were retained from a total of 13,960 genes. The normalized data together with the clinico-pathological variables were then used for further analysis. Pairwise Spearman correlations between GLUT3 and other available genes were calculated. Univariate and multivariate survival analyses were performed using proportional Cox regression (package ‘survival’, R). Kaplan-Meier figure was reported showing hazard ratio (HR), 95% confidence intervals, and Wald p_value from Cox regression model for a specific comparison of interest. The continuous GLUT3 expression was dichotomized in high and low risk levels by the median of the expression for visualization purposes. Multivariate analysis was performed with the following variables: GLUT3 continuous expression, stage, histology, gender, and age (dichotomized by the median of 62 years).
Results
GLUT3 expression correlates with EMT
To determine if the neuronal glucose transporter GLUT3 is expressed in lung tumor cells, we used a panel of ten human NSCLC cell lines. We noticed that GLUT3 expression, as determined by real-time PCR analysis, varied considerably between the cell lines, with five of them expressing high and the other five expressing low GLUT3 mRNA levels (Figure 1A). Several molecular markers can be used to classify human lung tumor cells according to their EMT status [26]. We monitored the epithelial or mesenchymal status in these cells by assessment of RNA levels of E-cadherin and the mature form of miR-200b (two markers of the epithelial state), and vimentin (a prototypical mesenchymal marker) using real-time PCR. These three markers clearly separated the cell lines into two groups: an ‘epithelial group’ (composed of H441, H727, H1944, H2009, and H2122 cells) that expresses E-cadherin and miR-200b, but almost no vimentin, and a ‘mesenchymal group’ (SW1573, H23, H460, A549, and Calu-6) that expresses high levels of vimentin, but very little E-cadherin and miR-200b. GLUT3 expression was elevated specifically in the mesenchymal group, whereas levels were very low in cell lines of the epithelial group (Figure 1A), strongly suggesting that this glucose transporter is up-regulated during or after an EMT. To determine if the correlation with EMT was specific for GLUT3, or if other transporters within this family had a similar expression pattern, we monitored the expression of additional GLUT family members that carry glucose (GLUT1, GLUT2, GLUT4, GLUT12, and GLUT14). Whereas GLUT2 and GLUT14 were undetectable across the cell lines (MM and EM, unpublished observations), GLUT1, GLUT4, and GLUT12 were expressed, but none of them showed any correlation with EMT (Additional file 1). Next, to confirm that high GLUT3 mRNA expression in mesenchymal cells reflected actual protein expression, we performed immunocytochemistry analyses. This demonstrated that GLUT3 protein was expressed and localized to the plasma membrane of mesenchymal cells, but was not detectable in the cells from the epithelial group (Figure 1B); once again, there was an inverse correlation between GLUT3 and E-cadherin expression.
Figure 1.
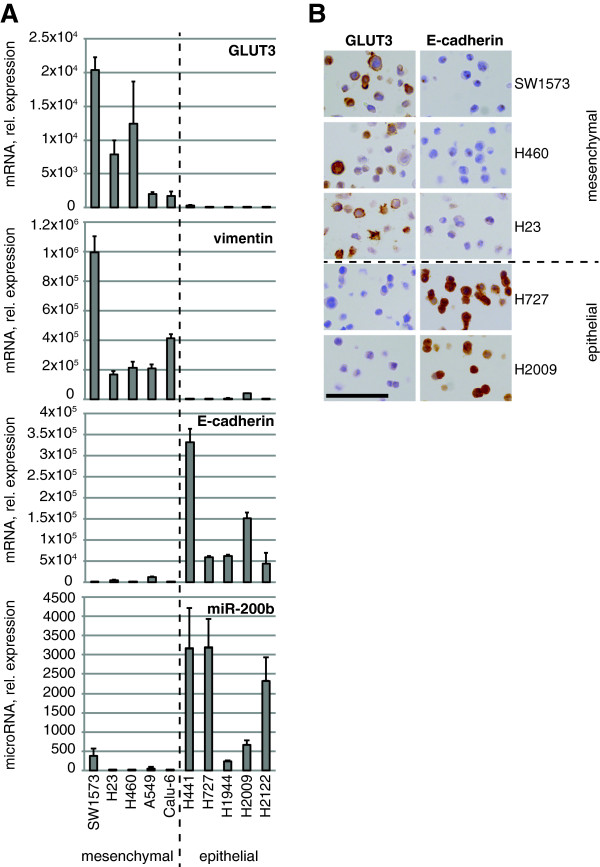
GLUT3 is strongly expressed in mesenchymal lung tumor cells. (A) The indicated cell lines were lysed for RNA preparation followed by reverse transcription. The cDNA was amplified by real-time PCR using probes specific for the indicated genes or internal controls. Data show means ± s.d. (n = 3) of mRNA or mature microRNA expression relative to the cell line expressing the least amount of the same gene (set to 1). (B) The indicated cells were stained to analyze the expression of GLUT3 or E-cadherin by immunocytochemistry. Scale bar, 100 μm. In (A) and (B), the dashed line indicates the separation between the mesenchymal and the epithelial groups of cells.
GLUT3 is induced during EMT
To test the hypothesis that GLUT3 is induced upon EMT, we first stimulated the H2122 epithelial cell line with TGF-β, a potent EMT-inducing factor. As expected, TGF-β treatment up-regulated the expression of the mesenchymal marker vimentin. More importantly, GLUT3 was concomitantly up-regulated in a temporal manner (Figure 2A). Because TGF-β is pleiotropic [27], we then directly tested the role of two known EMT-inducing transcription factors, SNAIL and ZEB1, in GLUT3 regulation. We transduced two low-GLUT3 expressing epithelial cell lines, H727 and H2009, with lentiviruses to generate cells stably expressing either mouse SNAIL or ZEB1 protein. As anticipated, each of H727-SNAIL, H727-ZEB1, H2009-SNAIL, and H2009-ZEB1 stable cell populations showed a downregulation of E-cadherin and an up-regulation of vimentin. Furthermore, there was a 5- to 27-fold, and a 132- to 354-fold up-regulation of GLUT3 in H727 and H2009 stable cells, respectively, compared to cells expressing a control plasmid (Figure 2B). Interestingly, and in agreement with previous findings [28], stable expression of SNAIL led to induction of endogenous ZEB1 (the reciprocal did not occur), raising the possibility that mouse SNAIL induces GLUT3 expression indirectly through ZEB1 (Figure 2C). Moreover, the extent of ZEB1 induction correlated to that of GLUT3: in H727 cells, mouse SNAIL overexpression resulted in a 2.5- and 27-fold induction of human ZEB1 and GLUT3 mRNA, respectively, whereas it was 9- and 354-fold in H2009 cells (Additional file 2). Next, to assess if the link between GLUT3 and EMT is specific for NSCLC, or whether it occurs in other tumor types, we analyzed GLUT3 expression in human cell lines derived from HCC. Based on marked differences in the expression of vimentin and E-cadherin, we could distinguish two groups of cells: an epithelial group (Huh-7, Hep3B, and Huh-1) and a mesenchymal group (HLF and HLE). Similar to NSCLC cell lines, GLUT3 but not GLUT1 expression was specifically elevated in the mesenchymal group, and very low in the epithelial group (Figure 3A). Additionally, TGF-β treatment of the low-GLUT3 expressing cell line, Hep3B, led to increased expression of GLUT3, whereas GLUT1 levels remained unchanged (Figure 3B). Altogether, these data demonstrate that the regulation of GLUT3 during EMT occurs in at least two different cancer types, NSCLC and HCC.
Figure 2.
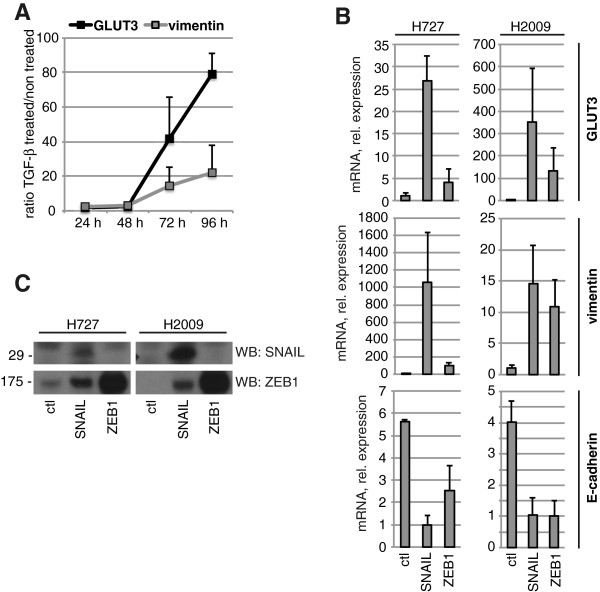
GLUT3 is induced upon EMT. (A) H2122 cells were stimulated with 10 ng/ml TGF-β for the indicated time points, after which the cells were lysed for RNA preparation followed by reverse transcription. The cDNA was amplified by real-time PCR using probes specific for the indicated genes or internal controls. Data show means ± s.d. (n = 4) of mRNA expression ratios between TGF-β treated and non-treated conditions. (B) H727 or H2009 cell populations stably expressing a control plasmid (ctl), SNAIL, or ZEB1 were lysed for RNA preparation followed by reverse transcription. The cDNA was amplified by real-time PCR using probes specific for the indicated genes or internal controls. Data show means ± s.d. (n = 3) of mRNA expression relative to the cell line expressing the least amount of the same gene (set to 1). (C) H727 or H2009 stable cell populations were lysed with RIPA buffer to prepare protein extracts and to analyze the expression of the indicated proteins by Western blot.
Figure 3.
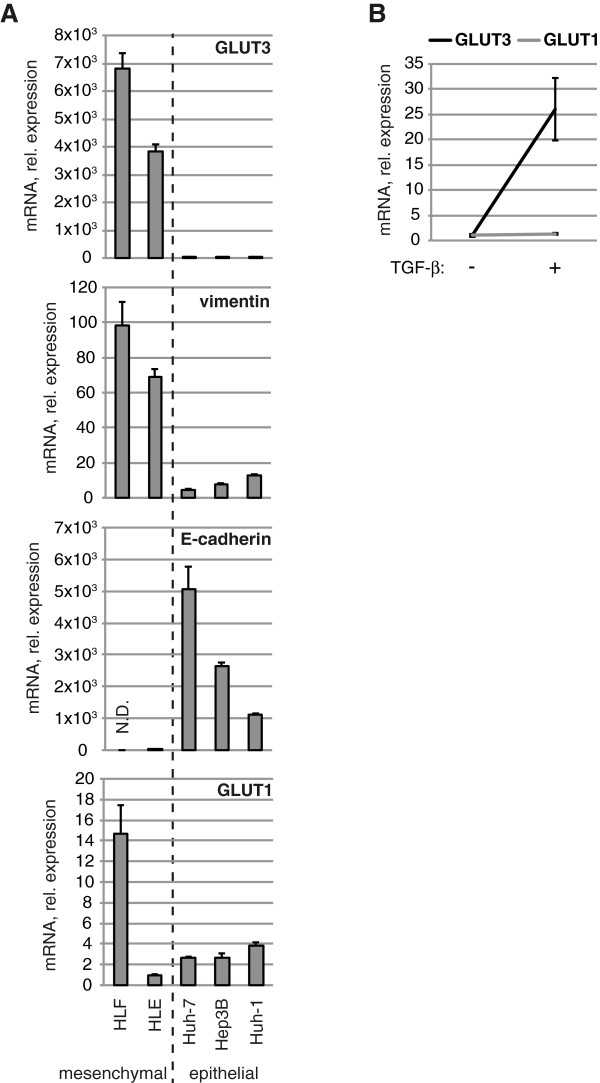
GLUT3 is strongly expressed in mesenchymal liver tumor cells. (A) The indicated cell lines were lysed for RNA preparation followed by reverse transcription. The cDNA was amplified by real-time PCR using probes specific for the indicated genes or internal controls. Data show means ± s.d. (n = 3) of mRNA expression relative to the cell line expressing the least amount of the same gene (set to 1). N.D., not detected. (B) Hep3B cells were stimulated with 10 ng/ml TGF-β for 72 h, after which the cells were lysed for RNA preparation followed by reverse transcription. The cDNA was amplified by real-time PCR using probes specific for the indicated genes or internal controls. Data show means ± s.d. (n = 3) of mRNA expression relative to the untreated conditions (set to 1).
ZEB1 induces GLUT3 through direct binding and transcriptional activation
Although ZEB1 is best characterized as a transcriptional repressor, depending on co-factor recruitment—and probably other parameters—it can activate transcription. For example, ZEB1 represses gene transcription when bound to the co-repressor C-terminal-binding protein (CtBP), whereas it activates target gene expression when bound to p300, P300/CBP-associated factor (P/CAF), or receptor-regulated (R)-SMADs [29-32]. Co-immunoprecipitation experiments performed on extracts from mesenchymal lung tumor cells revealed endogenous interactions between ZEB1 and CtBP, as well as ZEB1 and p300, suggesting ZEB1 forms different complexes to trigger target gene repression or activation in these cells (Additional file 3). To test the hypothesis that ZEB1 directly activates GLUT3 (SLC2A3) gene transcription, ZEB1 mRNA and protein expression was first monitored in the panel of lung tumor cells. This showed that ZEB1 was more strongly expressed in GLUT3-proficient, mesenchymal cells compared to cells from the epithelial group (Figure 4A and Additional file 1). Additionally, in response to TGF-β stimulation (see Figure 2A), there was an up-regulation of ZEB1, but not SNAIL, which preceded that of GLUT3 (Additional file 4), suggesting ZEB1 induction enables the activation of GLUT3 transcription. Next, we analyzed public data from lymphoblastoid cell extracts subjected to chromatin immunoprecipitation using anti-ZEB1, anti-RNA polymerase II or anti-histone H3 (acetyl K27) antibodies, followed by DNA sequencing (ChIP-seq) [16]. SLC2A3 was actively transcribed in these cells, as inferred from the occupancy of RNA polymerase II and the acetylation of histone H3 on Lysine 27 at the SLC2A3 locus (Figure 4B). Importantly, an interaction between ZEB1 and a region located within the second intron of SLC2A3 was uncovered, with a peak of interaction that localized to an E-box-like motif, composed of a CACCTC sequence (Figure 4B). Of note, ZEB1 has been reported to bind such DNA sequences to regulate transcription [33,34]. To determine if a similar ZEB1-SLC2A3 gene interaction occurred in GLUT3-expressing lung tumor cells, we performed ChIP-PCR analyses in extracts of SW1573 cells. As positive and negative controls, respectively, we amplified a sequence of E-cadherin (CDH1) known to bind ZEB1 and an intergenic sequence upstream of GAPDH[21,22]. ZEB1 bound to DNA sequences located within intron 2 of SLC2A3, but not to sequences from the promoter region (Figure 4C). Having established the localization of ZEB1 binding to the SLC2A3 gene, we cloned the DNA sequence of intron 2 or, for comparison, a 1-kb sequence preceding the SLC2A3 transcription start site into a luciferase reporter plasmid (Figure 4D). Upon transfection into 293 T cells, which express high amounts of endogenous ZEB1 (see Figure 4A), the basal luciferase activity was significantly stronger from the intron 2 construct than from the promoter construct. Also, luciferase activity from the intron 2 construct significantly decreased upon ZEB1 knockdown (Figure 4D). Furthermore, deletion of the E-box-like motif in intron 2 substantially reduced luciferase activity, revealing the CACCTC sequence as an important functional element in response to ZEB1 (Figure 4E). Collectively, these results demonstrate that GLUT3 is induced during EMT by a mechanism involving direct binding and transcriptional activation by ZEB1.
Figure 4.
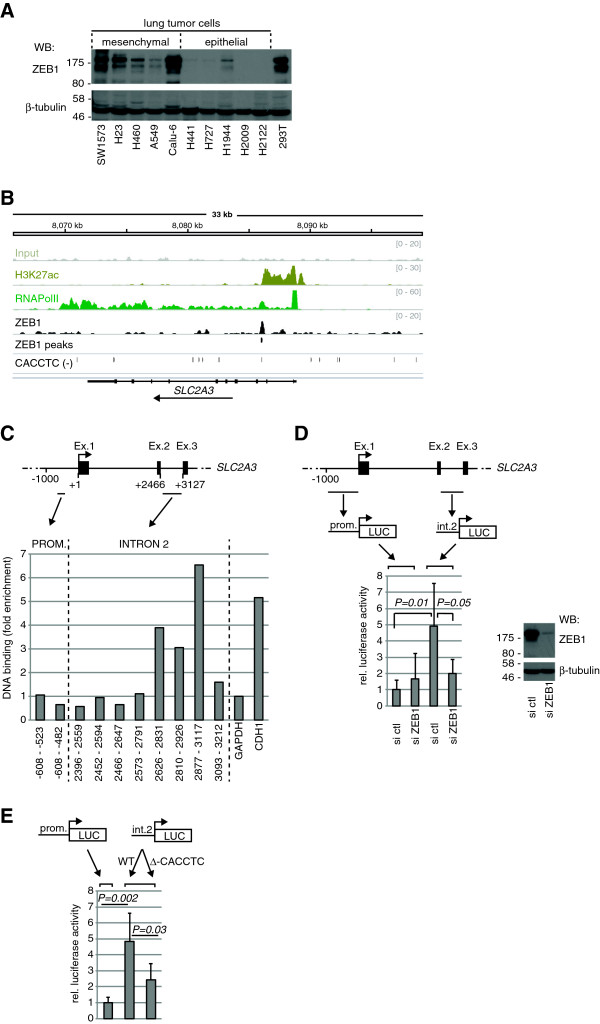
GLUT3 is a direct ZEB1-target gene. (A) The indicated cell lines were lysed with RIPA buffer to prepare protein extracts and to analyze the expression of ZEB1, or β-tubulin as control, by Western blot. (B) IGV Browser view of 33 kb around the SLC2A3 gene including ZEB1 (black), RNA Polymerase II (RNAPolII, green), the active histone mark H3K27ac (green-yellow) ChIP-seq, and Input (grey) tracks in human lymphoblastoid cell lines. ZEB1 peak calls and ‘CACCTC’ sequence matches on the reverse strand are also indicated (black). The orientation of SLC2A3 is indicated by an arrow. (C) Extracts from SW1573 cells were used for ZEB1 ChIP followed by real-time PCR. The coordinates of the PCR amplicons relative to the TSS are indicated. CDH1 and an untranscribed sequence upstream of GAPDH were used as positive and negative controls for ZEB1 binding, respectively. The data shown are representative of three independent experiments. (D) (Upper part) scheme showing the promoter region of 1,000 bp preceding the transcription start site (prom.) and the intron 2 region (int.2), which were amplified from the SLC2A3 gene and cloned into the luciferase reporter construct. Ex. exon, LUC luciferase. (Lower part) 293 T cells were transfected with the indicated luciferase reporter plasmids. Luciferase activity was analyzed 24 h later. Data show means ± s.d. (n = 5) of relative luciferase activity. (Lower right) 293 T cells were transfected with control (ctl) or ZEB1 siRNAs and were lysed 96 h later to prepare protein extracts and to analyze the expression of ZEB1, or β-tubulin as control, by Western blot. (E) 293 T cells were transfected with the indicated luciferase reporter plasmids. For the int.2 construct, either wild-type (WT) or a variant with a deletion of the E-box-like motif (Δ-CACCTC) was used. Luciferase activity was analyzed 24 h later. Data show means ± s.d. (n = 5) of relative luciferase activity.
GLUT3 promotes glucose uptake and proliferation of mesenchymal lung tumor cells
We next sought to determine the functional consequences of high GLUT3 expression in mesenchymal lung tumor cells using GLUT3 knockdown. Transfection of small interfering (si)RNAs to GLUT3 into SW1573 cells caused a reduction in the number of live cells, whereas GLUT1 knockdown did not or only slightly reduced cell number (Figure 5A and Additional file 5). The proportion of dead cells was not changed upon GLUT3 knockdown in SW1573 cells (Additional file 5), suggesting that GLUT3 primarily regulates proliferation of these cells, not survival. In contrast, H727, a low-GLUT3 expressor, was not affected by GLUT3 knockdown. Next, we transduced SW1573 cells with lentiviruses delivering short hairpin (sh)RNAs targeting GLUT3 to generate cells with stable GLUT3 knockdown (Additional file 5). We first used these cells to interrogate the contribution of GLUT3 to glucose import. Knockdown of GLUT3 with two independent shRNAs led to a decrease of more than 50% in the efficiency of radioactive 2-deoxyglucose incorporation, revealing that GLUT3 plays a prominent role in glucose uptake of mesenchymal cells (Figure 5B). Importantly, GLUT3 knockdown also led to a reduction in the number of colonies grown in anchorage-independent conditions (Figure 5C). Next, to determine if GLUT3 knockdown had an impact on the mesenchymal state of tumor cells, we monitored vimentin and E-cadherin expression. Interestingly, vimentin expression was substantially decreased upon GLUT3 knockdown (Additional file 6). Hence, although we did not observe any statistically significant variation in E-cadherin mRNA levels, these data suggest that GLUT3-dependent glucose uptake participates in the maintenance of a mesenchymal state. Finally, we wanted to determine if increased GLUT3 expression is sufficient to impact on the growth of lung tumor cells with epithelial traits. To do this, we generated H727 cells stably expressing human GLUT3 (Additional file 7). In high glucose concentrations, there was no difference in the proliferation of cells overexpressing GLUT3 and control cells (Additional file 7). However, we reasoned that because parental H727 cells have adapted to grow in high glucose levels without GLUT3, ectopic expression of this high-affinity glucose transporter might affect proliferation specifically when glucose concentrations are low. Indeed, the growth rate of parental cells was diminished by a reduction in glucose levels. Under these conditions, H727-GLUT3 cells grew faster than control cells, demonstrating that GLUT3 expression is sufficient to increase cancer cell proliferation in conditions where glucose concentrations are limiting (Figure 5D). Altogether, these results demonstrate an important contribution for GLUT3 in lung tumor cell proliferation.
Figure 5.
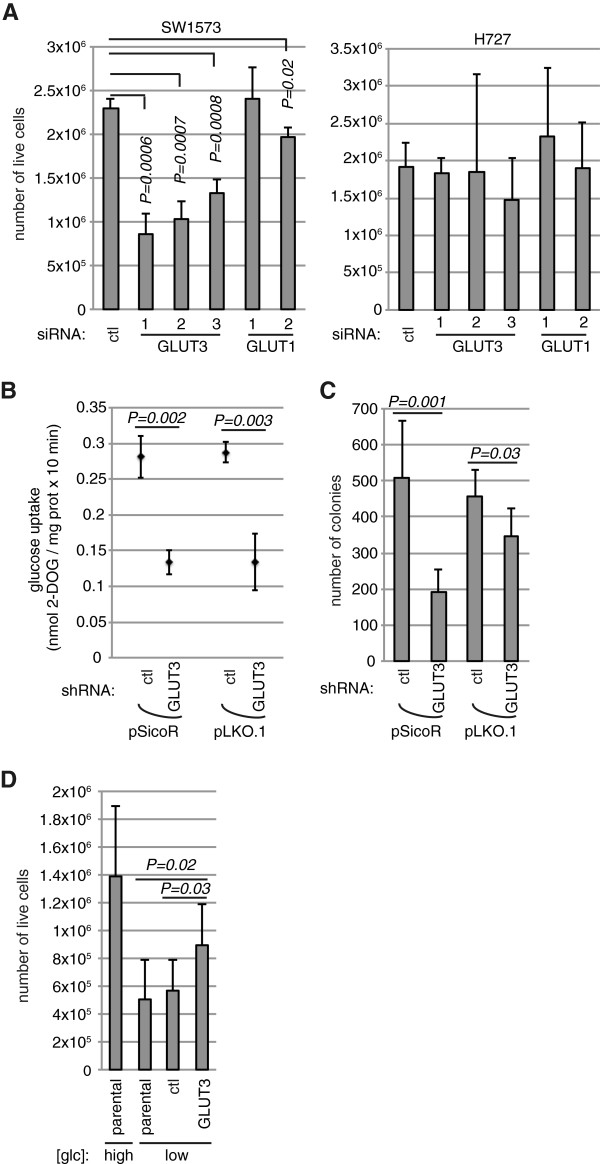
GLUT3 promotes glucose uptake and the proliferation of mesenchymal lung tumor cells. (A) Mesenchymal (SW1573) or epithelial (H727) cells were transfected with control (ctl) siRNA, each of three different siRNAs to decrease GLUT3, or each of two siRNAs to decrease GLUT1, as indicated. One hundred forty-four hours later, live cells were counted by trypan blue exclusion (n = 3 or 4). (B) 2-Deoxy-d-[3H]glucose (DOG) incorporation was measured in SW1573 cells stably expressing ctl shRNAs or shRNAs targeting GLUT3 (in pSicoR or pLKO.1 vectors, see ‘Methods’). Data show means ± s.d. (n = 3) of glucose uptake (nmol) measured for 10 min, normalized to protein concentration. (C) SW1573 cells stably expressing ctl shRNAs or shRNAs targeting GLUT3 (in pSicoR or pLKO.1 vectors) were prepared in soft agar for anchorage-independent growth. Three weeks later, the number of colonies was determined (n = 6). (D) H727 cells, either parental or stably expressing a control (ctl) plasmid or a GLUT3 cDNA, were cultured in high or low glucose (glc) concentrations for 4 days, after which live cells were counted by trypan blue exclusion (n = 7).
GLUT3 expression in human NSCLC correlates with poor overall survival and EMT signatures
To assess the relevance of our findings in vivo, we used a combination of five publicly available microarray datasets of human NSCLC. This yielded 462 samples with follow-up of overall survival. The analysis revealed that high GLUT3 expression was associated with a poor overall survival (Figure 6A), a finding consistent with previous studies [10,35]. Additionally, a multivariate analysis provided evidence for high GLUT3 expression as an independent predictor of poor overall survival (Table 1). Next, we made pairwise comparisons between each of SLC2A3, CDH1, and various EMT markers or inducers (VIM, SNAI1, SNAI2, ZEB1, ZEB2, and TWIST1). As expected, most mesenchymal genes had a positive correlation with each other and a negative correlation with CDH1. Importantly, all mesenchymal genes showed a statistically significant positive correlation with SLC2A3 expression (P < 0.001) (Figure 6B). SLC2A3 expression was also negatively correlated with CDH1, as expected, although these data were not significant (Figure 6B). Altogether, these in vivo findings support the in vitro data that position GLUT3 as an important factor in the process of EMT and lung tumor progression.
Figure 6.
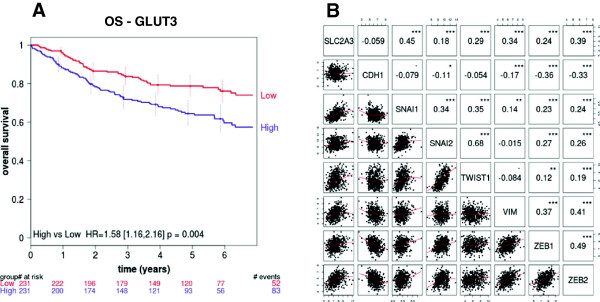
GLUT3 expression correlates with poor overall survival and EMT in human NSCLC. (A) Kaplan-Meier plot representing overall survival comparing tumor samples with high or low GLUT3 levels. The numbers of living patients are indicated for each year and group. HR hazard ratio, OS overall survival. (B) The correlations between each of GLUT3 (SLC2A3), E-cadherin (CDH1), SNAIL (SNAI1), SLUG (SNAI2), Twist (TWIST1), vimentin (VIM), ZEB1, and ZEB2 were assessed from the pooled dataset. Spearman correlation coefficients (indicated numbers) were calculated between the expression values of each pair. 0.1 < P < 0.05; *P < 0.05; **P < 0.01; ***P < 0.001.
Table 1.
Univariate and multivariate analyses of overall survival in NSCLC
| HR | Lower 0.95 | Upper 0.95 | P value (Wald) | |
|---|---|---|---|---|
| Univariate |
|
|
|
|
| SLC2A3 (continuous) |
1.224 |
1.047 |
1.432 |
1.14E − 02 |
| Stage (II vs. I) |
1.590 |
1.070 |
2.363 |
2.19E − 02 |
| Histology LCC vs. AD |
1.131 |
0.458 |
2.792 |
7.89E − 01 |
| Histology SCC vs. AD |
1.624 |
1.150 |
2.293 |
5.91E − 03 |
| Gender (F vs. M) |
0.610 |
0.429 |
0.868 |
6.06E − 03 |
| Age (>62 vs. ≤62) |
1.906 |
1.385 |
2.622 |
7.47E − 05 |
| Multivariate |
|
|
|
|
| SLC2A3 (continuous) |
1.194 |
1.016 |
1.404 |
3.16E − 02 |
| Stage (II vs. I) |
1.432 |
0.945 |
2.170 |
9.04E − 02 |
| Histology LCC vs. AD |
1.006 |
0.406 |
2.495 |
9.90E − 01 |
| Histology SCC vs. AD |
1.552 |
1.071 |
2.248 |
2.03E − 02 |
| Gender (F vs. M) |
0.702 |
0.484 |
1.020 |
6.33E − 02 |
| Age (>62 vs. ≤62) | 1.829 | 1.323 | 2.528 | 2.56E − 04 |
AD adenocarcinoma, F female, HR hazard ratio, LCC large cell carcinoma, M male, SCC squamous cell carcinoma.
Discussion
In this study, we have identified a unique role for glucose transporter GLUT3 in the proliferation of lung tumor cells with mesenchymal characteristics. We found that GLUT3 is strongly up-regulated during EMT and contributes to glucose uptake specifically in mesenchymal-like lung tumor cells. Furthermore, GLUT3 overexpression is sufficient to substantially enhance the proliferation of lung tumor cells with epithelial traits specifically in the context of low glucose. Interestingly, increased glucose consumption was shown to promote EMT via stabilization of SNAIL [36], suggesting that increased expression of GLUT family members promotes the induction and/or maintenance of EMT. In our experiments, we did not detect an EMT occurring in response to ectopic GLUT3 protein expression in epithelial cells (MM and EM, unpublished observations), but we found a decrease in vimentin expression upon GLUT3 knockdown in mesenchymal cells. Collectively, these data suggest the intriguing possibility that GLUT3 plays a role in the successful establishment or maintenance of the EMT, when tumor cells become more motile and invasive, or in tumor cell survival upon loss of cell-cell adhesion.
EMT in cancer is a complex phenomenon that is orchestrated by different transcription factors with partially overlapping gene targets. These transcription factors, like SNAIL or ZEB1, often act as transcriptional repressors. However, the mechanisms of target gene regulation are not fully elucidated. For example, ZEB1 inhibits the transcription of target genes when bound to CtBP [29,31]. In contrast, ZEB1 binding to R-SMADs and the acetyltransferases p300 and P/CAF promotes target gene transcription [30,32]. Likewise, in our study, we have demonstrated that ZEB1 can interact with each of CtBP and p300 in mesenchymal lung tumor cells and that it is a direct activator of SLC2A3 gene transcription. Of note, ZEB1 expression was higher in the mesenchymal, GLUT3-expressing cell lines compared to the epithelial cell lines, and ZEB1 was induced prior to GLUT3 in response to TGF-β (see Figures 2A and 4A, and Additional files 1 and 4). This is in agreement with a previous finding reporting that the SLC2A3 gene was one of the top 50 genes correlating positively with ZEB1 gene expression in a panel of human lung tumor cell lines [37]. In the future, it will be important to decipher all of the genes that are directly targeted—either repressed or induced—by ZEB1 in lung cancer and to determine if this includes additional genes that regulate tumor cell metabolism.
The increased GLUT3 expression observed in tumor cells upon EMT may have important implications for cancer treatment. Indeed, not only is EMT well recognized as a crucial process that increases invasive capacities of tumor cells and promotes the acquisition of stem cell characteristics [38], it may occur earlier in tumor development than previously anticipated. For example, in a K-ras LSL-G12D/+ ; p53 Flox/Flox mouse model of PDAC, EMT and invasiveness precede the detection of tumors in situ[39]. Interestingly, a molecular classification of human PDAC was performed recently, where three subtypes were defined: classical, exocrine-like, and quasi-mesenchymal (which has the worst prognosis of all three) [40]; SLC2A3 was one of a 20-gene signature of the quasi-mesenchymal subtype. This observation, together with our findings of a connection between GLUT3 and EMT in human liver cells from HCC, indicates that the regulation of GLUT3 by EMT extends beyond lung cancer.
Conclusions
By uncovering direct regulation of GLUT3 by the transcription factor ZEB1, our study provides evidence for a tight association between two central characteristics of carcinoma development: EMT and glucose metabolism. Because GLUT3 expression is mainly restricted to the brain of healthy individuals, the future development of small molecule compounds that selectively block GLUT3 and that do not cross the blood-brain barrier may become a viable strategy to treat patients with NSCLC and possibly other cancer types.
Abbreviations
EMT: Epithelial-mesenchymal transition; HCC: Hepatocellular carcinoma; NSCLC: Non-small cell lung cancer; PDAC: Pancreatic ductal adenocarcinoma; SLC2A3: Solute carrier family 2 (facilitated glucose transporter), member 3; ZEB1: Zinc finger E-box-binding homeobox 1.
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
MM participated in the design of the study, performed most of the experiments, and analyzed the data. JV performed several experiments and analyzed the data. SR did the bioinformatics analyses of the microarray datasets. SG carried out the immunoprecipitation experiments and analyzed the data. NS carried out the ZEB1 knockdown experiments and analyzed the data. PCS participated in the bioinformatics analyses of the ChIP-seq datasets. BD participated in the bioinformatics analyses of the ChIP-seq datasets. LEF carried out the initial real-time PCR experiments of the cell line panel and analyzed the data. JG, DM, and TGO analyzed the data. EM conceived the study, participated in its design and coordination, analyzed the data, and wrote the manuscript. All authors read and approved the final manuscript.
Supplementary Material
GLUT1, GLUT4, GLUT12, and ZEB1 expression in mesenchymal and epithelial lung tumor cells. The indicated cell lines were lysed for RNA preparation followed by reverse transcription. The cDNA was amplified by real-time PCR using probes specific for the indicated genes or internal controls. Data show means ± s.d. (n = 3) of mRNA expression relative to the cell line expressing the least amount of the same gene (set to 1). When there was no specific amplification after 40 cycles of PCR, the samples were called not determined (N.D.).
hZEB1 and hGLUT3 induction upon mSNAIL overexpression. H727 or H2009 cell populations stably expressing a control plasmid (ctl) or mouse (m) SNAIL were lysed for RNA preparation followed by reverse transcription. The cDNA was amplified by real-time PCR using probes specific for human ZEB1, GLUT3, or an internal control. Data show means ± s.d. (n = 3) of mRNA expression, represented as fold induction (mSNAIL/ctl) for each cell line. The arrows and numbers indicate the fold induction between samples.
ZEB1 interacts with CtBP and p300 in mesenchymal lung tumor cells. Protein extracts from the indicated cell lines were used for ZEB1 or control (IgG) immunoprecipitation, followed by Western blot using the indicated antibodies. The arrowheads indicate the position of each protein. XT, cell extract; ø, empty lane.
ZEB1 is induced rapidly upon TGF-β stimulation. H2122 cells were stimulated with 10 ng/ml TGF-β for the indicated time points, after which the cells were lysed for RNA preparation followed by reverse transcription. The cDNA was amplified by real-time PCR using probes specific for ZEB1, SNAIL, or an internal control. Data show means ± s.d. (n = 4) of mRNA expression ratios between TGF-β treated and non-treated conditions.
Efficiency of siRNA or shRNA knockdown. (A) Efficiency of GLUT3 or GLUT1 siRNA knockdown. SW1573 cells were transfected with one of three different siRNAs to target GLUT3, two to target GLUT1, or a control (ctl) siRNA, or were left untransfected (--). Seventy-two hours later, the cells were lysed for RNA preparation followed by reverse transcription. The cDNA was amplified by real-time PCR using probes specific for the indicated genes or GAPDH as an internal control. Data show the percentage of mRNA expression relative to the non-transfected condition (set to 100%). (B) Efficiency of GLUT3 siRNA knockdown. SW1573 cells were transfected with one of three different siRNAs to target GLUT3, a control (ctl) siRNA, or were left untransfected (--). Seventy-two hours later, the cells were lysed with RIPA buffer to prepare protein extracts and to analyze the expression of GLUT3 by Western blot. (C) GLUT3 or GLUT1 knockdown does not affect the number of dead cells. SW1573 cells were transfected with control (ctl) siRNA, each of three different siRNAs to decrease GLUT3, or each of two siRNAs to decrease GLUT1, as indicated. One hundred forty-four hours later, dead cells were counted by trypan blue exclusion (n = 3). (D) Efficiency of GLUT3 shRNA stable knockdown. SW1573 cells stably expressing ctl or GLUT3 shRNAs (in the indicated vectors) were lysed (left panel) with RIPA buffer to prepare protein extracts and to analyze the expression of GLUT3 by Western blot, or (right panel) for RNA preparation followed by reverse transcription. The cDNA was amplified by real-time PCR using probes specific for GLUT3 or GAPDH as an internal control. Data show the percentage of GLUT3 mRNA expression relative to the control (ctl) shRNA expressing cells (set to 100%). *n.s., non-specific.
GLUT3 knockdown leads to diminished vimentin expression. SW1573 cells stably expressing a control (ctl) or GLUT3 shRNA were lysed for RNA preparation followed by reverse transcription. The cDNA was amplified by real-time PCR using probes specific for the indicated genes or internal controls. Data show means ± s.d. (n = 3) of mRNA expression relative to the condition with the least amount of mRNA for each gene (set to 1).
Generation of H727 cells stably expressing GLUT3. (A) Stable expression of GLUT3. (Left) Parental H727 cells or H727 cells stably expressing a control (ctl) plasmid or a GLUT3 cDNA were lysed with RIPA buffer to prepare protein extracts and to analyze the expression of GLUT3 (or β-tubulin as loading control) by Western blot. (Right) Parental or GLUT3-expressing H727 cells were stained to analyze the expression of GLUT3 by immunocytochemistry (ICC). Scale bar, 100 μm. (B) Ectopic GLUT3 expression does not affect cell proliferation in high glucose concentrations. H727 cells, either parental or stably expressing a control (ctl) plasmid or a GLUT3 cDNA, were cultured in high glucose (glc) concentrations for 4 days, after which live cells were counted by trypan blue exclusion (n = 7).
Contributor Information
Mark Masin, Email: mark.masin@epfl.ch.
Jessica Vazquez, Email: jessica.vazquez@epfl.ch.
Simona Rossi, Email: simona.rossi@unil.ch.
Svenja Groeneveld, Email: svenja.groeneveld@epfl.ch.
Natasha Samson, Email: natasha.samson@epfl.ch.
Petra C Schwalie, Email: petra.schwalie@epfl.ch.
Bart Deplancke, Email: bart.deplancke@epfl.ch.
Laura E Frawley, Email: frawleyl@mit.edu.
Jérôme Gouttenoire, Email: jerome.gouttenoire@chuv.ch.
Darius Moradpour, Email: darius.moradpour@chuv.ch.
Trudy G Oliver, Email: trudy.oliver@hci.utah.edu.
Etienne Meylan, Email: etienne.meylan@epfl.ch.
Acknowledgements
We thank Anja Irmisch and Ute Koch (ISREC, EPFL) for their help on the ChIP-PCR and 2-DOG uptake experiments, respectively, and the EPFL SV Histology Core Facility for histological sectioning. We thank Tyler Jacks (Massachusetts Institute of Technology) for insightful comments, and Mikael J. Pittet (Harvard Medical School) and Inder M. Verma (The Salk Institute) for critical reading of the manuscript. This work was supported by the Swiss National Science Foundation (PP00P3_133661) and by a ‘Molecular Life Sciences’ grant from the ISREC Foundation. The funding bodies played no role in the design, collection, analysis, interpretation of data, writing, or decision to submit the manuscript for publication.
References
- Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646–674. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- Lunt SY, Vander Heiden MG. Aerobic glycolysis: meeting the metabolic requirements of cell proliferation. Annu Rev Cell Dev Biol. 2011;27:441–464. doi: 10.1146/annurev-cellbio-092910-154237. [DOI] [PubMed] [Google Scholar]
- Thorens B, Mueckler M. Glucose transporters in the 21st century. Am J Physiol Endocrinol Metab. 2010;298:E141–145. doi: 10.1152/ajpendo.00712.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ganapathy V, Thangaraju M, Prasad PD. Nutrient transporters in cancer: relevance to Warburg hypothesis and beyond. Pharmacol Ther. 2009;121:29–40. doi: 10.1016/j.pharmthera.2008.09.005. [DOI] [PubMed] [Google Scholar]
- Yun J, Rago C, Cheong I, Pagliarini R, Angenendt P, Rajagopalan H, Schmidt K, Willson JK, Markowitz S, Zhou S, Diaz LA Jr, Velculescu VE, Lengauer C, Kinzler KW, Vogelstein B, Papadopoulos N. Glucose deprivation contributes to the development of KRAS pathway mutations in tumor cells. Science. 2009;325:1555–1559. doi: 10.1126/science.1174229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ying H, Kimmelman AC, Lyssiotis CA, Hua S, Chu GC, Fletcher-Sananikone E, Locasale JW, Son J, Zhang H, Coloff JL, Yan H, Wang W, Chen S, Viale A, Zheng H, Paik JH, Lim C, Guimaraes AR, Martin ES, Chang J, Hezel AF, Perry SR, Hu J, Gan B, Xiao Y, Asara JM, Weissleder R, Wang YA, Chin L, Cantley LC. et al. Oncogenic Kras maintains pancreatic tumors through regulation of anabolic glucose metabolism. Cell. 2012;149:656–670. doi: 10.1016/j.cell.2012.01.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simpson IA, Dwyer D, Malide D, Moley KH, Travis A, Vannucci SJ. The facilitative glucose transporter GLUT3: 20 years of distinction. Am J Physiol Endocrinol Metab. 2008;295:E242–253. doi: 10.1152/ajpendo.90388.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsukioka M, Matsumoto Y, Noriyuki M, Yoshida C, Nobeyama H, Yoshida H, Yasui T, Sumi T, Honda K, Ishiko O. Expression of glucose transporters in epithelial ovarian carcinoma: correlation with clinical characteristics and tumor angiogenesis. Oncol Rep. 2007;18:361–367. [PubMed] [Google Scholar]
- Younes M, Lechago LV, Somoano JR, Mosharaf M, Lechago J. Immunohistochemical detection of Glut3 in human tumors and normal tissues. Anticancer Res. 1997;17:2747–2750. [PubMed] [Google Scholar]
- Flavahan WA, Wu Q, Hitomi M, Rahim N, Kim Y, Sloan AE, Weil RJ, Nakano I, Sarkaria JN, Stringer BW, Day BW, Li M, Lathia JD, Rich JN, Hjelmeland AB. Brain tumor initiating cells adapt to restricted nutrition through preferential glucose uptake. Nat Neurosci. 2013;16:1373–1382. doi: 10.1038/nn.3510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ha TK, Her NG, Lee MG, Ryu BK, Lee JH, Han J, Jeong SI, Kang MJ, Kim NH, Kim HJ, Chi SG. Caveolin-1 increases aerobic glycolysis in colorectal cancers by stimulating HMGA1-mediated GLUT3 transcription. Cancer Res. 2012;72:4097–4109. doi: 10.1158/0008-5472.CAN-12-0448. [DOI] [PubMed] [Google Scholar]
- Kawauchi K, Araki K, Tobiume K, Tanaka N. p53 regulates glucose metabolism through an IKK-NF-kappaB pathway and inhibits cell transformation. Nat Cell Biol. 2008;10:611–618. doi: 10.1038/ncb1724. [DOI] [PubMed] [Google Scholar]
- Thiery JP, Acloque H, Huang RY, Nieto MA. Epithelial-mesenchymal transitions in development and disease. Cell. 2009;139:871–890. doi: 10.1016/j.cell.2009.11.007. [DOI] [PubMed] [Google Scholar]
- Yang J, Weinberg RA. Epithelial-mesenchymal transition: at the crossroads of development and tumor metastasis. Dev Cell. 2008;14:818–829. doi: 10.1016/j.devcel.2008.05.009. [DOI] [PubMed] [Google Scholar]
- Canto C, Suarez E, Lizcano JM, Grino E, Shepherd PR, Fryer LG, Carling D, Bertran J, Palacin M, Zorzano A, Guma A. Neuregulin signaling on glucose transport in muscle cells. J Biol Chem. 2004;279:12260–12268. doi: 10.1074/jbc.M308554200. [DOI] [PubMed] [Google Scholar]
- Consortium EP, Bernstein BE, Birney E, Dunham I, Green ED, Gunter C, Snyder M. An integrated encyclopedia of DNA elements in the human genome. Nature. 2012;489:57–74. doi: 10.1038/nature11247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, Salzberg SL. Fast gapped-read alignment with Bowtie 2. Nat Methods. 2012;9:357–359. doi: 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu H, Handoko L, Wei X, Ye C, Sheng J, Wei CL, Lin F, Sung WK. A signal-noise model for significance analysis of ChIP-seq with negative control. Bioinformatics. 2010;26:1199–1204. doi: 10.1093/bioinformatics/btq128. [DOI] [PubMed] [Google Scholar]
- Thorvaldsdottir H, Robinson JT, Mesirov JP. Integrative Genomics Viewer (IGV): high-performance genomics data visualization and exploration. Brief Bioinform. 2013;14:178–192. doi: 10.1093/bib/bbs017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raghav SK, Deplancke B. Genome-wide profiling of DNA-binding proteins using barcode-based multiplex Solexa sequencing. Methods Mol Biol. 2012;786:247–262. doi: 10.1007/978-1-61779-292-2_15. [DOI] [PubMed] [Google Scholar]
- Cieply B, Farris J, Denvir J, Ford HL, Frisch SM. Epithelial-mesenchymal transition and tumor suppression are controlled by a reciprocal feedback loop between ZEB1 and Grainyhead-like-2. Cancer Res. 2013;73:6299–6309. doi: 10.1158/0008-5472.CAN-12-4082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roche J, Nasarre P, Gemmill R, Baldys A, Pontis J, Korch C, Guilhot J, Ait-Si-Ali S, Drabkin H. Global decrease of histone H3K27 acetylation in ZEB1-induced epithelial to mesenchymal transition in lung cancer cells. Cancer. 2013;5:334–356. doi: 10.3390/cancers5020334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shedden K, Taylor JM, Enkemann SA, Tsao MS, Yeatman TJ, Gerald WL, Eschrich S, Jurisica I, Giordano TJ, Misek DE, Chang AC, Zhu CQ, Strumpf D, Hanash S, Shepherd FA, Ding K, Seymour L, Naoki K, Pennell N, Weir B, Verhaak R, Ladd-Acosta C, Golub T, Gruidl M, Sharma A, Szoke J, Zakowski M, Rusch V, Kris M, Viale A. et al. Gene expression-based survival prediction in lung adenocarcinoma: a multi-site, blinded validation study. Nat Med. 2008;14:822–827. doi: 10.1038/nm.1790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu CQ, Ding K, Strumpf D, Weir BA, Meyerson M, Pennell N, Thomas RK, Naoki K, Ladd-Acosta C, Liu N, Pintilie M, Der S, Seymour L, Jurisica I, Shepherd FA, Tsao MS. Prognostic and predictive gene signature for adjuvant chemotherapy in resected non-small-cell lung cancer. J Clin Oncol. 2010;28:4417–4424. doi: 10.1200/JCO.2009.26.4325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolstad BM, Irizarry RA, Astrand M, Speed TP. A comparison of normalization methods for high density oligonucleotide array data based on variance and bias. Bioinformatics. 2003;19:185–193. doi: 10.1093/bioinformatics/19.2.185. [DOI] [PubMed] [Google Scholar]
- Singh A, Greninger P, Rhodes D, Koopman L, Violette S, Bardeesy N, Settleman J. A gene expression signature associated with “K-Ras addiction” reveals regulators of EMT and tumor cell survival. Cancer Cell. 2009;15:489–500. doi: 10.1016/j.ccr.2009.03.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Massague J. TGFbeta signalling in context. Nat Rev Mol Cell Biol. 2012;13:616–630. doi: 10.1038/nrm3434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guaita S, Puig I, Franci C, Garrido M, Dominguez D, Batlle E, Sancho E, Dedhar S, De Herreros AG, Baulida J. Snail induction of epithelial to mesenchymal transition in tumor cells is accompanied by MUC1 repression and ZEB1 expression. J Biol Chem. 2002;277:39209–39216. doi: 10.1074/jbc.M206400200. [DOI] [PubMed] [Google Scholar]
- Furusawa T, Moribe H, Kondoh H, Higashi Y. Identification of CtBP1 and CtBP2 as corepressors of zinc finger-homeodomain factor deltaEF1. Mol Cell Biol. 1999;19:8581–8590. doi: 10.1128/mcb.19.12.8581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Postigo AA. Opposing functions of ZEB proteins in the regulation of the TGFbeta/BMP signaling pathway. EMBO J. 2003;22:2443–2452. doi: 10.1093/emboj/cdg225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Postigo AA, Dean DC. ZEB represses transcription through interaction with the corepressor CtBP. Proc Natl Acad Sci U S A. 1999;96:6683–6688. doi: 10.1073/pnas.96.12.6683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Postigo AA, Depp JL, Taylor JJ, Kroll KL. Regulation of Smad signaling through a differential recruitment of coactivators and corepressors by ZEB proteins. EMBO J. 2003;22:2453–2462. doi: 10.1093/emboj/cdg226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikeda K, Kawakami K. DNA binding through distinct domains of zinc-finger-homeodomain protein AREB6 has different effects on gene transcription. FEBS. 1995;233:73–82. doi: 10.1111/j.1432-1033.1995.073_1.x. [DOI] [PubMed] [Google Scholar]
- Remacle JE, Kraft H, Lerchner W, Wuytens G, Collart C, Verschueren K, Smith JC, Huylebroeck D. New mode of DNA binding of multi-zinc finger transcription factors: deltaEF1 family members bind with two hands to two target sites. EMBO J. 1999;18:5073–5084. doi: 10.1093/emboj/18.18.5073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Younes M, Brown RW, Stephenson M, Gondo M, Cagle PT. Overexpression of Glut1 and Glut3 in stage I nonsmall cell lung carcinoma is associated with poor survival. Cancer. 1997;80:1046–1051. doi: 10.1002/(sici)1097-0142(19970915)80:6<1046::aid-cncr6>3.0.co;2-7. [DOI] [PubMed] [Google Scholar]
- Park SY, Kim HS, Kim NH, Ji S, Cha SY, Kang JG, Ota I, Shimada K, Konishi N, Nam HW, Hong SW, Yang WH, Roth J, Yook JI, Cho JW. Snail1 is stabilized by O-GlcNAc modification in hyperglycaemic condition. EMBO J. 2010;29:3787–3796. doi: 10.1038/emboj.2010.254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gemmill RM, Roche J, Potiron VA, Nasarre P, Mitas M, Coldren CD, Helfrich BA, Garrett-Mayer E, Bunn PA, Drabkin HA. ZEB1-responsive genes in non-small cell lung cancer. Cancer Lett. 2011;300:66–78. doi: 10.1016/j.canlet.2010.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polyak K, Weinberg RA. Transitions between epithelial and mesenchymal states: acquisition of malignant and stem cell traits. Nat Rev Cancer. 2009;9:265–273. doi: 10.1038/nrc2620. [DOI] [PubMed] [Google Scholar]
- Rhim AD, Mirek ET, Aiello NM, Maitra A, Bailey JM, McAllister F, Reichert M, Beatty GL, Rustgi AK, Vonderheide RH, Leach SD, Stanger BZ. EMT and dissemination precede pancreatic tumor formation. Cell. 2012;148:349–361. doi: 10.1016/j.cell.2011.11.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collisson EA, Sadanandam A, Olson P, Gibb WJ, Truitt M, Gu S, Cooc J, Weinkle J, Kim GE, Jakkula L, Feiler HS, Ko AH, Olshen AB, Danenberg KL, Tempero MA, Spellman PT, Hanahan D, Gray JW. Subtypes of pancreatic ductal adenocarcinoma and their differing responses to therapy. Nat Med. 2011;17:500–503. doi: 10.1038/nm.2344. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
GLUT1, GLUT4, GLUT12, and ZEB1 expression in mesenchymal and epithelial lung tumor cells. The indicated cell lines were lysed for RNA preparation followed by reverse transcription. The cDNA was amplified by real-time PCR using probes specific for the indicated genes or internal controls. Data show means ± s.d. (n = 3) of mRNA expression relative to the cell line expressing the least amount of the same gene (set to 1). When there was no specific amplification after 40 cycles of PCR, the samples were called not determined (N.D.).
hZEB1 and hGLUT3 induction upon mSNAIL overexpression. H727 or H2009 cell populations stably expressing a control plasmid (ctl) or mouse (m) SNAIL were lysed for RNA preparation followed by reverse transcription. The cDNA was amplified by real-time PCR using probes specific for human ZEB1, GLUT3, or an internal control. Data show means ± s.d. (n = 3) of mRNA expression, represented as fold induction (mSNAIL/ctl) for each cell line. The arrows and numbers indicate the fold induction between samples.
ZEB1 interacts with CtBP and p300 in mesenchymal lung tumor cells. Protein extracts from the indicated cell lines were used for ZEB1 or control (IgG) immunoprecipitation, followed by Western blot using the indicated antibodies. The arrowheads indicate the position of each protein. XT, cell extract; ø, empty lane.
ZEB1 is induced rapidly upon TGF-β stimulation. H2122 cells were stimulated with 10 ng/ml TGF-β for the indicated time points, after which the cells were lysed for RNA preparation followed by reverse transcription. The cDNA was amplified by real-time PCR using probes specific for ZEB1, SNAIL, or an internal control. Data show means ± s.d. (n = 4) of mRNA expression ratios between TGF-β treated and non-treated conditions.
Efficiency of siRNA or shRNA knockdown. (A) Efficiency of GLUT3 or GLUT1 siRNA knockdown. SW1573 cells were transfected with one of three different siRNAs to target GLUT3, two to target GLUT1, or a control (ctl) siRNA, or were left untransfected (--). Seventy-two hours later, the cells were lysed for RNA preparation followed by reverse transcription. The cDNA was amplified by real-time PCR using probes specific for the indicated genes or GAPDH as an internal control. Data show the percentage of mRNA expression relative to the non-transfected condition (set to 100%). (B) Efficiency of GLUT3 siRNA knockdown. SW1573 cells were transfected with one of three different siRNAs to target GLUT3, a control (ctl) siRNA, or were left untransfected (--). Seventy-two hours later, the cells were lysed with RIPA buffer to prepare protein extracts and to analyze the expression of GLUT3 by Western blot. (C) GLUT3 or GLUT1 knockdown does not affect the number of dead cells. SW1573 cells were transfected with control (ctl) siRNA, each of three different siRNAs to decrease GLUT3, or each of two siRNAs to decrease GLUT1, as indicated. One hundred forty-four hours later, dead cells were counted by trypan blue exclusion (n = 3). (D) Efficiency of GLUT3 shRNA stable knockdown. SW1573 cells stably expressing ctl or GLUT3 shRNAs (in the indicated vectors) were lysed (left panel) with RIPA buffer to prepare protein extracts and to analyze the expression of GLUT3 by Western blot, or (right panel) for RNA preparation followed by reverse transcription. The cDNA was amplified by real-time PCR using probes specific for GLUT3 or GAPDH as an internal control. Data show the percentage of GLUT3 mRNA expression relative to the control (ctl) shRNA expressing cells (set to 100%). *n.s., non-specific.
GLUT3 knockdown leads to diminished vimentin expression. SW1573 cells stably expressing a control (ctl) or GLUT3 shRNA were lysed for RNA preparation followed by reverse transcription. The cDNA was amplified by real-time PCR using probes specific for the indicated genes or internal controls. Data show means ± s.d. (n = 3) of mRNA expression relative to the condition with the least amount of mRNA for each gene (set to 1).
Generation of H727 cells stably expressing GLUT3. (A) Stable expression of GLUT3. (Left) Parental H727 cells or H727 cells stably expressing a control (ctl) plasmid or a GLUT3 cDNA were lysed with RIPA buffer to prepare protein extracts and to analyze the expression of GLUT3 (or β-tubulin as loading control) by Western blot. (Right) Parental or GLUT3-expressing H727 cells were stained to analyze the expression of GLUT3 by immunocytochemistry (ICC). Scale bar, 100 μm. (B) Ectopic GLUT3 expression does not affect cell proliferation in high glucose concentrations. H727 cells, either parental or stably expressing a control (ctl) plasmid or a GLUT3 cDNA, were cultured in high glucose (glc) concentrations for 4 days, after which live cells were counted by trypan blue exclusion (n = 7).