

Abstract
Melatonin has a cellular protective effect in cerebrovascular and neurodegenerative diseases. Protection of brain endothelial cells against hypoxia and oxidative stress is important for treatment of central nervous system (CNS) diseases, since brain endothelial cells constitute the blood brain barrier (BBB). In the present study, we investigated the protective effect of melatonin against oxygen-glucose deprivation, followed by reperfusion- (OGD/R-) induced injury, in bEnd.3 cells. The effect of melatonin was examined by western blot analysis, cell viability assays, measurement of intracellular reactive oxygen species (ROS), and immunocytochemistry (ICC). Our results showed that treatment with melatonin prevents cell death and degradation of tight junction protein in the setting of OGD/R-induced injury. In response to OGD/R injury of bEnd.3 cells, melatonin activates Akt, which promotes cell survival, and attenuates phosphorylation of JNK, which triggers apoptosis. Thus, melatonin protects bEnd.3 cells against OGD/R-induced injury.
1. Introduction
Stroke is the third most frequent worldwide cause of adult death [1, 2]. Specifically, about 80% of all strokes are ischemic, resulting from arterial occlusion in the brain [1]. Reperfusion after occlusion results in serious brain injury, due to overproduction of reactive oxygen species (ROS), calcium overload [3, 4], and blood-brain barrier (BBB) injury [5]. Finally, in ischemic stroke, the brain is damaged because of hypoxia and oxidative stress [6–10]. Reactive oxygen species (ROS) play a key role in the pathogenesis of many diseases, including central nervous system (CNS) diseases [11–14]. During ischemic stroke, the excessive generation of ROS leads to inflammation and cell apoptosis [15–21] and induces mitogen-activated protein kinase (MAPK) signaling [22–24]. c-Jun N-terminal kinase (JNK), one of the MAPKs, is activated by a variety of cell stresses, including hyperosmotic shock, hypoxia, and ROS [25, 26]. JNK plays key roles in apoptosis and inflammation [27, 28]. JNK signaling is activated by inflammatory cytokines and promotes neuronal cell death [29]. Endothelial cells are also damaged by activation of JNK signaling, in response to oxidative stress [30]. Several studies have demonstrated that, in hypoxia and a state of reoxygenation, cells induce apoptotic signaling through JNK and p38 MAPK [31, 32]. The BBB controls the exchange of materials between blood and the brain and plays an important role in the homeostatic regulation of the brain microenvironment [33]. The tight junctions between capillary endothelial cells, which form an essential structural component of the BBB [34], include membrane proteins like occludin [35] and claudins [36, 37]. Several studies have suggested that hypoxia causes alterations of the tight junction proteins Claudin 5, occludin, ZO-1, and ZO-2, which affect BBB permeability [38, 39]. In addition, vascular endothelial growth factor (VEGF) is an inducer of vascular leakage [40] and is also known as vascular permeability enhancing factor [41, 42]. During ischemia, VEGF interacts with receptors for VEGF on the ischemic vessels and contributes to disruption of the BBB [43, 44]. Zhang el al. demonstrated that inhibition of VEGF reduces BBB permeability [43]. Melatonin is synthesized in the pineal gland and has been known to function as an antioxidant [45]. Melatonin reduces the cellular toxicity of ROS in ischemia and reperfusion (I/R) brain injury [46]. In an in vivo cerebral ischemia model, several researches have demonstrated that melatonin treatment reduces brain damage in the setting of ischemia or hypoxia-induced injury [47, 48]. In vitro, melatonin protects primary neuronal cells from apoptotic death [49] and enhances survival of human neuroblastoma cells [50] in the setting of oxygen-glucose deprivation- (OGD-) induced injury. Furthermore, melatonin suppresses VEGF expression in cancer cells [51, 52] and inhibits serum VEGF levels in patients [53]. In the present study, we investigate whether melatonin protects brain endothelial cells against oxygen-glucose deprivation followed by reperfusion- (OGD/R-) induced injury. We show that melatonin reduces the generation of ROS, prevents disruption of the BBB by stabilizing expression of tight junction proteins and suppressing VEGF expression, and attenuates phosphorylation of JNK, a mediator of cellular apoptosis. Therefore, our results suggest that melatonin is important in protecting the BBB against cerebral ischemic damage.
2. Materials and Methods
2.1. Cell Culture
Murine brain endothelial cells (bEnd.3 cells; ATCC, VA, USA) were purchased from ATCC and cultured in Dulbecco's modified Eagle's medium (DMEM, Hyclone Laboratories, UT, USA), supplemented with 10% (v/v) fetal bovine serum (FBS, Hyclone Laboratories, UT, USA) and 100 units/mL of penicillin/streptomycin (Hyclone Laboratories, UT, USA), at 37°C in a humidified atmosphere in the presence of 5% CO2 [54]. bEND.3 cells were used at 13 passages in this study.
2.2. Oxygen-Glucose Deprivation (OGD) and Reperfusion
Confluent cells were transferred to an anaerobic chamber (Forma Scientific, OH, USA) (O2 tension, 0.1%) and washed three times with PBS. Then, culture medium was replaced with deoxygenated, glucose-free balanced salt solution, and cells were incubated for 6 h. Following oxygen-glucose deprivation (OGD) injury, cells were incubated for 18 h under normal growth conditions, with or without drug treatment [55].
2.3. Drug Treatment
Melatonin was purchased from Sigma (Sigma, MO, USA) and dissolved in ethanol. An equivalent volume of ethanol (final: 0.01%) or water was added to control and all melatonin-containing wells. bEnd.3 cells were exposed to 1–100 nM melatonin for 24 h before OGD/R injury. The present study consisted of four groups: (1) normal control (NC), bEnd.3 cells cultured with normal media without OGD injury; (2) experimental control (EC), bEnd.3 cells cultured in nontreated medium for 18 h after 6 h of OGD injury; (3) 10 nM melatonin (Mel 10 nM), bEnd.3 cells treated with 10 nM melatonin for 24 h before 6 h of OGD injury; these cells were then cultured in nontreated medium for 18 h; (4) 100 nM melatonin (Mel 100 nM): bEnd.3 cells were also treated with 100 nM melatonin (100 nM melatonin group) for 24 h before 6 h of OGD injury. These cells were then cultured in nontreated medium for 18 h. In Akt inhibitor groups, we treated 100 nM Akt inhibitor (Sigma, MO, USA) together with melatonin.
2.4. Hoechst 33258 and Propidium Iodide (PI) Staining
Cell viability was evaluated by staining bEnd.3 cells with Hoechst 33258 dye (Sigma, MO, USA) and propidium iodide (PI; Sigma, MO, USA). Hoechst dye was added to the culture medium (2-3 μg/mL) and samples were then incubated at 37.8°C for 30 min. PI solution was then added (2–5 μg/mL) just before cells were observed with a microscope (BX51; Olympus) equipped with epifluorescence and a UV filter block. PI-positive cells were counted as dead cells [56].
2.5. Cell Viability Assay
bEnd.3 cells (2 × 105 cells/mL) were seeded in 98-well plates to monitor all experiment conditions, including pretreatment, OGD injury, and reperfusion. Next, cells were rinsed twice with phosphate-buffered saline (PBS), and culture medium was replaced with serum-free medium and 100 μL 3-[4,5-dimethylthiazol-2-yl]-2,5-diphenyl tetrasodium bromide (MTT) (Sigma, MO, USA) solution (5 mg/mL in PBS) per well. After 1 h of incubation, medium was removed and dimethyl sulfoxide (DMSO) was added to solubilize the purple formazan product of MTT treatment. The supernatant from each well was analyzed using an ELISA plate reader (Labsystems Multiskan MCC/340; Fisher Scientific, PA, USA) at a wavelength of 570 nm, with background subtraction at 650 nm. All experiments were repeated at least three times. Cell viability in the control medium, without any treatment, was represented as 100%. Cell viability was reported as a relative value, compared to the control group.
2.6. Lactate Dehydrogenase (LDH) Assay
Cytotoxicity in all treatment groups was quantified by measuring the amount of LDH released into the culture medium from OGD/R-injured cells [57, 58]. LDH release (cytotoxicity %) was calculated by dividing the value at the experimental time point by the maximum value. The maximum LDH release was measured after freezing each culture at −70°C overnight, followed by rapid thawing, which induced nearly complete cell damage.
2.7. Determination of Intracellular ROS
The level of intracellular ROS in each treatment group was measured using a fluorescent probe, 2′,7′-dichlorodihydrofluorescein diacetate (DCF-DA; Invitrogen, CA, USA), as previously described [59]. Cells were plated at a density of 1 × 106 cells/mL and treated with melatonin for 24 h. After melatonin pretreatment, OGD injury and reperfusion were conducted. Then, bEND.3 cells were treated with 5 μM DCF-DA for 30 min at 37°C. After washing with PBS, fluorescence was measured with a microscope (Nikon TS100-F ECLIPSE) equipped with a CCD camera (Hamamatsu Photonics) [54].
2.8. Western Blot Analysis
After pretreatment, OGD injury, and restoration, cells were washed rapidly with ice-cold PBS, scraped, and collected. Cell pellets were lysed with ice-cold RIPA buffer (Sigma, MO, USA). The lysates were centrifuged at 13,200 rpm for 1 h at 4°C to produce whole-cell extracts. Protein content was quantified using the BCA method (Pierce, IL, USA). Protein (20 μg) was separated on a 10% SDS-polyacrylamide (PAGE) gel and transferred onto a polyvinylidene difluoride (PVDF) membrane. After blocking with 5% bovine serum albumin, prepared in Tris-buffered saline/Tween (TBS-T; 20 nM Tris (pH 7.2); 150 mM NaCl; 0.1% Tween 20), for 1 h at RT, immunoblots were incubated overnight at 4°C with primary antibodies that specifically detect Akt (1 : 2000, Cell Signaling, MA, USA), p-Akt (1 : 2000, Cell Signaling, MA, USA), JNK (1 : 2000, Cell Signaling, MA, USA), p-JNK (1 : 2000, Cell Signaling, MA, USA), Claudin 5 (1 : 1000, Santa Cruz, CA, USA), VEGF (1 : 1000, Millipore, MA, USA), Bax (1 : 2000, Cell Signaling, MA, USA), or β-actin (1 : 2000, Cell Signaling, MA, USA). Next, blots were incubated with HRP-linked anti-mouse and -rabbit IgG antibodies purchased from Abcam (Cambridge, MA, USA) for 1 h at RT. Enhanced chemiluminescence was performed by ECL (Pierce, IL, USA) [54].
2.9. Immunocytochemistry (ICC)
The expression of VEGF and Claudin 5 in bEnd.3 cells was confirmed by immunocytochemistry. Cells in all experimental groups were washed three times with PBS, fixed with 4% paraformaldehyde for 3 h, and then washed with PBS. bEnd.3 cells were permeabilized with 0.025% Triton X-100 and blocked for 1 h at RT with dilution buffer (Invitrogen, CA, USA). Primary anti-rabbit VEGF (1 : 500, Millipore, MA, USA) and anti-rabbit Claudin 5 (1 : 500, Santa Cruz, CA, USA) antibodies were prepared in dilution buffer, added to samples, and incubated for 3 h at RT. Primary antibody was then removed and cells were washed three times for 3 min each with PBS. Later, samples were incubated with FITC-conjugated goat, anti-rabbit (1 : 200, Jackson Immunoresearch, PA, USA) or Rhodamine-conjugated donkey, or anti-rabbit secondary antibodies (1 : 500, Millipore, MA, USA) for 2 h at RT. Cells were washed again three times for 3 min each with PBS and stained with 1 μg/mL 4′,6-diamidino-2-phenylindole (DAPI) (1 : 100, Invitrogen, CA, USA) for 10 min at RT. Fixed samples were imaged using a Zeiss LSM 700 confocal microscope (Carl Zeiss, NY, USA).
2.10. Statistical Analysis
Statistical comparisons were performed using independent t-tests for two groups. SPSS software was used for all analyses. Data were expressed as mean ± S.E.M. of three independent experiments. Differences were considered significant at # P < 0.1, ∗P < 0.05, and ∗∗P < 0.001.
3. Results
3.1. Melatonin Attenuates the Cell Death of bEND.3 Cells after OGD/R-Induced Injury
To confirm the protective effect of melatonin on OGD/R-induced injury, we first conducted an MTT assay to check cell viability in all treatment groups (Figure 1(a)). Cell viability showed that the OGD/R injury exposed group exhibited decreased cell viability, compared to the normal control group (100% cell viability in the normal control group; 39% cell viability in the OGD/R injury exposed group). We checked the cell viability by pretreatment with melatonin 1 nM to 100 nM. Cell viability in 1 nM and 5 nM melatonin pretreatment group was almost not different from the OGD/R injury exposed group. Treatment with 10 nM melatonin also did not change cell viability compared to the OGD/R injury exposed group (48% cell viability in the Mel 10 nM group). However, treatment with 100 nM melatonin obviously increased cell viability after OGD/R-induced injury, compared to the normal control group (62% cell viability in the Mel 100 nM group) (Figure 1(a)). In addition, we evaluated cytotoxicity in bEND.3 cells following OGD/R injury using an LDH assay (Figure 1(b)). Cytotoxicity was 12% in the normal control group but was 28% in the OGD/R injury exposed group. Cytotoxicity in 1 nM and 5 nM melatonin pretreatment group was not largely different from the OGD/R injury exposed group. Treating cells with 10 nM melatonin resulted in 21% cytotoxicity and treating cells with 100 nM melatonin resulted in 18% cytotoxicity (Figure 1(b)). Considering cell viability and cytotoxicity data, we decided two concentrations of melatonin (10 nM melatonin concentration (among the low concentrations: 1 nM, 5 nM, and 10 nM) and 100 nM melatonin concentration (among the high concentrations: 50 nM, 100 nM)) to compare the effect of melatonin easily. We also conducted Hoechst/PI staining to check the dead cells in all groups (Figure 1(c)). Hoechst/PI staining images showed that only melatonin treatment groups were almost not different from the normal control group. PI-positive cells (dead cells) evidently were increased in the OGD/R injury exposed group, compared to the normal control group. 10 nM and 100 nM melatonin treatment promoted cell survival and inhibited cell death against OGD/R-induced injury. In the 100 nM melatonin treatment group, the protective effect of melatonin against OGD/R injury death in bEND.3 cells was more obvious than in the 10 nM melatonin treatment group (Figure 1(c)). Taken together, these findings suggest that melatonin attenuates OGD/R-induced damage in brain endothelial cells.
Figure 1.
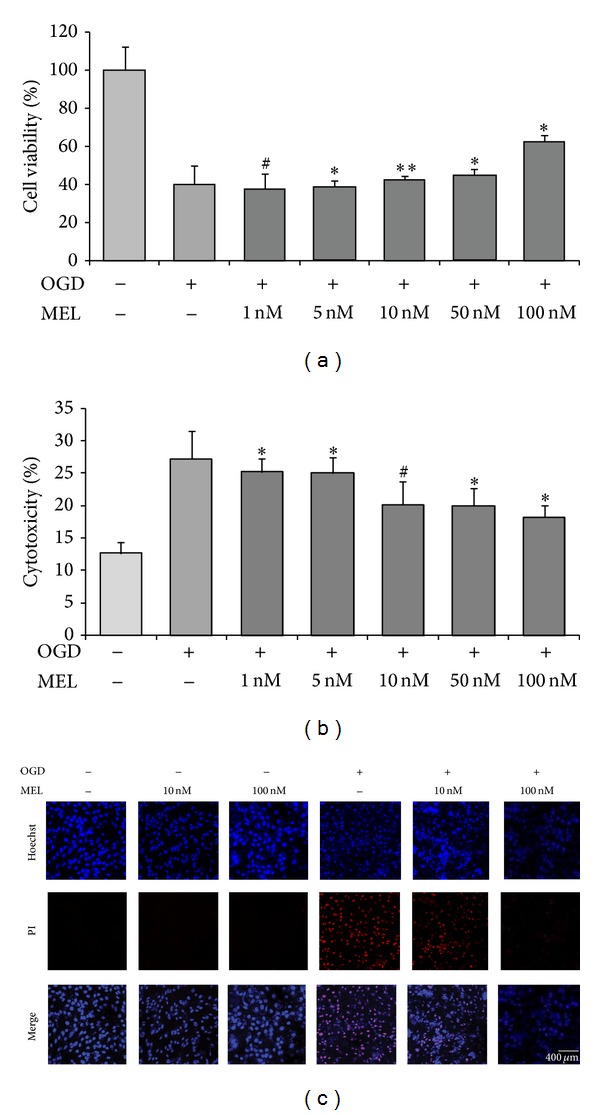
The measurement of brain endothelial cell viability after OGD/R-induced injury. (a) A 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay shows that bEND.3 cells in the OGD/R injury exposed group exhibited decreased viability compared to cells in the normal control group. Cell viability of bEND.3 cells in 1 nM and 5 nM melatonin pretreatment groups was not largely different form OGD/R injury exposed group. bEND.3 cells in 10 nM, 50 nM, and especially 100 nM melatonin pretreatment groups exhibited increased cell viability compared to OGD/R injury exposed group. Data are expressed as mean ± S.E.M. (# P < 0.1, ∗P < 0.05, and ∗∗P < 0.001). (b) Cytotoxicity (%) was measured using an LDH assay. Cytotoxicity increased in OGD/R injury exposed group compared to the normal control group. Melatonin treatment (especially 100 nM melatonin pretreatment) reduced cytotoxicity after OGD/R injury. Data are expressed as mean ± S.E.M. (# P < 0.1, ∗P < 0.05, and ∗∗P < 0.001). (c) Dead and live cells were measured by Hoechst/PI staining. PI-positive cells (red) are regarded as the dead cells. PI-positive cells were higher in OGD/R injury exposed group than in the normal control group. Melatonin treatment groups (both in 10 nM and in 100 nM melatonin groups) exhibited reduced PI-positive cells compared to the OGD/R injury exposed group. Hoechst: Hoechst 33342 (blue color) and PI: propidium iodide (red color). Scale bar = 400 µm.
3.2. Melatonin Decreases OGD/R-Induced ROS Production
We measured ROS levels using DCF-DA reagent, a fluorescent dye that visualizes ROS. DCF-DA-positive cells increased after OGD/R. ROS levels in melatonin pretreatment groups (10 nM, 100 nM melatonin) were not largely different from ROS levels in the normal control group. In the OGD/R injury exposed group, ROS levels were evidently increased compared to the normal control group. This was partially blocked by pretreatment with 10 nM melatonin (Figures 2(a) and 2(b)). 100 nM melatonin pretreatment clearly decreased the number of DCF-DA-positive cells, compared to the OGD/R injury exposed group. This result suggests that melatonin inhibits OGD/R-induced ROS production in brain endothelial cells.
Figure 2.
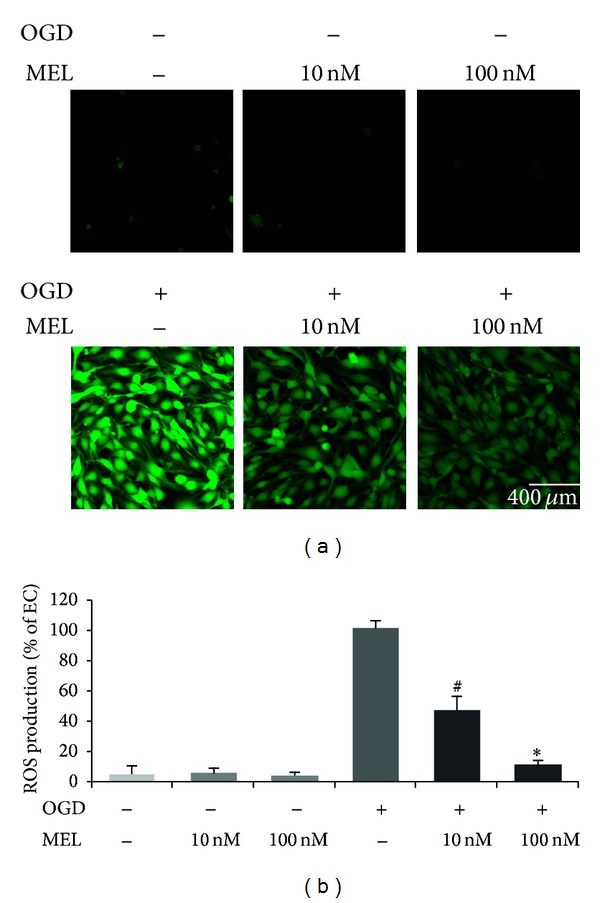
Immunocytochemistry to measure ROS generation in bEND.3 cells after OGD/R-induced injury. bEND.3 cells were treated with melatonin for 24 h before OGD/R injury. ROS levels were measured using DCF-DA. (a) ROS levels in only melatonin treatment groups (both 10 nM and 100 nM melatonin pretreatment groups) were the same as the normal control group. ROS levels in bEND.3 cells were increased in OGD/R injury exposed group. Under OGD/R injury, ROS levels in the melatonin pretreatment group were decreased compared to OGD/R injury exposed group. Melatonin decreased the OGD/R-induced increase in DCF-DA-positive cells (green). (b) ROS production was calculated by measuring the intensity of ROS. This graph shows relative intensity as a percentage of OGD/R injury exposed group. Data are expressed as mean ± S.E.M. (# P < 0.1 and ∗P < 0.05). 2′,7′-Dichlorodihydrofluorescein diacetate (DCF-DA): green. Scale bar = 400 µm.
3.3. Melatonin Prevents Degradation of Tight Junction Proteins against OGD/R Injury
To check the protective effect of melatonin on the integrity of tight junctions during OGD/R, we measured the level of Claudin 5, a tight junction protein, by immunocytochemistry (Figure 3(a)) and western blot analysis (Figure 3(b)). OGD/R stress obviously decreased the expression of Claudin 5 in the bEND.3 cells compared to the normal control (NC) group. The expression of Claudin 5 did not nearly change in the 10 nM melatonin treatment group, compared to the experimental control (EC) group which in exposed OGD/R injury. The expression of Claudin 5 was evidently attenuated by treatment with 100 nM melatonin (Figures 3(a) and 3(b)). This result shows that melatonin pretreatment protects degradation of Claudin 5 following OGD/R injury. Namely, melatonin may prevent deterioration of tight junctions in response to OGD/R-induced injury.
Figure 3.
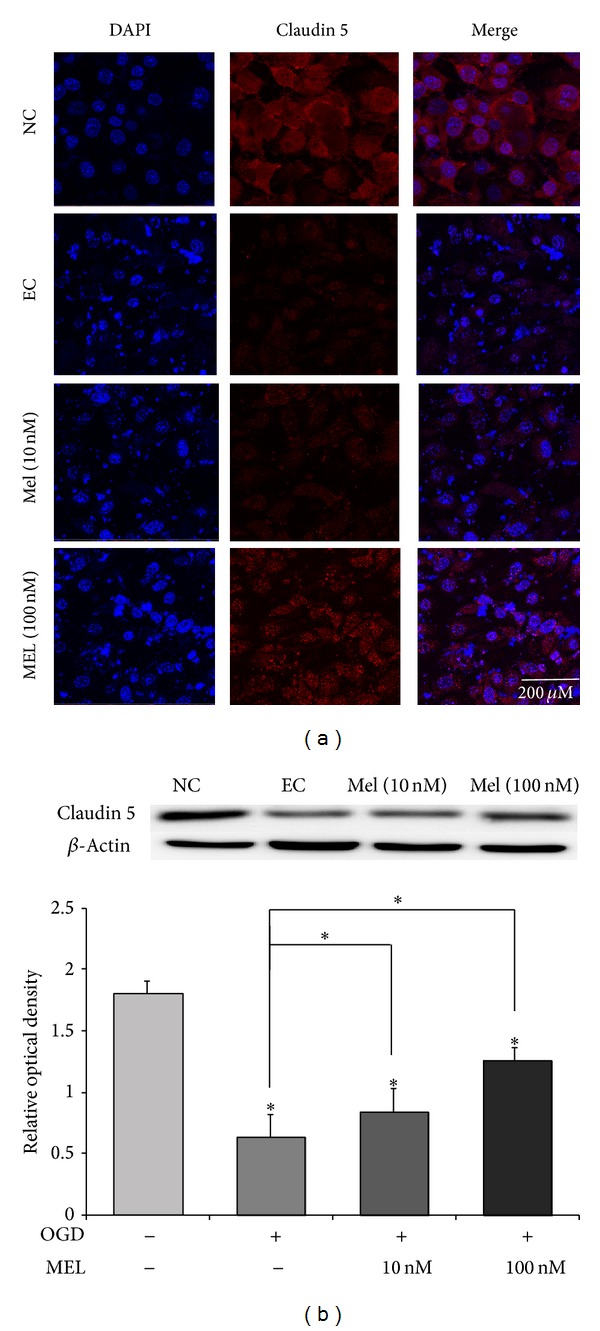
The measurement of the tight junction protein in bEND.3 cells after OGD/R-induced injury. (a) The level of Claudin 5, a tight junction protein, was evaluated by immunocytochemistry. This image shows that expression of Claudin 5 in the experimental control (EC) group decreased compared to the normal control (NC) group. Melatonin increased the expression of Claudin 5 under OGD/R injury (green). In the Mel (10 nM) and Mel (100 nM) groups, the expression of Claudin 5 was higher than in the EC group. Claudin 5 was preserved in the melatonin treatment group, following OGD/R-induced injury. Scale bar: 200 µm, Claudin 5: red, and 4′,6-diamidino-2-phenylindole (DAPI): blue. (b) Western blotting showed that the relative protein level of Claudin 5 was reduced in EC compared to the NC group. The relative level of Claudin 5 was increased in Mel (10 nM) and Mel (100 nM) groups, compared to the EC group. The bar graph shows the quantification of Claudin 5 protein in all groups. β-Actin was used as an internal control. Data are expressed as mean ± S.E.M. (∗P < 0.05). (i) Normal control (NC): bEnd.3 cells cultured with normal media without OGD injury, (ii) experimental control (EC): bEnd.3 cells cultured in nontreated medium for 18 h after 6 h of OGD injury, and (iii) 10 nM melatonin (Mel 10 nM): bEnd.3 cells treated with 10 nM melatonin for 24 h before 6 h of OGD injury. These cells were then cultured in nontreated medium for 18 hr. (iv) 100 nM melatonin (Mel 100 nM): bEnd.3 cells were also treated with 100 nM melatonin (100 nM melatonin group) for 24 h before 6 h of OGD injury. These cells were then cultured in nontreated medium for 18 h.
3.4. Melatonin Attenuates the Expression of VEGF after OGD/R-Induced Injury
We conducted immunocytochemistry (Figures 4(a) and 4(b)) and western blot analysis (Figure 4(c)) to confirm the expression of VEGF in all treatment groups. This result indicated that the expression of VEGF became considerably elevated after OGD/R injury in the bEND.3 cells. However, the expression of VEGF was reduced by melatonin treatment (both 10 nM and 100 nM melatonin pretreatment) (Figures 4(a) and 4(b)). This finding suggests that melatonin attenuates the expression of VEGF in brain endothelial cells following OGD/R-induced injury.
Figure 4.
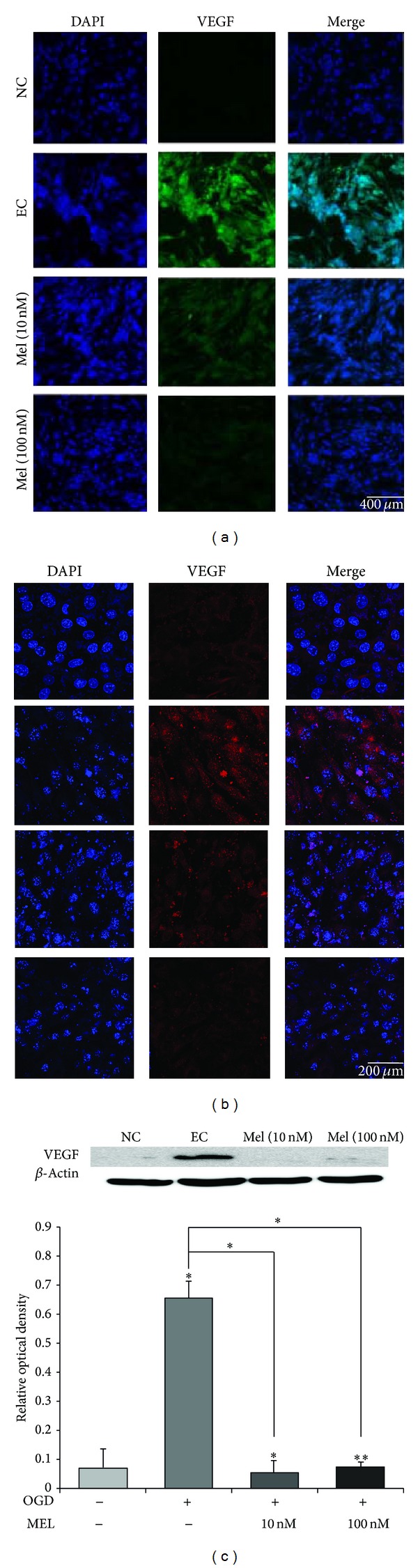
The measurement of VEGF expression in bEND.3 cells after OGD/R-induced injury. (a) The level of VEGF was evaluated by immunocytochemistry. This image shows that the expression of VEGF in the experimental control (EC) group was increased compared to the normal control (NC) group. Melatonin attenuated the OGD/R-induced increase in the number of VEGF-positive cells. In Mel (10 nM) and Mel (100 nM) groups, the expression of VEGF was lower than in the EC group. VEGF expression was attenuated in the melatonin treatment group under OGD/R-induced injury. Scale bar: 400 µm. (b) Scale bar: 200 µm, vascular endothelial growth factor (VEGF): green, red, and 4′,6-diamidino-2-phenylindole (DAPI): blue. (c) Western blotting showed that the protein level of VEGF was evidently increased in EC compared to the NC group. The protein level of VEGF was attenuated in both Mel (10 nM) and Mel (100 nM) groups, compared to the EC group. The bar graph shows the quantification of VEGF protein in all groups. β-Actin was used as an internal control. Data are expressed as mean ± S.E.M. (∗P < 0.05 and ∗∗P < 0.001).
3.5. Melatonin Protects bEND.3 Cells via Akt Activation and JNK Suppression
To investigate whether Akt signaling was activated in OGD/R-induced stress, we first measured the phosphorylation status of Akt by western blot analysis (Figure 5(a)). Phosphorylation of Akt is associated with activation of Akt signaling and cell survival. Our result suggests that the protein expression of phosphor-Akt/Akt in the EC group is attenuated compared to the NC group. Expression of phosphor-Akt in the 10 nM melatonin treatment group did not nearly change compared to the EC group. However, expression of phosphor-Akt in the 100 nM melatonin treatment group was higher than in the EC group (Figure 5(a)). Next, we also examined the phosphorylation status of JNK by western blot analysis (Figure 5(b)), because the phosphorylation of JNK correlates with activation of apoptosis signaling. The expression of phosphor-JNK was decreased by melatonin treatment after OGD/R-induced injury. Pretreatment with 100 nM melatonin resulted in the obvious inhibition of JNK signaling whereas JNK activation in 10 nM melatonin pretreatment group was not largely different from the EC group (Figure 5(b)). These results suggest that melatonin 100 nM increases Akt activation and suppresses JNK activation. To confirm the relationship between melatonin and Akt signaling, we checked the expression of Bax by western blot analysis (Figure 5(c)). We confirmed that the protein expression of Bax in the bEND.3 cells was increased under OGD/R injury compared to the NC group. Also, 10 nM and 100 nM melatonin treatment reduced the protein expression of Bax under OGD/R injury. When we checked the expression of Bax in OGD/R injured bEND.3 cells with Akt inhibitor and melatonin pretreatment, we confirmed that Akt inhibitor pretreatment did not reduce the expression of Bax in melatonin pretreatment groups (Figure 5(d)). These findings indicate that melatonin may promote Akt signaling and suppress JNK signaling. Specifically, melatonin may attenuate the expression of Bax, known as an apoptotic protein through Akt activation in brain endothelial cells following OGD/R stress.
Figure 5.
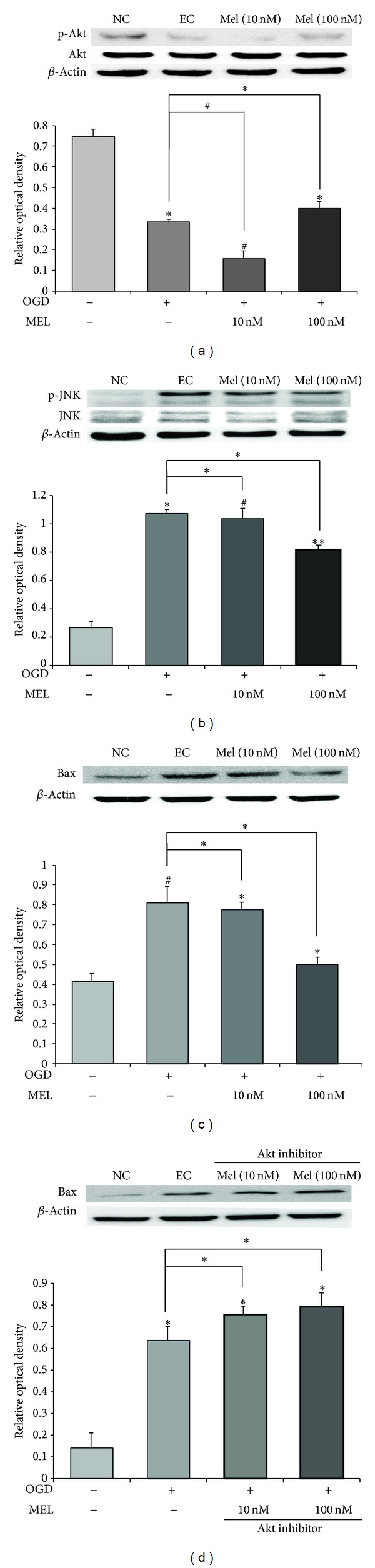
The measurement of JNK, Akt, and Bax expression in brain endothelial cells after OGD/R-induced injury. (a) Western blotting showed that the relative protein level of phosphor-Akt was reduced in EC compared to the NC group. The protein level of phosphor-Akt was increased in Mel (100 nM) groups, compared to the EC group. The bar graph shows the quantification of phosphor-Akt/Akt protein in all groups. (b) Western blotting showed that the relative protein expression of phosphor-JNK increased in the EC group, compared to the NC group. The relative level of phosphor-JNK decreased in Mel 10 nM and Mel 100 nM groups, compared to the EC group. The bar graph shows the quantification of phosphor-JNK/JNK protein in all groups. (c) Western blotting showed that the relative protein expression of Bax increased in the EC group, compared to the NC group. The protein level of Bax decreased in Mel 10 nM and Mel 100 nM groups, compared to the EC group. The bar graph shows the quantification of Bax protein in all groups. (d) Western blotting showed the relative protein expression of Bax by melatonin and 100 nM Akt inhibitor pretreatment under OGD/R injury. The expression of Bax was increased in the EC treatment group, compared to the NC group. The protein level of Bax was increased in Mel 10 nM and Mel 100 nM groups with 100 nM Akt inhibitor copretreatment, compared to the EC group. The bar graph shows the quantification of Bax in all groups. β-Actin was used as an internal control. Data are expressed as mean ± S.E.M. (# P < 0.1, ∗P < 0.05, and ∗∗P < 0.001). Protein kinase B (Akt), phosphorylated Akt (p-Akt), c-Jun N-terminal kinases (JNK), and phosphorylated JNK (p-JNK).
4. Discussion
Ischemic stroke causes oxidative stress in the brain as well as various neuropathological impairments [60]. BBB disruption is commonly observed in stroke patients [61, 62]. BBB damage is aggravated by reperfusion after ischemia [63]. ROS are generated during cerebral ischemia-reperfusion injury and lead to severe brain damage by promoting the cell apoptosis pathway [64, 65]. Also, ROS cause BBB hyperpermeability, brain edema, hemorrhage, and inflammation [66]. In the present study, we induced OGD/R injury, which is known as an appropriate in vitro model of stroke [67, 68], in brain endothelial cells to investigate the effect of ischemia-reperfusion injury. Recent research suggests that antioxidants attenuate oxidative damage induced by ischemia-reperfusion injury by decreasing mechanisms of ROS production [69]. Previous researches have suggested that antioxidants preserve BBB disruption and attenuate ROS generation after cerebral ischemia reperfusion in vivo [70–72] and in vitro [73, 74]. Melatonin is known as an antioxidant [75], a powerful free radical scavenger [76–78], and the cellular protector against various oxidative stress-associated diseases [79, 80]. Several studies in animals have suggested that melatonin reduces cellular damage by decreasing ROS in ischemia-reperfusion injury [46, 81, 82] and ischemia-hypoxia injury [83]. In the present study, we confirmed that melatonin reduces OGD/R-induced ROS generation in brain endothelial cells and prevents cell death of brain endothelial cells following OGD/R injury. Hypoxia causes degradation of tight junction proteins, such as Claudin 3, ZO-1 and ZO-2, and occludin [38, 39]. Several studies have demonstrated that claudins are major proteins in tight junctions [84–87], which are essential structural components of the BBB [34]. And, Claudin 5 is an important molecule that promotes disruption of the BBB in hypoxic conditions [88]. Tao et al. have demonstrated that melatonin prevents degradation of ZO-1, a tight junction protein that protects against ischemic injury in endothelial cells [89]. To determine the protective effect of melatonin on impaired BBB function caused by ischemia reperfusion, we examined Claudin 5 protein expression in brain endothelial cells following OGD/R injury. Our findings suggest that melatonin may prevent BBB disruption during ischemia-reperfusion injury by inhibiting degradation of the Claudin 5 tight junction protein. Hypoxia results in increased paracellular permeability [38, 90–92], leading to formation of cerebral edema [93]. Hypoxia induces the expression of VEGF [94–97], which is considered as one of the most important factors that stimulates the formation of new blood vessels [94, 95]. VEGF increases the permeability of blood vessels [92, 98, 99] and leads to vasogenic edema [100–103]. Several studies have demonstrated that VEGF increases BBB permeability [99], while inhibition of VEGF reduces BBB permeability [43]. Melatonin protects BBB hyperpermeability and reduces brain edema in ischemic stroke [104, 105]. Also, recent research has shown that melatonin reduces expression of VEGF in hypoxic damage [53, 106–108]. In the present study, our results showed that melatonin reduced the expression of VEGF in brain endothelial cells following OGD/R-induced injury. In oxidative stress, ROS acts as an important mediator to activate the MAPK pathway [23, 24]. The phosphatidylinositol-3-kinase/protein kinase B (PI3K/Akt) signaling pathway is considered to be one of the cell survival pathways [109]. Many researches have demonstrated that Akt plays a major role in protection from cell death under oxidative stress [110–115] and attenuates ROS production, which protects cells [116]. In brain endothelial cells, Akt enhances cell survival and inhibits apoptosis [117–119]. Melatonin promotes Akt signaling to protect cells in response to stress [120]. In the present study, our result showed that melatonin enhanced Akt activation following OGD/R injury. This finding may indicate that melatonin protects brain endothelial cells via Akt activation in the setting of ischemia-reperfusion injury. In addition, Akt can protect cellular apoptosis by regulating a proapoptotic protein such as Bax [121–124]. Several studies demonstrated that melatonin may regulate the Bax expression and may be involved in the apoptosis signaling [125, 126]. In the present study, our results showed that melatonin may regulate the Bax expression through regulating Akt activation. Considering that Bax is the proapoptotic protein, melatonin may protect the apoptosis of brain endothelial cells through suppressing the expression of Bax in response to hypoxia and reperfusion stress. JNK signaling contributes to cellular apoptosis triggered by various stresses, including oxidized LDL, proinflammatory cytokines, or high glucose [127–129]. Specifically, excessive ROS generation is closely linked to JNK activation [130–132]. JNK activation triggers the mitochondrial apoptotic pathway [133, 134] and disrupts the BBB [135]. Several studies have shown that JNK inhibitors exert protective effects against ischemic injury in a rodent model [136–139]. In the present study, our findings suggest that melatonin attenuates JNK activation in OGD/R-exposed brain endothelial cells. This result indicates that melatonin may inhibit the death of brain endothelial cells via JNK suppression. In conclusion, melatonin protects brain endothelial cells against ischemic-reperfusion injury by reducing the production of ROS, by preserving tight junction proteins, by attenuating expression of VEGF, and by regulating Akt activation and JNK suppression. Hence, this study suggests that melatonin may play as the protector on brain endothelial cells under brain hypoxic injury such as stroke. For application to the patients with stroke, this study has many limitations because of confirmation only in vitro study. However, these findings may provide the basic data for the further study on stroke.
Acknowledgment
This research was supported by the Brain Research Program through the National Research Foundation of Korea (NRF), which is funded by the Ministry of Science, ICT & Future Planning (2012-0005827).
Conflict of Interests
The authors declare that there is no conflict of interests regarding the publication of this paper.
References
- 1.Flynn RWV, MacWalter RSM, Doney ASF. The cost of cerebral ischaemia. Neuropharmacology. 2008;55(3):250–256. doi: 10.1016/j.neuropharm.2008.05.031. [DOI] [PubMed] [Google Scholar]
- 2.Doyle KP, Simon RP, Stenzel-Poore MP. Mechanisms of ischemic brain damage. Neuropharmacology. 2008;55(3):310–318. doi: 10.1016/j.neuropharm.2008.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kuroda S, Siesjö BK. Reperfusion damage following focal ischemia: pathophysiology and therapeutic windows. Clinical Neuroscience. 1997;4(4):199–212. [PubMed] [Google Scholar]
- 4.Nakamura T, Minamisawa H, Katayama Y, et al. Increased intracellular Ca2+ concentration in the hippocampal CA1 area during global ischemia and reperfusion in the rat: a possible cause of delayed neuronal death. Neuroscience. 1999;88(1):57–67. doi: 10.1016/s0306-4522(98)00207-3. [DOI] [PubMed] [Google Scholar]
- 5.Abbott NJ, Patabendige AAK, Dolman DEM, Yusof SR, Begley DJ. Structure and function of the blood-brain barrier. Neurobiology of Disease. 2010;37(1):13–25. doi: 10.1016/j.nbd.2009.07.030. [DOI] [PubMed] [Google Scholar]
- 6.Lee J, Grabb MC, Zipfel GJ, Choi DW. Brain tissue responses to ischemia. The Journal of Clinical Investigation. 2000;106(6):723–731. doi: 10.1172/JCI11003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.MacManus JP, Buchan AM, Hill IE, Rasquinha I, Preston E. Global ischemia can cause DNA fragmentation indicative of apoptosis in rat brain. Neuroscience Letters. 1993;164(1-2):89–92. doi: 10.1016/0304-3940(93)90864-h. [DOI] [PubMed] [Google Scholar]
- 8.Kihara S, Shiraishi T, Nakagawa S, Toda K, Tabuchi K. Visualization of DNA double strand breaks in the gerbil hippocampal CA1 following transient ischemia. Neuroscience Letters. 1994;175(1-2):133–136. doi: 10.1016/0304-3940(94)91097-9. [DOI] [PubMed] [Google Scholar]
- 9.Yan GM, Irwin RP, Lin SZ, Weller M, Wood KA, Paul SM. Diphenylhydantoin induces apoptotic cell death of cultured rat cerebellar granule neurons. Journal of Pharmacology and Experimental Therapeutics. 1995;274(2):983–990. [PubMed] [Google Scholar]
- 10.Wilson FC, Harpur J, Watson T, Morrow JI. Adult survivors of severe cerebral hypoxia—case series survey and comparative analysis. NeuroRehabilitation. 2003;18(4):291–298. [PubMed] [Google Scholar]
- 11.Hsieh H-L, Yang C-M. Role of redox signaling in neuroinflammation and neurodegenerative diseases. BioMed Research International. 2013;2013:18 pages. doi: 10.1155/2013/484613.484613 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Li J, Wuliji O, Li W, Jiang ZG, Ghanbari HA. Oxidative stress and neurodegenerative disorders. International Journal of Molecular Sciences. 2013;14(12):24438–24475. doi: 10.3390/ijms141224438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wang X, Wang W, Li L, Perry G, Lee HG, Zhu X. Oxidative stress and mitochondrial dysfunction in Alzheimer's disease. Biochimica et Biophysica Acta. 2013;1842(8):1240–1247. doi: 10.1016/j.bbadis.2013.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yan MH, Wang X, Zhu X. Mitochondrial defects and oxidative stress in Alzheimer disease and Parkinson disease. Free Radical Biology and Medicine. 2013;62:90–101. doi: 10.1016/j.freeradbiomed.2012.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Paschen W. Role of calcium in neuronal cell injury: which subcellular compartment is involved? Brain Research Bulletin. 2000;53(4):409–413. doi: 10.1016/s0361-9230(00)00369-5. [DOI] [PubMed] [Google Scholar]
- 16.Chan PH. Reactive oxygen radicals in signaling and damage in the ischemic brain. Journal of Cerebral Blood Flow and Metabolism. 2001;21(1):2–14. doi: 10.1097/00004647-200101000-00002. [DOI] [PubMed] [Google Scholar]
- 17.Iadecola C, Alexander M. Cerebral ischemia and inflammation. Current Opinion in Neurology. 2001;14(1):89–94. doi: 10.1097/00019052-200102000-00014. [DOI] [PubMed] [Google Scholar]
- 18.Petty MA, Wettstein JG. Elements of cerebral microvascular ischaemia. Brain Research Reviews. 2001;36(1):23–34. doi: 10.1016/s0165-0173(01)00062-5. [DOI] [PubMed] [Google Scholar]
- 19.Noh HS, Hah Y, Nilufar R, et al. Acetoacetate protects neuronal cells from oxidative glutamate toxicity. Journal of Neuroscience Research. 2006;83(4):702–709. doi: 10.1002/jnr.20736. [DOI] [PubMed] [Google Scholar]
- 20.Halliwell B. Free radicals, antioxidants, and human disease: curiosity, cause, or consequence? The Lancet. 1994;344(8924):721–724. doi: 10.1016/s0140-6736(94)92211-x. [DOI] [PubMed] [Google Scholar]
- 21.Sugawara T, Chan PH. Reactive oxygen radicals and pathogenesis of neuronal death after cerebral ischemia. Antioxidants and Redox Signaling. 2003;5(5):597–607. doi: 10.1089/152308603770310266. [DOI] [PubMed] [Google Scholar]
- 22.Millar TM, Phan V, Tibbles LA. ROS generation in endothelial hypoxia and reoxygenation stimulates MAP kinase signaling and kinase-dependent neutrophil recruitment. Free Radical Biology and Medicine. 2007;42(8):1165–1177. doi: 10.1016/j.freeradbiomed.2007.01.015. [DOI] [PubMed] [Google Scholar]
- 23.McCubrey JA, LaHair MM, Franklin RA. Reactive oxygen species-induced activation of the MAP kinase signaling pathways. Antioxidants and Redox Signaling. 2006;8(9-10):1775–1789. doi: 10.1089/ars.2006.8.1775. [DOI] [PubMed] [Google Scholar]
- 24.Torres M, Forman HJ. Redox signaling and the MAP kinase pathways. BioFactors. 2003;17(1–4):287–296. doi: 10.1002/biof.5520170128. [DOI] [PubMed] [Google Scholar]
- 25.Bogoyevitch MA, Gillespie-Brown J, Ketterman AJ, et al. Stimulation of the stress-activated mitogen-activated protein kinase subfamilies in perfused heart: p38/RK mitogen-activated protein kinases and c-Jun N-terminal kinases are activated by ischemia/reperfusion. Circulation Research. 1996;79(2):162–173. doi: 10.1161/01.res.79.2.162. [DOI] [PubMed] [Google Scholar]
- 26.Laderoute KR, Webster KA. Hypoxia/reoxygenation stimulates Jun kinase activity through redox signaling in cardiac myocytes. Circulation Research. 1997;80(3):336–344. doi: 10.1161/01.res.80.3.336. [DOI] [PubMed] [Google Scholar]
- 27.Chen Z, Gibson TB, Robinson F, et al. MAP kinases. Chemical Reviews. 2001;101(8):2449–2476. doi: 10.1021/cr000241p. [DOI] [PubMed] [Google Scholar]
- 28.Kyriakis JM, Avruch J. Sounding the alarm: protein kinase cascades activated by stress and inflammation. The Journal of Biological Chemistry. 1996;271(40):24313–24316. doi: 10.1074/jbc.271.40.24313. [DOI] [PubMed] [Google Scholar]
- 29.Rockwell P, Martinez J, Papa L, Gomes E. Redox regulates COX-2 upregulation and cell death in the neuronal response to cadmium. Cellular Signalling. 2004;16(3):343–353. doi: 10.1016/j.cellsig.2003.08.006. [DOI] [PubMed] [Google Scholar]
- 30.Li DJ, Zhao T, Xin RJ, Wang YY, Fei YB, Shen FM. Activation of alpha7 nicotinic acetylcholine receptor protects against oxidant stress damage through reducing vascular peroxidase-1 in a JNK signaling-dependent manner in endothelial cells. Cellular Physiology and Biochemistry. 2014;33(2):468–478. doi: 10.1159/000358627. [DOI] [PubMed] [Google Scholar]
- 31.Harper SJ, Lograsso P. Signalling for survival and death in neurones: the role of stress-activated kinases, JNK and p38. Cellular Signalling. 2001;13(5):299–310. doi: 10.1016/s0898-6568(01)00148-6. [DOI] [PubMed] [Google Scholar]
- 32.Nozaki K, Nishimura M, Hashimoto N. Mitogen-activated protein kinases and cerebral ischemia. Molecular Neurobiology. 2001;23(1):1–19. doi: 10.1385/MN:23:1:01. [DOI] [PubMed] [Google Scholar]
- 33.Abbott NJ. Astrocyte-endothelial interactions and blood-brain barrier permeability. Journal of Anatomy. 2002;200(6):629–638. doi: 10.1046/j.1469-7580.2002.00064.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ballabh P, Braun A, Nedergaard M. The blood-brain barrier: an overview: Structure, regulation, and clinical implications. Neurobiology of Disease. 2004;16(1):1–13. doi: 10.1016/j.nbd.2003.12.016. [DOI] [PubMed] [Google Scholar]
- 35.Furuse M, Hirase T, Itoh M, Nagafuchi A, Yonemura S, Tsukita S. Occludin: a novel integral membrane protein localizing at tight junctions. Journal of Cell Biology. 1993;123(6):1777–1788. doi: 10.1083/jcb.123.6.1777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Furuse M, Fujita K, Hiiragi T, Fujimoto K, Tsukita S. Claudin-1 and -2: novel integral membrane proteins localizing at tight junctions with no sequence similarity to occludin. Journal of Cell Biology. 1998;141(7):1539–1550. doi: 10.1083/jcb.141.7.1539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Furuse M, Sasaki H, Tsukita S. Manner of interaction of heterogeneous claudin species within and between tight junction strands. Journal of Cell Biology. 1999;147(4):891–903. doi: 10.1083/jcb.147.4.891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mark KS, Davis TP. Cerebral microvascular changes in permeability and tight junctions induced by hypoxia-reoxygenation. American Journal of Physiology: Heart and Circulatory Physiology. 2002;282(4):H1485–H1494. doi: 10.1152/ajpheart.00645.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Witt KA, Mark KS, Hom S, Davis TP. Effects of hypoxia-reoxygenation on rat blood-brain barrier permeability and tight junctional protein expression. American Journal of Physiology—Heart and Circulatory Physiology. 2003;285(6):H2820–H2831. doi: 10.1152/ajpheart.00589.2003. [DOI] [PubMed] [Google Scholar]
- 40.Mark KS, Burroughs AR, Brown RC, Huber JD, Davis TP. Nitric oxide mediates hypoxia-induced changes in paracellular permeability of cerebral microvasculature. The American Journal of Physiology: Heart and Circulatory Physiology. 2004;286(1):H174–H180. doi: 10.1152/ajpheart.00669.2002. [DOI] [PubMed] [Google Scholar]
- 41.Croll SD, Goodman JH, Scharfman HE. Vascular Endothelial Growth Factor (VEGF) in seizures: a double-edged sword. Advances in Experimental Medicine and Biology. 2004;548:57–68. doi: 10.1007/978-1-4757-6376-8_4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Dobrogowska DH, Lossinsky AS, Tarnawski M, Vorbrodt AW. Increased blood-brain barrier permeability and endothelial abnormalities induced by vascular endothelial growth factor. Brain Cell Biology. 1998;27(3):163–173. doi: 10.1023/a:1006907608230. [DOI] [PubMed] [Google Scholar]
- 43.Zhang ZG, Zhang L, Jiang Q, et al. VEGF enhances angiogenesis and promotes blood-brain barrier leakage in the ischemic brain. Journal of Clinical Investigation. 2000;106(7):829–838. doi: 10.1172/JCI9369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Dvorak HF, Nagy JA, Feng D, et al. Vascular permeability factor/vascular endothelial growth factor and the significance of microvascular hyperpermeability in angiogenesis. Current Topics in Microbiology and Immunology. 1999;237:97–132. doi: 10.1007/978-3-642-59953-8_6. [DOI] [PubMed] [Google Scholar]
- 45.Poeggeler B, Saarela S, Reiter RJ, et al. Melatonin—a highly potent endogenous radical scavenger and electron donor: new aspects of the oxidation chemistry of this indole accessed in vitro. Annals of the New York Academy of Sciences. 1994;738:419–420. doi: 10.1111/j.1749-6632.1994.tb21831.x. [DOI] [PubMed] [Google Scholar]
- 46.Dupuis F, Régrigny O, Atkinson J, et al. Impact of treatment with melatonin on cerebral circulation in old rats. The British Journal of Pharmacology. 2004;141(3):399–406. doi: 10.1038/sj.bjp.0705629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wakatsuki A, Okatani Y, Izumiya C, Ikenoue N. Melatonin protects against ischemia and reperfusion-induced oxidative lipid and DNA damage in fetal rat brain. Journal of Pineal Research. 1999;26(3):147–152. doi: 10.1111/j.1600-079x.1999.tb00576.x. [DOI] [PubMed] [Google Scholar]
- 48.Ginsberg MD, Busto R. Rodent models of cerebral ischemia. Stroke. 1989;20(12):1627–1642. doi: 10.1161/01.str.20.12.1627. [DOI] [PubMed] [Google Scholar]
- 49.Harms C, Lautenschlager M, Bergk A, et al. Melatonin is protective in necrotic but not in caspase-dependent, free radical-independent apoptotic neuronal cell death in primary neuronal cultures. The FASEB Journal. 2000;14(12):1814–1824. doi: 10.1096/fj.99-0899com. [DOI] [PubMed] [Google Scholar]
- 50.Pei Z, Cheung RTF. Melatonin protects SHSY5Y neuronal cells but not cultured astrocytes from ischemia due to oxygen and glucose deprivation. Journal of Pineal Research. 2003;34(3):194–201. doi: 10.1034/j.1600-079x.2003.00026.x. [DOI] [PubMed] [Google Scholar]
- 51.Lv D, Cui P, Yao S, Xu Y, Yang Z. Melatonin inhibits the expression of vascular endothelial growth factor in pancreatic cancer cells. Chinese Journal of Cancer Research. 2012;24(4):310–316. doi: 10.3978/j.issn.1000-9604.2012.09.03. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Dai M, Cui P, Yu M, Han J, Li H, Xiu R. Melatonin modulates the expression of VEGF and HIF-1α induced by CoCl2 in cultured cancer cells. Journal of Pineal Research. 2008;44(2):121–126. doi: 10.1111/j.1600-079X.2007.00498.x. [DOI] [PubMed] [Google Scholar]
- 53.Lissoni P, Rovelli F, Malugani F, Bucovec R, Conti A, Maestroni GJM. Anti-angiogenic activity of melatonin in advanced cancer patients. Neuroendocrinology Letters. 2001;22(1):45–47. [PubMed] [Google Scholar]
- 54.Jung H-J, Jeon Y-H, Bokara KK, et al. Agmatine promotes the migration of murine brain endothelial cells via multiple signaling pathways. Life Sciences. 2013;92(1):42–50. doi: 10.1016/j.lfs.2012.10.018. [DOI] [PubMed] [Google Scholar]
- 55.Mei ZY, Chin CM, Yoon JC, et al. Agmatine inhibits matrix metalloproteinase-9 via endothelial nitric oxide synthase in cerebral endothelial cells. Neurological Research. 2007;29(7):749–754. doi: 10.1179/016164107X208103. [DOI] [PubMed] [Google Scholar]
- 56.Bokara KK, Kwon KH, Nho Y, Lee WT, Park KA, Lee JE. Retroviral expression of arginine decarboxylase attenuates oxidative burden in mouse cortical neural stem cells. Stem Cells and Development. 2011;20(3):527–537. doi: 10.1089/scd.2010.0312. [DOI] [PubMed] [Google Scholar]
- 57.Hong S, Kim CY, Lee JE, Seong GJ. Agmatine protects cultured retinal ganglion cells from tumor necrosis factor-alpha-induced apoptosis. Life Sciences. 2009;84(1-2):28–32. doi: 10.1016/j.lfs.2008.10.006. [DOI] [PubMed] [Google Scholar]
- 58.Ahn SK, Hong S, Park YM, Lee WT, Park KA, Lee JE. Effects of agmatine on hypoxic microglia and activity of nitric oxide synthase. Brain Research. 2011;1373:48–54. doi: 10.1016/j.brainres.2010.12.002. [DOI] [PubMed] [Google Scholar]
- 59.Royall JA, Ischiropoulos H. Evaluation of 2',7'-dichlorofluorescin and dihydrorhodamine 123 as fluorescent probes for intracellular H2O2 in cultured endothelial cells. Archives of Biochemistry and Biophysics. 1993;302(2):348–355. doi: 10.1006/abbi.1993.1222. [DOI] [PubMed] [Google Scholar]
- 60.Nyakas C, Buwalda B, Luiten PGM. Hypoxia and brain development. Progress in Neurobiology. 1996;49(1):1–51. doi: 10.1016/0301-0082(96)00007-x. [DOI] [PubMed] [Google Scholar]
- 61.Zhou J, Kong H, Hua X, Xiao M, Ding J, Hu G. Altered blood-brain barrier integrity in adult aquaporin-4 knockout mice. NeuroReport. 2008;19(1):1–5. doi: 10.1097/WNR.0b013e3282f2b4eb. [DOI] [PubMed] [Google Scholar]
- 62.Rubin LL, Staddon JM. The cell biology of the blood-brain barrier. Annual Review of Neuroscience. 1999;22:11–28. doi: 10.1146/annurev.neuro.22.1.11. [DOI] [PubMed] [Google Scholar]
- 63.del Zoppo GJ, von Kummer R, Hamann GF. Ischaemic damage of brain microvessels: inherent risks for thrombolytic treatment in stroke. Journal of Neurology Neurosurgery and Psychiatry. 1998;65(1):1–9. doi: 10.1136/jnnp.65.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Hardeland R. Antioxidative protection by melatonin: multiplicity of mechanisms from radical detoxification to radical avoidance. Endocrine. 2005;27(2):119–130. doi: 10.1385/endo:27:2:119. [DOI] [PubMed] [Google Scholar]
- 65.Wu G, Xue RL, Lv JR, Li W, Lei XM. Fas and TNFR1 expressions after cerebral ischemia and reperfusion in rats: association with cell apoptosis and the effects of Bcl-2 overexpression. Journal of Southern Medical University. 2011;31(8):1298–1303. [PubMed] [Google Scholar]
- 66.Li V, Brustovetsky T, Brustovetsky N. Role of cyclophilin D-dependent mitochondrial permeability transition in glutamate-induced calcium deregulation and excitotoxic neuronal death. Experimental Neurology. 2009;218(2):171–182. doi: 10.1016/j.expneurol.2009.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Lipton P. Ischemic cell death in brain neurons. Physiological Reviews. 1999;79(4):1431–1568. doi: 10.1152/physrev.1999.79.4.1431. [DOI] [PubMed] [Google Scholar]
- 68.Andjelkovic AV, Stamatovic SM, Keep RF. The protective effects of preconditioning on cerebral endothelial cells in vitro. Journal of Cerebral Blood Flow and Metabolism. 2003;23(11):1348–1355. doi: 10.1097/01.WCB.0000091762.61714.FE. [DOI] [PubMed] [Google Scholar]
- 69.Cervantes MI, de Oca Balderas PM, de Jesús Gutiérrez-Baños J, et al. Comparison of antioxidant activity of hydroethanolic fresh and aged garlic extracts and their effects on cerebral ischemia. Food Chemistry. 2013;140(1-2):343–352. doi: 10.1016/j.foodchem.2013.02.053. [DOI] [PubMed] [Google Scholar]
- 70.Wang X-C, Zhang Y-C, Chatterjie N, Grundke-Iqbal I, Iqbal K, Wang J. Effect of melatonin and melatonylvalpromide on β-amyloid and neurofilaments in N2a cells. Neurochemical Research. 2008;33(6):1138–1144. doi: 10.1007/s11064-007-9563-y. [DOI] [PubMed] [Google Scholar]
- 71.Li J, Yin H, Gu X, Zhou Y, Zhang W, Qin Y. Melatonin protects liver from intestine ischemia reperfusion injury in rats. World Journal of Gastroenterology. 2008;14(48):7392–7396. doi: 10.3748/wjg.14.7392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Wang J, Shen J, Gao Q, et al. Ischemic postconditioning protects against global cerebral ischemia/reperfusion-induced injury in rats. Stroke. 2008;39(3):983–990. doi: 10.1161/STROKEAHA.107.499079. [DOI] [PubMed] [Google Scholar]
- 73.Li YC, Lin HJ, Yang JH, et al. Baicalein-induced apoptosis via endoplasmic reticulum stress through elevations of reactive oxygen species and mitochondria dependent pathway in mouse-rat hybrid retina ganglion cells (N18) Neurochemical Research. 2009;34(3):418–429. doi: 10.1007/s11064-008-9799-1. [DOI] [PubMed] [Google Scholar]
- 74.Ozbal S, Ergur BU, Erbil G, Tekmen I, Bagryank A, Cavdar Z. The effects of α-lipoic acid against testicular ischemia-reperfusion injury in rats. The Scientific World Journal. 2012;2012:8 pages. doi: 10.1100/2012/489248.489248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Reiter RJ. Antioxidant actions of melatonin. Advances in pharmacology. 1997;38:103–117. doi: 10.1016/s1054-3589(08)60981-3. [DOI] [PubMed] [Google Scholar]
- 76.Tütünculer F, Eskiocak S, Başaran ÜN, Ekuklu G, Ayvaz S, Vatansever Ü. The protective role of melatonin in experimental hypoxic brain damage. Pediatrics International. 2005;47(4):434–439. doi: 10.1111/j.1442-200x.2005.02085.x. [DOI] [PubMed] [Google Scholar]
- 77.Tan D, Reiter RJ, Manchester LC, et al. Chemical and physical properties and potential mechanisms: melatonin as a broad spectrum antioxidant and free radical scavenger. Current Topics in Medicinal Chemistry. 2002;2(2):181–197. doi: 10.2174/1568026023394443. [DOI] [PubMed] [Google Scholar]
- 78.Çelik Ö, Naziroğlu M. Melatonin modulates apoptosis and TRPM2 channels in transfected cells activated by oxidative stress. Physiology & Behavior. 2012;107(3):458–465. doi: 10.1016/j.physbeh.2012.09.013. [DOI] [PubMed] [Google Scholar]
- 79.Ray PD, Huang BW, Tsuji Y. Reactive oxygen species (ROS) homeostasis and redox regulation in cellular signaling. Cellular Signalling. 2012;24(5):981–990. doi: 10.1016/j.cellsig.2012.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Kubo E, Miyoshi N, Fukuda M, Akagi Y. Cataract formation through the polyol pathway is associated with free radical production. Experimental Eye Research. 1999;68(4):457–464. doi: 10.1006/exer.1998.0624. [DOI] [PubMed] [Google Scholar]
- 81.Lim YJ, Zheng S, Zuo Z. Morphine preconditions purkinje cells against cell death under in vitro simulated ischemia-reperfusion conditions. Anesthesiology. 2004;100(3):562–568. doi: 10.1097/00000542-200403000-00015. [DOI] [PubMed] [Google Scholar]
- 82.Alonso-Vale MIC, Andreotti S, Peres SB, et al. Melatonin enhances leptin expression by rat adipocytes in the presence of insulin. American Journal of Physiology-Endocrinology and Metabolism. 2005;288(4):E805–E812. doi: 10.1152/ajpendo.00478.2004. [DOI] [PubMed] [Google Scholar]
- 83.Hartman RE, Lee JM, Zipfel GJ, Wozniak DF. Characterizing learning deficits and hippocampal neuron loss following transient global cerebral ischemia in rats. Brain Research. 2005;1043(1-2):48–56. doi: 10.1016/j.brainres.2005.02.030. [DOI] [PubMed] [Google Scholar]
- 84.Morita K, Sasaki H, Fujimoto K, Furuse M, Tsukita S. Claudin-11/OSP-based tight junctions of myelin sheaths in brain and Sertoli cells in testis. Journal of Cell Biology. 1999;145(3):579–588. doi: 10.1083/jcb.145.3.579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Liebner S, Fischmann A, Rascher G, et al. Claudin-1 and claudin-5 expression and tight junction morphology are altered in blood vessels of human glioblastoma multiforme. Acta Neuropathologica. 2000;100(3):323–331. doi: 10.1007/s004010000180. [DOI] [PubMed] [Google Scholar]
- 86.Liebner S, Kniesel U, Kalbacher H, Wolburg H. Correlation of tight junction morphology with the expression of tight junction proteins in blood-brain barrier endothelial cells. European Journal of Cell Biology. 2000;79(10):707–717. doi: 10.1078/0171-9335-00101. [DOI] [PubMed] [Google Scholar]
- 87.Lippoldt A, Kniesel U, Liebner S, et al. Structural alterations of tight junctions are associated with loss of polarity in stroke-prone spontaneously hypertensive rat blood-brain barrier endothelial cells. Brain Research. 2000;885(2):251–261. doi: 10.1016/s0006-8993(00)02954-1. [DOI] [PubMed] [Google Scholar]
- 88.Koto T, Takubo K, Ishida S, et al. Hypoxia disrupts the barrier function of neural blood vessels through changes in the expression of claudin-5 in endothelial cells. The American Journal of Pathology. 2007;170(4):1389–1397. doi: 10.2353/ajpath.2007.060693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Tao R, Huang J, Shao X, et al. Ischemic injury promotes Keap1 nitration and disturbance of antioxidative responses in endothelial cells: a potential vasoprotective effect of melatonin. Journal of Pineal Research. 2013;54(3):271–281. doi: 10.1111/jpi.12009. [DOI] [PubMed] [Google Scholar]
- 90.Abbruscato TJ, Davis TP. Protein expression of brain endothelial cell E-cadherin after hypoxia/aglycemia: influence of astrocyte contact. Brain Research. 1999;842(2):277–286. doi: 10.1016/s0006-8993(99)01778-3. [DOI] [PubMed] [Google Scholar]
- 91.Chryssanthou C, Palaia T, Goldstein G, Stenger R. Increase in blood-brain barrier permeability by altitude decompression. Aviation Space and Environmental Medicine. 1987;58(11):1082–1086. [PubMed] [Google Scholar]
- 92.Schoch HJ, Fischer S, Marti HH. Hypoxia-induced vascular endothelial growth factor expression causes vascular leakage in the brain. Brain. 2002;125, part 11:2549–2557. doi: 10.1093/brain/awf257. [DOI] [PubMed] [Google Scholar]
- 93.Hayashi K, Nakao S, Nakaoke R, Nakagawa S, Kitagawa N, Niwa M. Effects of hypoxia on endothelial/pericytic co-culture model of the blood-brain barrier. Regulatory Peptides. 2004;123(1–3):77–83. doi: 10.1016/j.regpep.2004.05.023. [DOI] [PubMed] [Google Scholar]
- 94.Battegay EJ. Angiogenesis: mechanistic insights, neovascular diseases, and therapeutic prospects. Journal of Molecular Medicine. 1995;73(7):333–346. doi: 10.1007/BF00192885. [DOI] [PubMed] [Google Scholar]
- 95.Milkiewicz M, Ispanovic E, Doyle JL, Haas TL. Regulators of angiogenesis and strategies for their therapeutic manipulation. International Journal of Biochemistry and Cell Biology. 2006;38(3):333–357. doi: 10.1016/j.biocel.2005.10.006. [DOI] [PubMed] [Google Scholar]
- 96.Semenza GL. Hydroxylation of HIF-1: oxygen sensing at the molecular level. Physiology. 2004;19(4):176–182. doi: 10.1152/physiol.00001.2004. [DOI] [PubMed] [Google Scholar]
- 97.Jośko J, Mazurek M. Transcription factors having impact on vascular endothelial growth factor (VEGF) gene expression in angiogenesis. Medical Science Monitor. 2004;10(4):RA89–RA98. [PubMed] [Google Scholar]
- 98.Mayhan WG. VEGF increases permeability of the blood-brain barrier via a nitric oxide synthase/cGMP-dependent pathway. American Journal of Physiology—Cell Physiology. 1999;276(5, part 1):C1148–C1153. doi: 10.1152/ajpcell.1999.276.5.C1148. [DOI] [PubMed] [Google Scholar]
- 99.Young PP, Fantz CR, Sands MS. VEGF disrupts the neonatal blood-brain barrier and increases life span after non-ablative BMT in a murine model of congenital neurodegeneration caused by a lysosomal enzyme deficiency. Experimental Neurology. 2004;188(1):104–114. doi: 10.1016/j.expneurol.2004.03.007. [DOI] [PubMed] [Google Scholar]
- 100.Pichiule P, Chávez JC, Xu K, Lamanna JC. Vascular endothelial growth factor upregulation in transient global ischemia induced by cardiac arrest and resuscitation in rat brain. Molecular Brain Research. 1999;74(1-2):83–90. doi: 10.1016/s0169-328x(99)00261-2. [DOI] [PubMed] [Google Scholar]
- 101.Sinor AD, Irvin SM, Cobbs CS, Chen J, Graham SH, Greenberg DA. Hypoxic induction of vascular endothelial growth factor (VEGF) protein in astroglial cultures. Brain Research. 1998;812(1-2):289–291. doi: 10.1016/s0006-8993(98)00976-7. [DOI] [PubMed] [Google Scholar]
- 102.Walter R, Maggiorini M, Scherrer U, Contesse J, Reinhart WH. Effects of high-altitude exposure on vascular endothelial growth factor levels in man. European Journal of Applied Physiology. 2001;85(1-2):113–117. doi: 10.1007/s004210100419. [DOI] [PubMed] [Google Scholar]
- 103.Bates DO, Harper SJ. Regulation of vascular permeability by vascular endothelial growth factors. Vascular Pharmacology. 2002;39(4-5):225–237. doi: 10.1016/s1537-1891(03)00011-9. [DOI] [PubMed] [Google Scholar]
- 104.Ersahin M, Toklu HZ, Çetinel Ş, Yüksel M, Yèen BÇ, Şener G. Melatonin reduces experimental subarachnoid hemorrhage-induced oxidative brain damage and neurological symptoms. Journal of Pineal Research. 2009;46(3):324–332. doi: 10.1111/j.1600-079X.2009.00664.x. [DOI] [PubMed] [Google Scholar]
- 105.Chen H-Y, Chen T-Y, Lee M-Y, et al. Melatonin decreases neurovascular oxidative/nitrosative damage and protects against early increases in the blood-brain barrier permeability after transient focal cerebral ischemia in mice. Journal of Pineal Research. 2006;41(2):175–182. doi: 10.1111/j.1600-079X.2006.00351.x. [DOI] [PubMed] [Google Scholar]
- 106.Kaur C, Sivakumar V, Lu J, Tang FR, Ling EA. Melatonin attenuates hypoxia-induced ultrastructural changes and increased vascular permeability in the developing hippocampus. Brain Pathology. 2008;18(4):533–547. doi: 10.1111/j.1750-3639.2008.00156.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Zhang Y, Liu Q, Wang F, et al. Melatonin antagonizes hypoxia-mediated glioblastoma cell migration and invasion via inhibition of HIF-1α . Journal of Pineal Research. 2013;55(2):121–130. doi: 10.1111/jpi.12052. [DOI] [PubMed] [Google Scholar]
- 108.Kaur C, Sivakumar V, Lu J, Ling EA. Increased vascular permeability and nitric oxide production in response to hypoxia in the pineal gland. Journal of Pineal Research. 2007;42(4):338–349. doi: 10.1111/j.1600-079X.2007.00424.x. [DOI] [PubMed] [Google Scholar]
- 109.Maddika S, Ande SR, Panigrahi S, et al. Cell survival, cell death and cell cycle pathways are interconnected: implications for cancer therapy. Drug Resistance Updates. 2007;10(1-2):13–29. doi: 10.1016/j.drup.2007.01.003. [DOI] [PubMed] [Google Scholar]
- 110.Wang B, Shravah J, Luo H, Raedschelders K, Chen DDY, Ansley DM. Propofol protects against hydrogen peroxide-induced injury in cardiac H9c2 cells via Akt activation and Bcl-2 up-regulation. Biochemical and Biophysical Research Communications. 2009;389(1):105–111. doi: 10.1016/j.bbrc.2009.08.097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Kimura R, Okouchi M, Fujioka H, et al. Glucagon-like peptide-1 (GLP-1) protects against methylglyoxal-induced PC12 cell apoptosis through the PI3K/Akt/mTOR/GCLc/redox signaling pathway. Neuroscience. 2009;162(4):1212–1219. doi: 10.1016/j.neuroscience.2009.05.025. [DOI] [PubMed] [Google Scholar]
- 112.Lee S, Shin J, Hong Y, et al. Beneficial effects of melatonin on stroke-induced muscle atrophy in focal cerebral ischemic rats. Laboratory Animal Research. 2012;28(1):47–54. doi: 10.5625/lar.2012.28.1.47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Jiang T, Chang Q, Zhao Z, et al. Melatonin-mediated cytoprotection against hyperglycemic injury in Müller cells. PLoS ONE. 2012;7(12) doi: 10.1371/journal.pone.0050661.e50661 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Lee SH, Chun W, Kong PJ, et al. Sustained activation of Akt by melatonin contributes to the protection against kainic acid-induced neuronal death in hippocampus. Journal of Pineal Research. 2006;40(1):79–85. doi: 10.1111/j.1600-079X.2005.00283.x. [DOI] [PubMed] [Google Scholar]
- 115.Kong P, Byun J, Lim S, et al. Melatonin induees Akt phosphorylation through Melatonin receptor- and PI3K-dependent pathways in primary astrocytes. Korean Journal of Physiology and Pharmacology. 2008;12(2):37–41. doi: 10.4196/kjpp.2008.12.2.37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Manning BD, Cantley LC. AKT/PKB signaling: navigating downstream. Cell. 2007;129(7):1261–1274. doi: 10.1016/j.cell.2007.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Zhou H, Liu L, Zhang Y, et al. Glutathione prevents free fatty acids-induced oxidative stress and apoptosis in human brain vascular endothelial cells through Akt pathway. CNS Neuroscience and Therapeutics. 2013;19(4):252–261. doi: 10.1111/cns.12068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Shiojima I, Walsh K. Role of Akt signaling in vascular homeostasis and angiogenesis. Circulation Research. 2002;90(12):1243–1250. doi: 10.1161/01.res.0000022200.71892.9f. [DOI] [PubMed] [Google Scholar]
- 119.Song G, Ouyang G, Bao S. The activation of Akt/PKB signaling pathway and cell survival. Journal of Cellular and Molecular Medicine. 2005;9(1):59–71. doi: 10.1111/j.1582-4934.2005.tb00337.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Bai J, Dong L, Song Z, et al. The role of melatonin as an antioxidant in human lens epithelial cells. Free Radical Research. 2013;47(8):635–642. doi: 10.3109/10715762.2013.808743. [DOI] [PubMed] [Google Scholar]
- 121.Zhang Y, Wang X, Yang H, et al. Kinase AKT controls innate immune cell development and function. Immunology. 2013;140(2):143–152. doi: 10.1111/imm.12123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Teicher BA. Molecular targets and cancer therapeutics: discovery, development and clinical validation. Drug Resistance Updates. 2000;3(2):67–73. doi: 10.1054/drup.2000.0123. [DOI] [PubMed] [Google Scholar]
- 123.Huang Y, Wu D, Fan W. Protection of ginsenoside Rg1 on chondrocyte from IL-1beta-induced mitochondria-activated apoptosis through PI3K/Akt signaling. Molecular and Cellular Biochemistry. 2014;392(1-2):249–257. doi: 10.1007/s11010-014-2035-1. [DOI] [PubMed] [Google Scholar]
- 124.Zhang X, Bi L, Ye Y, Chen J. Formononetin induces apoptosis in PC-3 prostate cancer cells through enhancing the Bax/Bcl-2 ratios and regulating the p38/Akt pathway. Nutrition and Cancer. 2014;66(4):656–661. doi: 10.1080/01635581.2014.894098. [DOI] [PubMed] [Google Scholar]
- 125.Xu C, Wu A, Zhu H, et al. Melatonin is involved in the apoptosis and necrosis of pancreatic cancer cell line SW-1990 via modulating of Bcl-2/Bax balance. Biomedicine and Pharmacotherapy. 2013;67(2):133–139. doi: 10.1016/j.biopha.2012.10.005. [DOI] [PubMed] [Google Scholar]
- 126.Mohseni M, Mihandoost E, Shirazi A, Sepehrizadeh Z, Bazzaz JT, Ghazi-Khansari M. Melatonin may play a role in modulation of bax and bcl-2 expression levels to protect rat peripheral blood lymphocytes from gamma irradiation-induced apoptosis. Mutation Research—Fundamental and Molecular Mechanisms of Mutagenesis. 2012;738-739(1):19–27. doi: 10.1016/j.mrfmmm.2012.08.006. [DOI] [PubMed] [Google Scholar]
- 127.Ho FM, Liu SH, Liau CS, Huang PJ, Lin-Shiau SY. High glucose-induced apoptosis in human endothelial cells is mediated by sequential activations of c-JUN NH2-terminal kinase and caspase-3. Circulation. 2000;101(22):2618–2624. doi: 10.1161/01.cir.101.22.2618. [DOI] [PubMed] [Google Scholar]
- 128.Takabe W, Li R, Ai L, Yu F, Berliner JA, Hsiai TK. Oxidized low-density lipoprotein-activated c-Jun NH2-terminal kinase regulates manganese superoxide dismutase ubiquitination: implication for mitochondrial redox status and apoptosis. Arteriosclerosis, Thrombosis, and Vascular Biology. 2010;30(3):436–441. doi: 10.1161/ATVBAHA.109.202135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Garin G, Abe J, Mohan A, et al. Flow antagonizes TNF-α signaling in endothelial cells by inhibiting caspase-dependent PKCζ processing. Circulation Research. 2007;101(1):97–105. doi: 10.1161/CIRCRESAHA.107.148270. [DOI] [PubMed] [Google Scholar]
- 130.Shen HM, Liu ZG. JNK signaling pathway is a key modulator in cell death mediated by reactive oxygen and nitrogen species. Free Radical Biology and Medicine. 2006;40(6):928–939. doi: 10.1016/j.freeradbiomed.2005.10.056. [DOI] [PubMed] [Google Scholar]
- 131.Cai B, Li X, Wang Y, et al. Apoptosis of bone marrow mesenchymal stem cells caused by homocysteine via activating jnk signal. PLoS ONE. 2013;8(5) doi: 10.1371/journal.pone.0063561.e63561 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Choi WS, Yoon SY, Oh TH, Choi EJ, O’Malley KL, Oh YJ. Two distinct mechanisms are involved in 6-hydroxydopamine- and MPP+-induced dopaminergic neuronal cell death: role of caspases, ROS, and JNK. Journal of Neuroscience Research. 1999;57(1):86–94. doi: 10.1002/(SICI)1097-4547(19990701)57:1<86::AID-JNR9>3.0.CO;2-E. [DOI] [PubMed] [Google Scholar]
- 133.Chang K, Hsu C, Liu S, et al. Cadmium induces apoptosis in pancreatic beta-cells through a mitochondria-dependent pathway: the role of oxidative stress-mediated c-Jun N-terminal kinase activation. PLoS ONE. 2013;8(2) doi: 10.1371/journal.pone.0054374.e54374 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Shaulian E, Karin M. AP-1 as a regulator of cell life and death. Nature Cell Biology. 2002;4(5):E131–E136. doi: 10.1038/ncb0502-e131. [DOI] [PubMed] [Google Scholar]
- 135.Rosenberg GA. Matrix metalloproteinases in neuroinflammation. Glia. 2002;39(3):279–291. doi: 10.1002/glia.10108. [DOI] [PubMed] [Google Scholar]
- 136.Gao Y, Signore AP, Yin W, et al. Neuroprotection against focal ischemic brain injury by inhibition of c-Jun N-terminal kinase and attenuation of the mitochondrial apoptosis-signaling pathway. Journal of Cerebral Blood Flow and Metabolism. 2005;25(6):694–712. doi: 10.1038/sj.jcbfm.9600062. [DOI] [PubMed] [Google Scholar]
- 137.Guan Q-H, Pei D-S, Zong Y-Y, Xu T-L, Zhang G-Y. Neuroprotection against ischemic brain injury by a small peptide inhibitor of c-Jun N-terminal kinase (JNK) via nuclear and non-nuclear pathways. Neuroscience. 2006;139(2):609–627. doi: 10.1016/j.neuroscience.2005.11.067. [DOI] [PubMed] [Google Scholar]
- 138.Guan QH, Pei DS, Liu XM, Wang XT, Xu TL, Zhang GY. Neuroprotection against ischemic brain injury by SP600125 via suppressing the extrinsic and intrinsic pathways of apoptosis. Brain Research. 2006;1092(1):36–46. doi: 10.1016/j.brainres.2006.03.086. [DOI] [PubMed] [Google Scholar]
- 139.Borsellol T, Clarkel PGH, Hirt L, et al. A peptide inhibitor of c-Jun N-terminal kinase protects against excitotoxicity and cerebral ischemia. Nature Medicine. 2003;9(9):1180–1186. doi: 10.1038/nm911. [DOI] [PubMed] [Google Scholar]