

Abstract
While specific mutations in the tyrosine kinase domain of epidermal growth factor receptor (EGFR) identify tumors that are responsive to EGFR tyrosine kinase inhibitors (TKIs), these genetic alterations are present in only a minority of patients. Patients with tumors expressing wild-type (wt) EGFR lack reliable predictive markers of their clinical response to EGFR TKIs. Although epithelial-mesenchymal transition (EMT) has been inversely correlated with the response of cancers to EGFR-targeted therapy, the precise molecular mechanisms underlying this association have not been defined and no specific EMT-associated biomarker of clinical benefit has been identified. Here we show that during transforming growth factor-β (TGFβ)-mediated EMT, inhibition of the microRNAs 200 (miR200) family results in upregulated expression of mitogen-inducible gene 6 (Mig6), a negative regulator of EGFR. The Mig6-mediated reduction of EGFR occurs concomitantly with a TGFβ-induced EMT-associated kinase switch of tumor cells to an AKT-activated EGFR-independent state. In a panel of 25 cancer cell lines of different tissue origins, we find that the ratio of the expression levels of Mig6 and miR200c is highly correlated with EMT and resistance to erlotinib. Analyses of primary tumor xenografts of patient-derived lung and pancreatic cancers carrying wild type EGFR showed that the tumor Mig6(mRNA)/miR200 ratio was inversely correlated with response to erlotinib in vivo. Our data demonstrate that the TGFβ-miR200-Mig6 network orchestrates the EMT-associated kinase switch that induces resistance to EGFR inhibitors, and identify a low ratio of Mig6 to miR200 as a promising predictive biomarker of the response of tumors to EGFR TKIs.
Introduction
The sensitivity of some tumors to EGFR inhibitors can be explained by the presence of mutations in the EGFR tyrosine kinase domain (1, 2). However, such mutations are rare in tumors other than non-small cell lung carcinoma (NSCLC) (3–6). There is a need to elucidate the mechanisms underlying the differential drug response of cancer cells with wild type EGFR in order to identify those patients who could respond and clinically benefit from TKIs, and to develop new therapeutic strategies to circumvent the de novo or acquired resistance of tumors to EGFR inhibitors.
The response to EGFR-targeted agents is inversely correlated with epithelial-mesenchymal transition (EMT) in multiple types of tumors without known EGFR mutations, including NSCLC, head and neck (H&N), bladder, colorectal, pancreas and breast carcinomas (7–11). Notably, epithelial tumor cells have been shown to be significantly more sensitive to EGFR inhibitors than tumor cells which have undergone an EMT-like transition and acquired mesenchymal characteristics (11). These data suggest that EMT is a common denominator of tumors that are resistant to EGFR inhibitors. However, the precise molecular mechanisms underlying this association have not been defined and no specific EMT-associated biomarker of clinical benefit has been identified.
EMT is driven by a network of transcriptional repressors which include SNAIL1, SNAIL2 (SLUG), ZEB1 (zinc-finger E-box binding factor), ZEB2, and TWIST (12). TGFβ-activated SMAD3/4 stimulates the expression of SNAIL1 and TWIST1, which cooperate with SMAD proteins to repress the expression of epithelial genes such as CDH1 (which encodes E-cadherin) (12, 13). These transcriptional effects of TGFβ cooperate with TGFBR2-mediated phosphorylation of partitioning defective 6 (PAR6) to trigger EMT (12, 14). Whereas TGFβ stimulates EMT, bone morphogenetic protein (BMP) signaling through SMAD1/4 induces expression of pro-epithelial microRNAs (miR200 and miR205) that oppose EMT (12, 15). The miR200 family consists of five members localized on two genomic clusters that can be further divided into two subgroups according to their seed sequences—subgroup I: miR141 and miR-200a; subgroup II: miR200b, miR200c and miR429 (16). During TGFβ-induced EMT, miR200 family and miR205, but not the other microRNAs, are greatly downregulated to facilitate this transition (10, 16, 17).
Members of the miR200 family not only inhibit EMT, but also influence sensitivity to EGFR inhibitors (10, 17–19). miR200c may directly inhibit the expression of Mig6 (also known as RALT, ERRFI1 or Gene 33) (10), a negative regulator of EGFR, which plays an important role in signal attenuation of the EGFR network by blocking the formation of the activating dimer interface through interaction with the kinase domains of EGFR and ERBB2 (20–23). We recently reported that EGFR activity was markedly decreased during acquired resistance to the EGFR TKI erlotinib, with a concomitant increase of Mig6 through the activation of the PI3K-AKT pathway. A low Mig6/EGFR ratio was highly correlated with erlotinib sensitivity in a panel of cancer cell lines and early passage xenografts of human tumors with wild type EGFR (24).
In the current study we report that in response to tumor cell-autonomous expression of TGFβ, erlotinib-sensitive tumor cells undergo EMT-associated suppression of the miR200 family and subsequent upregulation of Mig6 expression. We show that the Mig6-mediated reduction of EGFR occurs concomitantly with a TGFβ-induced EMT-associated kinase switch of tumor cells to an AKT-activated state, thereby leading to an EGFR-independent phenotype that is refractory to EGFR TKI. In a panel of 25 cancer cell lines of different tissue origins, we find that the ratio of the expression levels of Mig6 and miR200c is highly correlated with EMT and resistance to erlotinib. Moreover, analyses of primary tumor xenografts of patient-derived lung and pancreatic cancers carrying wild type EGFR showed that the tumor Mig6(mRNA)/miR200 ratio is inversely correlated with response to erlotinib in vivo. Our data demonstrate that the TGFβ-miR200-Mig6 network orchestrates the EMT-associated kinase switch that induces resistance to EGFR inhibitors, and identify the ratio of Mig6 to miR200 as a promising predictive biomarker of the response of tumors to EGFR TKIs.
Materials and Methods
Compounds and reagents
Erlotinib was purchased from Johns Hopkins Pharmacy. LY294002 and U0126 were obtained from Cell Signaling (Beverly, MA). TGFβ and TGFβ RII/Fc were purchased from R&D Systems (Minneapolis, MN). All other chemicals were purchased from Sigma (St. Louis, MO). All reagents were dissolved according to the manufacturer’s recommendations.
Cell lines
Human NSCLC cell lines (H226, H292, H358, H1838, A549, Calu6, H460, H1703, H1915, H1299, Calu3, H1437, H23), human bladder cancer cell lines (5637, SCaBER, UMUC-3, T24, HT-1376, BFTC-905, J82) and human H&N cell-line FaDu were obtained from American Type Culture Collection (ATCC). The cell lines were freshly ordered and used within 6 month of order date.
Establishment of acquired resistance to erlotinib
Drug resistant cell-lines were generated via a process of slowly escalating exposure to erlotinib, as reported previously (24). SCC-S is used to designate the parental UM-SCC1 cells exposed to DMSO, and SCC-R refers to the erlotinib resistant clone.
Antibodies and immunoblot analysis
Pelleted cells were lysed on ice with RIPA lysis buffer (Thermo Scientific, Rockford, IL) supplemented with protease/phosphatase inhibitors (Roche, Basel, Switzerland). Protein concentrations were determined by the BCA method and lysates diluted in SDS sample buffer (Bio-Rad, Hercules, CA) prior to SDS-PAGE. Anti-Mig6 antibody was a gift from Dr. Ferby (25). β-actin was obtained from Abcam (Cambridge, MA). All other antibodies were obtained from Cell Signaling (Beverly, MA). Secondary horseradish peroxidase (HRP)-conjugated antibodies were from KPL (Gaithersburg, MD) and signals developed using West-Pico chemiluminescence substrate (Thermo Scientific). ImageJ software was used to quantify immunoblot signals on exposed films.
Reverse transcription and real-time PCR
RNA was extracted using Trizol (Invitrogen, Carlsbad, CA) followed by RNeasy kit cleanup (Qiagen, Valencia, CA). RNA was reverse transcribed to cDNA using Superscript III (Invitrogen) which was then used as a template for real-time PCR. Gene products were amplified using iTaq SYBR green Supermix with Rox dye (Bio-Rad Laboratories, Hercules, CA). All reactions were performed in triplicate and relative quantity was calculated after normalizing to GAPDH expression.
Quantitative real-time PCR for miRNAs
RNA from cultured cells was extracted using the mirVana™ Kit (Ambion, Austin, TX). Total RNA from fresh frozen tumors was isolated using the Trizol reagent (Invitrogen, Carlsbad, CA). Specific quantitative real-time PCR was carried out using TaqMan MicroRNA Assays for miR200a, miR200b, miR200c, miR205 and control RNU6b (Applied Biosystems, Foster City, CA) on a 7900HT detector (Applied Biosystems, Foster City, CA).
Cell viability assay
Relative cell viability was determined using an Alamar Blue assay as outlined by the manufacturer (AbDSerotec, Raleigh, NC). New media containing 1/10 volume of Alamar Blue reagent was added to the wells and cells were incubated at 37°C for 1 hour. Fluorescence (545 nm excitation, 590 nm emission wavelengths) was measured using a SpectraMax-Plus384 fluorometer (Sunnyvale, CA). Cell viability was calculated relative to an untreated culture of cells incubated in parallel.
Measurement of TGFβ in tumor cell supernatants
1 × 106 cells were plated in media containing 0.1% FBS. Tumor cell supernatants were amount of TGFβ expressed by 1 × 106 cells per 24 hours.
Xenograft generation
The xenografts were generated and erlotinib treatment was performed as published previously (26, 27). Relative tumor growth inhibition (TGI) in response to Erlotinib (35 mg/kg) was calculated as the relative tumor growth of treated mice divided by relative tumor growth of control mice (T/C). The animals were maintained in accordance to guidelines of the American Association of Laboratory Animal Care and the research protocol was approved by the Johns Hopkins University Animal Use and Care Committee.
Statistical analysis
Student t-tests were used for statistical analysis between two groups. The significance level was defined as 0.05. All statistical analyses were performed using SPSS. IC50 was generated using GraphPad Prism software (La Jolla, CA).
Results
The Erlotinib-resistant tumor phenotype is associated with a kinase switch that enables EGFR-independent activation of AKT
To identify the molecular mechanisms underlying the resistance of tumor cells to EGFR TKI, we examined tumor cell expression and activity of EGFR and alternative receptor tyrosine kinases (RTKs) that lead to EGFR-independent AKT activation. We evaluated pairs of cancer cell lines with wild type EGFR that were either sensitive or resistant to the EGFR TKI, erlotinib; lung carcinoma (H358/H1703 and Calu3/Calu6) and H&N cancer (SCC-S/SCC-R and JHU011/JHU028). Erlotinib-resistant (SCC-R) and erlotinib-sensitive (SCC-S) isogenic cell lines were generated by chronic exposure of human H&N squamous cell carcinoma UM-SCC1 cells to either erlotinib or DMSO (vehicle control) (24). The other three pairs of cell lines (JHU011/JHU028, H358/H1703 and Calu3/Calu6) are intrinsically erlotinib-sensitive or erlotinib-resistant. For every sensitive/resistant cell line pair tested, the IC50 of the resistant cells was at least 10 times higher than that of their sensitive counterparts (Figure 1A). Comparison of the expression and activity of EGFR family members in resistant and sensitive cell lines revealed that the levels of phosphorylated EGFR, HER2 and HER3 were markedly decreased in resistant cells (Figure 1B). In resistant cells, low activity of EGFR family kinases was associated with a significantly higher expression of the endogenous EGFR family negative regulator, Mig6. Consistent with the observed upregulation of Mig6 expression by PI3K-dependent pathways (24, 28), the resistant cell lines exhibited higher AKT phosphorylation levels compared to their sensitive counterparts. (Figure 1B). In accordance with their increased AKT phosphorylation despite low activity of the EGFR family members, erlotinib-resistant cells exhibited a switch from EGFR to activation of an alternative tumor cell-specific RTKs (PDGFR, FGFR, VEGFR, and/or IGFR) (Figure 1C).
Figure 1. Erlotinib-resistant phenotype is associated with a kinase switch that enables EGFR-independent activation of AKT.

A. Two pairs (sensitive/resistant) of lung (H358/H1703 and Calu3/Calu6) and two pairs of H&N (SCC-S/SCC-R and JHU011/JHU028) cancer cell lines were treated with the indicated concentrations of erlotinib and cell viability was assayed. Values were set at 100% for untreated controls. B. Cells were subjected to immunoblot analysis with antibodies specific for phosphorylated and total EGFR, HER2, HER3, AKT and total Mig6. β-actin was used a control. C. Western blot analysis demonstrates expression and activation levels of the indicated RTKs in four pairs of erlotinib resistant/sensitive call lines.
Increased production of TGFβ induces an EMT-associated kinase switch that promotes erlotinib-resistance of tumor cells
The non-receptor focal adhesion kinase (FAK) plays an important role in TGFβ induced EMT progression (29) and up-regulation of mesenchymal markers (30). We tested FAK phosphorylation and total expression level in our four pairs of erlotinib resistant and sensitive cell lines and found that FAK activity is significantly higher in erlotinib-resistant cells from lung and H&N origin (Supplemental Figure 1). To evaluate whether resistance to erlotinib is associated with features of EMT, we tested levels of E-cadherin and vimentin in the panel of 25 erlotinib-sensitive or erlotinib-resistant cell lines with wild type EGFR from lung, H&N and bladder cancer origin (24). While erlotinib-sensitive cells displayed characteristics of typical epithelial cells, including expression of E-cadherin and absence of vimentin, the majority of resistant cells displayed a mesenchymal phenotype manifested by loss of E-cadherin and acquisition of vimentin (Figure 2A). To determine whether erlotinib sensitivity correlates with levels of tumor cell expression of TGFβ (31), we measured the amount of TGFβ produced in cell supernatants of each of the 25 tumor cell lines. Erlotinib-resistant, mesenchymal-like tumor cell lines produced higher levels of TGFβ compared to the erlotinib-sensitive, epithelial-like tumor cells (Figure 2B). To examine whether TGFβ induces the EMT-associated kinase switch responsible for resistance to erlotinib, erlotinib-sensitive epithelial cell lines were exposed to TGF-β1 or TGF-β3. These cell lines included one H&N (SCC-S) and two lung (H358 and H292) cancer cell lines. Serial examination of EMT markers (loss of E-cadherin and upregulation of vimentin) in a time course (1–21 days) showed that TGFβ treatment resulted in complete EMT by day 14 (Figure 2C and Supp. Figure 2A). Strikingly, both total EGFR and phospho-EGFR were reduced with this transition and was accompanied by elevated expression of Mig6 in cells with a mesenchymal phenotype (Figure 2C and Supp. Figure 2A). Concomitant with these molecular alterations, the mesenchymal-like cells acquired a relative resistance to erlotinib (Figure 2D and Supp. Figure 2B). The acquisition of an erlotinib-resistant EMT phenotype in response to TGFβ was associated with a significant increase in AKT activity (Figure 2C and Supp. Figure 2A). To confirm the causal role of AKT in upregulating Mig6 in tumor cells that have acquired resistance to erlotinib, we treated H358, H358/TGFβ1–day 21, and H358/TGFβ3-day 21 cells with LY294002 (PI3K inhibitor), U0126 (MEK inhibitor) or erlotinib (Figure 2E). Whereas all three inhibitors reduced basal expression of Mig6 in H358 cells, only LY294002 resulted in significant inhibition of Mig6 in the erlotinib-resistant H358/TGFβ1-day 21 and H358/TGFβ3-day 21 cells. These data indicate that basal EGFR activity induces an autoregulatory expression of Mig6 in epithelial cells, and that TGFβ-induced activation of AKT coopts this activity in mesenchymal cells (Figure 2E). Together with the data shown in Figure 1C, these data suggested that Mig6 elevation in EMT cells is due to activation of AKT by EGFR-independent tyrosine kinases. To test whether TGFβ can promote this kinase switch, levels of phospho IGFR, PDGFR, FGFR and FAK kinases were assessed in response to treatment of erlotinib-sensitive cells (H358, H292 and SCC-S) with TGFβ1 for 21 days. These kinases showed significantly greater activity in TGFβ1-treated cells when compared to the untreated counterparts (Figure 2F and Supp. Figure 2C). These data indicate that TGFβ-mediated activation of AKT via alternative kinases may substitute for the loss of EGFR activity in a cell-specific manner and contribute to the acquisition of an erlotinib-resistant phenotype.
Figure 2. Increased production of TGFβ induces an EMT-associated kinase switch which promotes erlotinib-resistance of tumor cells.

A. Protein lysates were extracted from indicated cell lines with known sensitivity to erlotinib. Immunoblot analysis was performed with antibodies against E-cadherin, vimentin and β-actin. B. Tumor cell supernatants of 25 cancer cell lines shown in A. were collected and differential levels of TGFβ production were analyzed by ELISA. C. Erlotinib-sensitive lung cancer cell line H358 was treated with TGFβ1/TGFβ3 (4ng/ml) or control vehicle for 21 days. Cells were collected at different time points and Immunoblot analysis was performed with indicated antibodies. D. Parental and TGFβ-induced H358 cells were treated with erlotinib for 72 hours at indicated time points and cell viability was assayed. E. Cells treated with TGFβ1/TGFβ3 or control vehicle for 21 day were exposed to LY294002, U0126, or erlotinib for 24 hours. Immunoblot analysis was performed with antibodies against Mig6 and β-actin. F. Protein lysates were extracted from H358 cells treated with TGFβ1 or control vehicle for 21 days and immunoblot analysis was performed with antibodies against indicated RTKs. β-actin used as a loading control.
TGFβ-induced EMT and erlotinib resistance is associated with decreased levels of the miR200 family and increased Mig6 expression
Since the miR-200 family of microRNAs is downregulated to facilitate EMT (10, 17), we used RT-PCR to assess the level of expression of miR200 in 3 sensitive cell lines (SCC-S, H358 and H292) in response to exposure to TGFβ for 21 days. In all tested cell lines, expression of the miR200 family members (200a, 200b, 200c and 205) was significantly reduced upon TGFβ treatment (Figure 3A). Consistent with the observed ability of miR200c to directly inhibit expression of Mig6 (10), the loss of miR200 family in response to TGFβ was attended with elevation in Mig6 expression during EMT-associated resistance to erlotinib (Figure 2C and Supp. Figure 2A). We next examined changes in miR200 levels in erlotinib-sensitive (SCC-S) and erlotinib-resistant (SCC-R) isogenic H&N cell lines. We found that parental erlotinib-sensitive SCC-S cells displayed significantly higher levels of miR200 family members than the resistant, mesenchymal like, SCC-R cells (Figure 3B). The same pattern was observed in the other three intrinsically sensitive/resistant cell lines pairs (JHU011/JHU028, H358/H1703 and Calu3/Calu6). Finally, examination of the 25 H&N, bladder, and lung cancer cell lines used in this study demonstrated a clear inverse correlation of miR200 levels and erlotinib sensitivity (Figure 3C). Notably, the levels of miR200 family members were also inversely correlated with the expression of Mig6. While erlotinib-sensitive cells demonstrated a high level of miR200 and a low level of Mig6, most of the erlotinib-resistant cells showed decreased levels of miR200 microRNAs and elevated Mig6 expression (Figure 3C and Supp. Figure 3). Taken together, these data indicate that TGFβ-induced repression of miR200 family unleashes the expression of Mig6 in tumor cells during their EMT-associated conversion to an erlotinib-resistant phenotype.
Figure 3. TGFβ-induced EMT and erlotinib resistance is associated with decreased levels of the miR200 family and increased Mig6 expression.

A. Erlotinib-sensitive cell lines H358, H292 and SCC-S were exposure to TGFβ for 21 days. RNA was extracted and expression levels of miR200a, miR200b, miR200c and miR205 were quantified by real-time PCR. B. RNA was extracted from four sensitive/resistant cancer cell lines pairs. Levels of miR200a, miR200b, miR200c and miR205 were measured and relative expression is presented as average fold change of each miRNA in erlotinib-sensitive cell lines relatively to that in resistant cells (ΔΔCt). C. qRT-PCR analysis of miR200a, miR200b, miR200c and miR205 in a panel of 25 human cancer cell lines with known erlotinib sensitivity. Relative quantification of miRNA expression was performed by using RNU6b as an internal control. The results are presented as expression average of each miRNA in erlotinib-sensitive cell lines relatively to that in erlotinib-resistant cells.
Inhibition of TGFβ signaling results in upregulation of miR200c and miR205, decrease in Mig6 levels, and increased erlotinib sensitivity
Autocrine or paracrine TGFβ signaling is required for the maintenance of the mesenchymal state. Blockage of this signaling can inhibit or reverse EMT by upregulating miR200 and subsequently downregulating ZEB1/2 (31–34). As a corollary to this observation, over-expression of miR-200c restores the sensitivity of resistant NSCLC cells to the anti-EGFR antibody cetuximab (35). To determine whether inhibition of TGFβ can restore miR200 expression and reverse the erlotinib-resistant phenotype, we blocked TGFβ signaling in two erlotinib-resistant cell lines of lung (H1703) and H&N (JHU028) origin with SB-431542, a potent inhibitor of the activin receptor-like kinase (ALK) receptors family. Tumor cells were cultured with TGFβ alone or in combination with TGFβ-inhibitor for 7 days, and then treated with 1μM erlotinib for an additional 72 hours. In both cell lines, exposure to TGFβ-inhibitor resulted in a significant increase in miR200c and miR205 levels, and concurrent downregulation of AKT phosphorylation and Mig6 expression (Figure 4A, B). Treatment with SB-431542 increased the sensitivity of tumor cells to erlotinib (Figure 4C). Likewise, cells incubated with TGFβ RII/Fc (recombinant TGFβ receptor II, which binds to and inhibits TGF-β1, TGF-β3, and TGF-β5), displayed a similar increase in erlotinib sensitivity (Figure 4C).
Figure 4. Inhibition of TGFβ signaling results in upregulation of miR200c and miR205, decrease in Mig6 levels, and increased erlotinib sensitivity.
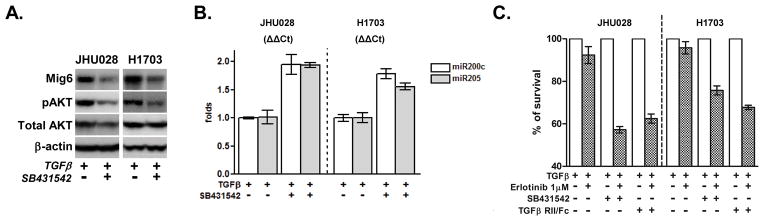
Two erlotinib-resistant cell lines (JHU028 and H1703) were treated with TGFβ (2ng/ml) alone or in combination with SB-431542 (10μM) for 7 days. A. Cell lysates were collected and subjected to immunoblot analysis with indicated antibodies. B. Levels of miR200c and miR205 were measured and relative expression is presented as ΔΔCt. C. Cells were incubated with TGFβ (2ng/ml) alone or in combination with either SB-431542 (10μM) or TGFβ-RII/Fc (20ng/ml) for 7 days and then were treated with 1μM of erlotinib for an additional 72 hours. Cell viability was assayed and values were set at 100% for untreated controls.
Elevated ratio of Mig6(mRNA)/miR200 expression is associated with erlotinib resistance in cancer cell lines of different tissue origins
We observed a strong correlation between Mig6 mRNA and protein levels in 25 tumor cell lines (Figure 5A). Akin to the Mig6 protein (24), Mig6 mRNA expression was considerably lower in erlotinib-sensitive cell lines. Next we tested whether the ratio between Mig6 mRNA and miR200 levels is a reliable predictor of tumor cells response to erlotinib. We found that across the panel of 25 cancer cell lines, the ratio of Mig6 mRNA to each one of the miR200 family members tested appeared to be a reliable predictor of tumor cell responsiveness to erlotinib (Figure 5B and Supp. Figure 4A). Given the strong correlation between Mig6 mRNA and protein levels (Figure 5A), the ability of Mig6(mRNA)/miR200 ratios to predict erlotinib sensitivity was equal to the predictive value of the Mig6/miR200 protein expression ratio (Figure 5C). Interestingly, the ability of Mig6(mRNA)/miR200 ratio to predict erlotinib sensitivity in cancer cell lines was even better than the predictive value of the Mig6/EGFR protein expression ratio (Figure 5D and Supp. Figure 4B).
Figure 5. Elevated ratio of Mig6(mRNA)/miR200 expression is associated with erlotinib resistance in cancer cell lines of different tissue origins.

A. Levels of Mig6 protein (gray bars) or mRNA transcript (white bars) were measured in the panel of 25 human cancer cell lines and plotted on a single graph. Scatter plot showing the ratio between Mig6 mRNA B. or Mig6 protein levels C. and each one of the tested microRNAs (log2 scale) plotted against the IC50 of the corresponding cell line. D. The exposure density of both EGFR and Mig6 blotted on the same membrane were quantified by densitometry and the values of Mig6/EGFR (log2 scale) were plotted against IC50.
Mig6(mRNA)/miR200 ratio predicts response to erlotinib in directly xenografted primary human lung and pancreatic tumors
We obtained 18 human NSCLCs, and 27 pancreatic tumors that were directly xenografted into nude mice (27). Tumor characteristics, including KRAS, NRAS and p53 mutation status, are summarized in Supplemental Table 1. No erlotinib-sensitizing mutations in EGFR were detected in any of these tumors and there was no correlation of KRAS mutation with erlotinib response. For all models tested, miR200 levels were measured by quantitative RT-PCR and mRNA levels of Mig6 and EGFR were determined by Affymetrix expression array. Relative tumor growth inhibition (TGI) in response to Erlotinib (35 mg/kg) was calculated as the relative tumor growth of treated mice divided by relative tumor growth of control mice. We next plotted the Mig6(mRNA)/miR200 ratio against erlotinib responsiveness, with the more resistant tumors clustered to the left and the more sensitive models clustered on the right. Lung and pancreatic tumors that display a high Mig6(mRNA)/miR200 ratio tended to cluster on the left side of the chart, indicating that they were more resistant to erlotinib (Figure 6A and 6B). Lung models with a TGI higher than 40% and pancreatic models with TGI greater than 50%, were associated with significantly lower Mig6(mRNA)/miR200a, Mig6(mRNA)/miR200b or Mig6(mRNA)/miR200c ratios and greater miR200 expression (Figure 6). Our data showed that expression of miR200c (Supp. Figure 5A) and subsequently the Mig6(mRNA)/miR200c ratio (Figure 6) showed the strongest correlation with erlotinib response compared to miR200b and miR200a, suggesting that miR200c might play a more dominant role in regulating Mig6. Supporting this observation, an inverse correlation between miR200c and Mig6 expression levels was noted across the pancreatic models (Supp. Figure 5B). In lung models, tumors with higher erlotinib sensitivity displayed a similar pattern of low Mig6(mRNA)/miR200c ratio. Of note, four erlotinib-resistant lung tumors with low EGFR and Mig6 expression (CTG-0167, CTG-0502, CTG-0199 and CTG-0157) (Supp. Figure 5C) exhibited even lower levels of miR200c (Supp. Figure 5B). Unlike the limited predictive ability of the Mig6/EGFR ratio in such tumors with low EGFR expression (24, 27), the Mig6(mRNA)/miR200c ratio was still able to correctly identify three out of four of these lung tumors with low EGFR mRNA as erlotinib-resistant. Therefore, the ratio of Mig6 to miR200 was a reliable predictive biomarker of the primary tumors response to EGFR TKIs regardless of their EGFR status.
Figure 6. Mig6(mRNA)/miR200 ratio predicts response to erlotinib in directly xenografted primary human lung and pancreatic tumors.

RNA was extracted from A. 18 human NSCLCs and B. 27 pancreatic directly xenografted low passage tumors. Levels of miR200 family members were measured by quantitative RT-PCR and mRNA levels of Mig6 were determined by Affymetrix expression array. The ratios of Mig6(mRNA)/miR200a, Mig6(mRNA)/miR200b and Mig6(mRNA)/miR200c were plotted against erlotinib responsiveness, with the more resistant tumors clustered to the left and the more sensitive models clustered on the right.
Discussion
TGFβ, a multi-functional cytokine that regulates cell growth and differentiation, is frequently upregulated in many human cancers (31, 36–38). Although TGFβ exerts a suppressive effect on normal epithelial cells, tumor cells frequently become refractory to the growth-inhibitory effect of TGFβ and acquire an ability to increase expression and secretion of TGFβ (31–34). This switch enables tumor cells to leverage the tumor-promoting effects of TGFβ in the tumor microenvironment to facilitate tumor progression, invasion, and metastasis (31–39). Previous studies have demonstrated that TGFβ plays a key role in promoting EMT, a switch of epithelial cells into a mesenchymal migratory phenotype that is driven by a network of transcriptional repressors that include SNAIL1, ZEB1, ZEB2, and TWIST (12, 39, 40). TGFβ employs both, canonical and non-canonical signaling pathways to engineer this kinase switch. TGFβ-activated SMAD3/4 stimulates the expression of SNAIL1 and TWIST1, which repress the expression of epithelial genes, such as CDH1 (which encodes E-cadherin) (13). TGFβ also inhibits the expression of pro-epithelial microRNAs (miR200 and miR205) that inhibit ZEB1/2 and oppose EMT (17, 18, 32, 41). Besides promoting EMT, TGFβ engages SMAD-independent pathways to activate PI3K-AKT, such as TACE-mediated secretion of EGFR ligands (42).
In the current study we report that TGFβ induces tumor cells to undergo an EMT-associated kinase switch that renders them resistant to EGFR inhibitors. TGFβ-mediated suppression of the miR200 family not only facilitates EMT, but also enables upregulation of Mig6, a negative regulator of EGFR whose expression is held in check by miR200c. In addition to curtailing EGFR activity via upregulation of Mig6, TGFβ promotes EGFR-independent activation of alternative RTKs and PI3K-AKT signaling. We find that the net effect of TGFβ-signaling is the loss of EGFR activity with a concomitant EMT-associated kinase switch of tumor cells to an AKT-activated state, thereby leading to an EGFR-independent mesenchymal phenotype that is refractory to EGFR TKI. Our study demonstrates that the molecular signature of this resistant tumor phenotype is an elevated Mig6/miR200 ratio.
Our data demonstrate that the TGFβ-miR200-Mig6 network orchestrates the EMT-associated kinase switch that induces resistance to EGFR inhibitors (Figure 7). As such, the autonomous production of TGFβ by tumor cells may be a frequent mechanism by which cancers induce an erlotinib-resistant phenotype. Our study provides the following lines of evidence to support this conclusion. In a panel of 25 cancer cell lines of different tissue origins (H&N, bladder, and lung), erlotinib-resistant, mesenchymal-like cells produced higher levels of TGFβ than the epithelial-like, erlotinib-sensitive cells, suggesting that increased autocrine exposure to TGFβ may be a driving force behind the erlotinib-resistant phenotype. In the same panel, resistance to erlotinib was highly correlated with EMT and an elevated Mig6/miR200c ratio. Besides the high TGFβ expression and elevated Mig6/miR200 ratio exhibited by de novo erlotinib-resistant cell lines, this phenotype was also exhibited by SCC-R tumor cells that had acquired erlotinib-resistance by culturing erlotinib-sensitive SCC-S cells in the presence of escalating concentrations of erlotinib. SCC-R cells expressed more than 10-fold higher levels of TGFβ compared with SCC-S cells (31), and this was associated with reduction of miR200 family members (200a, 200b, 200c and 205) and concomitant increase in Mig6 expression. Furthermore, these cells showed evidence of EMT and manifested a kinase switch involving reduced activity of the EGFR kinase family and activation of alternative RTKs (pPDGFR, pFGFR, pVEGFR, and pIGFR) and AKT. In support of the causal association of tumor cell expression of TGFβ with an elevated Mig6/miR200 ratio and erlotinib-resistance, exposure of various erlotinib-sensitive epithelial tumor cells to exogenous TGFβ resulted in their EMT-associated conversion to an erlotinib-resistant phenotype with an attendant reduction of miR200, increase in Mig6 expression, decrease in EGFR activity, and activation of AKT. Conversely, blockade of TGFβ signaling in erlotinib-resistant, mesenchymal–like cell lines resulted in a concurrent increase of miR200c and miR205 transcripts, downregulation of AKT activity and Mig6 levels, and a significant increase in erlotinib sensitivity.
Figure 7. Evolution of resistance to erlotinib.

The 25 H&N, bladder, and lung cancer cell lines used in this study showed an inverse correlation between the expression levels of Mig6 and miR200. Whereas erlotinib-sensitive cells displayed a low Mig6/miR200 ratio, erlotinib-resistant cells exhibited a high Mig6/miR200 ratio. A similar pattern was noted during TGFβ-induced EMT, wherein downregulation of miR200 family members was paralleled by upregulation of Mig6. By performing PicTar, TargetScan, miRanda and miRBase searches to predict miRNA-mRNA interactions on the Mig6 3′UTR region, we found that the 3′UTR of Mig6 contains conserved potential binding sites for miR-200 family members. Additionally, recent work indicates that miR200c can directly bind to the 3′UTR region of Mig6 mRNA and downregulate its expression (10). In line with this data, the Mig6(mRNA)/miR200c ratio showed the strongest association with erlotinib sensitivity in cancer cell lines as well as primary human tumor xenografts in vivo. These data suggest that TGFβ-mediated suppression of the miR200 family unleashes expression of Mig6, which in turn quenches EGFR activity. The elevation of Mig6 following TGFβ-induced EMT is sustained by EGFR-independent activation of AKT since this is reduced by PI3K inhibitors, but not by erlotinib. Therefore, a high Mig6/miR200c ratio is a sequel of TGFβ-induced EMT and a signature of the EMT-associated kinase switch responsible for resistance to EGFR TKI. Consistent with these observations, our analyses of patient-derived primary tumor xenografts of 18 NSCLC and 27 pancreatic cancers carrying wild type EGFR showed that the tumor Mig6/miR200 ratio is inversely correlated with response to erlotinib in vivo.
Our study demonstrates that an elevated Mig6/miR200 ratio is a molecular signature that characterizes the erlotinib-resistant tumor phenotype. These observations have important clinical implications for the treatment of patients with EGFR inhibitors. The tumor Mig6/miR200 ratio may have clinical value as a predictive biomarker to discern patients who are likely to benefit from EGFR inhibitors from those who are unlikely to respond to such therapy. Our findings further suggest that inhibition of the molecular determinants of the EMT-associated kinase switch, such as TGFβ, may prevent or reverse the de novo or acquired resistance of cancers to EGFR inhibitors.
Supplementary Material
Acknowledgments
Grant Support: This work was supported by NIH grants SPORE P50 DE019032, EDRN U01 CA084986, R37DE012588 and FAMRI-funded 072017_YCSA.
Footnotes
The authors disclose no potential conflicts of interest.
References
- 1.Lynch TJ, Bell DW, Sordella R, Gurubhagavatula S, Okimoto RA, Brannigan BW, et al. Activating mutations in the epidermal growth factor receptor underlying responsiveness of non-small-cell lung cancer to gefitinib. N Engl J Med. 2004;350:2129–39. doi: 10.1056/NEJMoa040938. [DOI] [PubMed] [Google Scholar]
- 2.Paez JG, Janne PA, Lee JC, Tracy S, Greulich H, Gabriel S, et al. EGFR mutations in lung cancer: correlation with clinical response to gefitinib therapy. Science. 2004;304:1497–500. doi: 10.1126/science.1099314. [DOI] [PubMed] [Google Scholar]
- 3.Tsao MS, Sakurada A, Cutz JC, Zhu CQ, Kamel-Reid S, Squire J, et al. Erlotinib in lung cancer - molecular and clinical predictors of outcome. N Engl J Med. 2005;353:133–44. doi: 10.1056/NEJMoa050736. [DOI] [PubMed] [Google Scholar]
- 4.Lemos-Gonzalez Y, Paez de la Cadena M, Rodriguez-Berrocal FJ, Rodriguez-Pineiro AM, Pallas E, Valverde D. Absence of activating mutations in the EGFR kinase domain in Spanish head and neck cancer patients. Tumour Biol. 2007;28:273–9. doi: 10.1159/000110425. [DOI] [PubMed] [Google Scholar]
- 5.Tzeng CW, Frolov A, Frolova N, Jhala NC, Howard JH, Buchsbaum DJ, et al. Epidermal growth factor receptor (EGFR) is highly conserved in pancreatic cancer. Surgery. 2007;141:464–9. doi: 10.1016/j.surg.2006.09.009. [DOI] [PubMed] [Google Scholar]
- 6.van den Bent MJ, Brandes AA, Rampling R, Kouwenhoven MC, Kros JM, Carpentier AF, et al. Randomized phase II trial of erlotinib versus temozolomide or carmustine in recurrent glioblastoma: EORTC brain tumor group study 26034. J Clin Oncol. 2009;27:1268–74. doi: 10.1200/JCO.2008.17.5984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yauch RL, Januario T, Eberhard DA, Cavet G, Zhu W, Fu L, et al. Epithelial versus mesenchymal phenotype determines in vitro sensitivity and predicts clinical activity of erlotinib in lung cancer patients. Clin Cancer Res. 2005;11:8686–98. doi: 10.1158/1078-0432.CCR-05-1492. [DOI] [PubMed] [Google Scholar]
- 8.Thomson S, Petti F, Sujka-Kwok I, Epstein D, Haley JD. Kinase switching in mesenchymal-like non-small cell lung cancer lines contributes to EGFR inhibitor resistance through pathway redundancy. Clin Exp Metastasis. 2008;25:843–54. doi: 10.1007/s10585-008-9200-4. [DOI] [PubMed] [Google Scholar]
- 9.Thomson S, Buck E, Petti F, Griffin G, Brown E, Ramnarine N, et al. Epithelial to mesenchymal transition is a determinant of sensitivity of non-small-cell lung carcinoma cell lines and xenografts to epidermal growth factor receptor inhibition. Cancer Res. 2005;65:9455–62. doi: 10.1158/0008-5472.CAN-05-1058. [DOI] [PubMed] [Google Scholar]
- 10.Adam L, Zhong M, Choi W, Qi W, Nicoloso M, Arora A, et al. miR-200 expression regulates epithelial-to-mesenchymal transition in bladder cancer cells and reverses resistance to epidermal growth factor receptor therapy. Clin Cancer Res. 2009;15:5060–72. doi: 10.1158/1078-0432.CCR-08-2245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Barr S, Thomson S, Buck E, Russo S, Petti F, Sujka-Kwok I, et al. Bypassing cellular EGF receptor dependence through epithelial-to-mesenchymal-like transitions. Clin Exp Metastasis. 2008;25:685–93. doi: 10.1007/s10585-007-9121-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Massague J. TGFbeta signalling in context. Nat Rev Mol Cell Biol. 2012;13:616–30. doi: 10.1038/nrm3434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Vincent T, Neve EP, Johnson JR, Kukalev A, Rojo F, Albanell J, et al. A SNAIL1-SMAD3/4 transcriptional repressor complex promotes TGF-beta mediated epithelial-mesenchymal transition. Nat Cell Biol. 2009;11:943–50. doi: 10.1038/ncb1905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ozdamar B, Bose R, Barrios-Rodiles M, Wang HR, Zhang Y, Wrana JL. Regulation of the polarity protein Par6 by TGFbeta receptors controls epithelial cell plasticity. Science. 2005;307:1603–9. doi: 10.1126/science.1105718. [DOI] [PubMed] [Google Scholar]
- 15.Samavarchi-Tehrani P, Golipour A, David L, Sung HK, Beyer TA, Datti A, et al. Functional genomics reveals a BMP-driven mesenchymal-to-epithelial transition in the initiation of somatic cell reprogramming. Cell Stem Cell. 2010;7:64–77. doi: 10.1016/j.stem.2010.04.015. [DOI] [PubMed] [Google Scholar]
- 16.Korpal M, Lee ES, Hu G, Kang Y. The miR-200 family inhibits epithelial-mesenchymal transition and cancer cell migration by direct targeting of E-cadherin transcriptional repressors ZEB1 and ZEB2. J Biol Chem. 2008;283:14910–4. doi: 10.1074/jbc.C800074200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gregory PA, Bert AG, Paterson EL, Barry SC, Tsykin A, Farshid G, et al. The miR-200 family and miR-205 regulate epithelial to mesenchymal transition by targeting ZEB1 and SIP1. Nat Cell Biol. 2008;10:593–601. doi: 10.1038/ncb1722. [DOI] [PubMed] [Google Scholar]
- 18.Tryndyak VP, Beland FA, Pogribny IP. E-cadherin transcriptional down-regulation by epigenetic and microRNA-200 family alterations is related to mesenchymal and drug-resistant phenotypes in human breast cancer cells. Int J Cancer. 2010;126:2575–83. doi: 10.1002/ijc.24972. [DOI] [PubMed] [Google Scholar]
- 19.Burk U, Schubert J, Wellner U, Schmalhofer O, Vincan E, Spaderna S, et al. A reciprocal repression between ZEB1 and members of the miR-200 family promotes EMT and invasion in cancer cells. EMBO Rep. 2008;9:582–9. doi: 10.1038/embor.2008.74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fiorentino L, Pertica C, Fiorini M, Talora C, Crescenzi M, Castellani L, et al. Inhibition of ErbB-2 mitogenic and transforming activity by RALT, a mitogen-induced signal transducer which binds to the ErbB-2 kinase domain. Molecular and cellular biology. 2000;20:7735–50. doi: 10.1128/mcb.20.20.7735-7750.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hackel PO, Gishizky M, Ullrich A. Mig-6 is a negative regulator of the epidermal growth factor receptor signal. Biol Chem. 2001;382:1649–62. doi: 10.1515/BC.2001.200. [DOI] [PubMed] [Google Scholar]
- 22.Anastasi S, Fiorentino L, Fiorini M, Fraioli R, Sala G, Castellani L, et al. Feedback inhibition by RALT controls signal output by the ErbB network. Oncogene. 2003;22:4221–34. doi: 10.1038/sj.onc.1206516. [DOI] [PubMed] [Google Scholar]
- 23.Zhang X, Pickin KA, Bose R, Jura N, Cole PA, Kuriyan J. Inhibition of the EGF receptor by binding of MIG6 to an activating kinase domain interface. Nature. 2007;450:741–4. doi: 10.1038/nature05998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chang X, Izumchenko E, Solis LM, Kim MS, Chatterjee A, Ling S, et al. The relative expression of Mig6 and EGFR is associated with resistance to EGFR kinase inhibitors. PLoS One. 8:e68966. doi: 10.1371/journal.pone.0068966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ferby I, Reschke M, Kudlacek O, Knyazev P, Pante G, Amann K, et al. Mig6 is a negative regulator of EGF receptor-mediated skin morphogenesis and tumor formation. Nat Med. 2006;12:568–73. doi: 10.1038/nm1401. [DOI] [PubMed] [Google Scholar]
- 26.Harsha HC, Jimeno A, Molina H, Mihalas AB, Goggins MG, Hruban RH, et al. Activated epidermal growth factor receptor as a novel target in pancreatic cancer therapy. J Proteome Res. 2008;7:4651–8. doi: 10.1021/pr800139r. [DOI] [PubMed] [Google Scholar]
- 27.Jimeno A, Tan AC, Coffa J, Rajeshkumar NV, Kulesza P, Rubio-Viqueira B, et al. Coordinated epidermal growth factor receptor pathway gene overexpression predicts epidermal growth factor receptor inhibitor sensitivity in pancreatic cancer. Cancer Res. 2008;68:2841–9. doi: 10.1158/0008-5472.CAN-07-5200. [DOI] [PubMed] [Google Scholar]
- 28.Fiorini M, Ballaro C, Sala G, Falcone G, Alema S, Segatto O. Expression of RALT, a feedback inhibitor of ErbB receptors, is subjected to an integrated transcriptional and post-translational control. Oncogene. 2002;21:6530–9. doi: 10.1038/sj.onc.1205823. [DOI] [PubMed] [Google Scholar]
- 29.Nakamura K, Yano H, Schaefer E, Sabe H. Different modes and qualities of tyrosine phosphorylation of Fak and Pyk2 during epithelial-mesenchymal transdifferentiation and cell migration: analysis of specific phosphorylation events using site-directed antibodies. Oncogene. 2001;20:2626–35. doi: 10.1038/sj.onc.1204359. [DOI] [PubMed] [Google Scholar]
- 30.Cicchini C, Laudadio I, Citarella F, Corazzari M, Steindler C, Conigliaro A, et al. TGFbeta-induced EMT requires focal adhesion kinase (FAK) signaling. Exp Cell Res. 2008;314:143–52. doi: 10.1016/j.yexcr.2007.09.005. [DOI] [PubMed] [Google Scholar]
- 31.Bedi A, Chang X, Noonan K, Pham V, Bedi R, Fertig EJ, et al. Inhibition of TGF-beta enhances the in vivo antitumor efficacy of EGF receptor-targeted therapy. Mol Cancer Ther. 2012;11:2429–39. doi: 10.1158/1535-7163.MCT-12-0101-T. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gregory PA, Bracken CP, Smith E, Bert AG, Wright JA, Roslan S, et al. An autocrine TGF-beta/ZEB/miR-200 signaling network regulates establishment and maintenance of epithelial-mesenchymal transition. Mol Biol Cell. 2011;22:1686–98. doi: 10.1091/mbc.E11-02-0103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Xu Q, Wang L, Li H, Han Q, Li J, Qu X, et al. Mesenchymal stem cells play a potential role in regulating the establishment and maintenance of epithelial-mesenchymal transition in MCF7 human breast cancer cells by paracrine and induced autocrine TGF-beta. Int J Oncol. 2012;41:959–68. doi: 10.3892/ijo.2012.1541. [DOI] [PubMed] [Google Scholar]
- 34.Bryant JL, Britson J, Balko JM, Willian M, Timmons R, Frolov A, et al. A microRNA gene expression signature predicts response to erlotinib in epithelial cancer cell lines and targets EMT. Br J Cancer. 2012;106:148–56. doi: 10.1038/bjc.2011.465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ceppi P, Mudduluru G, Kumarswamy R, Rapa I, Scagliotti GV, Papotti M, et al. Loss of miR-200c expression induces an aggressive, invasive, and chemoresistant phenotype in non-small cell lung cancer. Mol Cancer Res. 2010;8:1207–16. doi: 10.1158/1541-7786.MCR-10-0052. [DOI] [PubMed] [Google Scholar]
- 36.Derynck R, Goeddel DV, Ullrich A, Gutterman JU, Williams RD, Bringman TS, et al. Synthesis of messenger RNAs for transforming growth factors alpha and beta and the epidermal growth factor receptor by human tumors. Cancer Res. 1987;47:707–12. [PubMed] [Google Scholar]
- 37.Dickson RB, Kasid A, Huff KK, Bates SE, Knabbe C, Bronzert D, et al. Activation of growth factor secretion in tumorigenic states of breast cancer induced by 17 beta-estradiol or v-Ha-ras oncogene. Proc Natl Acad Sci U S A. 1987;84:837–41. doi: 10.1073/pnas.84.3.837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Xu J, Lamouille S, Derynck R. TGF-beta-induced epithelial to mesenchymal transition. Cell Res. 2009;19:156–72. doi: 10.1038/cr.2009.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Nieto MA. The ins and outs of the epithelial to mesenchymal transition in health and disease. Annual review of cell and developmental biology. 2011;27:347–76. doi: 10.1146/annurev-cellbio-092910-154036. [DOI] [PubMed] [Google Scholar]
- 40.Thiery JP, Acloque H, Huang RY, Nieto MA. Epithelial-mesenchymal transitions in development and disease. Cell. 2009;139:871–90. doi: 10.1016/j.cell.2009.11.007. [DOI] [PubMed] [Google Scholar]
- 41.Brabletz S, Brabletz T. The ZEB/miR-200 feedback loop--a motor of cellular plasticity in development and cancer? EMBO reports. 2010;11:670–7. doi: 10.1038/embor.2010.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wang SE, Xiang B, Guix M, Olivares MG, Parker J, Chung CH, et al. Transforming growth factor beta engages TACE and ErbB3 to activate phosphatidylinositol-3 kinase/Akt in ErbB2-overexpressing breast cancer and desensitizes cells to trastuzumab. Molecular and cellular biology. 2008;28:5605–20. doi: 10.1128/MCB.00787-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.