

Abstract
Nicotinic acetylcholine receptor (nAChR)-mediated neuroprotection has been implicated in the treatment of neurodegenerative disorders such as Alzheimer’s, Parkinson’s and hypoxic ischemic events, as well as other diseases hallmarked by excitotoxic and apoptotic neuronal death. Several modalities of nicotinic neuroprotection have been reported. However, although this process generally involves α4β2 and α7 subtypes, the underlying mechanisms are largely unknown. Interestingly, both activation and inhibition of α7 nAChRs have been reported to be neuroprotective. We have shown that inhibition of α7 nAChRs protects the function of acute hippocampal slices against excitotoxicity in a α4β2-dependent manner. Neuroprotection was assessed as the prevention of the NMDA-dependent loss of the area of population spikes (PSs) in the CA1 area of acute hippocampal slices. Our results support a model in which α7 AChRs control the release of GABA. Blocking either α7 or GABAA receptors reduces the inhibitory tone on cholinergic terminals, thereby promoting α4β2 activation, which in turn mediates neuroprotection. These results shed light on how α7 nAChR inhibition can be neuroprotective through a mechanism mediated by activation of α4β2 nAChRs.
Keywords: Nicotinic receptors, NMDA toxicity, Bicuculline, CA1 pyramidal neurons, GABAA receptors
Introduction
Many neurodegenerative diseases have in common pathological hallmarks that include excitotoxic neuronal death, apoptosis and inflammation. This shared pathology is underscored by the fact that symptoms of Alzheimer’s disease and Parkinson’s disease are mitigated or prevented by modulation of nicotinic acetylcholine receptors (nAChRs) (Buckingham et al. 2009; Liu et al. 2007; Quik and Wonnacott 2011). In the hippocampus, nAChRs are expressed mainly on GABAergic interneurons which most prominently express the homomeric α7 and the heteromeric α4β2 isoforms (Albuquerque et al. 1997; Alkondon and Albuquerque 2004; Alkondon et al. 2000; Lawrence 2008). The mechanisms of neuroprotection by nicotinic ligands remain to be elucidated. Several modalities of nicotinic neuroprotection can be manifested, depending on the subtypes of nAChRs involved. Stimulation of α7 and α4β2 nAChRs triggers a Ca2+ dependent intracellular cascade involving neuroprotective cell signaling kinases (Akaike et al. 2010; Kihara et al. 2001; Ueda et al. 2008). The signaling cascades involved are the PI3K/AKT and in some cases Raf/MEK/ERK and JAK2/STAT3 (Kawamata et al. 2011). It is likely that, through these pathways, nAChRs regulate brain plasticity (Morishita et al. 2010) and inflammation (Mabley et al. 2009; Pavlov and Tracey 2006; Wang et al. 2003).
Nicotine has been reported to be neuroprotective against N-methyl-D-aspartate (NMDA) induced excitotoxicity in neuronal cultures in a α7 nAChR-dependent manner (Dajas-Bailador et al. 2000; Gahring et al. 2003). However, reduction of the α7 nAChR activity has also been reported to be neuroprotective (Dziewczapolski et al. 2009; Escubedo et al. 2009; Eterovic et al. 2011; Ferchmin et al. 2005; Hu et al. 2007; Hu et al. 2008; Martin et al. 2004; Woodcock 2010). Clearly, a unifying hypothesis to account for the different modes of nicotinic neuroprotection has yet to emerge. The two α7 antagonists tested here were the classical α7 antagonist methyllycaconitine (MLA) (Davies et al. 1999) and the novel antagonist of α7, the cembranoid (1S,2E,4R,6R,7E,11E)-cembra-2,7,11-triene-4,6-diol. Since α7 nAChRs are excitatory receptors expressed mainly on GABAergic interneurons (Alkondon et al. 2000; Buhler and Dunwiddie 2002; Frazier et al. 1998; Lawrence 2008), inhibition of α7 nAChRs may decrease the inhibitory tone on cholinergic terminals in the hippocampus and increase the release of acetylcholine (Giorgetti et al. 2000; Materi and Semba 2001). In this light, the present study tested the hypothesis that bicuculline methiodide (BMI), a GABAA receptor antagonist, directly decreases the inhibitory tone on cholinergic afferents, and mimics the downstream effect observed with inhibition of α7 nAChRs to increase acetylcholine release, enhance α4β2 nAChR activation, and trigger neuroprotection. We propose that neuroprotection by inhibition of α7 nAChRs and by BMI occur via the same mechanism.
Materials and Methods
Reagents
LY294002, methyllycaconitine, and Ro 31-820 were purchased from Calbiochem (La Jolla, CA). Unless specified otherwise, all chemicals were purchased from Sigma–Aldrich (St. Louis, MO). Fetal bovine serum (SH 30071.03) was from HyClone, DMEM 30-2002 and F12K 30-2004 were from ATTC, Pen/Strep 15140 was from GIBCO. The tobacco cembranoid (1S,2E,4R,6R,7E,11E)-cembra-2,7,11-triene-4,6-diol (4R) was either purified form a crude tobacco extract by A.D. Rodriquez (Department of Chemistry, University of Puerto Rico, Rio Piedras, Puerto Rico or from tobacco leaves by K. El Sayed (School of Pharmacy, University of Louisiana, Monroe, LA). 4R stock solution was prepared in 100% dimethylsulfoxide (DMSO) and diluted in buffer the day of the experiment.
Pretesting of compounds used in field potential determinations in slices
The compounds applied to slices were tested for untoward effects as previously reported (Ferchmin et al. 2005; Ferchmin et al. 2003). The drugs were superfused alone, washed off, and 2 hours later, there was no observable effect on the field potentials. BMI and vesamicol were used for the first time in this work; therefore, they were tested as described in results (Figs 2, 3B, 6B). Table 1 presents the drugs used in the present study, their pharmacological effects, and concentrations used.
Fig. 2.

Pre-exposure to 0.1 μM BMI protected against NMDA mediated excitotoxicity. Initial PSs and final PSs after the indicated treatments are shown. A) Initial PS recorded from stratum pyramidale of CA1. The final PS was recorded from the same slice and site after application of 0.5 mM NMDA for 10 min. B) Initial PS as in A. The final PS was recorded after superfusion for 1 hour with 0.1 μM BMI, washout with ACSF followed by NMDA as in A. C) Initial PS and final PS after superfusion with 0.1 μM BMI for 1 hour in the absence of NMDA. For details of experimental design see Fig 1.
Fig. 3.


Fig 3A: At 0.1 μM BMI decreases pair-pulse inhibition without secondary PSs. A cumulative dose response curve of BMI was done to determine the lowest concentration necessary to decrease paired-pulse inhibition. Schaffer collaterals were stimulated once per min with paired-pulses of 20 ms interpulse interval and the synaptically evoked PSs were recorded from stratum pyramidale. The concentration of BMI was incremented every 20 min. The recordings shown are the average of the last 5 responses for each concentration. The black arrow shows the primary PSs elicited by the second stimulus at 0.1 μM BMI. The white arrows show secondary PSs elicited by the first and the second stimuli.
Fig 3B: Preexposure to specific concentrations of BMI protected against NMDA excitotoxicity. Slices were pre-exposed for 1 hour to either ACSF or to the various BMI concentrations. BMI was removed by superfusion with ACSF for another hour after which the standard NMDA toxic stimulus was applied. After NMDA the slices were superfused for 1 hour with ACSF and the final PSs were recorded. The recovery of PSs in slices preincubated with 0.1 and 0.5 μM BMI was significantly larger than in NMDA controls exposed only to NMDA (white bar) or to 0.05 or 1 μM BMI (* p<0.05). The number of slices per condition was 42, 21, 90, 21, 14, and 21, respectively. The black line on top indicates the experimental groups exposed to 0.5 mM NMDA for 10 min. The gray bar below indicates the groups exposed to BMI. The hatched bar shows the lack of effect of superfusion for 1 hour with 0.1 μM BMI on the PSs. See Fig 1 for details of design.
Fig. 6.


Fig 6A: Synaptic release of acetylcholine is necessary for neuroprotection by BMI, 4R and MLA. Vesamicol (50 μM) coapplied with BMI, 4R or MLA blocked the neuroprotection. The white bars represent the NMDA controls showing the recovery of PSs after 10 min of 0.5 mM NMDA. I) The light gray bar shows the neuroprotection by 0.1 μM BMI pretreatment (N=21;*** p<0.001). The dark bar shows the inhibition of BMI neuroprotection by coapplication with vesamicol. II) Neuroprotection by 2 μM 4R (N=14;*** p<0.001) was nullified by coapplication with vesamicol. III) 10 nM MLA preapplied for 1 hour protected (N=14; *** p<0.001). Coapplication of 10 μM MLA with vesamicol inhibited the neuroprotective effect. See Fig 1 for design.
Fig 6B: Vesamicol per se did not affect NMDA neurotoxicity or the rundown of PS in the absence of NMDA. To estimate a possible enhancement of NMDA toxicity or neuroprotection by vesamicol, 0.2 mM NMDA was used. The white bar shows the decrease of PSs after application of 0.2 mM NMDA for 10 min. The gray bar reflects the effect of 1 hour application of vesamicol followed by 1 hour superfusion with ACSF and application of 0.2 mM NMDA. The comparison of the white and gray bar shows that vesamicol did not affect the toxicity of NMDA. The hatched bar shows a near complete recovery of PSs after 1 hour exposure to 50 μM vesamicol alone without later application of NMDA. The recovery of PSs in slices exposed only to vesamicol were significantly larger than in slices exposed to 0.2 mM NMDA without or with vesamicol preapplication (N=14; *; p<0.05). For design, see Fig 1.
Table I.
Pharmacological tools, concentrations used and targets.
| Compound | Abbreviation | Concentration | Target |
|---|---|---|---|
| N-Methyl-D-aspartic acid | NMDA | 0.5 mM | NMDA receptors agonist |
| Cembratriene-4R-diol | 4R | 2 μM | α7 antagonist |
| Bicuculline methiodide | BMI | 0.1 μM | GABAA antagonist |
| Dihydro-β-erythroidine | DHβE | 1 μM | α4β2 receptors |
| Ly294002 | 10 μM | PI3-kinase inhibitor | |
| PD98059 | 10 μM | MEK-1,2 inhibitor | |
| Ro-31-8220 | 100 nM | selective PKC inhibitor | |
| Vesamicol | 50 μM | cholinergic vesicles | |
| Methyllycaconitine | MLA | 10 nM | α7 antagonist |
The compounds used previously were tested alone for untoward effects on the size or shape of the PSs and on NMDA toxicity (Ferchmin et al., 2003; Ferchmin et al., 2005). BMI and vesamicol were used here for the first time and they were tested as described in results. BMI was routinely used at 0.1 μM; however, for the cumulative dose response curve (Figs. 2 and 3) it was used in the range of 0.01 to 1 μM. The following compounds were dissolved in DMSO: 4R, PD98059, Ro-31-8220, vesamicol, and Ly294002. At the concentrations used (<0.1% v/v), DMSO had no detectable effect. In every experiment, the slices in each condition were exposed to the same DMSO concentration for the same length of time.
Slice Preparation and Electrophysiological Recordings
All experimental procedures involving vertebrate animals have been reviewed and approved by the Institutional Animal Care and Use Committee of Universidad Central del Caribe. The methods for the dissection of hippocampi and the preparation of acute slices from male Sprague-Dawley rats (120–200 g) were performed as previously (Eterovic et al. 2011; Ferchmin et al. 1995). For dissection and incubation, a standard artificial cerebrospinal fluid (ACSF) saturated with 95% O2, 5% CO2 was used (in mM): 125 NaCl, 3.3 KCl, 1.25 NaH2PO4, 2 MgSO4, 2 CaCl2, 25 NaHCO3, and 10 glucose. The dissections were performed on ice. Transverse slices (400 μm thickness) were cut with a manual slicer and transferred immediately to a recording chamber. The chamber contained three lanes with independent perfusion lines exposed to humidified 95% O2, 5% CO2. The slices were kept over a nylon mesh, at the interface between ACSF and warmed and humidified 95% O2, 5% CO2 at 34±1°C. A bipolar electrode placed in the stratum radiatum was used to stimulate the Shaffer collaterals with a constant current in the range of 20 to 40 μA for 0.2 ms. The stimulus-evoked population spike (PS) was recorded in stratum pyramidale with a glass electrode filled with 2 M NaCl and impedance 1 to 5 MΩ.
Procedure for Testing Neurotoxicity and Neuroprotection
About 30 slices from the hippocampi of two rats were distributed equally among the three lanes of the chamber. A maximum of seven slices were analyzed per lane for each individual experiment. On average 21 slices per condition were tested for each experimental condition in three independent experiments. One hour after dissection, each slice received a stimulus twice the strength required to elicit a threshold PS. This initial response was recorded as initial PS area (ms x mv). Two hours later, excitotoxicity was induced by superfusing with 0.5 mM NMDA for 10 min in the presence of 95% O2, 5% CO2, and 10 mM glucose. After washout of the NMDA with ACSF for one hour, each slice was stimulated in the same area and intensity used to determine the initial (pre-NMDA exposure) PS. The PS areas obtained before and after NMDA exposure were compared, and the percentage of the initial response remaining at the end of the experiment was calculated and used as a measure of electrophysiological recovery.
The experimental conditions were set to allow for an average 20% recovery of PSs in slices treated only with NMDA (Ferchmin et al. 2000).
Neurotoxicity and neuroprotection was not quantified by measuring neuronal death, instead, the loss and recovery of the area of the PSs was used as an indirect measure of neurotoxicity and neuroprotection. See discussion for more details about the method.
Experimental Protocol
The experimental protocol used for hippocampal slices throughout this work is shown in Fig 1. There were 3 experimental categories: a) NMDA controls, slices treated for 10 min with 0.5 mM NMDA. After 1 hour of ACSF superfusion, the final PSs were recorded. b) Slices pretreated with BMI, MLA or 4R for 1 hour and washed for another hour before application of NMDA and recording of final PSs. c) Slices pretreated with BMI, MLA, or 4R but in the presence of drugs postulated to modify their effect. These drugs were applied for 15 min before, during the hour of and for 15 min after the application of BMI, MLA, or 4R. After 1 hour of washout, NMDA was applied. Overall, this protocol allowed BMI or other compounds sufficient time to trigger a neuroprotective program that would counteract the NMDA-induced excitotoxicity two hours after the drugs were washed out.
Fig. 1.
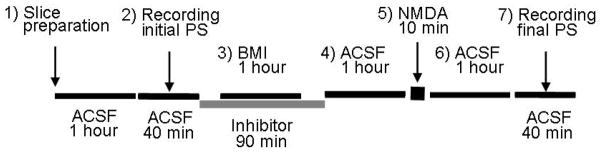
The experimental design shows the seven steps of the neuroprotection assay. 1) Recovery from slice preparation. 2) Recording of initial PSs. 3) BMI or other neuroprotective drug was applied for 1 hour alone or in the presence of an inhibitor. The inhibitors were present 15 min before, during, and for 15 min after BMI application. 4) Drugs were removed by superfusing with ACSF for 1 hour. 5) The excitotoxic stimulus, 0.5 mM NMDA, was superfused for 10 min. 6) NMDA was removed by washing with ACSF for 1 hr during which the slices, depending on the treatment, either recovered the PSs or failed to recover. 7) Recording of final PSs.
Data acquisition and statistical analysis
The areas of PSs were acquired and analyzed with the Labman program (gift from Dr. T. J. Teyler, WWAMI Medical Education Program, University of Idaho, Moscow, ID). The data were statistically analyzed with SigmaPlot, version 11.0 (Systat Software, Inc.). One-way analysis of variance was used whenever the data were distributed normally. In some experiments, a large proportion of slices treated with NMDA had zero recovery and the data failed the normality test. In these cases, the less powerful nonparametric Kruskal-Wallis one-way analysis of variance on ranks was used. The choice of the post-hoc test was based on the advisory statistics suite of SigmaPlot. Data were expressed as mean±SEM. Wherever appropriate, statistical significance of the differences with NMDA controls or other experimental conditions are indicated on the figure legends.
Cell culture of α7 expressing SHSY5Y cells
α7 nAChR expressed in SHSY5Y cells were obtained from Novartis Pharma AG. The cells were cultured in 1:1 DMEM/F12K, 10% FBS, 100μg/mL G418 and 100units/mL Penicillin and 100ug/mL Streptomycin. They were plated for culture maintenance in 75cm2 tissue culture flasks and in glass cover-slips inside 35mm Petri dishes for experiments in three or four days. They were split when 80–90% confluence was reached (about 4–5 days). Briefly the media containing the floating cells was transferred to a 50mL centrifuge tube and then 5mL of a solution containing 1X Trypsin diluted in 1X PBS was added for five to ten minutes to detach the cells from the flask. Then the cells were pooled with the ones that were already in the conical tube and centrifuged for three minutes at 1000 rpm. The pooled cells were then plated and incubated at 37°C and 5% CO2.
Whole-cell patch-clamp recording
Whole-cell current was measured using an EPC9 (HEKA) amplifier controlled by Pulse software. The current traces were low-pass filtered at 2 kHz and digitized at 10 kHz. The cells were clamped at −100 mV as previously described (Charpantier et al. 2005). The external buffer contained (in mM): 130 NaCl, 5 KCl, 2 MgCl2, 2 CaCl2, 10 HEPES; pH adjusted to 7.4 with 1M NaOH solution. The pipette solution contained (in mM): 130 K-D gluconate, 5 NaCl, 2 MgCl2, 5 EGTA, 10 HEPES, pH 7.4. The pipettes were made from borosilicate capillary tubes pulled to have a resistance of 2–4 MΩ. Agonists and modulators were applied with a 16-channel Dynaflow chip (Cellectricon, Inc. Sweden). 4R was dissolved in 100% DMSO and diluted immediately before the experiment with the bath solution to 0.1% DMSO; all agonist solutions also contained 0.1% DMSO. All experiments were carried out at room temperature (22–24°C). The analysis of whole-cell currents was carried out using the MiniAnalysis software (Synaptosoft Inc.)
Results
Pre exposure to BMI protects against NMDA excitotoxicity
To test the hypothesis that inhibition of α7 nAChR causes a decrease of GABAergic tone, which triggers neuroprotection (Ferchmin et al. 2005; Ferchmin et al. 2003), we pre-treated slices with 0.1 μM BMI for 1 hour following the experimental protocol shown in Fig 1. A model of this hypothesis is shown in Fig 8. As illustrated in Fig 2, pre-exposure to BMI reversed the excitotoxic effect of NMDA on PSs.
Fig. 8. Proposed model for the neuroprotection by MLA, 4R, and BMI.
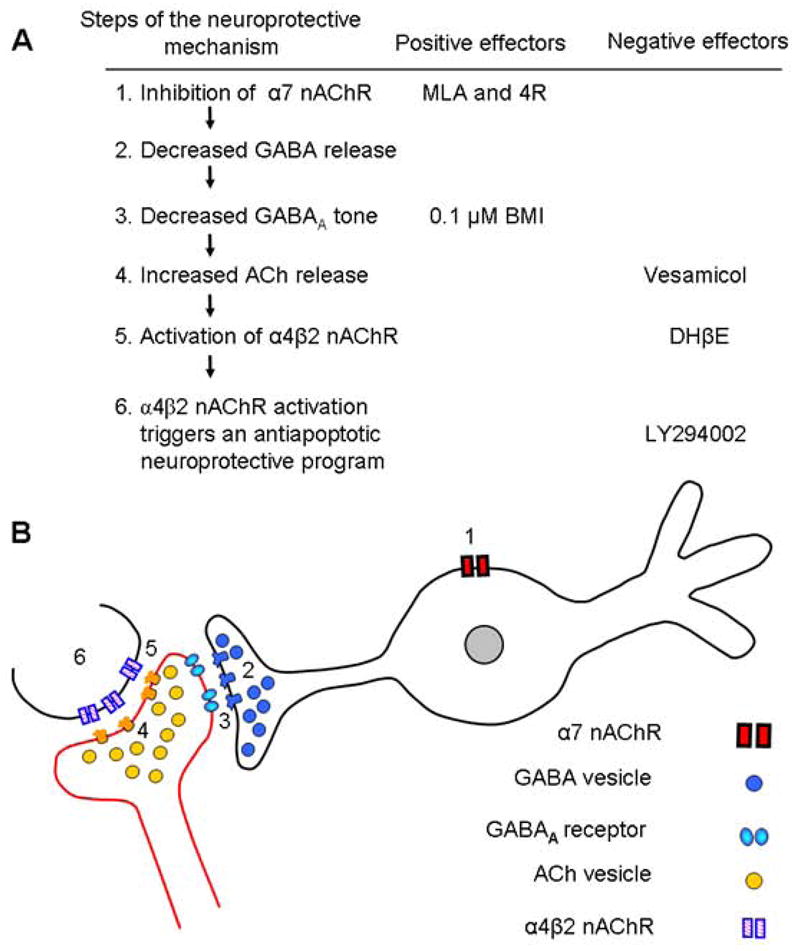
A) Proposed sequence of steps and the inhibitors used to explore them. 1) MLA and 4R inhibit α7 nAChR on GABAergic interneurons or septal afferents. 2) Decreased α7 activity reduces GABA release. 3) The hyperpolarizing tone decreases either because of reduced GABA release or because of the presence of the antagonist BMI. 4) Reduced inhibition on cholinergic terminals allows for more ACh release. Vesamicol blocks this pathway by blocking the synaptic release of ACh. 5) The enhanced ACh release activates the α4β2 nACh receptors. DHβE, a selective antagonist of α4β2 blocks this step. 6) Activation of α4β2 receptors triggers the phosphoinositide 3-kinase neuroprotective cascade, which is blocked by LY294002. Positive or negative effector refers to compounds that cause or reverse the neuroprotection.
B) Proposed localization of receptors and synaptic elements. Inhibition of α7 receptors (1) on GABAergic interneurons or GABAergic terminals attenuates GABA release (2) and decreases the GABAergic tone (3) on incoming cholinergic afferents. The decreased GABAergic tone is mimicked by BMI, a GABAA antagonist. Weaker GABAergic tone (3) disinhibits the synaptic release of ACh (4) and increases the activation of α4β2 (5) on terminals innervating pyramidal neurons or directly on pyramidal neurons. The icons representing receptors and synaptic vesicles are presented in the top to bottom sequence in which they are presumed to act in this mechanism.
Interestingly, BMI protection took place when NMDA was applied 1 hour after superfusion with ACSF to remove BMI. This shows that BMI per se is not neuroprotective but a neuroprotective metabolic consequence of the presence of BMI persisted after BMI was removed. In the same conditions but without NMDA application, BMI did not affect the PSs (Fig 2C) suggesting that the metabolic effect of BMI only protects against excitotoxicity but does not enhance the PSs per se.
Dose dependence of BMI neuroprotection
BMI, a water soluble analogue of bicuculline (DeFeudis et al. 1975), is an unlikely candidate to cause neuroprotection because it is a convulsant and, consequently, induces seizures with ensuing excitotoxic neuronal death (Olney et al. 1986). To find the optimal BMI concentration likely to induce neuroprotection without causing excitotoxic seizures, a cumulative dose response (0.1–1.0 μM) versus paired-pulse inhibition in CA1 was done (Fig 3). The paired-pulse paradigm is appropriate to determine the strength of GABAergic inhibition in CA1 (Ferchmin et al. 1995; Nathan et al. 1990). Paired-pulse stimulation of slices superfused with ACSF showed a PS only after the first stimulus. There was no PS after the second stimulus because of recurrent GABAergic inhibition. An incipient PS was elicited by the second stimulus at 0.05 μM BMI, suggesting a decrease in GABAergic inhibition. At 0.1 μM BMI the second stimulus evoked a small but robust PS without secondary PSs. A further increase in BMI concentration up to 0.5 μM produced a secondary PS after the first stimulus and the response to the second stimulus showed no paired-pulse inhibition (Fig 3A). At 1 μM BMI the disinhibition was significantly increased displaying large secondary spikes after the first and second stimuli indicative of a potentially excitotoxic effect of BMI. At higher BMI concentrations (>1 μM; results not shown) epileptiform waves were recorded. Overall, BMI was neuroprotective within the concentration range from 0.1 to 0.5 μM. Controls for the effect of BMI without NMDA application indicated that 0.1 μM BMI by itself did not affect the PS areas (Fig 3B). Since BMI was maximally neuroprotective at 0.1 μM, this concentration was used in all the remaining experiments.
4R is an antagonist of α7 nAChRs
The first step of the mechanism of neuroprotection by 4R and MLA is inhibition of α7 nAChR (Fig 8). MLA is a classic antagonist of the α7 receptors (Davies et al. 1999) but the direct effect of the 4R on α7 nAChRs was not described untill now. Here we show that 4R inhibited ACh evoked currents from SHSY5Y cells transfected with human α7 nAChR. Fig 4 illustrates that application of 300 μM ACh evoked robust inward currents, which desensitized rapidly and were inhibited by 10 nM MLA (data not shown), as expected for the α7 nAChR (Charpantier et al. 2005). The ACh evoked current was robustly but reversibly inhibited in the presence of 25 μM 4R. The inhibition required preincubation with 4R alone. In the absence of ACh, 25 μM 4R did not evoke any current, which indicated that 4R is not an agonist of α7 nAChR. These results were reproduced with 3 cells and the observed inhibition was 82% ± 18% (mean ± S.E.M.).
Fig. 4.
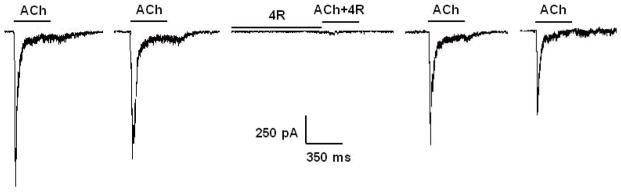
4R inhibits ACh-evoked currents from the α7 nAChR. ACh-evoked currents were recorded from SHSY5Y cells transfected with human α7 nAChR, using the patch-clamp technique as explained in Methods. The cells were exposed to 300 μM ACh for 350 ms; 90s washouts with the external buffer separated successive applications of the agonist. 300 μM ACh evoked robust inward currents (first two recordings to the left). The cells were then exposed to 25 μM 4R in the absence of ACh for 120s; this was followed by application of 25 μM 4R in the presence of 300 μM ACh for 350 ms. 25 μM 4R produced a profound inhibition of the ACh-evoked current with full recovery upon washout as shown by the last two recordings.
Pharmacological profile of BMI, MLA and 4R mediated neuroprotection
The purpose of the next experiments was to compare the pharmacological profile of the neuroprotection by BMI with the neuroprotection by MLA and 4R. These comparisons were made either with new data presented in this work or with experiments published previously. The compounds used for this purpose were tested for untoward effects on the health of the slices, and the size and shape of the recordings. The results of these control experiments were either published before (Ferchmin et al. 2005; Ferchmin et al. 2003) or, in the case of BMI and vesamicol, were tested here (see Fig 2C, 3B and 6B).
To determine whether BMI and MLA neuroprotection was mediated by the α4β2 nAChR, DHβE was coapplied with both compounds. DHβE, at 1 μM, is a selective α4β2 nAChR antagonist (Raggenbass and Bertrand 2002). Figure 5 shows that coapplication of 1 μM DHβE with either BMI or MLA completely abolished the neuroprotection confirming the nicotinic mechanism. Previously, we showed that DHβE prevented the neuroprotection by 4R without affecting NMDA toxicity or the size or shape of PSs (Ferchmin et al. 2005). Taken together, the results are consistent with neuroprotection by BMI, MLA and 4R, all being mediated by a nicotinic mechanism.
Fig. 5.

α4β2 nAChRs are required for BMI and MLA mediated neuroprotection. The white bars, NMDA control, show the remaining PSs in slices treated with 0.5 mM NMDA for 10 min. A) The light gray bar shows the recovery of slices treated for 1 hour with 0.1 μM BMI, washed for another hour with ACSF before treatment with NMDA. The dark gray bar shows the effect of 1 μM DHβE coapplied with 0.1 μM BMI. BMI significantly increased the recovery of PSs over NMDA controls and over BMI coapplied with DHβE (N=21;*, p<0.05). B) The light gray bar shows the neuroprotection by 10 nM MLA applied for 1 hour. Dark gray bar shows that 1 μM DHβE coapplied with 10 nM MLA prevented the neuroprotection (N=14;*p<0.05). See Fig 1 for design.
According to our hypothesis, inhibition of α7 nAChRs on GABAergic interneurons or direct inhibition of GABAA receptors will increase synaptic release of acetylcholine and activate α4β2 nAChRs. The role of synaptic release of acetylcholine on neuroprotection by BMI, 4R and MLA was tested by coapplication with vesamicol (50 μM). Vesamicol indirectly prevents vesicular release of ACh (Auld et al. 2000; Auld et al. 2001; Cobb et al. 1999). Coapplication of vesamicol inhibited the neuroprotection by BMI, 4R and MLA (Fig 6A) without affecting the PSs or the toxicity of 0.2 mM NMDA (Fig 6B). We have shown that neuroprotection by 4R is not dependent on the Raf-MEK-ERK cascade since it was not prevented by PD98059, a specific inhibitor of ERK-1,2 activation by MEK-1,2 (Ferchmin et al. 2005). PD98059 is a specific inhibitor of ERK-1,2 activation by MEK-1,2 (Alessi et al. 1995; Davies et al. 2000). Figure 7A shows that, like 4R, neuroprotection by BMI and by MLA are resistant to PD98059. We have shown previously that, at the concentrations tested (10–50 μM), PD98059 by itself did not affect the PS areas (Ferchmin et al. 2003). Similarly, the PKC inhibitor Ro31-8220 did not inhibit the neuroprotection by BMI and MLA (Fig 7B,C), nor did it affect the neuroprotection by 4R (Ferchmin et al. 2005). By contrast, Ly294002, a selective inhibitor of PI3K, inhibited the neuroprotection by BMI and MLA (Fig 7D, E) as was shown previously for 4R-induced neuroprotection (Ferchmin et al. 2005).
Fig. 7.

Cell signaling underlying BMI and MLA neuroprotection. White bars represent unprotected NMDA controls. A) 10 μM PD98059 did not inhibit the neuroprotection by 0.1 μM BMI (gray bar), or 10 nM MLA (hatched gray bar) (N=21; *** p<0.01). B) The neuroprotective effect of BMI was not affected by 100 nM Ro 31-8220 (N=21; *p<0.050). C) The neuroprotective effect of MLA was not affected by 100 nM Ro 31-8220 (N=21; *p<0.05). D) Neuroprotection by BMI was inhibited by 10 μM Ly294002 (N=21; *, p<0.05). E) 10 μM Ly294002 inhibited the neuroprotective effect of 10 nM MLA (N=21; *, p<0.05). See Fig 1 for design.
Discussion
The hypothesis tested here is that inhibition of α7 nAChRs causes indirectly a decrease in GABAergic tone, which can be produced directly by the GABAA antagonist BMI. The decreased inhibitory tone causes an increase in ACh release, which activates the α4β2 nAChRs responsible for the neuroprotection. A model of this hypothesis is presented in Fig 8.
According to this hypothesis, a mild reduction of GABAergic tone would mimic the effect of α7 inhibition. This was supported by our finding that within the narrow concentration range of 0.1 to 0.5 μM BMI was robustly neuroprotective (Fig 3). Higher BMI concentrations failed to protect either by causing excitotoxicity or by affecting other receptors or ion channels as is discussed below. The lowest BMI concentration that produced a small PS after the second stimulus in a paired pulse paradigm was the most neuroprotective (Fig 3A and B). Our assumption that a decrease in GABAergic tone increases ACh release agrees with reports that inhibition of GABAA receptors by bicuculline enhances synaptic ACh release (Giorgetti et al. 2000; Moor et al. 1998; Roland and Savage 2009; Vazquez and Baghdoyan 2004).
The model predicts that the mechanism of protection by either 4R, MLA or the GABAA antagonist will be identical. Consistent with this prediction is the observations that neuroprotection by 0.1 μM BMI and by 10 nM MLA was inhibited by DHβE proving the involvement of the α4β2 nAChRs (Fig 5). At 1 μM DHβE is a selective inhibitor of the α4β2 nAChRs (Alessi 1997; Khiroug et al. 2004; Raggenbass and Bertrand 2002). We have reported that the neuroprotection by 4R is inhibited by DHβE (Ferchmin et al. 2005) and here we present evidence that 4R is an antagonist of the α7 nAChR (Fig 4).
Since the protection by BMI, 4R, and MLA was abolished in the presence of vesamicol (Fig 6A), the synaptic release of ACh must participate in the pathway as proposed in the model (Fig 8A and B, step 4). The synaptically released ACh could act either on nicotinic or muscarinic ACh receptors. The fact that the selective inhibitor of α4β2 nAChR, 1 μM DHβE, completely blocked the protective effect of BMI and MLA (Fig 5) shows that a nicotinic receptor is involved (Fig. 8A, Step 5).
Therefore, although muscarinic receptors interact with GABAergic transmission (Fellous and Sejnowski 2000; Konopacki and Golebiewski 1993), they do not seem to have a significant effect on BMI neuroprotection.
The neuroprotection by BMI, MLA and 4R was effective two hours later when these compounds were already washed off (Fig 1). This suggests that BMI, MLA, and 4R are neuroprotective because of their outlasting effect on the cell signaling processes that confers the resistance to the excitotoxic stimulus.
Additional evidence that BMI neuroprotection was mediated by the same mechanism as MLA is supported by the pharmacological study of inhibitors of cell signaling protein kinases on the efficacy of both compounds (Fig 7). The protein kinase inhibitors had the same effect on neuroprotection by BMI and MLA. In addition, we have previously reported a similar pharmacological profile for 4R (Ferchmin et al. 2005).
The PS is an appropriate parameter for assessing early neurotoxicity and neuroprotection because it is the sum of the axon potentials of a population of neurons and it is directly proportional to the number of functionally active pyramidal neurons (Andersen et al. 1971). This variable was successfully used to assess neurotoxicity and neuroprotection against NMDA, anoxia, oxygen and glucose deprivation, excitotoxic amino acids and paraoxon on hippocampal neurons (Eterovic et al. 2011; Ferchmin et al. 2005; Ferchmin et al. 2000; Ferchmin et al. 2003; Fountain et al. 1992; Martins et al. 2012; Schurr et al. 1995a; Schurr et al. 1995b; Schurr and Rigor 1989). Agreement between histological markers of neurodegeneration and PSs (Monette et al. 1998; Small et al. 1996) or intracellular action potentials (Wang et al. 1999) was demonstrated. Recently, we showed that a cell permeable inhibitor of caspase 9 and SB-216763 an inhibitor of the proapoptotic enzyme GSK-3β prevented the loss of PSs in slices treated with excitotoxic concentration of NMDA. This suggests that the loss of PSs is caused by an early step of apoptosis (Martins et al. 2012).
There are several concerns about the mechanism of action and the selectivity of BMI in our paradigm. The low concentration of BMI (0.1 μM) found by us to be neuroprotective was seldom used. However, this concentration agrees with the literature considering that only a slight inhibition of GABAA receptors was needed to trigger neuroprotection (Fig 3A). Bicuculline was reported to inhibit specific GABA binding to brain membranes with an ED50 of 5 μM (Frere et al. 1982). BMI inhibited GABA evoked currents recorded from HEK293 cells expressing a recombinant GABA receptor with IC50=1.9 μM (McCartney et al. 2007). The above reports show that 0.1 μM bicuculline could inhibit 10 to 15% of GABAA receptors.
It is predictable that excitotoxicity will be ameliorated by activation of inhibitory GABA receptors (DeFazio et al. 2009; Sommer et al. 2003; Wei et al. 2012; Xu et al. 2008). Therefore, our finding that a weak inhibition of GABAA receptors is neuroprotective apparently contradicts the reports that enhancement of GABAergic transmission is neuroprotective. The difference between our experimental design and theirs is that we preapplied the GABAergic antagonist (Fig 1) whereas they enhanced the GABAergic transmission during and after the excitotoxic episode.
Exposure to 0.1 μM BMI could be considered a preconditioning treatment against toxic concentrations of NMDA by synaptic activation of NMDA receptors triggering neuroprotection as described before (Soriano et al. 2006). However, the protection described here is produced by 0.1 μM BMI in acute slices while Soriano et al. used 50 μM bicuculline in cultured neurons. In our experiments, BMI concentrations higher than 0.5 μM were not neuroprotective and the inhibition of BMI-mediated neuroprotection by vesamicol and DHβE proves that it is a novel mode of cholinergic and nicotinic neuroprotection.
Another concern about bicuculline and BMI selectivity is that they block nAChRs (Demuro et al. 2001; Zhang and Feltz 1991), the apamin-sensitive Ca2+-activated K+ current (Johansson et al. 2001; Johnson and Seutin 1997) or the small-conductance calcium-activated potassium channels (SK) (Khawaled et al. 1999). However, these effects were observed at concentrations 10 to 100 times higher than the 0.1 μM BMI used here. In addition, in our paradigm, increase in BMI concentration did not enhance the neuroprotective effect (Fig 3B).
It is possible that at higher concentration BMI activated other processes that impaired the neuroprotection. It is noteworthy that the neuroprotection assessed in this work was the recovery of the capability of the CA1 pyramidal neurons to produce synaptically elicited axon potentials after exposure to excitotoxic treatment with NMDA. As mentioned above, this modality of neuroprotection correlates with neuroprotection determined by other methods in which cell death was the end point measure. In conclusion, our data support the notion that neuroprotection by antagonists of α7 nAChRs and by mild inhibition of GABAA receptors are mediated by the same mechanism that involves synaptic interactions and activation of the α4β2 nAChRs. This work sheds light on the complexity of nicotinic neuroprotection mediated by inhibition of α7 nAChR.
Acknowledgments
Supported by the CounterACT Program, Office of the Director, National Institutes of Health (OD) and the National Institute of Neurological Disorders and Stroke (NINDS), Grant Number 1UO1NS063555, U54 NS39408-SNRP and core facilities supported by NIH Grant RCMI 2G12RR03035
We acknowledge the careful reviewing, corrections and advice provided by H. Yeh, R. Andrade, P. Lein and W. Sulikowski. This work was supported by NINDS ConterAct UO1 NS63555; NINDS, NCRR (SNRP) NS39408) and by NIGMS grantS06GM50695. Core facilities were supported by NCRR (RCMI) (grant G-12RR03035).
Literature cited
- Akaike A, Takada-Takatori Y, Kume T, Izumi Y. Mechanisms of neuroprotective effects of nicotine and acetylcholinesterase inhibitors: role of alpha4 and alpha7 receptors in neuroprotection. J Mol Neurosci. 2010;40(1–2):211–216. doi: 10.1007/s12031-009-9236-1. [DOI] [PubMed] [Google Scholar]
- Albuquerque EX, Alkondon M, Pereira EF, Castro NG, Schrattenholz A, Barbosa CT, Bonfante-Cabarcas R, Aracava Y, Eisenberg HM, Maelicke A. Properties of neuronal nicotinic acetylcholine receptors: pharmacological characterization and modulation of synaptic function. J Pharmacol Exp Ther. 1997;280(3):1117–1136. [PubMed] [Google Scholar]
- Alessi DR. The protein kinase C inhibitors Ro 318220 and GF 109203X are equally potent inhibitors of MAPKAP kinase-1beta (Rsk-2) and p70 S6 kinase. FEBS Lett. 1997;402(2–3):121–123. doi: 10.1016/s0014-5793(96)01510-4. [DOI] [PubMed] [Google Scholar]
- Alessi DR, Cuenda A, Cohen P, Dudley DT, Saltiel AR. PD 098059 is a specific inhibitor of the activation of mitogen-activated protein kinase kinase in vitro and in vivo. The Journal of biological chemistry. 1995;270(46):27489–27494. doi: 10.1074/jbc.270.46.27489. [DOI] [PubMed] [Google Scholar]
- Alkondon M, Albuquerque EX. The nicotinic acetylcholine receptor subtypes and their function in the hippocampus and cerebral cortex. Prog Brain Res. 2004;145:109–120. doi: 10.1016/S0079-6123(03)45007-3. [DOI] [PubMed] [Google Scholar]
- Alkondon M, Braga MF, Pereira EF, Maelicke A, Albuquerque EX. alpha7 nicotinic acetylcholine receptors and modulation of gabaergic synaptic transmission in the hippocampus. Eur J Pharmacol. 2000;393(1–3):59–67. doi: 10.1016/s0014-2999(00)00006-6. [DOI] [PubMed] [Google Scholar]
- Andersen P, Bliss TV, Skrede KK. Unit analysis of hippocampal polulation spikes. Exp Brain Res. 1971;13(2):208–221. doi: 10.1007/BF00234086. [DOI] [PubMed] [Google Scholar]
- Auld DS, Day JC, Mennicken F, Quirion R. Pharmacological Characterization of Endogenous Acetylcholine Release from Primary Septal Cultures. J Pharmacol Exp Ther. 2000;292(2):692–697. [PubMed] [Google Scholar]
- Auld DS, Mennicken F, Day JC, Quirion R. Neurotrophins differentially enhance acetylcholine release, acetylcholine content and choline acetyltransferase activity in basal forebrain neurons. J Neurochem 2001. 2001 Apr;77(1):253–262. doi: 10.1046/j.1471-4159.2001.t01-1-00234.x. [DOI] [PubMed] [Google Scholar]
- Buckingham SD, Jones AK, Brown LA, Sattelle DB. Nicotinic acetylcholine receptor signalling: roles in Alzheimer’s disease and amyloid neuroprotection. Pharmacol Rev. 2009;61(1):39–61. doi: 10.1124/pr.108.000562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buhler AV, Dunwiddie TV. alpha7 nicotinic acetylcholine receptors on GABAergic interneurons evoke dendritic and somatic inhibition of hippocampal neurons. J Neurophysiol. 2002;87(1):548–557. doi: 10.1152/jn.00316.2001. [DOI] [PubMed] [Google Scholar]
- Charpantier E, Wiesner A, Huh KH, Ogier R, Hoda JC, Allaman G, Raggenbass M, Feuerbach D, Bertrand D, Fuhrer C. Alpha7 neuronal nicotinic acetylcholine receptors are negatively regulated by tyrosine phosphorylation and Src-family kinases. J Neurosci. 2005;25(43):9836–9849. doi: 10.1523/JNEUROSCI.3497-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cobb SR, Bulters DO, Suchak S, Riedel G, Morris RGM, Davies CH. Activation of nicotinic acetylcholine receptors patterns network activity in the rodent hippocampus. J Physiol (Lond) 1999;518(1):131–140. doi: 10.1111/j.1469-7793.1999.0131r.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dajas-Bailador FA, Lima PA, Wonnacott S. The alpha7 nicotinic acetylcholine receptor subtype mediates nicotine protection against NMDA excitotoxicity in primary hippocampal cultures through a Ca(2+) dependent mechanism. Neuropharmacology. 2000;39(13):2799–2807. doi: 10.1016/s0028-3908(00)00127-1. [DOI] [PubMed] [Google Scholar]
- Davies ARL, Hardick DJ, Blagbrough IS, Potter BVL, Wolstenholme AJ, Wonnacott S. Characterisation of the binding of [3H]methyllycaconitine: a new radioligand for labelling [alpha]7-type neuronal nicotinic acetylcholine receptors. Neuropharmacology. 1999;38(5):679–690. doi: 10.1016/s0028-3908(98)00221-4. [DOI] [PubMed] [Google Scholar]
- Davies SP, Reddy H, Caivano M, Cohen P. Specificity and mechanism of action of some commonly used protein kinase inhibitors. Biochem J. 2000;351(Pt 1):95–105. doi: 10.1042/0264-6021:3510095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeFazio RA, Raval AP, Lin HW, Dave KR, Della-Morte D, Perez-Pinzon MA. GABA synapses mediate neuroprotection after ischemic and epsilonPKC preconditioning in rat hippocampal slice cultures. J Cereb Blood Flow Metab. 2009;29(2):375–384. doi: 10.1038/jcbfm.2008.126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeFeudis FV, Balfagon G, De Sagarra MR, Madtes P, Somoza E, Gervas-Camacho J. Action of N-methyl-bicuculline on the binding of gamma-aminobutyric acid to a synaptosomal fraction of rat cerebral cortex. Exp Neurol. 1975;49(2):497–505. doi: 10.1016/0014-4886(75)90104-1. [DOI] [PubMed] [Google Scholar]
- Demuro A, Palma E, Eusebi F, Miledi R. Inhibition of nicotinic acetylcholine receptors by bicuculline. Neuropharmacology. 2001;41(7):854–861. doi: 10.1016/s0028-3908(01)00137-x. [DOI] [PubMed] [Google Scholar]
- Dziewczapolski G, Glogowski CM, Masliah E, Heinemann SF. Deletion of the alpha 7 nicotinic acetylcholine receptor gene improves cognitive deficits and synaptic pathology in a mouse model of Alzheimer’s disease. J Neurosci. 2009;29(27):8805–8815. doi: 10.1523/JNEUROSCI.6159-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Escubedo E, Camarasa J, Chipana C, Garcia-Rates S, Pubill D. Involvement of nicotinic receptors in methamphetamine- and MDMA-induced neurotoxicity: pharmacological implications. Int Rev Neurobiol. 2009;88:121–166. doi: 10.1016/S0074-7742(09)88006-9. [DOI] [PubMed] [Google Scholar]
- Eterovic VA, Perez D, Martins AH, Cuadrado BL, Carrasco M, Ferchmin PA. A cembranoid protects acute hippocampal slices against paraoxon neurotoxicity. Toxicol In Vitro. 2011;25(7):1468–1474. doi: 10.1016/j.tiv.2011.04.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fellous JM, Sejnowski TJ. Cholinergic induction of oscillations in the hippocampal slice in the slow (0.5–2 Hz), theta (5–12 Hz), and gamma (35–70 Hz) bands. Hippocampus. 2000;10(2):187–197. doi: 10.1002/(SICI)1098-1063(2000)10:2<187::AID-HIPO8>3.0.CO;2-M. [DOI] [PubMed] [Google Scholar]
- Ferchmin PA, Eterovic VA, Rivera EM, Teyler TJ. Spermine increases paired-pulse facilitation in area CA1 of hippocampus in a calcium-dependent manner. Brain research. 1995;689(2):189–196. doi: 10.1016/0006-8993(95)00568-b. [DOI] [PubMed] [Google Scholar]
- Ferchmin PA, Hao J, Perez D, Penzo M, Maldonado HM, Gonzalez MT, Rodriguez AD, de Vellis J. Tobacco cembranoids protect the function of acute hippocampal slices against NMDA by a mechanism mediated by alpha4beta2 nicotinic receptors. J Neurosci Res. 2005;82(5):631–641. doi: 10.1002/jnr.20666. [DOI] [PubMed] [Google Scholar]
- Ferchmin PA, Perez D, Biello M. Spermine is neuroprotective against anoxia and N-methyl-D-aspartate in hippocampal slices. Brain research. 2000;859(2):273–279. doi: 10.1016/s0006-8993(00)01973-9. [DOI] [PubMed] [Google Scholar]
- Ferchmin PA, Perez D, Eterovic VA, de Vellis J. Nicotinic Receptors Differentially Regulate N-Methyl-D-aspartate Damage in Acute Hippocampal Slices. J Pharmacol Exp Ther. 2003;305(3):1071–1078. doi: 10.1124/jpet.102.048173. [DOI] [PubMed] [Google Scholar]
- Fountain SB, Ting YL, Teyler TJ. The in vitro hippocampal slice preparation as a screen for neurotoxicity. Toxicol In Vitro. 1992;6(1):77–87. doi: 10.1016/0887-2333(92)90088-9. [DOI] [PubMed] [Google Scholar]
- Frazier CJ, Rollins YD, Breese CR, Leonard S, Freedman R, Dunwiddie TV. Acetylcholine activates an alpha-bungarotoxin-sensitive nicotinic current in rat hippocampal interneurons, but not pyramidal cells. J Neurosci. 1998;18(4):1187–1195. doi: 10.1523/JNEUROSCI.18-04-01187.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frere RC, Macdonald RL, Young AB. GABA binding and bicuculline in spinal cord and cortical membranes from adult rat and from mouse neurons in cell culture. Brain Res. 1982;244(1):145–153. doi: 10.1016/0006-8993(82)90912-x. [DOI] [PubMed] [Google Scholar]
- Gahring LC, Meyer EL, Rogers SW. Nicotine-induced neuroprotection against N-methyl-D-aspartic acid or beta-amyloid peptide occur through independent mechanisms distinguished by pro-inflammatory cytokines. J Neurochem. 2003;87(5):1125–1136. doi: 10.1046/j.1471-4159.2003.02074.x. [DOI] [PubMed] [Google Scholar]
- Giorgetti M, Bacciottini L, Giovannini MG, Colivicchi MA, Goldfarb J, Blandina P. Local GABAergic modulation of acetylcholine release from the cortex of freely moving rats. Eur J Neurosci. 2000;12(6):1941–1948. doi: 10.1046/j.1460-9568.2000.00079.x. [DOI] [PubMed] [Google Scholar]
- Hu M, Schurdak ME, Puttfarcken PS, El Kouhen R, Gopalakrishnan M, Li J. High content screen microscopy analysis of A beta 1-42-induced neurite outgrowth reduction in rat primary cortical neurons: neuroprotective effects of alpha 7 neuronal nicotinic acetylcholine receptor ligands. Brain research. 2007;1151:227–235. doi: 10.1016/j.brainres.2007.03.051. [DOI] [PubMed] [Google Scholar]
- Hu M, Waring JF, Gopalakrishnan M, Li J. Role of GSK-3beta activation and alpha7 nAChRs in Abeta(1-42)-induced tau phosphorylation in PC12 cells. J Neurochem. 2008;106(3):1371–1377. doi: 10.1111/j.1471-4159.2008.05483.x. [DOI] [PubMed] [Google Scholar]
- Johansson S, Druzin M, Haage D, Wang MD. The functional role of a bicuculline-sensitive Ca2+-activated K+ current in rat medial preoptic neurons. J Physiol. 2001;532(Pt 3):625–635. doi: 10.1111/j.1469-7793.2001.0625e.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson SW, Seutin V. Bicuculline methiodide potentiates NMDA-dependent burst firing in rat dopamine neurons by blocking apamin-sensitive Ca2+-activated K+ currents. Neurosci Lett. 1997;231(1):13–16. doi: 10.1016/s0304-3940(97)00508-9. [DOI] [PubMed] [Google Scholar]
- Kawamata J, Suzuki S, Shimohama S. Enhancement of nicotinic receptors alleviates cytotoxicity in neurological disease models. Therapeutic Advances in Chronic Disease. 2011 doi: 10.1177/2040622310397691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khawaled R, Bruening-Wright A, Adelman JP, Maylie J. Bicuculline block of small-conductance calcium-activated potassium channels. Pflugers Arch. 1999;438(3):314–321. doi: 10.1007/s004240050915. [DOI] [PubMed] [Google Scholar]
- Khiroug SS, Khiroug L, Yakel JL. Rat nicotinic acetylcholine receptor alpha2beta2 channels: comparison of functional properties with alpha4beta2 channels in Xenopus oocytes. Neuroscience. 2004;124(4):817–822. doi: 10.1016/j.neuroscience.2004.01.017. [DOI] [PubMed] [Google Scholar]
- Kihara T, Shimohama S, Sawada H, Honda K, Nakamizo T, Shibasaki H, Kume T, Akaike A. alpha 7 nicotinic receptor transduces signals to phosphatidylinositol 3-kinase to block A beta-amyloid-induced neurotoxicity. J Biol Chem. 2001;276(17):13541–13546. doi: 10.1074/jbc.M008035200. [DOI] [PubMed] [Google Scholar]
- Konopacki J, Golebiewski H. Theta-like activity in hippocampal formation slices: cholinergic-GABAergic interaction. Neuroreport. 1993;4(7):963–966. doi: 10.1097/00001756-199307000-00032. [DOI] [PubMed] [Google Scholar]
- Lawrence JJ. Cholinergic control of GABA release: emerging parallels between neocortex and hippocampus. Trends Neurosci. 2008;31(7):317–327. doi: 10.1016/j.tins.2008.03.008. [DOI] [PubMed] [Google Scholar]
- Liu Q, Zhang J, Zhu H, Qin C, Chen Q, Zhao B. Dissecting the signaling pathway of nicotine-mediated neuroprotection in a mouse Alzheimer disease model. Faseb J. 2007;21(1):61–73. doi: 10.1096/fj.06-5841com. [DOI] [PubMed] [Google Scholar]
- Mabley JG, Pacher P, Szabo C. Activation of the cholinergic antiinflammatory pathway reduces ricin-induced mortality and organ failure in mice. Mol Med. 2009;15(5–6):166–172. doi: 10.2119/molmed.2008.00105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin SE, de Fiebre NE, de Fiebre CM. The alpha7 nicotinic acetylcholine receptor-selective antagonist, methyllycaconitine, partially protects against beta-amyloid1-42 toxicity in primary neuron-enriched cultures. Brain research. 2004;1022(1–2):254–256. doi: 10.1016/j.brainres.2004.07.016. [DOI] [PubMed] [Google Scholar]
- Martins AH, Alves JM, Perez D, Carrasco M, Torres-Rivera W, Eterović VA, Ferchmin PA, Ulrich H. Kinin-B2 Receptor Mediated Neuroprotection after NMDA Excitotoxicity Is Reversed in the Presence of Kinin-B1 Receptor Agonists. PLoS ONE. 2012;7(2):e30755. doi: 10.1371/journal.pone.0030755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Materi LM, Semba K. Inhibition of synaptically evoked cortical acetylcholine release by intracortical glutamate: involvement of GABAergic neurons. Eur J Neurosci. 2001;14(1):38–46. doi: 10.1046/j.0953-816x.2001.01619.x. [DOI] [PubMed] [Google Scholar]
- McCartney MR, Deeb TZ, Henderson TN, Hales TG. Tonically active GABAA receptors in hippocampal pyramidal neurons exhibit constitutive GABA-independent gating. Molecular pharmacology. 2007;71(2):539–548. doi: 10.1124/mol.106.028597. [DOI] [PubMed] [Google Scholar]
- Monette R, Small DL, Mealing G, Morley P. A fluorescence confocal assay to assess neuronal viability in brain slices. Brain Res Brain Res Protoc. 1998;2(2):99–108. doi: 10.1016/s1385-299x(97)00020-2. [DOI] [PubMed] [Google Scholar]
- Moor E, DeBoer P, Westerink BH. GABA receptors and benzodiazepine binding sites modulate hippocampal acetylcholine release in vivo. Eur J Pharmacol. 1998;359(2–3):119–126. doi: 10.1016/s0014-2999(98)00642-6. [DOI] [PubMed] [Google Scholar]
- Morishita H, Miwa JM, Heintz N, Hensch TK. Lynx1, a cholinergic brake, limits plasticity in adult visual cortex. Science. 2010;330(6008):1238–1240. doi: 10.1126/science.1195320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nathan T, Jensen MS, Lambert JD. GABAB receptors play a major role in paired-pulse facilitation in area CA1 of the rat hippocampus. Brain research. 1990;531(1–2):55–65. doi: 10.1016/0006-8993(90)90757-3. [DOI] [PubMed] [Google Scholar]
- Olney JW, Collins RC, Sloviter RS. Excitotoxic mechanisms of epileptic brain damage. Adv Neurol. 1986;44:857–877. [PubMed] [Google Scholar]
- Pavlov VA, Tracey KJ. Controlling inflammation: the cholinergic anti-inflammatory pathway. Biochem Soc Trans. 2006;34(Pt 6):1037–1040. doi: 10.1042/BST0341037. [DOI] [PubMed] [Google Scholar]
- Quik M, Wonnacott S. alpha6beta2* and alpha4beta2* nicotinic acetylcholine receptors as drug targets for Parkinson’s disease. Pharmacological reviews. 2011;63(4):938–966. doi: 10.1124/pr.110.003269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raggenbass M, Bertrand D. Nicotinic receptors in circuit excitability and epilepsy. J Neurobiol. 2002;53(4):580–589. doi: 10.1002/neu.10152. [DOI] [PubMed] [Google Scholar]
- Roland JJ, Savage LM. Blocking GABA-A receptors in the medial septum enhances hippocampal acetylcholine release and behavior in a rat model of diencephalic amnesia. Pharmacol Biochem Behav. 2009;92(3):480–487. doi: 10.1016/j.pbb.2009.01.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schurr A, Payne RS, Heine MF, Rigor BM. Hypoxia, excitotoxicity, and neuroprotection in the hippocampal slice preparation. J Neurosci Methods. 1995a;59(1):129–138. doi: 10.1016/0165-0270(94)00203-s. [DOI] [PubMed] [Google Scholar]
- Schurr A, Payne RS, Rigor BM. Protection by MK-801 against hypoxia-, excitotoxin-, and depolarization-induced neuronal damage in vitro. Neurochem Int. 1995b;26(5):519–525. doi: 10.1016/0197-0186(94)00148-n. [DOI] [PubMed] [Google Scholar]
- Schurr A, Rigor BM. Cerebral ischemia revisited: new insights as revealed using in vitro brain slice preparations. Experientia. 1989;45(8):684–695. doi: 10.1007/BF01974560. [DOI] [PubMed] [Google Scholar]
- Small DL, Monette R, Chakravarthy B, Durkin J, Barbe G, Mealing G, Morley P, Buchan AM. Mechanisms of 1S,3R-ACPD-induced neuroprotection in rat hippocampal slices subjected to oxygen and glucose deprivation. Neuropharmacology. 1996;35(8):1037–1048. doi: 10.1016/s0028-3908(96)00028-7. [DOI] [PubMed] [Google Scholar]
- Sommer C, Fahrner A, Kiessling M. Postischemic neuroprotection in the ischemia-tolerant state gerbil hippocampus is associated with increased ligand binding to inhibitory GABA(A) receptors. Acta Neuropathol. 2003;105(3):197–202. doi: 10.1007/s00401-002-0632-7. [DOI] [PubMed] [Google Scholar]
- Soriano FX, Papadia S, Hofmann F, Hardingham NR, Bading H, Hardingham GE. Preconditioning Doses of NMDA Promote Neuroprotection by Enhancing Neuronal Excitability. 2006:4509–4518. doi: 10.1523/JNEUROSCI.0455-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ueda M, Iida Y, Kitamura Y, Kawashima H, Ogawa M, Magata Y, Saji H. 5-Iodo-A-85380, a specific ligand for alpha 4 beta 2 nicotinic acetylcholine receptors, prevents glutamate neurotoxicity in rat cortical cultured neurons. Brain Res. 2008;1199:46–52. doi: 10.1016/j.brainres.2007.10.107. [DOI] [PubMed] [Google Scholar]
- Vazquez J, Baghdoyan HA. GABAA receptors inhibit acetylcholine release in cat pontine reticular formation: implications for REM sleep regulation. J Neurophysiol. 2004;92(4):2198–2206. doi: 10.1152/jn.00099.2004. [DOI] [PubMed] [Google Scholar]
- Wang H, Yu M, Ochani M, Amella CA, Tanovic M, Susarla S, Li JH, Yang H, Ulloa L, Al-Abed Y, Czura CJ, Tracey KJ. Nicotinic acetylcholine receptor alpha7 subunit is an essential regulator of inflammation. Nature. 2003;421(6921):384–388. doi: 10.1038/nature01339. [DOI] [PubMed] [Google Scholar]
- Wang T, Raley-Susman KM, Wang J, Chambers G, Cottrell JE, Kass IS. Thiopental attenuates hypoxic changes of electrophysiology, biochemistry, and morphology in rat hippocampal slice CA1 pyramidal cells. Stroke. 1999;30(11):2400–2407. doi: 10.1161/01.str.30.11.2400. [DOI] [PubMed] [Google Scholar]
- Wei XW, Yan H, Xu B, Wu YP, Li C, Zhang GY. Neuroprotection of co-activation of GABA receptors by preventing caspase-3 denitrosylation in KA-induced seizures. Brain Res Bull. 2012;88(6):617–623. doi: 10.1016/j.brainresbull.2012.05.008. [DOI] [PubMed] [Google Scholar]
- Woodcock TM. Modulation of the Alpha-7 Nicotinic Acetylcholine Receptor Following Experimental Rat Brain Injury Improves Cellular and Behavioral Outcomes. Lexington: University of Kentucky; 2010. [Google Scholar]
- Xu J, Li C, Yin XH, Zhang GY. Additive neuroprotection of GABA A and GABA B receptor agonists in cerebral ischemic injury via PI-3K/Akt pathway inhibiting the ASK1-JNK cascade. Neuropharmacology. 2008;54(7):1029–1040. doi: 10.1016/j.neuropharm.2008.01.014. [DOI] [PubMed] [Google Scholar]
- Zhang ZW, Feltz P. Bicuculline blocks nicotinic acetylcholine response in isolated intermediate lobe cells of the pig. Br J Pharmacol. 1991;102(1):19–22. doi: 10.1111/j.1476-5381.1991.tb12125.x. [DOI] [PMC free article] [PubMed] [Google Scholar]