

Abstract
This study used video supplemented by parent-report data to describe mobility in girls and women with Rett syndrome (n=99) and to investigate the effects of age, genotype, scoliosis and hand stereotypies on mobility. Most subjects were able to sit, slightly less than half were able to walk and a minority were able to transfer without assistance. Factor analysis enabled the calculation of general mobility and complex motor skills scores. General mobility declined with age and was poorer in those who had surgically treated scoliosis, but was not significantly related to the presence of conservatively managed scoliosis. Complex motor skills were better in those without scoliosis. Those who had a p.R133C, p.R294X, p.R306C or C terminal deletion mutation had better complex motor skills when younger than 13 years, and better general mobility and complex motor skills when 13 years or older. Neither set of motor skills scores was related to the frequency of hand stereotypies. The factor analysis and strong correlation between factor and WeeFIM scores supported the validity of the video assessment. Motor impairment is a fundamental but variable component of the Rett syndrome phenotype. General motor abilities declined with age whilst complex motor skills were more dependent on genotype. This information is useful for the clinician and family when planning support strategies and interventions.
Keywords: Rett syndrome, gross motor function, mobility, MECP2 mutation, phenotype
1. INTRODUCTION
The onset of Rett syndrome (RTT) in early childhood is characterised by a gradual or sudden loss of speech and hand function followed by a slow decrease in acquired gross motor skills[1–3] with subsequent severe functional dependence.[4] RTT has an incidence of 1:8500 girls by the age of 15 years [5] and is associated primarily with mutations in the MECP2 gene.[6] Despite sharing neurological features, subjects with RTT display considerable clinical variability.[1, 4] With time, the effects of genotype on the presentation of RTT are becoming more obvious. Mutations affecting the nuclear localisation signal (NLS) of the transcription repression domain appear to lead to more severe [7, 8] and late truncating mutations to milder clinical presentations.[7, 9, 10] Clinicians caring for children with RTT need detailed information about the clinical course and neurodevelopmental consequences to assist with identification and management of key issues.[11] Families also need relevant information to help them understand and manage their child’s needs.
In cross-sectional studies of subjects with RTT various neurobehavioural domains have been described using the observations of experienced professionals during clinic visits[11, 12] or parent-report measures.[8, 13] These studies suggest that most can sit independently, [11] approximately half can walk, [11, 13, 14] motor abilities improve until adolescence and decline thereafter, [11, 12] and there are difficulties with the performance of transitional movements.[11–13] Whilst the current literature describes some motor skills in RTT, details on levels of assistance required for completion of these tasks are not clear. In particular, the association between RTT genotype and specific aspects of mobility has not been clarified. Further, information on the relationships between motor abilities and other features such as scoliosis, and hand stereotypies are not available. Thus, there is a need for additional analysis of the motor skills of girls and young women with RTT at different ages.
We recently developed a video assessment tool to assess multiple features of the RTT phenotype, including motor skills.[15] The video assessment tool was broadly based on activities of daily living as conceptualised in the WeeFIM measure.[16] In this study, we characterised in detail gross motor function in naturalistic settings in a series of girls and young women with RTT using the aforementioned tool. We evaluated variability of motor function in RTT, and its relationship with age and MECP2 mutation, and examined the associations between motor function and other features of RTT such as scoliosis, frequency of hand stereotypies and functional status (WeeFIM scores).
2. METHOD
The Australian Rett Syndrome Database (ARSD) is a population-based register of confirmed RTT cases with data collected from family and clinician questionnaires.[17] In 2004, all families whose children were subjects in the ARSD were invited to participate in the video study.[15] Families who agreed to participate were sent a filming protocol, a demonstration video, a blank video and a parent-report checklist. Activities to be recorded on video demonstrated multiple aspects of function in daily living. This paper describes the gross motor mobility items.
A physiotherapist (JD) coded the highest ability demonstrated for each item of sitting on the floor, sitting on a chair with and without a back, standing, walking, side stepping, turning 180°, walking along a slope, stepping over an obstacle, and running. Transfers included sitting to standing, standing up from the floor, and bending down to touch or pick up an object on the floor and returning to standing. Each item was coded as performed with no assistance; minimal assistance (a requirement for light support or the holding of one hand to perform the activity); moderate assistance (requiring two-hand or trunk support to perform the activity); or maximal assistance (requiring a composite of support to balance, transfer weight and move legs appropriately for the task or if unable to perform the skill). Information contained in the parent-report checklist was used to replace missing video data.[15]
The molecular techniques used to identify pathogenic mutations in the MECP2 gene have been described previously.[18] We initially categorised the genotype of subjects as those with no identified mutation; eight separate individual common mutations (ie those with a frequency in the study of ≥ 5 cases); and a combination of all remaining identified mutations. Because of the small sample size, for further analysis the individual common mutations were subsequently grouped into three categories according to clinical severity. Subjects with a R133C, R294X, R306C or C terminal deletion mutation were considered likely to be milder in severity;[7, 9, 10, 18–20] those with a R255X or R270X mutation to be the most severe;[7, 9, 14, 21] and those with a T158M or R168X mutation to be mixed in severity.[22]
The ARSD 2004 follow-up questionnaire was used as an additional data source [23] to describe scoliosis, hand stereotypies and overall functional status using WeeFIM ratings. The score developed by Kerr et al [24] and modified by Colvin et al [4] was used to describe the severity of scoliosis, with scores of 0 denoting no scoliosis, 1 denoting scoliosis without surgery, and 2 denoting scoliosis treated with surgery. The frequency of hand stereotypies was assessed on a 9-point Likert scale ranging from never to all the time. WeeFIM scores were used to describe independent function.[16]
Ethical approval for the study was provided by the Ethics Committee of the Women’s and Children’s Health Services in Western Australia.
Data analysis
Age was grouped into four categories representing the pre-school and early school years, primary school, adolescent and adult years respectively. Gamma tests of association were used to assess the relationship between age category and level of assistance required for each skill on data provided by the video and parent-report checklist. Missing data were imputed using a linear regression model [25] for factor analysis. Principal Components Analysis was performed to reduce the set of interrelated motor variables (15 sitting, standing, transfer and walking items) into a smaller number of independent variables with extraction of factors having eigenvalues greater than one. After varimax rotation, Z-scores for the extracted factors representing different aspects of mobility were calculated for each subject and the internal consistency of the factors was analysed with Cronbach’s α. The relationships between mobility scores and other variables were analysed with linear regression, Spearman correlation, t-test and ANOVA as appropriate. All analyses were undertaken (by AB and PJ) using Stata 9.[26]
3. RESULTS
Subjects
Video data were available for 99 girls/women (median age 14.1 years; range 1.5 – 27.9 years). Twenty-seven girls were younger than 8 years, 15 were 8<13 years and 28 were 13<19 years. There were 29 women ≥ 19 years. More subjects presented with classical (79.8%) than atypical RTT and a mutation was identified in 73 of the 96 (76.0%) subjects who had genetic testing (Table 1). The mean WeeFIM score 29.02 (95% CI 26.80, 31.24) out of a total possible score of 126. For thirty four (35.1%) subjects scoliosis was not reported, whilst 39 (40.2%) were reported to have conservatively managed scoliosis and 24 (24.7%) to have had surgically treated scoliosis.
Table 1.
Distribution of MECP2 mutations in Rett syndrome subjects
| Mutation | Number (%) |
|---|---|
| p.R133C | 5 (5.2%) |
| p.R294X | 10 (10.4%) |
| p.R306C | 4 (4.2%) |
| C terminal deletions | 7 (7.3%) |
| p.T158M | 6 (6.2%) |
| p.R168X | 8 (8.3%) |
| p.R255X | 5 (5.2%) |
| p.R270X | 7 (7.3%) |
| p.R106W | 3 (3.1%) |
| p.R306H | 2 (2.1%) |
| Large deletions | 5 (5.2%) |
| Other mutations | 11 (11.5%) |
| No mutation | 23 (24.0%) |
Description of motor skills
Performance on 15 motor skills was coded from the video observations and the parent-report checklist (with an average of over 70% of data provided from the video for each item).
Ability to sit
Most subjects could sit on the floor (74%), a chair with a back (62%) and on a stool (71%) with no assistance, and typically, performed the skill with no assistance or could not perform the skill at all. Sitting in a wheelchair was coded as moderate assistance. The level of assistance required to sit on the floor (p < 0.001) and sit on a stool (p = 0.01) increased with age-group.
Ability to stand
For 3, 10 and 20 seconds respectively, a small proportion (38%, 34% and 30%) were able to stand without assistance and 26%, 31% and 38% were unable to stand. A smaller proportion could stand with no assistance than could sit with no assistance, and a higher proportion could not stand at all than could not sit. The required level of assistance increased with age-group for standing for 3 seconds (p = 0.001) for 10 seconds (p = 0.005) and for 20 seconds (p = 0.07).
Ability to transfer
Only small proportions were able to transfer from sitting to standing (20%), bend down to touch the floor and return to standing (14%), and get up from the floor into standing (9%) respectively without assistance. Between a quarter and a third were unable to perform these transfers. Many could however be assisted to transfer from sit to stand or floor to stand, although assistance to bend to touch the floor and return to standing was infrequently seen. The required level of assistance increased with age-group for transferring from sitting to standing (p = 0.001), bending to touch the floor and returning to standing (p = 0.02) and being able to get up from the floor into standing (p < 0.001).
Ability to walk
Nearly half (43%) were able to walk independently and could also side step and turn 180 degrees. Under a third (30%) of the group was unable to walk. Approximately a quarter were able to walk up a slope or step over an obstacle without assistance but only 14% were able to run. The level of assistance required to walk (p = 0.02), side step (p = 0.01), turn 180° (p = 0.01), step over an obstacle (p = 0.002), walk along a slope (p = 0.03) and run (p = 0.001) increased with age-group.
Development of motor profiles
Principal Components Analysis of 15 items resulted in the extraction of two factors that accounted for 74.5% of total data variance (Table 2). Ten items describing sitting, standing, walking, side stepping, turning 180 degrees and sit to stand loaded strongly on factor one. This factor was named ‘General Mobility’ because it related to a range of mobility skills. Five items of moving from the floor to standing, picking up an object from the floor from standing, stepping over an obstacle, walking along a slope, and running loaded strongly on factor two. These motor skills are more difficult and the factor was named ‘Complex Motor Skills’. The Cronbach’s α coefficient for ‘General Mobility’ was 0.96 and for ‘Complex Motor Skills’ was 0.89.
Table 2.
Factor loadings for items included in the factor analysis
| Factor one | Factor two | |
|---|---|---|
| Sitting on the floor | .727 | .043 |
| Sitting on a chair | .805 | .193 |
| Sitting on a stool | .688 | .183 |
| Transfer sitting to standing | .669 | .511 |
| Standing 3 seconds | .834 | .453 |
| Standing 10 seconds | .831 | .380 |
| Standing 20 seconds | .673 | .404 |
| Walking | .801 | .520 |
| Side stepping | .724 | .577 |
| Turning 180 degrees | .752 | .562 |
| Transfer floor to standing | .452 | .684 |
| Bending to pick up object from floor and returning to stand | .144 | .838 |
| Stepping over an obstacle | .538 | .709 |
| Walking up or down a slope | .583 | .689 |
| Running | .120 | .851 |
General mobility and complex motor skills factor scores
For each skill, the need for no assistance was scored 3 while maximal assistance or inability to perform the skill was scored 0, and Z-scores for each factor are described in Table 3. General mobility scores in girls who were younger than 13 years were better than those 13 years or older (p = 0.007), but there was no significant difference between the complex motor skills scores of girls who were younger than 13 years compared with those who were 13 years or older (p = 0.34).
Table 3.
General mobility and complex motor skills Z-scores by age-group
| < 8 years (n = 27) | 8 < 13 years (n = 15) | 13 < 19 years (n = 28) | ≥ 19 years (n = 29) | |
|---|---|---|---|---|
| General Mobility | ||||
| Mean ± SEM | 0.25 ± 0.15 | 0.40 ± 0.28 | −0.04 ± 0.18 | −0.40 ± 0.21 |
| Complex Motor Skills | ||||
| Mean ± SEM | 0.12 ±0.23 | 0.10 ± 0.23 | −0.08 ± 0.21 | −0.09 ± 0.14 |
General mobility scores of women 19 years or older were lower than of girls younger than 8 years (p = 0.01) or between 8 and 13 years (p = 0.03). The scores of girls younger than 8 years were not significantly different from those between 8 and 13 years (p = 0.60) or between 13 and 19 years (p = 0.21), and scores of girls between 13 and 19 years were not significantly different from women 19 years or older (p = 0.19). There was no significant difference in the complex motor skills score across the age-groups (p = 0.61).
Relationship between motor profiles and genotype
General mobility and complex motor skills Z-scores corrected for age are shown for the main individual mutations (Figures 1a and 1b) and for the mutation groups (Figures 2a, 2b, 2c and 2d). Those with individual mutations p.R133C, p.R294X or p.R255X have better motor abilities overall than those with p.R270X or large deletions (Figures 1a and 1b).
Figure 1.

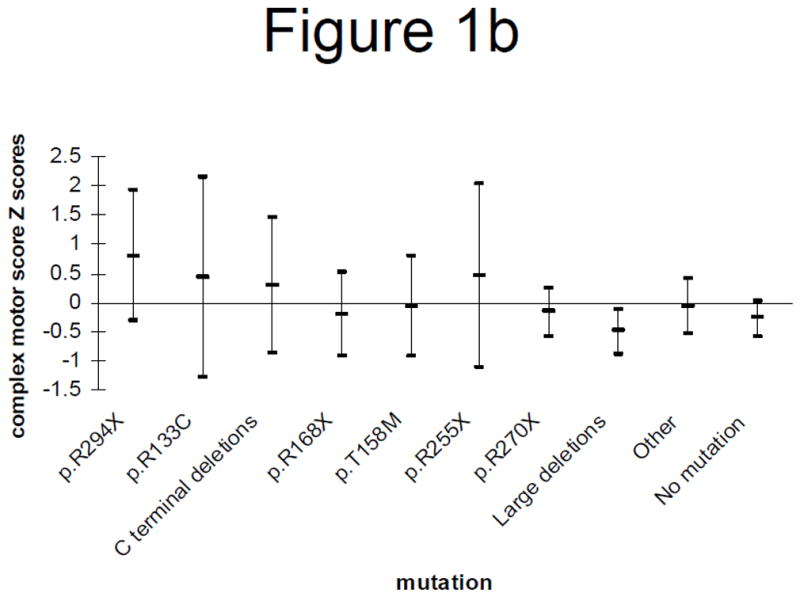
Figure 1a: Age-adjusted general mobility Z-scores (mean and 95% confidence interval) by genotype
Figure 1b: Age-adjusted complex motor skills Z-scores (mean and 95% confidence interval) by genotype
Figure 2.

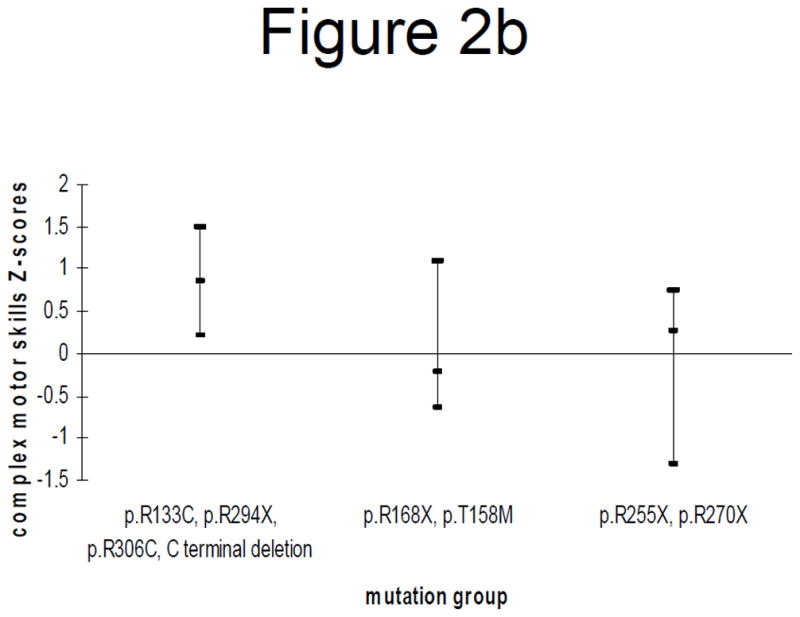

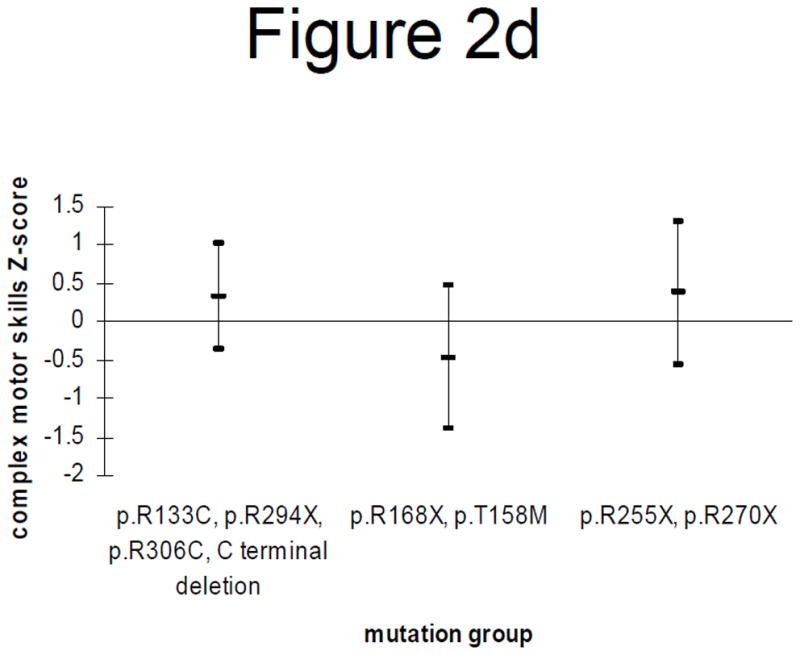
Figure 2a: General mobility Z-scores (mean and 95% confidence interval) by mutation category in those < 13 years
Figure 2b: Complex motor skills Z-scores (mean and 95% confidence interval) by mutation category in those < 13 years
Figure 2c: General mobility Z-scores (mean and 95% confidence interval) by mutation category in those > 13 years
Figure 2d: Complex motor skills Z-scores (mean and 95% confidence interval) by mutation category in those > 13 years
Grouping of mutations and stratification by age produced the following results. In individuals younger than 13 years, those with p.R133C, p.R294X, p.R306C or C terminal deletions had significantly better complex motor skills than those with p.R255X or p.R270X (p = 0.02) but there were no significant differences in relation to the general motor skills (Figures 2a and 2b). However in individuals 13 years or older, those with p.R133C, p.R294X, p.R306C or C terminal deletions had significantly better general mobility than those with p.R255X or p.R270X (p = 0.03). They also had better complex motor skills than those with p.R168X or p.T158M (p = 0.06), as did those with p.R255X or p.R270X (p = 0.05) (Figures 2c and 2d).
Relationships between mobility profiles and other factors
After adjusting for age, general mobility scores in those who had scoliosis surgery were significantly worse compared to those without scoliosis (p < 0.001) but not compared to those with conservatively managed scoliosis (p = 0.10). In contrast, complex motor skills were significantly worse both in the presence of conservatively treated scoliosis (p<0.001) and surgically treated scoliosis (p = 0.001) compared to those without scoliosis.
There was no significant correlation between the frequency of hand stereotypies and the general mobility score (rs = −0.167, p = 0.11) or the complex motor skills score (rs = 0.123, p = 0.24). However, poorer WeeFIM scores correlated with poorer general mobility (rs = 0.494, p < 0.001) and poorer complex motor skills (rs = 0.328, p = 0.001) scores.
4. DISCUSSION
Using a video assessment tool, motor function was examined in a large cohort of girls/women with RTT. Most subjects were able to sit, slightly less than half were able to walk or stand and a minority were able to transfer without assistance. In general, the level of assistance required increased with age and a higher percentage could achieve standing or walking with assistance than could transfer with assistance. Consistent with other literature, [2, 11, 13, 14] a broad range of abilities was found. It appears that basic postural control (sitting balance) is a strength in RTT, that some individuals walk despite voluntary motor control difficulties, and that difficulty in changing positions reflects the dramatic impact that RTT has on carrying out complex movements.
The use of factor analysis enabled us to develop two distinct gross motor profiles and to examine relationships between these profiles and other variables. Of particular interest is our finding that complex motor skills did not decrease with age. It is possible that these skills are either retained over time or because these scores are so low, there may be a floor effect and changes with age were not detectable. General mobility skills were poorer in those who had had scoliosis surgery but were not significantly related to the presence of conservatively managed scoliosis. General mobility skills were best for those with p.R294X mutation and close to significantly different from those with p.R270X or large deletions. Complex motor skills were better in those without scoliosis and in those with a C terminal deletion or with a p.R133C, p.R294X, p.R306C, or, unexpectedly, a R255X mutation. Both general mobility and complex motor skills were unrelated to the frequency of hand stereotypies.
The video footage collected for this study allowed us to examine motor function over a range of ages, clinical and genotypic variability. Our sample was sourced from a population-based register and was broadly representative of the Australian RTT population, [15] therefore minimising bias against particular groups. The number of subjects was of an equivalent order to previously performed studies.[11–14] The assessed items were comprehensive and relevant to daily living. By including detail of motor function not previously described, we have been better able to characterise the motor phenotype. Further, factor analysis enabled the calculation of composite motor scores that facilitated the investigation of relationships with other variables. The factor analysis supported the content and construct validity, and the internal reliability of our previously developed mobility profile, [15] and the strong correlation between the factor scores and WeeFIM scores supports its concurrent validity.
Despite the aforementioned strengths, there was limited power to be able to analyse specific mutations individually and we elected to group them into categories of severity based on previous literature. Because of the cross-sectional nature of the study, there is the possibility of bias towards survivors with the most severely affected dying earlier. Ideally, the progression of a disorder is best captured through longitudinal data. Therefore, our study may have provided a slightly more optimistic profile of the population than actually exists.
Using the Chailey sitting levels which measures sitting abilities on the floor and on a box (equivalent to our sitting on a stool), Cass et al [11] found that 82.5% of their group could sit independently. However they did not specify the exact sitting position used so that our findings may or may not be consistent with theirs. Our finding that slightly less than half of the subjects could walk with no assistance was however in keeping with their results.[11] A smaller proportion of our subjects could stand with no assistance compared with walking, representing the subjects who walked on their toes and, although upright, were unable to stand on a stable base of support. Not previously described in the literature, fewer girls/women could walk on a slope, step over an obstacle or run, skills which require greater planning, balance and co-ordination.
The difficulties with transitions that we identified may illustrate the motor impairment of dyspraxia as well as altered muscle tone and poor balance and coordination. [27] It is likely that dyspraxia becomes more evident with complex tasks such as transitions, and such difficulties are likely to represent an important burden to families. The majority of children require assistance and some require lifting when moving to a different room, getting in and out of a chair at mealtime and getting up from the floor to a chair or to an upright position. Further, poor hand function in RTT precludes self-operation of a wheel chair. Cass et al [11] found approximately 62% could sit to stand with light or no assistance and the discrepancy with our findings (33.7%) could illustrate the differences between clinical assessment and performance in the home setting. Larsson et al [13] found that nearly half of their group of girls and adults could complete the transitional movement of sitting to standing, [13] but the level of assistance was not specified. While there is some inconsistency in the literature, it is clear that transitional movements are difficult for girls/women with RTT, even in those who retain walking skills. Rehabilitation of transitional skills has been recommended [28, 29] and systematic examination of this treatment could represent a promising area for future intervention.
Consistent with the findings of Cass et al [11] and the described stages of RTT, [2] general mobility decreased with increasing age. Additional to previous studies, this study assessed complex motor skills and found that abilities to perform these skills did not deteriorate with age. For example, if a child can bend to touch the floor or run, then they might well retain those skills through to adulthood. This would be important information for families. However, the caveat is that our study, like those to which we previously referred, is cross-sectional. The findings should be confirmed in a prospective study with video data collected longitudinally.
Although it was difficult to assess for statistically significant differences between individual mutations because of power limitations, mutations p.R294X and p.R133C had the highest scores both for the general and complex domains. When we grouped these two along with p.R306C and C terminal deletions, the complex motor scores for this “mild” group were significantly better than for the “severe” group (the combination of p.R255X and p.R270X) in those younger than 13 years. However in the older group, it was with general mobility that the “milder” group demonstrated better scores than the most “severe” group. With relation to complex motor skills, the “severe” group fared better than expected. In this analysis, because of small numbers, we based our criteria for assignment of severity on the accumulated body of knowledge relating to genotype phenotype relationships in RTT.[30] The mutation p.R133C has already been shown to be a mild mutation in a number of studies.[9, 18, 20, 31] Our own group would be one of the first to have demonstrated a similar milder phenotype with p.R294X.[7] However, what we had previously noted was that gross motor function is a particular strength for those with this mutation and that there would appear to be a protective effect against the development of scoliosis.[32] Schanen et al., [10] Charman et al.[9] and Kerr et al.[20] have demonstrated the milder effects with p.R306C and in describing the profile of the C terminal mutations, Smeets et al.[19] have shown that they also have a mild phenotype. Thus, this study is showing that when these particular mutations (or groups of mutations) are combined together, their profile for complex motor skills is better than for the combination of p.R255X and p.R270X in girls younger than 13 years. Because the two mutations in the NLS region have generally been deemed to be severe [8] we grouped them together. Interestingly, we found that when viewed individually p.R255X actually had reasonably good scores. We also showed that in those 13 years and over, those we had assigned to the most “severe” group had better complex skills than those in the “mixed” group. It is possible that in the older group we are seeing a survivor effect with those most severely affected by NLS mutations already having died.[33] It did require considerable effort and a major contribution from families to obtain video material filmed according to a specific protocol on almost 100 Australian cases. However when attempting to disentangle the effects of individual mutations an even larger sample would be preferable.
Those who had surgery for scoliosis had lower motor scores although we do not know if the poorer motor skills preceded or followed the surgical procedure, and longitudinal data is required to determine any relationships. The characteristic of hand stereotypies can be dramatic in RTT and it is reassuring that this behaviour does not affect the development of motor skills.
Our descriptive and exploratory study has provided a rich bank of information, and we recommend that this be used as a base for the development of interventions to improve mobility and quality of life in RTT. Strategies that assist families in facilitating transitions or promote walking abilities using a modified treadmill could prove fruitful.[34] Gross motor skills are currently represented in the general severity scales by Kerr, [24] Pineda [35] and Percy [10] by one, two and three items respectively. It is possible that additional or alternative description could provide greater sensitivity to phenotypic variation in those instruments.
The use of video material was a novel aspect of this study. In future research, we plan to investigate the relationships between motor abilities and the other domains illustrated on the videotapes including feeding, communication and hand function. With the inclusion of data relating to genetic characteristics, this will have the potential for greater characterisation and understanding of RTT.
Acknowledgments
SOURCES OF SUPPORT:
NIH grant (HD24448) (W.E.K.).
The authors would like to acknowledge the funding of the video component of Australian Rett Syndrome program by the National Medical and Health Research Council (NHMRC) under project grant 303189 and the National Institutes of Health (1 R01 HD43100-01A1) for funding the major aspects of the program. HL is funded by NHMRC program grant 353514. Special thanks to Carol Philippe who was responsible for much of the video data collection. We would also like to acknowledge the molecular work of Linda Weaving and Sarah Williamson (under the guidance of Professor John Christodoulou and Dr Bruce Bennetts) in Sydney and Mark Davis in Perth. We would especially like to express our sincere gratitude to all the families who have contributed to the study in providing video material and completing questionnaires; the Australian Paediatric Surveillance Unit (APSU) and the Rett Syndrome Association of Australia who facilitated case ascertainment in Australia. The APSU is a Unit of the Division of Paediatrics, Royal Australasian College of Physicians and is funded by the Department of Health and Ageing and the Faculty of Medicine of the University of Sydney.
Contributor Information
Jennepher Anne DOWNS, Telethon Institute for Child Health Research, Centre for Child Health Research, The University of Western Australia, West Perth, Western Australia.
Ami BEBBINGTON, Telethon Institute for Child Health Research, Centre for Child Health Research, The University of Western Australia, West Perth, Western Australia.
Peter JACOBY, Telethon Institute for Child Health Research, Centre for Child Health Research, The University of Western Australia, West Perth, Western Australia.
Michael MSALL, University of Chicago Comer and LaRabida Children’s Hospitals, Kennedy Center, Institute of Molecular Pediatric Sciences & Section of Developmental and Behavioral Pediatrics, 5841 S. Maryland Ave., MC0900, Chicago, Illinois 60637.
Orla MCILROY, Telethon Institute for Child Health Research, Centre for Child Health Research, The University of Western Australia, West Perth, Western Australia.
Sue FYFE, School of Public Health, Curtin University of Technology, Perth, Western Australia.
Nadia BAHI-BUISSON, Pediatric Neurology, Necker Enfants Malades Hospital Paris, F-75015 France - University Paris V Rene Descartes Paris F-75005 France, Inserm, U663, Paris, F-75015 France; University Rene Descartes, Paris V, F-75005.
Walter E. KAUFMANN, Center for Genetic Disorders of Cognition & Behavior, Kennedy Krieger Institute and Johns Hopkins University School of Medicine, Baltimore, Maryland, USA.
Helen LEONARD, Telethon Institute for Child Health Research, Centre for Child Health Research, The University of Western Australia, West Perth, Western Australia.
References
- 1.Hagberg B. Rett syndrome: Long-term clinical follow-up experiences over four decades. J Child Neurol. 2005;20:722–7. doi: 10.1177/08830738050200090401. [DOI] [PubMed] [Google Scholar]
- 2.Hagberg B, Romell M. Rett females: Patterns of characteristic side-asymmetric neuroimpairments at long-term follow-up. Neuropediatrics. 2002;33:324–6. doi: 10.1055/s-2002-37083. [DOI] [PubMed] [Google Scholar]
- 3.Witt Engerstrom I. Rett syndrome: A retrospective pilot study on potential early predictive symptomatology. Brain Dev. 1987;9:481–6. doi: 10.1016/s0387-7604(87)80069-4. [DOI] [PubMed] [Google Scholar]
- 4.Colvin L, Fyfe S, Leonard S, Schiavello T, Ellaway C, de Klerk N, et al. Describing the phenotype in Rett syndrome using a population database. Arch Dis Child. 2003;88:38–43. doi: 10.1136/adc.88.1.38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Laurvick CL, de Klerk N, Bower C, Christodoulou J, Ravine D, Ellaway C, et al. Rett syndrome in Australia: a review of the epidemiology. J Pediatr. 2006;148:347–52. doi: 10.1016/j.jpeds.2005.10.037. [DOI] [PubMed] [Google Scholar]
- 6.Amir RE, Van den Veyver IB, Wan M, Tran CQ, Francke U, Zoghbi HY. Rett syndrome is caused by mutations in X-linked MECP2, encoding methyl-CpG-binding protein 2. Nat Genet. 1999;23:185–8. doi: 10.1038/13810. [DOI] [PubMed] [Google Scholar]
- 7.Colvin L, Leonard H, de Klerk N, Davis M, Weaving L, Williamson S, et al. Refining the phenotype of common mutations in Rett syndrome. J Med Genet. 2004;41:25–30. doi: 10.1136/jmg.2003.011130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Huppke P, Held M, Handefeld F, Engel W, Laccone F. Influence of Mutation Type and Location on Phenotype in 123 Patients with Rett Syndrome. Neuropediatrics. 2002;33:63–8. doi: 10.1055/s-2002-32365. [DOI] [PubMed] [Google Scholar]
- 9.Charman T, Neilson TCS, Mash V, Archer H, Gardiner MT, Knudsen GPS, et al. Dimensional phenotypic analysis and functional categorisation of mutations reveal novel genotype-phenotype associations in Rett syndrome. Eur J Hum Genet. 2005;13:1121–30. doi: 10.1038/sj.ejhg.5201471. [DOI] [PubMed] [Google Scholar]
- 10.Schanen C, Houwink EJ, Dorrani N, Lane J, Everett R, Feng A, et al. Phenotypic manifestations of MECP2 mutations in classical and atypical Rett syndrome. Am J Med Genet A. 2004;126A:129– 40. doi: 10.1002/ajmg.a.20571. [DOI] [PubMed] [Google Scholar]
- 11.Cass H, Reilly S, Owen L, Wisbeach A, Weekes L, Slonims V, et al. Findings from a multidisciplinary clinical case series of females with Rett syndrome. Dev Med Child Neurol. 2003;45:325–37. doi: 10.1017/s0012162203000616. [DOI] [PubMed] [Google Scholar]
- 12.Bashina VM, Simashkova NV, Grachev VV, Gorbchevskaya NL. Speech and motor disturbances in Rett syndrome. Neurosci Behav Physiol. 2002;32:323–7. doi: 10.1023/a:1015886123480. [DOI] [PubMed] [Google Scholar]
- 13.Larsson G, Lindstrom B, Witt Engerstrom I. Rett syndrome from a family perspective: The Swedish Rett Center survey. Brain Dev. 2005;27:S14–S9. doi: 10.1016/j.braindev.2005.03.015. [DOI] [PubMed] [Google Scholar]
- 14.Huppke P, Held M, Laccone F, Hanefeld F. The spectrum of phenotypes in females with Rett Syndrome. Brain Dev. 2003;25:346–51. doi: 10.1016/s0387-7604(03)00018-4. [DOI] [PubMed] [Google Scholar]
- 15.Fyfe S, Downs J, McIlroy O, Burford B, Lister J, Reilly S, et al. Development of a video-based evaluation tool in Rett syndrome. J Autism Dev Disord. doi: 10.1007/s10803-006-0293-9. epub ahead of print. [DOI] [PubMed] [Google Scholar]
- 16.Msall ME, DiGaudio K, Rogers BT, LaForest S, Catanzaro NL, Campbell J, et al. The Functional Independence Measure for Children (WeeFIM). Conceptual basis and pilot use in children with developmental disabilities. Clin Pediatr. 1994;33:421–30. doi: 10.1177/000992289403300708. [DOI] [PubMed] [Google Scholar]
- 17.Leonard H, Bower C, English D. The prevalence and incidence of Rett syndrome in Australia. Eur Child Adolesc Psychiatr. 1997;6:8–10. [PubMed] [Google Scholar]
- 18.Leonard H, Colvin L, Christodoulou J, Schiavello T, Williamson S, Davis M, et al. Patients with the R133C mutation: Is their phenotype different from patients with Rett syndrome with other mutations? J Med Genet. 2003;40:e52. doi: 10.1136/jmg.40.5.e52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Smeets E, Terhal P, Casaer P, Peters A, Midro A, Schollen E, et al. Rett syndrome in females with CTS hot spot deletions: a disorder profile. Am J Med Genet A. 2005;132:117–20. doi: 10.1002/ajmg.a.30410. [DOI] [PubMed] [Google Scholar]
- 20.Kerr AM, Prescott RJ. Predictive value of the early clinical signs in Rett disorder. Brain Dev. 2005;27 (Suppl 1):S20–4. doi: 10.1016/j.braindev.2004.10.007. [DOI] [PubMed] [Google Scholar]
- 21.Auranen M, Vanhala R, Vosman M, Levander M, Varilo T, Hietala M, et al. MECP2 gene analysis in classical Rett syndrome and in patients with Rett-like features. Neurology. 2001;56:611–7. doi: 10.1212/wnl.56.5.611. [DOI] [PubMed] [Google Scholar]
- 22.Archer HL, Evans J, Leonard H, Colvin L, Ravine D, Christodoulou J, et al. Correlation between clinical severity in Rett syndrome patients with a p.R168X or p. T158M MECP2 mutation and the direction and degree of skewing of X chromosome inactivation. J Med Genet. 2006;140A:691–4. doi: 10.1136/jmg.2006.045260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jian L, Nagarajan L, de Klerk N, Ravine D, Bower C, Anderson A, et al. Predictors of seizure onset in Rett syndrome. J Pediatr. 2006;149:542–7. doi: 10.1016/j.jpeds.2006.06.015. [DOI] [PubMed] [Google Scholar]
- 24.Kerr AM, Nomura Y, Armstrong D, Anvret M, Belichenko PV, Budden S, et al. Guidelines for reporting clinical features in cases with MECP2 mutations. Brain Dev. 2001;23:208–11. doi: 10.1016/s0387-7604(01)00193-0. [DOI] [PubMed] [Google Scholar]
- 25.Tabachnik BG, Fidell LS. Using Multivariate Statistics. 3. New York: Harper Collins College Publishers; 1996. [Google Scholar]
- 26.StataCorp. Stata Statistical Software. College Station, Texas: Stata Corporation; 2005. 9.0 R, editor. [Google Scholar]
- 27.Hagberg B. Clinical manifestations and stages of Rett syndrome. Ment Retard Dev Disabil Res Rev. 2002;8:61–5. doi: 10.1002/mrdd.10020. [DOI] [PubMed] [Google Scholar]
- 28.Hanks SB. Motor disabilities in the Rett syndrome and physical therapy strategies. Brain Dev. 1990;12:157–61. doi: 10.1016/s0387-7604(12)80201-4. [DOI] [PubMed] [Google Scholar]
- 29.Larsson G, Witt Engerstrom I. Gross motor ability in Rett syndrome - the power of expectation, motivation and planning. Brain Dev. 2001;23:S77–S81. doi: 10.1016/s0387-7604(01)00334-5. [DOI] [PubMed] [Google Scholar]
- 30.Ham AL, Kumar A, Deeter R, Schanen C. Does genotype predict phenotype in Rett syndrome. J Child Neurol. 2005;20:768–78. doi: 10.1177/08830738050200091301. [DOI] [PubMed] [Google Scholar]
- 31.Yamashita Y, Kondo I, Fukuda T, Morishima R, Kusaga A, Iwanaga R, et al. Mutation analysis of the methyl-CpG-binding protein 2 gene (MECP2) in Rett patients with preserved speech. Brain Dev. 2001;23 (Suppl 1):S157–60. doi: 10.1016/s0387-7604(01)00378-3. [DOI] [PubMed] [Google Scholar]
- 32.Ager S, Fyfe S, Christodoulou J, Jacoby P, Schmitt L, Leonard H. Predictors of scoliosis in Rett syndrome. J Child Neurol. 2006;21:809–13. doi: 10.1177/08830738060210091501. [DOI] [PubMed] [Google Scholar]
- 33.Jian L, Archer HL, Ravine D, Kerr A, de Klerk N, Christodoulou J, et al. p. R270X MECP2 mutation and mortality in Rett syndrome. Eur J Hum Genet. 2005;13:1235–8. doi: 10.1038/sj.ejhg.5201479. [DOI] [PubMed] [Google Scholar]
- 34.Lotan M, Isakov E, Merrick J. Improving functional skills and physical fitness in children with Rett syndrome. J Intellect Disabil Res. 2004;48:730–5. doi: 10.1111/j.1365-2788.2003.00589.x. [DOI] [PubMed] [Google Scholar]
- 35.Monros E, Armstrong J, Aibar E, Poo P, Canos I, Pineda M. Rett syndrome in Spain: mutation analysis and clinical correlations. Brain Dev. 2001;23 (Suppl 1):S251–3. doi: 10.1016/s0387-7604(01)00374-6. [DOI] [PubMed] [Google Scholar]