

Abstract
Purpose
17-(demethoxy), 17-allylamino geldanamycin (17-AAG) suppresses growth in some cancers by inhibiting Heat shock protein 90 (Hsp90). We examined the effects of 17-AAG-mediated Hsp90 inhibition on human hepatocellular carcinoma (HCC) growth in vitro and in vivo.
Methods
Human HCC cell lines, Hep3B and HuH7, were exposed to 17-AAG and cell viabilities and apoptosis were determined. Cell cycle profiles were analyzed and the G2/M cell cycle checkpoint proteins cdc2 and cyclin B1 were examined. Studies were performed to determine whether 17-AAG-mediated cdc2 decrease was due to altered gene expression, transcription, or protein degradation. The effects of 17-AAG on Hep3B and HuH7 xenograft growth in athymic nude mice were also examined.
Results
Hep3B and HuH7 treated with 17-AAG versus untreated controls showed decreased cell viability and increased apoptosis. Cells treated with 17-AAG also showed an increased fraction in G2/M phase and an associated decrease in cdc2 through protein degradation rather than through other mechanisms. Hsp90 inhibition by 17-AAG also decreased HCC xenograft growth in association with decreased cdc2 expression.
Conclusions
17-AAG-mediated inhibition of Hsp90 abrogates human HCC cell growth in vitro and in vivo through cdc2 decrease, which in turn induces G2/M cell cycle arrest and apoptosis. Hsp90 is a mediator of HCC growth and survival and its inhibition may serve as a potential treatment.
Keywords: Hepatocellular cancer, Hsp90, 17-AAG, Xenograft, cdc2
Introduction
Hepatocellular carcinoma (HCC) is the third most common cancer worldwide and over 5,00,000 people per year succumb to this late-presenting and difficult-to-treat cancer [1]. As only 10–20% of HCC cases can be cured by surgical means, effective non-surgical treatments are desperately needed. The development of new therapies requires a better understanding and novel approach to hepatocarcinogenesis at the molecular level.
One such approach may involve investigation of molecular chaperones such as heat shock protein 90 (Hsp90), a crucial mediator of cellular trafficking of proteins [2]. Hsp90 is constitutively expressed, makes up 1–2% of the cellular protein, and can be upregulated during stress [3]. Initial studies have shown that Hsp90 is strongly expressed in human HCC in both cell lines and patient specimens [4, 5]. Hsp90 forms complexes with co-chaperones (Hsp70, Hsp40, CDC37/p50, p23, AHA1) and accessory molecules [immunophilin, Hsp-interacting protein (HIP) and Hsp-organizing protein (HOP)] to stabilize client proteins [6]. The Hsp90 chaperone complex acts on client proteins to: (1) prevent protein aggregation, (2) facilitate crossing of cell membranes, (3) stabilize conformations for further activation (ie. ligand binding, phosphorylation, or incorporation into signaling complexes), and (4) target clients for degradation. Many of these client proteins are conformationally labile signaling molecules involved in cell growth and survival [7].
Increased chaperone activity in cancer is a stress response to the peri-tumoral conditions of acidosis, hypoxia, and nutrient-deprivation [8]. Interactions with client proteins enable Hsp90 to promote cancer cell growth by supporting proliferative or anti-apoptotic mechanisms [9]. In some cancers such as breast and ovarian, Hsp90 has been shown to support cell growth and survival through enhanced cell cycle efficiency and subsequent proliferation [8, 10]. Hsp90 protein expression is increased in some tumors as compared to corresponding normal tissue such as glioblastomas, breast cancer and lung cancer [11–13].
To date, few studies have investigated the role of Hsp90 in human HCC. In one study analyzing tissue samples of HCC and dysplastic nodules in patients with active hepatitis B virus, the expression of Hsp90 was increased. In addition, Hsp90, GRP78 and GRP94 expression was associated with aggressive HCC tumor characteristics such as vascular invasion and intrahepatic metastases [14]. Others have also shown increased Hsp90 expression in HCC tumors with poorer prognosis [15].
Geldanamycin is a naturally occurring benzoquinone ansamycin that specifically inhibits Hsp90 function by binding the N-terminal ATP binding domain [16]. Because geldanamycin and related semi-synthetic molecules bind the N-terminus of Hsp90 with greater affinity than ATP, these inhibitors prevent Hsp90 from cycling between its ADP-and ATP-bound conformations. 17-(demethoxy), 17-allylamino geldanamycin (17-AAG) is a less toxic analog of geldanamycin with enhanced efficacy against Hsp90 [17]. In some cancer cells, 17-AAG facilitates the degradation of client proteins that mediate proliferation, cell cycle progression and survival. As a consequence of degradation of pro-growth and survival proteins, these cells develop apoptosis by inhibiting oncogenic proteins such as N-ras, ki-ras, Akt, and p185erbB2 [18]. However, the potential effects of 17-AAG on human HCC cells has not been examined.
Although the inhibition of ubiquitous proteins such as Hsp90 raises concerns of side effects, this protein is well suited for targeting due to its increased expression and 100-fold greater affinities for the inhibitor in cancer cells [19, 20]. In this study, we showed that Hsp90 inhibition by 17-AAG decreased human HCC cell growth both in vitro and in vivo by decreasing cdc2, which in turn induced both G2/M cell cycle arrest and apoptosis. These data delineate an important role of Hsp90 in promoting human HCC growth and show that 17-AAG is an effective anti-cancer compound in the liver.
Materials and methods
Chemicals
17-allylamino-17demethoxygeldanamycin (17-AAG) (Sigma, St. Louis, MO) was dissolved in dimethylsulphoxide (DMSO) at a range of concentrations to determine IC50’s at 72 h of treatment for each cell line. Cycloheximide (10 μg/mL), lactacystin (1–10 μM), 3-methyladenine (10 mM) [21], roscovitine (50 μM) [22] and actinomycin D (10 μM) were purchased from Sigma and each dissolved in DMSO.
Cell culture
Human hepatocellular cancer cell lines Hep3B and HuH7 were cultured in Dulbecco’s modified Eagle’s medium (DMEM) (Mediatech, Herndon, VA) supplemented with 10% heat-inactivated fetal bovine serum (Mediatech) and penicillin-streptomycin solution (Mediatech) at 37°C in 5% CO2 atmosphere. For all experiments, the cells were plated overnight at initial cell densities as follows: 3 × 105 cells/60 mm culture dish, 1 × 105 cells/well for 6-well plates and 5 × 103 cells/well for 96-well plates. HCC cells were then incubated with fresh medium containing either DMSO alone or 17-AAG. In separate cells were pre-treated with cycloheximide, lactacystin, or 3-methyladenine (3-MA) for 20 min before 17-AAG treatment.
Cell viability and toxicity assay
Cells were initially cultured in 96-well microplates overnight, then incubated with 100 μl of fresh medium containing DMSO vehicle alone or 17-AAG at concentrations ranging from 1 nM to 10 μM for 72 h to calculate 50% inhibitory concentration values (IC50) for cell viability at 72 h. The cell viability and toxicity were measured by MTT assay, in which cells were incubated with 10 μl of 3-(4,5-dimethyl-thiazole-2-yl)-2,5-biphenyl tetrazolium (MTT, Sigma) dissolved in PBS (5 mg/ml) for 4 h and then exposed to 100 μl of 0.4 N HCl in isopropanol. MTT incorporation in viable cells was then measured with a microplate reader (Molecular Devices Corporation, Sunnyvale, CA) at 570 nm, and results were shown as ratios of viability in the treated over control (DMSO alone) groups [23]. In a separate set of experiments, dose and time-dependent responses were determined by incubating Hep3B and HuH7 cells in DMSO vehicle alone or 17-AAG at IC50’s for 24, 48 and 72 h, then expressed as relative cell viabilities as above.
Direct cell counting
Cells were incubated in six-well culture dishes overnight, then incubated with fresh medium containing DMSO vehicle alone or 17-AAG at IC50’s for 24, 48, and 72 h. Floating cells were collected from media, centrifuged at 600g, and counted by light microscopy using a hemacytometer. Adherent cells were separately resuspended after Trypsin (Mediatech) digestion, counted as above, and viabilities were determined by trypan blue (Sigma) exclusion test.
Western blot analysis
Both adherent and floating cells were collected from culture dishes and lysed in RIPA buffer [50 mM Tris–HCl (pH 8.0), 150 mM NaCl, 1% (v/v) NP-40, 0.5% (w/v) deoxycolic acid, 0.1% (w/v) SDS, 10 mM NaF, 1 mM sodium orthovanadate and 1× protease inhibitors (Sigma)], and protein concentrations were determined by BCA protein assay kit (Pierce, Rockford, IL). Equal amounts (10 μg) of protein were separated by electrophoresis through 4–12% polyacrylamide gels (InVitrogen, Carlsbad, CA). The blotted PVDF membranes (Millipore, Billerica, MA) were blocked with 5% non-fat milk in TBS-T [Tris–HCl (pH 8.0), 150 mM NaCl, 0.1% (v/v) Tween-20] and then incubated with primary antibodies at 4°C overnight. In order to assess the potential impact of 17-AAG on apoptosis, anti-caspase- 3, anti-cleaved caspase-3 and anti-PARP antibodies were immunoblotted. All antibodies were purchased from cell signaling (Danvers, MA) and used at 1:1,000 dilutions. Anti-β-actin antibody (Sigma, 1:10,000) was used to evaluate equal loading of protein samples. In order to assess the potential impact of 17-AAG on the G2/M checkpoint pathway, expression of phosphorylated cdc2 (Tyr15), cdc2, cyclin B1, phosphorylated cdc25C (Ser216), cdc25C, Chk1 and Chk2 was determined. All antibodies were purchased from cell signaling and used at 1:1,000 dilutions. After incubation with secondary antibodies [horseradish peroxidase-conjugated rabbit anti-mouse IgG (1:2,000) or goat anti-rabbit IgG (1:1,500) (Dako, Glostrup, Denmark)], protein expression was visualized using chemiluminescence (Amersham, Piscataway, NJ). Densitometry was performed using NIH Image 1.62 (NIH, Bethesda, MD).
Cell cycle analysis
Cells were incubated in 60 mm culture dishes overnight, then incubated with fresh medium containing DMSO vehicle alone or 17-AAG at IC50’s for 24 h. Cells were then detached from plates using trypsin (Mediatech), washed twice with PBS and fixed in 70% ethanol. The fixed cells were then washed once with PBS and incubated with PBS containing 100 μg/ml RNase A (Sigma) and 40 μg/ml propidium iodide (Sigma) for 30 min at 37°C. The samples were examined on an FACS caliber (BD Biosciences, San Jose, CA) and the acquired data were analyzed using ModFit LT Ver3.1 software (Venity software house, Topsham, ME).
Fluorescence microscopy
To examine apoptotic cell death, cells were incubated in the four-well-chambered slides (2.5 × 104 cells/well) (Nalge Nunc, Naperville, NY), in the presence or absence of 17-AAG at respective IC50 for 72 h. To morphologically evaluate apoptotic cell death, cells were stained with Hoechst 33342 (5 μM, final concentration) (Sigma) and propidium iodide (6 μM) (Sigma). Apoptotic cell death was determined by chromatin condensation and nuclear fragmentation. Necrotic cell death was determined by counting PI-stained nuclei without apoptotic nuclear morphology. Images were collected using fluorescence microscopy (Olympus, New York, NY). Apoptotic, necrotic and total dead cells (apoptotic plus necrotic) were expressed as percentages of total cell numbers.
RT-PCR
In order to detect potential changes in cdc2 mRNA, cells were exposed to DMSO or 17-AAG at respective IC50 for 24 h. Total RNA from cells was isolated with Trizol (Invitrogen) and 100 ng of RNA was applied for RT-PCR using SuperScript III One-Step RT-PCR System (Invitrogen) with Eppendorf Mastercycler (Hamburg, Germany). The temperature profile was at 55°C for 30 min for cDNA synthesis and 94°C for 2 min for denaturation. Thermal cycling conditions were 94°C for 15 s, then the respective annealing temperatures for 30 s, 68°C for 30 s, and 68°C for 5 min for final extension. The primers used were 5′-TGGGGTCAGCTCGTTACTCA-3′ (forward) and 5′-CACTTCTGGCCACACTTCATTTA-3′ (reverse) for cdc2 (annealing temperature; 59°C, 25 cycles) [24], 5′-ACCAAAATACCTACTGGGTCGG-3′ (forward) and 5′-GCATGAACCGATCAATAATGG-3′ (reverse) for cyclin B1 (55°C, 27 cycles) [25], and 5′-ACCATGGATGATGATATCGCC-3′ (forward) and 5′-GCCTTGCACATGCCGG-3′ (reverse) for β-actin (58°C, 30 cycles) [26]. The RT-PCR products were separated by electrophoresis and the gels were stained with Ethidium Bromide solution. The gels were imaged with Eagle Eye II still video system (Stratagene, La Jolla, CA).
Hsp90 inhibition in HCC xenografts
Hep3B and HuH7 cells were used to create HCC xenografts subcutaneously by injecting 5 × 106 cells into the flanks of adult female athymic nude (nu/nu) mice. Tumor dimensions were measured daily with calipers for up to 4 weeks, and volumes were calculated using the formula [tumor volume = (a × b2)/2], where ‘a’ is the largest dimension and ‘b’ is the perpendicular dimension to ‘a’ [27]. Relative tumor volumes were calculated with respect to the volume at day 0.
The effect of 17-AAG on HCC xenograft growth were assessed after tumors reached a volume 100 mm3 or larger (day 0). Animals were randomly assigned to receive 17-AAG (80 mg/kg per day by intraperitoneal injection on days 1–4 and 8–11), DMSO (vehicle only, same time schedule) or no treatment. The 17-AAG conversion of an in vitro IC50 of 500–3,000 nM to an in vivo dose of 80 mg/kg/day intraperitoneal injection over a 4 day period was derived from the pharmacokinetic and pharmacodynamic studies performed by Banerji et al. [28] in which a similar conversion was used to correlate an in vitro IC50 value to a dose and schedule for their nude mouse ovarian xenograft model. 17-AAG was prepared in 10% volume of DMSO and 90% volume of PBS containing 0.05% Tween-80 [29]. Each mouse was euthanized when the largest diameter of the tumor reached 15 mm. No animals became ill or expired before being euthanized. The time of euthanasia was the endpoint for all animals. Because this endpoint was solely determined by the tumor reaching a certain size, it was the event used to determine the “tumor-related survival” for Kaplan-Meier analysis and log rank testing.
In order to analyze the potential effects of 17-AAG on cdc2 protein expression in HCC xenografts, HuH7 xenografts from mice euthanized at day 14 in each of the three groups (17-AAG, DMSO, and no treatment) were snap frozen in liquid nitrogen, then homogenized with RIPA buffer with protease and phosphatase inhibitors. These whole tissue extracts were then analyzed for total and phospho (Tyr15)-cdc2 by Western blots as described above. All experiments were IACUC-approved and the “principles of laboratory animal care” (NIH publication No. 85–23, revised 1985) were followed or all experiments complied with standards equivalent to the IKCCCR guidelines for the welfare of animals in experimental neoplasia.
Statistical analysis
All experiments were performed in triplicate. All values are expressed as averages ±1 standard deviation. Continuous data were assessed using unpaired two-tailed Student’s t test. Statistical significance was defined as a P value <0.05. Statistical analysis was performed using SPSS version 12.0 (SPSS Inc., Chicago, IL).
Results
17-AAG inhibits human HCC cell growth
MTT assay showed that the viability of Hep3B and HuH7 cells decreased with treatment of 17-AAG from 1 nM to 10 μM for 72 h, suggesting that Hsp90 inhibition decreases HCC growth (Fig. 1a). In addition, 17-AAG induced cell death at all time points in a time- and dose–dependent fashion (Fig. 1b). The apparent IC50 values of 17-AAG for Hep3B and HuH7 at 72 h were 2,600 and 430 nM, respectively. Subsequent experiments were then performed using approximate or “experimental” IC50 values of 3,000 nM for Hep3B, and 500 nM for HuH7.
Fig. 1.

Viability effects of 17-AAG on human HCC cells. a Hep3B and HuH7 cells were incubated with a range of 17-AAG concentrations for 72 h, viabilities were measured by MTT assay and expressed as relative viabilities to DMSO control cells. b Hep3B and HuH7 cells were incubated with or without a range of 17-AAG concentrations (including IC50) for 24, 48, and 72 h, then viabilities were measured by MTT assay. The results are shown as relative cell viability as compared to controls (DMSO alone) at various concentrations of 17-AAG for up to 72 h and the values are expressed as the mean ± SD of three independent experiments * P < 0.05
Direct counting of cells revealed that 17-AAG treatment for up to 72 h caused a marked loss of adherent cell increase that was found in the DMSO control groups (Figure S1A and A′). Trypan blue staining of adherent cells demonstrated that 95% viability in all cell lines (data not shown). The percentage of floating cells (calculated as floating cells/sum of floating and adherent cells) at 72 h for 17-AAG-treated versus untreated cells were 72% versus 21% for Hep3B, and 48% versus 4% for HuH7 (Figures S1B and B′). The total cell numbers of 17-AAG treated Hep3B and HuH7 cells were 72 and 22% of their untreated cell numbers, respectively. These results indicated that 17-AAG decreased cell proliferation (total liver cell numbers) and increased cell death (floating cells).
17-AAG promotes apoptosis in human HCC cells
Fluorescence microscopy with Hoechst 33342 and PI-stained cells showed that at 72 h, 17-AAG increased apoptosis from 4.5 ± 1.4% (untreated cells) to 63.7 ± 18.2% (treated cells) (P < 0.05) in Hep3B, and from 6.6 ± 7.6 to 23.6 ± 6.6% (P < 0.05) in HuH7 (Fig 2a). Immunoblots showed that 17-AAG induced apoptosis in both caspase-3 and/or caspase-7 (PARP cleavage)-dependent manners (Fig. 2b). Caspase-3 activation (Hep3B) and PARP cleavage (Hep3B and HuH7) were detected at 48 h after 17-AAG treatment. We used 20 ng/ml of TNF-α and 200 ng/ml of Actinomycin D (ActD) as positive control. Activation of caspase-3 was abrogated by the pan-caspase inhibitor, z-VAD-fmk, whereas PARP cleavage was substantially suppressed by this inhibitor. These results indicate that 17-AAG caused apoptosis in both cell types.
Fig. 2.

17-AAG induces apoptosis and decreases the expression of cell cycle-related protein. a Apoptotic and necrotic cells were counted by fluorescence microscopy of Hep3B and HuH7 cells after live cell staining with Hoechst 33342 and PI. Dead cells were the sum of apoptotic and necrotic cells. Each of the cell counts were expressed as a percentage of the total cell number, and significant differences in percent cell number between 17-AAG treated and untreated groups were indicated (* P < 0.05). b Hep3B and HuH7 cells were treated with 17-AAG (AAG) at respective IC50, 20 μM z-VAD-fmk or vehicle alone (DMSO) for up to 72 h. Cell lysates were analyzed by immunoblots for uncleaved and cleaved caspase-3, PARP and β-actin. As a positive control, the cells treated with 20 ng/ml TNF-α (TNFα) and 200 ng/ml Actinomycin D (ActD) for 24 h were used. c Hep3B and HuH7 cells were incubated with 17-AAG at respective IC50 for up to 24 h. Protein extracts were then analyzed by immunoblots for cdc2 (total and phosphorylated at Tyr15), cyclin B1, cdc25C (total and phosphorylated at Ser216), Chk1, Chk2 and β-actin
17-AAG causes cell cycle arrest in human HCC cells
As cell cycle progression is a critical determinant of growth, FACS analysis was performed to investigate whether Hsp90 inhibition arrests HCC cell cycle. As compared to untreated controls, HuH7 cells treated with 17-AAG increased the percentage of cells arrested in both G1 and G2/M phases at 24 h; whereas Hep3B treated with 17-AAG increased the percentage of cells in G2/M (Table 1). These data suggest that 17-AAG induced cell cycle arrest and prevented mitosis in human HCC cells.
Table 1.
Cell cycle profile of Hep3B and HuH7 cells treated with or without 17-AAG for 24 h
| (%) | Hep3B DMSO |
Hep3B 17-AAG |
HuH7 DMSO |
HuH7 17-AAG |
|---|---|---|---|---|
| G0/G1 | 50.9 ± 8.0 | 37.2 ± 5.6 | 44.4 ± 8.6 | 55.9 ± 7.1 |
| S | 23.4 ± 5.7 | 19.7 ± 6.7 | 36.8 ± 0.7 | 15.2 ± 5.8 |
| G2/M | 25.7 ± 11.5 | 43.2 ± 9.9 | 18.8 ± 7.9 | 28.9 ± 13.0 |
17-AAG mediates G2/M cell cycle arrest through its effects on the cdc2
To further investigate the mechanism of 17-AAG mediated G2/M cell cycle arrest, proteins involved in G2/M transition [cdc25C, Chk1, Chk2, cyclin-dependent kinase 1 (cdk1 or cdc2) and cyclin B1] were analyzed by immunoblotting. Since substantial G2/M arrest occurred within 24 h of 17-AAG treatment, we examined changes in these proteins in this time period. Immunoblots showed decreased phosphorylated and total cdc2 and cdc25C, and Chk1 during 24 h, whereas cyclinB1 and chk2 did not change (Fig. 2c). As cdc2 plays a major role in G2/M transition [30], these data suggest that 17-AAG-mediated G2/M arrest is associated with decreased cdc2 levels.
17-AAG-mediated cdc2 decrease is due to degradation through autophagy
A decrease in cdc2 levels can be attributed to either altered gene/protein expression or degradation. Real-time semi-quantitative reverse transcriptase (RT)-PCR was used to determine whether decreased cdc2 following 24 h exposure to 17-AAG was due to decreased gene expression. As shown in Fig. 3a, there were no differences in cdc2 mRNA levels between control and 17-AAG-treated cells, suggesting that Hsp90 inhibition decreased cdc2 by a mechanism other than decreased gene expression.
Fig. 3.

17-AAG mediates cdc2 decrease. a Hep3B and HuH7 cells were incubated with 17-AAG (AAG) (+) at respective IC50 concentrations or DMSO vehicle alone (−) for 24 h. Total mRNA were extracted then analyzed by RT-PCR for cdc2, cyclin B1 and β-actin mRNA. b Protein cell lysates were extracted from Hep3B and HuH7 cells treated for up to 6 h with 10 μg/ml of cycloheximide (CHX) in the presence or absence of respective IC50 concentration of 17-AAG (AAG), and then analyzed by immunoblots for cdc2 and β-actin. c Protein cell extracts were derived from HCC cells incubated for 24 h in the presence or absence of IC50 of 17-AAG (AAG) and/or 10 μM lactacystin (Lac). Immunoblots were probed for cdc2 and β-actin. d Each cell line was exposed for 24 h with DMSO (control), IC50 of 17-AAG (AAG), with or without 3-methyladenine (3-MA) at 10 mM. Cell lysates were analyzed by immunoblots for cdc2 and β-actin
In order to determine whether 17-AAG-mediated cdc2 decrease was through altered protein translation or degradation, cdc2 expression was measured in the presence or absence of cycloheximide (CHX) from 0 to 6 h. This time range was chosen since 17-AAG began decreasing cdc2 expression within 6 h, and the cdc2 half-life is about 18 h [30]. Immunoblots showed that in both HCC cell types, there was no change in cdc2 expression with cycloheximide alone. However, when cells were treated with both cycloheximide and 17-AAG, cdc2 expression decreased in both cell lines by 40 and 50% by densitometry at 6 h (Fig. 3b). These data suggest that decreased cdc2 following 17-AAG treatment is due to a protein degradation mechanism.
In order to examine whether protein degradation contributes to 17-AAG-dependent cdc2 loss, the effects of proteasome inhibitors lactacystin on cell viability at 24 h were examined in each HCC cell line using MTT assays. When all HCC cells were treated for up to 24 h with lactacystin at 10 μM, immunoblots showed that cdc2 expression in 17-AAG treated cells did not recover to control values (Fig. 3c). These data suggest that 17-AAG-mediated decrease in cdc2 is not due to proteasome-mediated degradation.
Autophagy, an alternative pathway for protein degradation, was investigated as another potential mechanism of cdc2 decrease by 17-AAG. In HuH7 cells treated with 17-AAG for 24 h showed increased cdc2 expression when exposed concurrently to 3-methyladenine (3-MA), a selective inhibitor of autophagy, as compared to 17-AAG-treated cells without 3-MA (Fig. 3d). The effect of 3-MA was not seen in Hep3B. These data suggest that 17-AAG mediated decrease in cdc2 in HuH7 cells may involve autophagy.
Decreased cdc2 results in apoptosis in HCC cells
In order to determine whether cdc2 inhibition can cause apoptosis in HCC cells, the cdc2 inhibitor roscovitine was used. HCC cells treated with 50 μM roscovitine showed not only a progressive decrease in phosphorylated and total cdc2, but also a progressive increase in cleaved caspase-3 and PARP at 48 and 72 h (Fig. 4). These data demonstrate that cdc2 inhibition results in apoptosis in HCC cells.
Fig. 4.
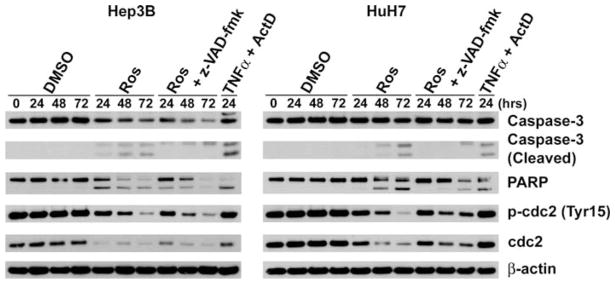
Inhibition of cdc2 induces apoptosis. Cell lysates of Hep3B and HuH7 cells treated with 50 μM Roscovitine (Ros), 20 μM z-VAD-fmk or vehicle alone (DMSO) for up to 72 h were analyzed by immunoblots for uncleaved and cleaved caspase-3, PARP, total and phosphorylated (Tyr15) cdc2, and β-actin. As a positive control, the cells treated with 20 ng/ml TNF-α (TNFα) and 200 ng/ml Actinomycin D (ActD) for 24 h were used
17-AAG inhibited human HCC xenograft growth in vivo
The effects of 17-AAG on HCC growth in vivo were examined using Hep3B and HuH7 xenografts in athymic nude mice. Hep3B xenografts in 17-AAG-treated animals showed decreased tumor volume at all time points as compared to both the DMSO and no treatment groups although these differences were not statistically significant (Fig. 5a). This lack of significance is likely related to one aberrant animal in each of the latter groups, which resulted in an increase in the tumor volume range (as demonstrated by the error bars). However, HuH7 xenografts in 17-AAG-treated animals did show significantly decreased tumor volume after day 4 as compared to both groups.
Fig. 5.

17-AAG decreases human HCC xenograft growth. a Once Hep3B and HuH7 xenografts reached 100 mm3 (day 0), each nude mouse received 17-AAG (80 mg/kg/day, days 1–4 and 8–11), DMSO (vehicle only, same days) or no treatment (n = 6 per group). The effect of 17-AAG on HCC growth was determined by measuring Hep3B and HuH7 xenografts dimensions, calculating tumor volumes and expressing these as a percentage of the day 0 volumes (relative tumor volume). The values are shown as the mean ± SD (* P < 0.05, vs. DMSO). b Each mouse was euthanized when the largest tumor diameter reached 15 mm, and this event was defined as “tumor-related death”. Tumor-related survival for mice with Hep3B and HuH7 xenografts were determined by Kaplan-Meier analysis, and log rank test was used to detect any differences between the three treatment groups. (* P < 0.05, vs. DMSO) c Protein extracts from HuH7 cell xenografts at day 14 were analyzed by immunoblots for cdc2 (total and phosphorylated at Tyr15) and β-actin
Tumor-related survival (defined as the time at which each animal was euthanized when its xenograft measured 15 mm) in animals with Hep3B xenografts treated with 17-AAG were not significantly different when compared to DMSO or no treatmnt mice (Fig. 5b). However, mice with HuH7 xenografts treated with 17-AAG showed a significantly greater tumor-related survival as compared to mice treated with DMSO or no treatment. In these experiments, there were no animal deaths, signs of toxicity or body weight loss due to drug administration among any groups.
In order to validate our findings in vitro through an in vivo model of HCC, cdc2, caspase-3 and PARP expression was examined in tissue lysates from the above HuH7 xenografts. Expression of phosphorylated (Tyr15) and total cdc2 markedly decreased in the 17-AAG-treated xenografts while no change was observed in untreated xenografts (Fig. 5c). There was no difference in the expression of caspase-3 and PARP in any groups (data not shown). These data confirm in vivo that 17-AAG suppresses human HCC xenograft growth in association with cdc2 decrease.
Discussion
Our results demonstrate that 17-AAG-mediated inhibition of Hsp90 decreases human HCC growth. This growth inhibition is achieved through (1) cdc2 decrease by protein degradation (Fig. 3), which in turn diminishes tumor cell replication by G2/M cell cycle arrest (Table 1) and (2) increased apoptosis (Fig. 2a, b). Furthermore, Hsp90 inhibition by 17-AAG inhibits human HCC xenografts growth in mice (Fig. 5a), and this growth inhibition is also associated with cdc2 decrease (Fig. 5c).
MTT viability assays showed that 17-AAG-mediated inhibition of Hsp90 decreased growth in Hep3B and HuH7 cell lines in a time and dose–dependent fashion. Relative cell viabilities (ratio of treated over untreated cells) significantly decreased for each cell line at their respective IC50’s (Fig. 1a). IC50 values of 17-AAG for Hep3B and HuH7 at 72 h were 2,600 and 430 nM, respectively, and reflect the greater anti-growth effect of 17-AAG on HuH7 versus Hep3B cells. Similarly wide ranges in 17-AAG IC50’s have been found in different cell lines within a given human cancer type [31, 32]. This difference is partly due to the inherently lower resilience and growth rate exhibited by control Hep3B cells in our experiments, which resulted in a less dramatic drop in relative cell viability with 17-AAG treatment compared to HuH7. In addition, the disparate IC50’s between the two cell lines may be due to the different p53 tumor suppressor gene status of each cell line. Hep3B has no p53 protein whereas HuH7 does. As p53 is a known client protein of Hsp90, its absence in Hep3B cells may cause resistance to Hsp90 inhibitors including 17-AAG. Chemoresistance in many other cancer cells lines has been correlated with mutant/absent p53 status [33, 34]. Other groups have observed differences in IC50’s among different cell lines of a given cancer type, and have suggested that it may be secondary to variable expression of the multidrug-resistance protein (MDR) or P-glycoprotein [35]. Despite this difference, the significant degree to which 17-AAG inhibits growth in both Hep3B and HuH7 suggests that Hsp90 inhibition may be an important strategy to treat HCC.
The number of living cells at any given time, as measured by MTT assays, is determined by the cumulative effects of cell division and cell death. Therefore, total, adherent (viable and dead) and floating (dead) cells were counted to determine the effects of 17-AAG on overall growth, cell division, and cell death (Figure S1). The total numbers of 17-AAG treated Hep3B and HuH7 at 72 h were 72 and 22% of their untreated controls, respectively. As the total cell number is mainly the result of cell division, these results suggest that 17-AAG inhibited HCC cell division. Ansamycins have been shown to impact growth by inhibiting cell division in other cancer cells such as glioblastomas [11], and cervical cancer [36].
A significant increase in HCC cell death following 17-AAG treatment was verified by fluorescence microscopy of nuclear morphology. Although apoptosis and necrosis occurred contributed to cell death by 17-AAG, apoptosis was the predominant cause of cell death. The level of cell death varied widely between cell lines as the percentage of apoptotic cells following respective IC50 17-AAG treatment was higher in Hep3B (64%) and lower in HuH7 (24%) (Fig. 2a). Immunoblot analysis of caspase-3 and PARP cleavage showed that 17-AAG gradually induced apoptosis for up to 72 h (Fig. 2b). Apoptosis has been shown by others to be the dominant process by which 17-AAG kills some cancer cells [31, 32]. Although apoptosis signaling generally occurs within hours, 17-AAG causes a slower development of apoptosis peaking in 48 to 72 h. However, such a slow apoptosis has been demonstrated in colon cancer [32] and Hodgkin’s lymphoma cells [37] upon treatment with ansamycins.
17-AAG-mediated Hsp90 inhibition induced G1 and G2/M arrest in HuH7 and G2/M in Hep3B cell lines at 24 h (Table 1). As G2/M arrest decreases cell division, this cell cycle arrest is one mechanism by which 17-AAG inhibits HCC proliferation. The G2/M transition is regulated by a heterodimer composed of cyclin-dependent kinase 1 (cdk1 or cdc2) and cyclin B1 which translocates to the nucleus to phosphorylate mitotic substrate proteins [38]. Cdc25C, a client protein of Hsp90, activates cdc2. Cdc25C is inactivated by chk1, also a client protein, and chk2 [39]. As 17-AAG decreased levels of cdc25C and cdc2 in these cell lines, its ability to induce G2/M arrest is through inhibition of the cdc2/cyclin B1 heterodimer. Since chk1 inhibits cdc25C, it is possible that its decrease following 17-AAG would cause an opposite increase in both cdc25C and cdc2 activity. However, since cdc25C is also a client protein, 17-AAG-mediated Hsp90 inhibition results in its decreased levels. The decrease in cdc2 levels following this signaling cascade ultimately leads to G2/M arrest, and this association has been confirmed by others [39]. Hsp90 inhibition by ansamycins has been shown to induce G2/M arrest by decreasing cdc2 in glioblastomas [11] and lung cancers [12].
One group observed that in two different types of malignant mesothelioma cell lines, 17-AAG caused G1 and G2/M arrest in one and only G1 arrest in the other. They concluded that the disparate cell cycle effects of 17-AAG were related to the level of Akt inhibition and the collective effect on proteins that regulate both G0–G1 and G2-M progression [40]. Similarly, we observed in immunoblots that 17-AAG decreased Akt expression in both Hep3B and HuH7 but to differing degrees (data not shown). As Hsp90 chaperones a number of cell cycle client proteins that regulate both G0/G1 and G2/M progression and that are themselves controlled by the Akt pathway, the overall effect of 17-AAG on the cell cycle of each cell line may depend on the collective effect of all these proteins. Then, the different degrees to which 17-AAG alters cell cycle in Hep3B and HuH7 may be due to its disparate inhibition of Akt in each of these cell lines.
As the second component of the cell cycle checkpoint heterodimer, cyclin B1 decrease has been linked to G2/M arrest. Although one group studying glioblastomas cells showed that geldanamycin induced G2 arrest by decreasing both cdc2 and cyclin B1 [41], another group studying the same cell lines found only a decrease in cdc2 with 17-AAG [11]. Our data failed to show in HCC cells either a decrease in cyclin B1 protein or mRNA expression even after 72 h of treatment with 17-AAG (data not shown).
Others have found that 17-AAG treatment of different cancer cells has been shown to decrease cdc2 levels [11, 12]. Although decreased transcription may cause lowered cdc2 levels in some pathologic states [39, 42], our data showed no changes in cdc2 mRNA levels in both types of HCC cells following 17-AAG treatment for 24 h (Fig. 3a).
Cdc2 expression decreased in 17-AAG-treated cells when examined from 0 to 6 h in the presence of cycloheximide (Fig. 3b), suggesting that cdc2 decrease was due to increased degradation rather than altered translation. Protein degradation is a known mechanism by which Hsp90 inhibition decreases cdc2 levels [30, 43]. Common mechanisms of protein degradation were then examined to determine how 17-AAG decreased cdc2. The recovery of cdc2 expression in 17-AAG-treated HuH7 using 3-MA suggests that this cdc2 degradation is partly through autophagy in this cell line (Fig. 3d). The absence of observed cdc2 recovery in Hep3B following 3-MA may be because 17-AAG decreases Akt expression to a far greater degree in Hep3B versus HuH7 cells (data not shown). As Akt inhibition increases autophagy [44], the marked 17-AAG-mediated Akt inhibition and autophagy promotion in Hep3B may overwhelm the autophagy-inhibitory effects of 3-MA. Although autophagy is a known mechanism of protein degradation [45], its role in cdc2 degradation has not been described previously.
Others have studied ansamycin-mediated cdc2 decrease in other cancer types. In one study of glioblastomas cells, proteasome was found to be the mechanisms of cdc2 degradation [46]. Others have shown that ansamycins decrease cdc2 through calpains [11]. We also investigated whether the proteasome (Fig. 3c) or calpains (data not shown) were involved in 17-AAG mediated cdc2 decrease in HCC but found no involvement of these potential mechanisms.
Our data demonstrate that 17-AAG inhibited HCC growth by both G2/M arrest and apoptosis in human HCC cells. Cell cycle arrest occurred by decreasing cdc2, a key promoter of G2/M transition. Although some have suggested that G2/M arrest may lead to apoptosis, this relationship has not been examined until now. We confirmed that inhibition of cdc2 was a key signaling event resulting in decreased HCC cell viability by MTT (data not shown). By demonstrating that cdc2 inhibition by roscovitine resulted in caspase cleavage in a fashion similar to that achieved by 17-AAG (Fig. 4), these data suggest for the first time that Hsp90 inhibition by 17-AAG induces apoptosis through its effects on cdc2-mediated G2/M arrest.
In a mouse xenograft model of human HCC, Hsp90 inhibition by 17-AAG decreased both Hep3B and HuH7 cell growth. This inhibition was cell type dependent as it was statistically significant only in HuH7 xenografts. This cell-specific inhibition has been reported by others [31] and parallels the greater decrease in HuH7 relative viability seen in vitro (Fig. 1a). The 17-AAG-treated Hep3B xenografts as compared to DMSO or no treatment groups were notably decreased during the treatment periods. Once treatments ended, 17-AAG treated Hep3B xenografts resumed a growth rate similar to DMSO and no treatment xenografts. This pattern of growth inhibition and recovery mediated by 17-AAG has been observed in breast [47] and ovarian cancer [28] xenografts. We therefore concluded that 17-AAG treatment inhibited human HCC xenograft growth in vivo in a cell type dependent fashion.
The decreased levels of cdc2 expression found in HuH7 xenografts of 17-AAG treated mice corroborate our in vitro data, and represent the first in vivo confirmation of cdc2 decrease associated with HCC growth inhibition. However, we could not confirm our in vitro findings of increased apoptosis-associated proteins, cleaved caspase-3 or PARP, in these xenograft lysates. The inability to detect these common indicators of apoptosis in vivo has been encountered by others, and may be the result of an effective housekeeping system that clears apoptotic bodies or an inadequate amount of these proteins.
Phase I studies have shown that 17-AAG has acceptable toxicity profiles while effectively inhibiting Hsp90 [48, 49]. The only published Phase II study in renal cell cancer showed no significant clinical response, but the authors caution that the study was not large enough to exclude an anticancer effect by slowing tumor growth [50]. No controlled studies have assessed the anti-cancer effects of 17-AAG or other geldanamycin analogs on human hepatocellular cancer.
We herein show for the first time that Hsp90 inhibition by 17-AAG decreases human HCC growth both in vitro and in vivo through cell cycle arrest and apoptosis. Ultimately, an understanding of the complex machinery regulated by Hsp90 may offer treatments that address the multiple components of HCC growth.
Supplementary Material
Footnotes
Electronic supplementary material The online version of this article (doi:10.1007/s00280-008-0888-2) contains supplementary material, which is available to authorized users.
References
- 1.Bosch FX, Ribes J, Diaz M, et al. Primary liver cancer: worldwide incidence and trends. Gastroenterology. 2004;127:S5–S16. doi: 10.1053/j.gastro.2004.09.011. [DOI] [PubMed] [Google Scholar]
- 2.Young JC, Moarefi I, Hartl FU. Hsp90: a specialized but essential protein-folding tool. J Cell Biol. 2001;154:267–273. doi: 10.1083/jcb.200104079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Welch WJ, Feramisco JR. Purification of the major mammalian heat shock proteins. J Biol Chem. 1982;257:14949–14959. [PubMed] [Google Scholar]
- 4.Luk JM, Lam CT, Siu AF, et al. Proteomic profiling of hepatocellular carcinoma in Chinese cohort reveals heat-shock proteins (Hsp27, Hsp70, GRP78) up-regulation and their associated prognostic values. Proteomics. 2006;6:1049–1057. doi: 10.1002/pmic.200500306. [DOI] [PubMed] [Google Scholar]
- 5.Calvisi DF, Pascale RM, Feo F. Dissection of signal transduction pathways as a tool for the development of targeted therapies of hepatocellular carcinoma. Rev Recent Clin Trials. 2007;2:217–236. doi: 10.2174/157488707781662715. [DOI] [PubMed] [Google Scholar]
- 6.Scheufler C, Brinker A, Bourenkov G, et al. Structure of TPR domain-peptide complexes: critical elements in the assembly of the Hsp70-Hsp90 multichaperone machine. Cell. 2000;101:199–210. doi: 10.1016/S0092-8674(00)80830-2. [DOI] [PubMed] [Google Scholar]
- 7.Pratt WB. The hsp90-based chaperone system: involvement in signal transduction from a variety of hormone and growth factor receptors. Proc Soc Exp Biol Med. 1998;217:420–434. doi: 10.3181/00379727-217-44252. [DOI] [PubMed] [Google Scholar]
- 8.Bagatell R, Whitesell L. Altered Hsp90 function in cancer: a unique therapeutic opportunity. Mol Cancer Ther. 2004;3:1021–1030. [PubMed] [Google Scholar]
- 9.Takayama S, Reed JC, Homma S. Heat-shock proteins as regulators of apoptosis. Oncogene. 2003;22:9041–9047. doi: 10.1038/sj.onc.1207114. [DOI] [PubMed] [Google Scholar]
- 10.Whitesell L, Lindquist SL. HSP90 and the chaperoning of cancer. Nat Rev Cancer. 2005;5:761–772. doi: 10.1038/nrc1716. [DOI] [PubMed] [Google Scholar]
- 11.Garcia-Morales P, Carrasco-Garcia E, Ruiz-Rico P, et al. Inhibition of Hsp90 function by ansamycins causes downregulation of cdc2 and cdc25c and G(2)/M arrest in glioblastoma cell lines. Oncogene. 2007;26(51):7185–7193. doi: 10.1038/sj.onc.1210534. [DOI] [PubMed] [Google Scholar]
- 12.Senju M, Sueoka N, Sato A, et al. Hsp90 inhibitors cause G2/M arrest associated with the reduction of Cdc25C and Cdc2 in lung cancer cell lines. J Cancer Res Clin Oncol. 2006;132:150–158. doi: 10.1007/s00432-005-0047-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Beliakoff J, Whitesell L. Hsp90: an emerging target for breast cancer therapy. Anticancer Drugs. 2004;15:651–662. doi: 10.1097/01.cad.0000136876.11928.be. [DOI] [PubMed] [Google Scholar]
- 14.Lim SO, Park SG, Yoo JH, et al. Expression of heat shock proteins (HSP27, HSP60, HSP70, HSP90, GRP78, GRP94) in hepatitis B virus-related hepatocellular carcinomas and dysplastic nodules. World J Gastroenterol. 2005;11:2072–2079. doi: 10.3748/wjg.v11.i14.2072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pascale RM, Simile MM, Calvisi DF, et al. Role of HSP90, CDC37, and CRM1 as modulators of P16(INK4A) activity in rat liver carcinogenesis and human liver cancer. Hepatology. 2005;42:1310–1319. doi: 10.1002/hep.20962. [DOI] [PubMed] [Google Scholar]
- 16.Whitesell L, Mimnaugh EG, De Costa B, et al. Inhibition of heat shock protein HSP90-pp60v-src heteroprotein complex formation by benzoquinone ansamycins: essential role for stress proteins in oncogenic transformation. Proc Natl Acad Sci USA. 1994;91:8324–8328. doi: 10.1073/pnas.91.18.8324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Schulte TW, Neckers LM. The benzoquinone ansamycin 17-allylamino-17-demethoxygeldanamycin binds to HSP90 and shares important biologic activities with geldanamycin. Cancer Chemother Pharmacol. 1998;42:273–279. doi: 10.1007/s002800050817. [DOI] [PubMed] [Google Scholar]
- 18.Clarke PA, Hostein I, Banerji U, et al. Gene expression pro- filing of human colon cancer cells following inhibition of signal transduction by 17-allylamino-17-demethoxygeldanamycin, an inhibitor of the hsp90 molecular chaperone. Oncogene. 2000;19:4125–4133. doi: 10.1038/sj.onc.1203753. [DOI] [PubMed] [Google Scholar]
- 19.Chiosis G, Huezo H, Rosen N, et al. 17AAG: low target binding affinity and potent cell activity—finding an explanation. Mol Cancer Ther. 2003;2:123–129. [PubMed] [Google Scholar]
- 20.Workman P. Altered states: selectively drugging the Hsp90 cancer chaperone. Trends Mol Med. 2004;10:47–51. doi: 10.1016/j.molmed.2003.12.005. [DOI] [PubMed] [Google Scholar]
- 21.Seglen PO, Gordon PB. 3-Methyladenine: specific inhibitor of autophagic/lysosomal protein degradation in isolated rat hepatocytes. Proc Natl Acad Sci USA. 1982;79:1889–1892. doi: 10.1073/pnas.79.6.1889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Meijer L, Borgne A, Mulner O, et al. Biochemical and cellular effects of roscovitine, a potent and selective inhibitor of the cyclin-dependent kinases cdc2, cdk2 and cdk5. Eur J Biochem. 1997;243:527–536. doi: 10.1111/j.1432-1033.1997.t01-2-00527.x. [DOI] [PubMed] [Google Scholar]
- 23.Gomez-Flores R, Gupta S, Tamez-Guerra R, et al. Determination of MICs for Mycobacterium avium-M. Intracellulare complex in liquid medium by a colorimetric method. J Clin Microbiol. 1995;33:1842–1846. doi: 10.1128/jcm.33.7.1842-1846.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Le Gac G, Esteve PO, Ferec C, et al. DNA damage-induced down-regulation of human Cdc25C and Cdc2 is mediated by cooperation between p53 and maintenance DNA (cytosine–5) methyltransferase 1. J Biol Chem. 2006;281:24161–24170. doi: 10.1074/jbc.M603724200. [DOI] [PubMed] [Google Scholar]
- 25.Jurchott K, Bergmann S, Stein U, et al. YB–1 as a cell cycle-regulated transcription factor facilitating cyclin A and cyclin B1 gene expression. J Biol Chem. 2003;278:27988–27996. doi: 10.1074/jbc.M212966200. [DOI] [PubMed] [Google Scholar]
- 26.Fujii T, Nomoto S, Koshikawa K, et al. Overexpression of pituitary tumor transforming gene 1 in HCC is associated with angiogenesis and poor prognosis. Hepatology. 2006;43:1267–1275. doi: 10.1002/hep.21181. [DOI] [PubMed] [Google Scholar]
- 27.Yang J, Yang JM, Iannone M, et al. Disruption of the EF-2 kinase/Hsp90 protein complex: a possible mechanism to inhibit glioblastoma by geldanamycin. Cancer Res. 2001;61:4010–4016. [PubMed] [Google Scholar]
- 28.Banerji U, Walton M, Raynaud F, et al. Pharmacokinetic-pharmacodynamic relationships for the heat shock protein 90 molecular chaperone inhibitor 17-allylamino, 17-demethoxygeldanamycin in human ovarian cancer xenograft models. Clin Cancer Res. 2005;11:7023–7032. doi: 10.1158/1078-0432.CCR-05-0518. [DOI] [PubMed] [Google Scholar]
- 29.Yin X, Zhang H, Burrows F, et al. Potent activity of a novel dimeric heat shock protein 90 inhibitor against head and neck squamous cell carcinoma in vitro and in vivo. Clin Cancer Res. 2005;11:3889–3896. doi: 10.1158/1078-0432.CCR-04-2272. [DOI] [PubMed] [Google Scholar]
- 30.Gannon JV, Nebreda A, Goodger NM, et al. A measure of the mitotic index: studies of the abundance and half-life of p34cdc2 in cultured cells and normal and neoplastic tissues. Genes Cells. 1998;3:17–27. doi: 10.1046/j.1365-2443.1998.00163.x. [DOI] [PubMed] [Google Scholar]
- 31.Burger AM, Fiebig HH, Stinson SF, et al. 17-(Allylamino)-17-demethoxygeldanamycin activity in human melanoma models. Anticancer Drugs. 2004;15:377–387. doi: 10.1097/00001813-200404000-00011. [DOI] [PubMed] [Google Scholar]
- 32.Hostein I, Robertson D, DiStefano F, et al. Inhibition of signal transduction by the Hsp90 inhibitor 17-allylamino-17-demethoxygeldanamycin results in cytostasis and apoptosis. Cancer Res. 2001;61:4003–4009. [PubMed] [Google Scholar]
- 33.Giovannetti E, Backus HH, Wouters D, et al. Changes in the status of p53 affect drug sensitivity to thymidylate synthase (TS) inhibitors by altering TS levels. Br J Cancer. 2007;96:769–775. doi: 10.1038/sj.bjc.6603639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bossi G, Sacchi A. Restoration of wild-type p53 function in human cancer: relevance for tumor therapy. Head Neck. 2007;29:272–284. doi: 10.1002/hed.20529. [DOI] [PubMed] [Google Scholar]
- 35.Kelland LR, Sharp SY, Rogers PM, et al. DT-Diaphorase expression and tumor cell sensitivity to 17-allylamino, 17-demethoxygeldanamycin, an inhibitor of heat shock protein 90. J Natl Cancer Inst. 1999;91:1940–1949. doi: 10.1093/jnci/91.22.1940. [DOI] [PubMed] [Google Scholar]
- 36.de Carcer G. Heat shock protein 90 regulates the metaphase-anaphase transition in a polo-like kinase-dependent manner. Cancer Res. 2004;64:5106–5112. doi: 10.1158/0008-5472.CAN-03-2214. [DOI] [PubMed] [Google Scholar]
- 37.Georgakis GV, Li Y, Rassidakis GZ, et al. Inhibition of heat shock protein 90 function by 17-allylamino-17-demethoxygeldanamycin in Hodgkin’s lymphoma cells down-regulates Akt kinase, dephosphorylates extracellular signal-regulated kinase, and induces cell cycle arrest and cell death. Clin Cancer Res. 2006;12:584–590. doi: 10.1158/1078-0432.CCR-05-1194. [DOI] [PubMed] [Google Scholar]
- 38.Wolf F, Sigl R, Geley S. ‘The end of the beginning’: cdk1 thresholds and exit from mitosis. Cell Cycle. 2007;6:1408–1411. [PubMed] [Google Scholar]
- 39.Castedo M, Perfettini JL, Roumier T, et al. Cyclin-dependent kinase-1: linking apoptosis to cell cycle and mitotic catastrophe. Cell Death Differ. 2002;9:1287–1293. doi: 10.1038/sj.cdd.4401130. [DOI] [PubMed] [Google Scholar]
- 40.Okamoto J, Mikami I, Tominaga Y, et al. Inhibition of Hsp90 leads to cell cycle arrest and apoptosis in human malignant pleural mesothelioma. J Thorac Oncol. 2008;3:1089–1095. doi: 10.1097/JTO.0b013e3181839693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Nomura M, Nomura N, Newcomb EW, et al. Geldanamycin induces mitotic catastrophe and subsequent apoptosis in human glioma cells. J Cell Physiol. 2004;201:374–384. doi: 10.1002/jcp.20090. [DOI] [PubMed] [Google Scholar]
- 42.Morla AO, Draetta G, Beach D, et al. Reversible tyrosine phosphorylation of cdc2: dephosphorylation accompanies activation during entry into mitosis. Cell. 1989;58:193–203. doi: 10.1016/0092-8674(89)90415-7. [DOI] [PubMed] [Google Scholar]
- 43.Welch PJ, Wang JY. Coordinated synthesis and degradation of cdc2 in the mammalian cell cycle. Proc Natl Acad Sci USA. 1992;89:3093–3097. doi: 10.1073/pnas.89.7.3093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Rubinsztein DC, Gestwicki JE, Murphy LO, et al. Potential therapeutic applications of autophagy. Nat Rev Drug Discov. 2007;6:304–312. doi: 10.1038/nrd2272. [DOI] [PubMed] [Google Scholar]
- 45.Klionsky DJ, Emr SD. Autophagy as a regulated pathway of cellular degradation. Science. 2000;290:1717–1721. doi: 10.1126/science.290.5497.1717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Nomura N, Nomura M, Newcomb EW, et al. Geldanamycin induces G2 arrest in U87MG glioblastoma cells through downregulation of Cdc2 and cyclin B1. Biochem Pharmacol. 2007;73:1528–1536. doi: 10.1016/j.bcp.2007.01.022. [DOI] [PubMed] [Google Scholar]
- 47.Bagatell R, Khan O, Paine-Murrieta G, et al. Destabilization of steroid receptors by heat shock protein 90-binding drugs: a ligand-independent approach to hormonal therapy of breast cancer. Clin Cancer Res. 2001;7:2076–2084. [PubMed] [Google Scholar]
- 48.Grem JL, Morrison G, Guo XD, et al. Phase I and pharmacologic study of 17-(allylamino)-17-demethoxygeldanamycin in adult patients with solid tumors. J Clin Oncol. 2005;23:1885–1893. doi: 10.1200/JCO.2005.12.085. [DOI] [PubMed] [Google Scholar]
- 49.Banerji U, O’Donnell A, Scurr M, et al. Phase I pharmacokinetic and pharmacodynamic study of 17-allylamino, 17-demethoxygeldanamycin in patients with advanced malignancies. J Clin Oncol. 2005;23:4152–4161. doi: 10.1200/JCO.2005.00.612. [DOI] [PubMed] [Google Scholar]
- 50.Ronnen EA, Kondagunta GV, Ishill N, et al. A phase II trial of 17-(Allylamino)-17-demethoxygeldanamycin in patients with papillary and clear cell renal cell carcinoma. Invest New Drugs. 2006;24:543–546. doi: 10.1007/s10637-006-9208-z. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.