

Abstract
From our investigations on phosphine-catalyzed [4+2] annulations between α-alkyl allenoates and activated olefins for the synthesis of cyclohexenes, we discovered a hexamethylphosphorous triamide (HMPT)-catalyzed [4+2] reaction between α-alkyl allenoates 1 and arylidene malonates or arylidene cyanoacetates 2 that provides highly functionalized cyclohexenes 3 and 4 in synthetically useful yields (30–89%), with moderate to exclusive regioselectivity, and reasonable diastereoselectivity. Interestingly, the [4+2] annulations between the a-alkyl allenoates 1 and the olefins 2 manifested a polarity inversion of the 1,4-dipole synthon 1, depending on the structure of the olefin, thus providing cyclohexenes 3 exclusively when using arylidene cyanoacetates. The polarity inversion of α-alkyl allenoates from a 1,4-dipole A to B under phosphine catalysis can be explained by an equilibrium between the phosphonium dienolate C and the phosphorous ylide D.
Keywords: alkenes, allenoates, annulation, phosphine catalysis, regioselectivity
Introduction
The prevalence of six-membered carbocycles in pharmaceutical agents and natural products renders this ring system among the most important chemical structural motifs.[1] Not surprisingly, therefore, a considerable amount of effort has been directed toward the development of new technologies to access these systems. The Diels–Alder reaction is probably the most efficient and widely used method for the rapid construction of these carbocycles.[2] Remarkably, relatively few other chemical methods are available for the preparation of cyclohexenes through intramolecular processes[3] and, even less commonly, intermolecular annulations.[4]
Nucleophilic phosphine catalysis has recently played an important role in the development of new reactions, thus providing access to a variety of molecular frameworks.[5] In particular, allenoates, in the presence of a suitable tertiary phosphine catalyst, react with a long list of activated electrophiles to form various carbocycles and heterocycles.[6] In this area, we have disclosed our findings on the use of ethyl 2-alkyl allenoates as a 1,4-dipole synthon and their reactions with activated imines to generate tetrahydropyridines under phosphine catalysis.[7] Subsequent investigations have led to the construction of cyclohexenes when ethyl 2-alkyl allenoates are treated with arylidene malononitriles.[8] Intriguingly, we observed the highly regioselective formation of ethyl 5,5-dicyano-4-arylcyclohex-1-enecarboxylate and ethyl 4,4-dicyano-5-arylcyclohex-1-enecarboxylate, depending on the catalyst employed. In this process, the ethyl 2-alkyl allenoates 1 serve as a 1,4-dipole synthon A in the presence of catalytic hexamethylphosphorous triamide (HMPT), but as a polarity-reversed 1,4-dipole B in the presence of electron-deficient triarylphosphines (Scheme 1). This polarity inversion from the 1,4-dipole A to the 1,4-dipole B appears to arise from a change in the equilibrium between the phosphonium dienolate C and the vinylogous ylide D. The resulting [4+2] adducts are valuable intermediates for further transformations, both in natural product synthesis[9] and diversity-oriented synthesis.[10] Herein, we provide a full account of our investigation into this [4+2] reaction using various classes of olefinic electrophiles.
Scheme 1.
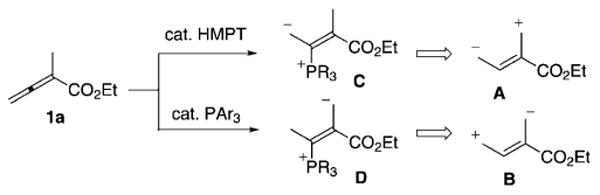
Ethyl 2-methylallenoate functioning as 1,4-dipoles.
Results and Discussion
Based on our previous investigations into tetrahydropyridine syntheses,[7] we initially hypothesized that ethyl 2-methyl allenoate could potentially react with alkenes to form cyclohexenes under the influence of an appropriate phosphine catalyst. Mechanistically, the first step involves the generation of a phosphonium dienolate C through conjugate addition of the phosphine to ethyl 2-methylallenoate 1a (Scheme 2). Michael addition of the dienolate C to an activated olefin 2 facilitates the formation of the zwitterionic intermediate E. Subsequent proton-transfer steps facilitate tautomerization of the vinylphosphonium zwitterion E to an allylphosphonium zwitterion F. The intermediate F undergoes an intramolecular 6-endo cyclization and β-elimination of the phosphine to furnish the cyclohexene 3.
Scheme 2.
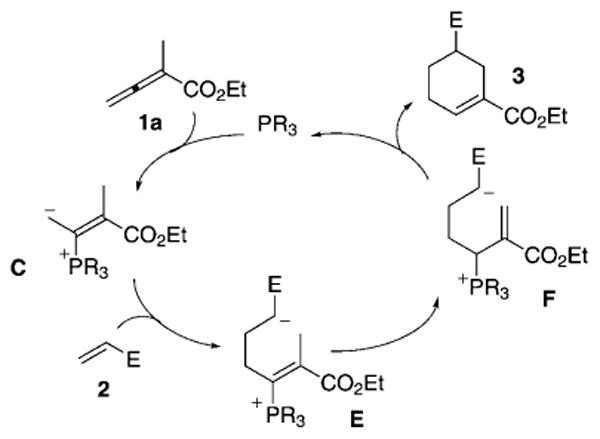
Proposed Mechanism for phosphine-catalyzed cyclohexene formation.
With this mechanism in mind, we set out to examine the effects of a range of activated alkenes, catalysts with varying degrees of nucleophilicity,[11] and solvents of various polarity[12] on the formation of cyclohexenes through the [4+2] annulations of 2-alkyl allenoates. Our initial attempts were made using ethyl 2-methyl allenoate and commercially available electrophiles, including ethyl and methyl acrylates, acrylonitrile, cyclohexenone, dimethyl maleate, dimethyl fumarate, ethyl maleimide, and ethyl cinnamate. Our preliminary results were disappointing: we could not detect any cyclohexene products under phosphine catalysis conditions when using triphenylphosphine or tributylphosphine as the catalyst in benzene or dichloromethane. We hypothesized that the incompatibility of these electrophiles for [4+2] annulation was due, in part, to inefficient isomerization of the zwitterion E to the intermediate F (Scheme 2), thus inhibiting the final cyclization step.[13] To assist this isomerization process, we envisioned that a longer-living zwitterion E might be required to facilitate the formation of the intermediate F. Therefore, we chose to employ benzylidene malonate and benzylidene cyanoacetate as the coupling electrophiles.
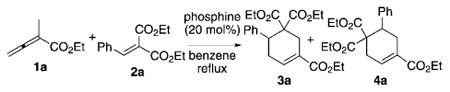
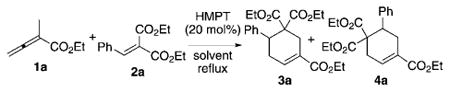
Promisingly, the expected cyclohexene 3a was formed when diethyl benzylidene malonate (2a, 1.0 mmol) was treated with ethyl 2-methyl allenoate (1a, 2.0 mmol) and triphenylphosphine (20 mol%) in benzene under reflux (Table 1, entry 1). Although product formation was apparent through 1H NMR spectroscopic analysis, the reaction proceeded at a slow rate and reached completion only after 96 hours, thereby providing a moderate isolated yield of 3a (32%). Triarylphosphines featuring benzene rings substituted with electron-rich or electron-poor groups did not yield any product after performing the reaction for one day (Table 1, entries 2–4). Electron-rich tertiary phosphines containing alkyl, aryl, alkoxy, or diethylamino substituents were ineffective at mediating cyclohexene formation (Table 1, entries 5–11). These catalysts led to oligomerization of the allenoate 1a instead of incorporation of the benzylidene malonate (Table 1, entries 2–11). Further examination of catalysts revealed that bis(diethylamino)phenylphosphine could induce cyclohexene formation with a moderate isolated yield (10%, Table 1, entry 12). This observation led us to explore HMPT as the catalyst. To our delight, we achieved an appreciable increased reaction efficiency, thus leading to higher mass recovery of the [4+2] adduct, with an isolated yield of 63%, albeit as a 28:72 mixture of 3a and 4a (Table 1, entry 13). Our assignments of the structures of compounds 3a and 4a are based on comparison with those of their dicyano counterparts derived from the benzylidene malononitrile.[8]
Table 1.
Survey of phosphine catalysts for [4+2] annulation of the allenoate 1a and the alkene 2a.[a]
| ||||
|---|---|---|---|---|
|
| ||||
| Entry | Phosphine | Reaction Time [h] | Yield [%][b] | 3a:4a[c] |
| 1 | PPh3 | 96 | 32 | >95:5 |
| 2 | (4-Me2NC6H4)3P | 24 | NR[d] | NA[e] |
| 3 | (4-MeOC6H4)3P | 24 | NR[d] | NA[e] |
| 4 | (4-FC6H4)3P | 24 | NR[d] | NA[e] |
| 5 | PBu3 | 48 | NR[d] | NA[e] |
| 6 | PEt2Ph | 48 | NR[d] | NA[e] |
| 7 | PEtPh2 | 48 | NR[d] | NA[e] |
| 8 | (EtO)3P | 48 | NR[d] | NA[e] |
| 9 | (EtO)2PhP | 48 | NR[d] | NA[e] |
| 10 | (MeO)2(Et2N)P | 48 | NR[d] | NA[e] |
| 11 | (MeO)(Et2N)2P | 48 | 2 | ND[f] |
| 12 | (Et2N)2PhP | 48 | 10 | 22:78 |
| 13 | HMPT | 24 | 63 | 28:72 |
Reaction conditions: 1a (1.4–2.0 mmol), 2a (1 mmol), and the phosphine (20 mol%) were heated under reflux in benzene (10 mL).
Yield of isolated product.
Regioisomeric ratio 3a:4a was determined through analysis of the 1H NMR spectrum of the crude reaction mixture.
No reaction.
Not applicable.
Not determined.
Having found the optimal catalyst, we further optimized the reaction parameters to improve the reaction yields and regioselectivities (Table 2). There appeared to be a trend toward better mass recovery and regioselectivity when less-polar solvents were used. For instance, aromatic solvents were optimal, with very little difference in yield or regioselectivity when using benzene, toluene, or xylene, with the notable exception of the poor performance in high boiling mesitylene (Table 2, entries 1–4).[14] While hexanes provided regioselectivity similar to those of the aromatic solvents (Table 2, entry 5), pentane exhibited excellent selectivity for 4a in good yield (Table 2, entry 6). Although pentane would have been an ideal solvent for the formation of cyclohexenes 4, the arylidene malonates other than diethyl benzylidene malonate did not dissolve in pentane and their reactions did not proceed. Speculating that the high regioselectivity might be due to the low reaction temperature, we employed dichloromethane and a series of dichloromethane/pentane mixtures, but without success (Table 2, entries 7 and 8). Polar aprotic solvents provided modest results (Table 2, entries 9–12). The use of freshly distilled diethyl benzyliden emalonate (2a) and HMPT, with slow addition (over 4 h) of ethyl 2-methyl allenoate (1a) to minimize oligomerization of the allenoate, improved the reaction yield dramatically (89%) with retention of regioselectivity (Table 2, entry 13). Use of a stoichiometric amount of HMPT provided a similar result (Table 2, entry 14). Although there are reports of nucleophilic phosphine catalyzed reactions of allenes being facilitated by water,[15–17] we found that the presence of water had a detrimental effect on cyclohexene formation.
Table 2.
Survey of solvents for [4+2] annulation of the allenoate 1a and the alkene 2a.[a]
| |||||
|---|---|---|---|---|---|
|
| |||||
| Entry | Solvent | Reaction Temp. [°C] | Reaction Time [h] | Yield [%][b] | 3a:4a[c] |
| 1 | benzene | 80 | 24 | 63 | 28:72 |
| 2 | toluene | 111 | 24 | 65 | 33:67 |
| 3 | xylene | 140 | 48 | 72 | 23:77 |
| 4 | mesitylene | 165 | 48 | trace | ND[d] |
| 5 | hexanes | 69 | 24 | 48 | 28:72 |
| 6 | pentane | 36 | 14 | 72 | >5:95 |
| 7 | CH2Cl2/pentane (1:1) | 36 | 24 | trace | 55:45 |
| 8 | CH2Cl2 | 39 | 48 | NR[e] | NA[f] |
| 9 | Et2O | 33 | 24 | 15 | 20:80 |
| 10 | THF | 66 | 24 | 64 | 55:45 |
| 11 | dioxane | 101 | 48 | 12 | 53:47 |
| 12 | MeCN | 81 | 15 | 22 | 60:40 |
| 13 | benzene | 80 | 24 | 89[g] | 28:72 |
| 14 | benzene | 80 | 24 | 90[h] | 28:72 |
Reaction conditions: 1a (1.4–2.0 mmol), 2a (1 mmol), and the phosphine (20 mol%) were heated under reflux in the designated solvent (10 mL).
Yield of isolated product.
Regioisomeric ratio 3a:4a was determined through analysis of the 1H NMR spectrum of the crude reaction mixture.
Not determined.
No reaction.
Not applicable.
All starting reagents were freshly distilled.
1 or 2 equivalents of HMPT was used.
With the optimized conditions for cyclohexene formation in hand, we performed experiments to probe the scope of this [4+2] annulation, and found out that a wide range of arylidene malonates was tolerated (Table 3). For example, arylidene malonates containing benzene rings with electron-withdrawing groups (e.g., nitro, cyano, halogen) at the para-position produced cyclohexenes in good yields with reasonable regioselectivity (favoring the cyclohexene 4; Table 3, entries 2–6). The formation of the cyclohexenes was much slower when the electron-rich para-methyl benzylidene malonate 2g was used, thus leading to lower mass recovery as a result of the competing oligomerization of the allenoate (Table 3, entry 7). Allene oligomerization was circumvented when the allenoate 1a was added over 12 hours to a mixture of dimethyl 4-methylbenzylidene malonate (2g) and HMPT (20 mol%) in benzene under reflux; here, the reaction yield improved to 84% (Table 3, entry 8). The reaction worked well with meta-methyl, bromo, and methoxy-substituted benzylidene malonates, albeit with poorer regioselectivities (Table 3, entries 9–11). Furthermore, the reaction was compatible with hetereoarylidene malonates: dimethyl 2-furylidene malonate (2k), dimethyl 2-thienylidene malonate (2l), and dimethyl N-Boc-2-indolylidene malonate (2m) were viable substrates that furnished cyclohexene products with good yields and regioselectivities (Table 3, entries 12–14).
Table 3.
Survey of arylidenemalonates for [4+2] annulations with the allenoate 1a.[a]
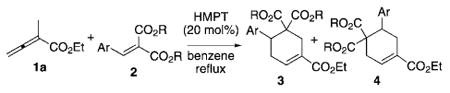
| ||||
|---|---|---|---|---|
|
| ||||
| Entry | Ar | R | Yield [%][b] | 3a/4a[c] |
| 1 | Ph (2a) | Et | 89 | 28:72 |
| 2 | 4-O2NC6H4 (2b) | Me | 69 | 23:77 |
| 3 | 4-NCC6H4 (2c) | Me | 75 | 24:76 |
| 4 | 4-BrC6H4 (2d) | Me | 84 | 21:79 |
| 5 | 4-ClC6H4 (2e) | Me | 86 | 30:70 |
| 6 | 4-FC6H4 (2f) | Me | 82 | 26:74 |
| 7 | 4-MeC6H4 (2g) | Me | 23 | 22:78 |
| 8 | 4-MeC6H4 (2g) | Me | 84[d] | 22:78 |
| 9 | 3-MeC6H4 (2h) | Me | 81 | 50:50 |
| 10 | 3-BrC6H4 (2i) | Me | 83 | 39:61 |
| 11 | 3-MeOC6H4 (2j) | Me | 81 | 33:66 |
| 12 | 2-furyl (2k) | Me | 72 | 16:84 |
| 13 | 2-thienyl (2l) | Me | 71 | 29:71 |
| 14 | N-Boc-2-indolyl (2m) | Me | 87 | 20:80 |
Reaction conditions: A solution of the allenoate 1a (1.4–3.0 mmol) in benzene (10 mL) was added to a solution of the malonate 2 (1 mmol) and HMPT (20 mol%) in benzene (5 mL) under reflux over 4 h and then the mixture was further heated under reflux in benzene until the disappearance (TLC) of the malonate.
Yield of isolated product.
Regioisomeric ratio 3a/4a was determined through analysis of the 1H NMR spectrum of the crude reaction mixture.
The allenoate 1a was added over 12 h.
Next, we examined (Table 4) the use of arylidene cyanoacetates as activated olefin partners for the [4+2] annulations of ethyl 2-methyl allenoate (1a). Unlike the situation with arylidene malonates, here we isolated only one cyclohexene regioisomer 3 when arylidene cyanoacetates were employed under otherwise identical conditions. Slightly higher yields resulted from electron-deficient arylidenes (Table 4, entries 1–3 and 8) compared with those from electron-rich arylidenes (Table 4, entries 4, 5, and 7). Product yields of the desired cyclohexenes 3 diminished when 2-substituted arylidenes were used, presumably because of steric hindrance and subsequent decreased reactivity at the β-carbon atom (Table 4, entries 7 and 8).[18] For a given substituent (e.g., chloro, bromo), the para-substituted arylidene provided a better reaction yield than its ortho or meta counterpart (Table 4, entries 2/8 and 3/9).
Table 4.
Survey of Arylidenecyanoacetates for [4 + 2] Annulations with the Allenoate 1a.[a]
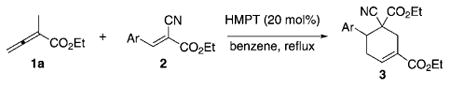
| |||
|---|---|---|---|
|
| |||
| Entry | Ar | Product | Yield [%][b] |
| 1 | Ph (2n) | 3n | 75 |
| 2 | 4-ClC6H4 (2o) | 3o | 75 |
| 3 | 4-BrC6H4 (2p) | 3p | 76 |
| 4 | 4-MeC6H4 (2q) | 3q | 68 |
| 5 | 4-MeOC6H4 (2r) | 3r | 65 |
| 6 | 3-MeOC6H4 (2s) | 3s | 63 |
| 7 | 2-MeOC6H4 (2t) | 3t | 30 |
| 8 | 2-ClC6H4 (2u) | 3u | 60 |
| 9 | 3-BrC6H4 (2v) | 3v | 63 |
Reaction conditions: A solution of the allenoate 1a (1.4–3.0 mmol) in benzene (10 mL) was added to a solution of the cyanoacetate 2 (1 mmol) and HMPT (20 mol%) in benzene (5 mL) under reflux over 4 h and then the mixture was further heated under reflux.
Yield of isolated product.
The overall mass recoveries for the [4+2] annulations with the arylidene cyanoacetates were lower than those of the corresponding arylidene malonates. 1H NMR spectroscopic analysis of the crude reaction mixtures revealed the presence of trace amounts (typically <15%) of the other isomer 4. All attempts to isolate and characterize these minor products 4 were unsuccessful. Further examination of the crude reaction mixture obtained from ortho-methoxybenzylidene cyanoacetate (2t) allowed the isolation of a new minor product in 15% yield; its structure was assigned as that of the [2+2] adduct 5.
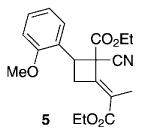
To further explore the substrate scope for the phosphine-catalyzed [4+2] cyclohexene formation, we investigated the effects of various α-alkyl allenoates (Table 5). This reaction tolerated a range of allenylic β′-substituents on the allenoate 1, including aryl, substituted aryl, and carboxylic ester moieties, thus providing cyclohexenes 3 in good isolated yields with reasonable diastereoselectivities (Table 5, entries 1–3). For these β′-substituted allenoates 1, only the cyclohexenes 3 were formed, in accordance with previous observations.[8] Notably, dimethyl benzylidene malonate (2a) and ethyl 2-benzylallenoate (1b) did not furnish any of their corresponding cyclohexenes 3, presumably because of steric hindrance and the diminished reactivities of these olefins.
Table 5.
Survey of allenoates 1 for [4+2] annulations with benzylidenecyanoacetate 2n.[a]
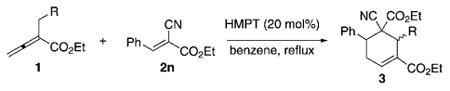
| ||||
|---|---|---|---|---|
|
| ||||
| Entry | R | Product | Yield [%][b] | cis/trans[c] |
| 1 | Ph (1b) | 3w | 81 | 88:12 |
| 2 | 4-ClC6H4 (1c) | 3x | 74 | 89:11 |
| 3 | CO2Et (1d) | 3y | 78 | 77:23 |
Reaction conditions: A solution of the allenoate 1 (1.4–3.0 mmol) in benzene (10 mL) was added to a solution of the olefin 2n (1 mmol) and HMPT (20 mol%) in benzene (5 mL) under reflux over 4 h and then the mixture was further heated under reflux.
Yield of isolated product.
Determined through analysis of the 1H NMR spectrum of the crude reaction mixture and by comparison with the spectrum of cis-ethyl 5,5-dicyano-4,6-diphenylcyclohex-1-enecarboxylate (for which a single-crystal X-ray structure was obtained). See Ref. [8].
Mechanistic Considerations
Based on these observations, we rationalize that the formation of 3 and 4 arises through the γ- or β′-addition of the phosphonium intermediate (C⇌D) to the activated olefin 2. With more reactive arylidene cyanoacetates and/or sterically congested 2-alkylallenoates with α-alkyl groups larger than methyl, the addition occurs at the γ-carbon atom of the phosphonium enolate C; the formation of regioisomer 3 ensues (Scheme 2). With less-reactive arylidene malonates, however, the phosphonium dienolate C isomerizes into the vinylogous ylide D, which adds to the olefin 2 and produces intermediate G. Consecutive proton transfers provide the deconjugated enoate H, which undergoes 6-endo cyclization to generate the cyclic ylide I. Finally, 1,2-proton transfer and β-elimination of the phosphine catalyst furnish the cyclohexene 4 (Scheme 3).
Scheme 3.
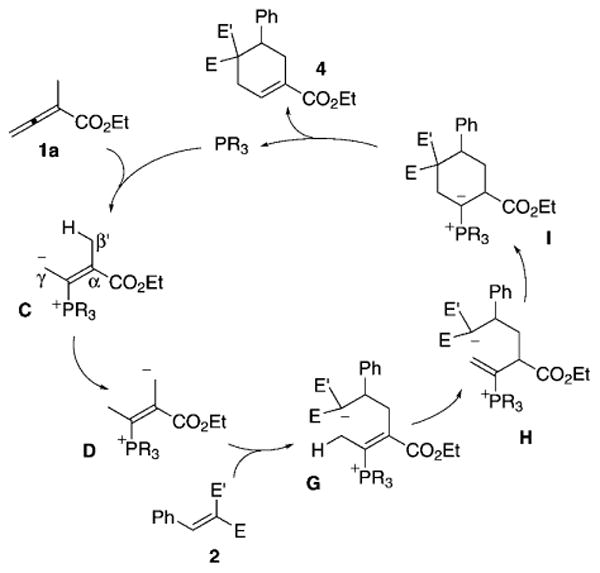
Mechanistic rationale for the formation of cyclohexenes 4.
Synthetic Application of Functionalized Cyclohexenes
Scheme 4 illustrates the synthetic utility of the gem-cyanoacetate adduct. Alkaline hydrolysis of the nitrile 3o using aqueous potassium carbonate and hydrogen peroxide furnished the carboxamide 6 in 84% yield without saponification of the ethyl carboxylic ester groups.
Scheme 4.
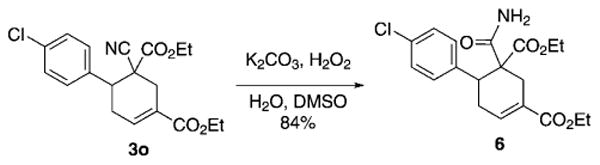
Hydrolysis of the nitrile 3o to the carboxamide 6. DMSO=dimethyl sulfoxide.
Scheme 5 further demonstrates the potential utility of this [4+2] annulation for the synthesis of biologically active natural products. We achieved rapid entry into the tetracyclic framework 7[19] of isoborreverine[20] through N-Boc deprotection of the cyclohexene triester 4m by using trifluoroacetic acid followed by the lactamization with the tert-butyl-magnesium chloride.
Scheme 5.
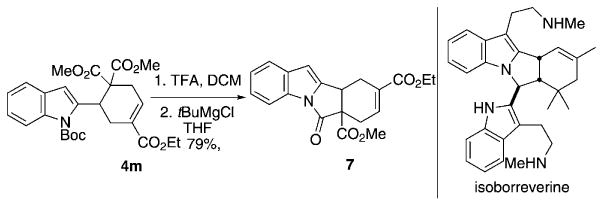
Pyrroloindolinone formation from the cyclohexene gem-diester 4m. TFA=trifluoroacetic acid.
Conclusions
We have further established that the novel phosphine-catalyzed [4+2] annulation between allenoates and activated alkenes is a valuable strategy for cyclohexene synthesis. Ethyl 2-alkyl allenoates can serve as either a 1,4-dipole A or an inverted dipole B by virtue of the steric environment around the allenoate or the reactivity of the activated alkene. Further manipulation of the cyclohexene adducts demonstrates the utility of the phosphine-catalyzed [4+2] annulations of allenoates with olefins.
Experimental Section
Materials and Methods
All reactions were performed under an atmosphere of argon with dry solvents and anhydrous conditions, unless otherwise noted. Benzene, toluene, and CH2Cl2 were freshly distilled from CaH2. All other reagents were used as received from commercial sources. All ethyl 2-substituted methylallenoates 1 were synthesized according to procedures reported previously. Arylidenene malonate and arylidene cyanoacetate 2 were synthesized through phosphine-catalyzed Knoevenagel condensation of the pertinent aldehyde and cyanoacetate, according to the procedure reported by Yadav,[21] or through piperidine-catalyzed condensation of the corresponding aldehyde and malonate. HMPT was distilled under vacuum in three fractions; only the middle fraction was used. The distilled HMPT was then stored under an atmosphere of argon in a dry glove box. Reactions were monitored through thin layer chromatography (TLC) on 0.25 mm Silicycle silica gel plates (TLG-R10011B-323), visualizing with UV light or staining with permanganate or anisaldehyde. Flash column chromatography was performed using Silicycle Silia-P gel (50 μm particle size, R12030B) and compressed air. Preparative HPLC was performed by using (R,R)-1,2-diaminocyclohexane(DACH)-3,5-dinitrobenzoyl(DNB)-modified silica gel. IR spectra were recorded using a Perkin–Elmer Paragon 1000 FTIR spectrometer. NMR spectra were obtained using Bruker Avance-500, ARX-500, or Avance-300 instruments and calibrated using CHCl3/CDCl3 as the internal reference (7.26 and 77.0 ppm for 1H and [13]C NMR spectra, respectively). Data for 1H NMR spectra are reported as follows: chemical shift (δ ppm), multiplicity, coupling constant (Hz), and integration. Data for 13C NMR spectra are reported in terms of chemical shift and multiplicities, with coupling constants (Hz) in the case of JCF coupling. The following abbreviations are used to explain the multiplicities: s, singlet; d, doublet; t, triplet; q, quartet; sept, septet; m, multiplet; br, broad; app, apparent. High-resolution electron ionization (EI) mass spectra were recorded after rapid thermal vaporization of samples deposited on a desorption ionization filament inserted directly into an EI (70 eV, 200°C) source of a triple-sector high-resolution instrument (VG/Micromass Autospec) tuned to 8000 static resolution (M/DM, 10% valley) using perfluorinated kerosene (formula weight 705, Lancaster Synthesis, Windham, NH) as the internal calibrant. High-resolution electrospray ionization (HRESI) mass spectra were recorded after flow injection of CHCl3 solutions into an ESI source attached to a 7.5-T FTMS (Ion Spec Ultima, Irvine, CA) instrument. Data were analyzed using the instrument-supplied software. Melting points (m.p.) are uncorrected; they were recorded using an electrothermal capillary melting point apparatus.
General Procedure for the Synthesis of Cyclohexenes 3 and 4
A solution of arylidene malonate or arylidene cyanoacetate (1.0 mmol) and HMPT (0.2 or 1.0 mmol) in benzene (5 mL) was heated under reflux in a 50 mL flame-dried round-bottom flask under an atmosphere of argon. The allenoate (3.0 mmol) in benzene (10 mL) was added dropwise over a period of at least 4 h. An orange-red solution was obtained; the mixture was stirred under reflux until TLC analysis revealed consumption of the olefin 2. The reaction mixture was concentrated and the residue purified through flash column chromatography (gradient eluent: 520% EtOAc in hexanes), followed by preparative HPLC (gradient eluent: 520% CH2Cl2 in hexanes), to provide the corresponding cyclohexene 3 and 4.
Supplementary Material
Acknowledgments
We thank the U.S. National Institutes of Health (O.K.: R01M071779, P41M081282) for financial support.
Footnotes
Dedicated to Professor Eun Lee on the occasion of his retirement and 65th birthday
Supporting information for this article is available on the WWW under http://dx.doi.org/10.1002/asia.201100190.
References
- 1.a) Myers RL. The 100 Most Important Chemical Compounds. Greenwood Press; Westport, Connecticut: 2007. [Google Scholar]; b) Buss AD, Butler MS, editors. Natural Product Chemistry for Drug Discovery. RSC Publishing; Cambridge, UK: 2010. [Google Scholar]; c) Gringauz A. Introduction to Medicinal Chemistry—How Drugs Act and Why. Wiley-VCH; New York: 1997. [Google Scholar]; d) Fischer J, Ganellin RC. Analogue-Based Drug Discovery. Wiley-VCH; Weinheim: 2006. [Google Scholar]; e) Ho TL. Carbocycle Construction in Terpene Synthesis. Wiley-VCH; Weinheim, Germany: 1998. [Google Scholar]
- 2.a) Fringuelli F, Taticchi A, editors. The Diels–Alder Reaction: Selected Practical Methods. Wiley; West Sussex, England: 2002. [Google Scholar]; b) Kobayashi S, Jorgensen KA, editors. Cyclo-addition Reactions in Organic Synthesis. Wiley-VCH; Weinheim, Germany: 2002. [Google Scholar]; c) Nicolaou KC, Snyder SA, Montagnon T, Vassilikogiannakis GE. Angew Chem. 2002;114:1742. doi: 10.1002/1521-3773(20020517)41:10<1668::aid-anie1668>3.0.co;2-z. [DOI] [PubMed] [Google Scholar]; Angew Chem Int Ed. 2002;41:1668. [Google Scholar]; d) Takao KI, Munakata R, Tadano KI. Chem Rev. 2005;105:4779. doi: 10.1021/cr040632u. [DOI] [PubMed] [Google Scholar]
- 3.a) Jung ME, Ho DG. Org Lett. 2007;9:375. doi: 10.1021/ol062980j. [DOI] [PubMed] [Google Scholar]; b) Wang LC, Luis AL, Agapiou K, Jang HY, Krische MJ. J Am Chem Soc. 2002;124:2402. doi: 10.1021/ja0121686. [DOI] [PubMed] [Google Scholar]; c) Frank SA, Mergott DJ, Roush WR. J Am Chem Soc. 2002;124:2404. doi: 10.1021/ja017123j. [DOI] [PubMed] [Google Scholar]; d) Krafft ME, Haxell TFN. J Am Chem Soc. 2005;127:10168. doi: 10.1021/ja052146+. [DOI] [PubMed] [Google Scholar]; e) Grubbs RH, Miller SJ, Fu GC. Acc Chem Res. 1995;28:446. [Google Scholar]; f) Kim H, Goble SD, Lee C. J Am Chem Soc. 2007;129:1030. doi: 10.1021/ja067336e. [DOI] [PubMed] [Google Scholar]
- 4.Enders D, Huttl MRM, Grondal C, Raabe G. Nature. 2006;441:861. doi: 10.1038/nature04820. [DOI] [PubMed] [Google Scholar]
- 5.For reviews, see: Lu X, Zhang C, Xu Z. Acc Chem Res. 2001;34:535. doi: 10.1021/ar000253x.; Valentine DH, Hillhouse JH. Synthesis. 2003;3:317.; Methot JL, Roush WR. Adv Synth Catal. 2004;346:1035.; Lu X, Du Y, Lu C. Pure Appl Chem. 2005;77:1985.; Nair V, Menon RS, Sreekanth AR, Abhilash N, Biji AT. Acc Chem Res. 2006;39:520. doi: 10.1021/ar0502026.; Denmark SE, Beutner GL. Angew Chem. 2008;120:1584. doi: 10.1002/anie.200604943.; Angew Chem Int Ed. 2008;47:1560.; Ye L, Zhou J, Tang Y. Chem Soc Rev. 2008;37:1140. doi: 10.1039/b717758e.; Kwong CKW, Fu MY, Lam CSL, Toy PH. Synthesis. 2008:2307.; Aroyan CE, Dermenci A, Miller SJ. Tetrahedron. 2009;65:4069.; Cowen BJ, Miller SJ. Chem Soc Rev. 2009;38:3102. doi: 10.1039/b816700c.; Marinetti A, Voituriez A. Synlett. 2010:174.; Beata K. Cent Eur J Chem. 2010:1147.
- 6.For selected examples of phosphine catalysis of allenes with electrophiles, see: Zhang C, Lu X. J Org Chem. 1995;60:2906.; Xu Z, Lu X. Tetrahedron Lett. 1997;38:3461.; Kumar K, Kapur A, Ishar MPS. Org Lett. 2000;2:787. doi: 10.1021/ol000007l.; Kumar K, Kapoor R, Kapur A, Ishar MPS. Org Lett. 2000;2:2023. doi: 10.1021/ol0000713.; Ishar MPS. Org Lett. 2000;2:2023. doi: 10.1021/ol0000713.; Zhu XF, Henry CE, Wang J, Dudding T, Kwon O. Org Lett. 2005;7:1387. doi: 10.1021/ol050203y.; Zhu XF, Schaffner AP, Li RC, Kwon O. Org Lett. 2005;7:2977. doi: 10.1021/ol050946j.; Zhu XF, Henry CE, Kwon O. Tetrahedron. 2005;61:6276.; Wallace DJ, Sidda RL, Reamer RA. J Org Chem. 2007;72:1051. doi: 10.1021/jo062170l.; Henry CE, Kwon O. Org Lett. 2007;9:3069. doi: 10.1021/ol071181d.; Creech GS, Kwon O. Org Lett. 2008;10:429. doi: 10.1021/ol702462w.; Xu S, Zhou L, Ma R, Song H, He Z. Chem Eur J. 2009;15:8698. doi: 10.1002/chem.200901276.; Guo H, Xu Q, Kwon O. J Am Chem Soc. 2009;131:6318. doi: 10.1021/ja8097349.; Wang T, Ye S. Org Lett. 2010;12:4168. doi: 10.1021/ol101762z.
- 7.a) Zhu XF, Lan J, Kwon O. J Am Chem Soc. 2003;125:4716. doi: 10.1021/ja0344009. [DOI] [PubMed] [Google Scholar]; b) Zhao GL, Shi M. Org Biomol Chem. 2005;3:3686. doi: 10.1039/b510572b. [DOI] [PubMed] [Google Scholar]; c) Baskar B, Dakas PY, Kumar K. Org Lett. 2011;13:1988. doi: 10.1021/ol200389p. [DOI] [PubMed] [Google Scholar]
- 8.Tran YS, Kwon O. J Am Chem Soc. 2007;129:12632. doi: 10.1021/ja0752181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.a) Wurz RP, Fu GC. J Am Chem Soc. 2005;127:12234. doi: 10.1021/ja053277d. [DOI] [PubMed] [Google Scholar]; b) Tran YS, Kwon O. Org Lett. 2005;7:4289. doi: 10.1021/ol051799s. [DOI] [PubMed] [Google Scholar]
- 10.a) Castellano S, Fiji HDG, Kinderman SS, Watanabe M, De Leon P, Tamanoi F, Kwon O. J Am Chem Soc. 2007;129:5843. doi: 10.1021/ja070274n. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Watanabe M, Fiji MHDF, Guo L, Chan L, Kinderman SS, Slamon DJ, Kwon O, Tamanoi F. J Biol Chem. 2008;283:9571. doi: 10.1074/jbc.M706229200. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Lu J, Chan L, Fiji HDF, Dahl R, Kwon O, Tamanoi F. Mol Cancer Ther. 2009;8:1218. doi: 10.1158/1535-7163.MCT-08-1122. [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Wang Z, Castellano S, Kinder-man SS, Argueta CE, Beshir AB, Fenteany G, Kwon O. Chem Eur J. 2011;17:649. doi: 10.1002/chem.201002195. [DOI] [PMC free article] [PubMed] [Google Scholar]; e) Cruz D, Wang Z, Kibbie J, Modlin R, Kwon O. Proc Natl Acad Sci U S A. 2011;108:6769. doi: 10.1073/pnas.1015254108. [DOI] [PMC free article] [PubMed] [Google Scholar]; f) Choi J, Mouillesseauex K, Wang Z, Fiji HDG, Kinderman SS, Otto GW, Geisler R, Kwon O, Chen JN. Development. 2011;138:1173. doi: 10.1242/dev.054049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.a) Ofial A, Mayr H. [accessed February 2011]; Reactivity Scales. http://www.cup.uni-muen-chen.de/oc/mayr/CDpublika.html.; b) Brotzel F, Kempf B, Singer T, Zipse H, Mayr H. Chem Eur J. 2007;13:336. doi: 10.1002/chem.200600941. [DOI] [PubMed] [Google Scholar]; c) see Ref. [5l].
- 12.Reichardt C. Angew Chem. 1979;91:119. [Google Scholar]; Angew Chem Int Ed Engl. 1979;18:98. [Google Scholar]
- 13.It is possible that the initial addition of the intermediate C to the olefin 2 is the stumbling block; it might also be aided by the use of more-electrophilic Michael acceptors, such as arylidene malononitriles and arylidene malonates.
- 14.With mesitylene as a solvent, only traces of cyclohexenes were formed, presumably because the high temperature of refluxing mesitylene led to decomposition of the allenoate 1a.
- 15.Unlike the cases for the [3+2] annulations of allenoates with alkenes or imines (see Refs. [16] and [17]), addition of water (25, 50, 100, or 400 mol%) under the optimized conditions for the [4+2] annulation of ethyl 2-methyl allenoate (1a) and the alkene 2a was detrimental to the reaction.
- 16.Fang YQ, Jacobsen EN. J Am Chem Soc. 2008;130:5660. doi: 10.1021/ja801344w. [DOI] [PubMed] [Google Scholar]
- 17.a) Xia Y, Liang Y, Chen Y, Wang M, Jiao L, Huang F, Liu S, Li Y, Yu ZX. J Am Chem Soc. 2007;129:3470. doi: 10.1021/ja068215h. [DOI] [PubMed] [Google Scholar]; b) Mercier E, Fonovic B, Henry CE, Kwon O, Dudding T. Tetrahedron Lett. 2007;48:3617. [Google Scholar]
- 18.This result is consistent with our previous findings for allenoate/arylimine [4+2] annulations. See Ref. [7].
- 19.Compound 7 was obtained as a single diastereoisomer. The relative stereochemistry of the two stereogenic centers in compound 7 was not established.
- 20.a) Fernandez LS, Buchanan MS, Carroll AR, Feng YJ, Quinn RJ, Avery VM. Org Lett. 2009;11:329. doi: 10.1021/ol802506n. [DOI] [PubMed] [Google Scholar]; b) Baldé AM, Pieters LA, Gergely A, Wray V, Claeys M, Vlietinck AJ. Phytochemistry. 1991;30:997. [Google Scholar]; c) Tillequin F, Koch M, Rabaron A. J Nat Prod. 1985;48:120. [Google Scholar]; d) Tillequin F, Rousselet R, Koch M, Bert M, Sevenet T. Ann Pharm Fr. 1979;37:543. [PubMed] [Google Scholar]; e) Tillequin F, Koch M, Bert M, Sevenet T. J Nat Prod. 1979;42:92. doi: 10.1021/np50001a003. [DOI] [PubMed] [Google Scholar]; f) Tillequin F, Koch M, Pousset JL, Cave A. J Chem Soc Chem Commun. 1978:826. [Google Scholar]
- 21.Yadav JS, Subba Reddy BV, Basak AK, Visali B, Narsaiah AV, Nagaih K. Eur J Org Chem. 2004:546. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.