

Abstract
The utility of bone marrow cells (BMCs) to regenerate cardiac myocytes is controversial. The present study examined the capacity of different types of BMCs to generate functional cardiac myocytes. Isolated c‐kit+ BMCs (BMSCs), c‐kit+ and crude BMCs from the adult feline femur were membrane stained with PKH26 dye or infected with a control enhanced green fluorescence protein transcript (EGFP)‐adenovirus prior to co‐culture upon neonatal rat ventricular myocytes (NRVM). Co‐cultured cells were immuno‐stained for c‐kit, α‐tropomyosin, α‐actinin, connexin 43 (C×43) and Ki67 and analyzed with confocal microscopy. Electrophysiology of BMSC derived myocytes were compared to NRVMs within the same culture dish. Gap junction function was analyzed by fluorescence recovery after photo‐bleaching (FRAP). BMCs proliferated and differentiated into cardiac myocytes during the first 48 hours of co‐culturing. These newly formed cardiac myocytes were able to contract spontaneously or synchronously with neighboring NRVMs. The myogenic rate of c‐kit+ BMSCs was significantly greater than c‐kit+ and crude BMCs (41.2 ± 2.1, 6.1 ± 1.2, and 17.1 ± 1.5%, respectively). The newly formed cardiac myocytes exhibited an immature electrophysiological phenotype until they became electrically coupled to NRVMs through functional gap junctions. BMSCs did not become functional myocytes in the absence of NRVMs. In conclusion, c‐kit+ BMSCs have the ability to transdifferentiate into functional cardiac myocytes.
Keywords: stem cell, cardiac myocyte, differentiation
Introduction
Cardiac diseases that ultimately lead to congestive heart failure involve the progressive loss of cardiac myocytes. Acute myocyte loss results from myocardial infarction. 1 Slow progressive myocyte loss, involving apoptosis and/or necrosis can result from the sequellae of chronic hypertension 2 or the hyper‐adrenergic state of congestive heart failure. 3 There is now strong evidence supporting the idea that heart failure progression involves a progressive reduction in cardiac myocyte number, due to myocyte apoptosis and necrosis.
Current congestive heart failure (CHF) therapies involve interruption of excessive adrenergic and angiotensin signaling cascades, 4 , 5 but these therapies do not replace the myocytes that have died during CHF development and progression. Recently there has been great interest in cell therapy to repair the damaged heart. 6 , 7 Many animal studies have been performed and most have shown that cell therapy can improve cardiac performance after myocardial infarction. 8 , 9 , 10 , 11 , 12 , 13 A number of small scale clinical trials have also been completed and some of these have produced promising results. 14 , 15 , 16
There are several major questions in the field of cell therapy for cardiac dysfunction. These include studying what cell types are best suited to repair the heart and whether the injected stem/progenitor cells induce improved function by differentiating into functional myocytes or through paracrine effects that reduce cardiac injury and/or enhance endogenous repair. 7 , 17 , 18
Bone marrow is a reservoir for stem/progenitor cells. The idea that these cells might be useful for cardiac cell therapy has been studied with mixed results. 8 , 9 , 10 , 18 , 19 Some groups have found in animal studies that bone marrow stem cells (BMSCs) can differentiate into functional myocytes when injected into the infarct border zone, 8 , 10 others have found that all injected cells die and there are no beneficial effects 9 , 19 and some have found improvement in function but through paracrine effects rather than stem cell differentiation into new cardiac myocytes. 18 These studies highlight the heated debate related to the ability of bone marrow derived stem/progenitor cells to differentiate into functional cardiac myocytes.
We have recently found that c‐kit+ stem/progenitor cells isolated from adolescent feline and human hearts (resident cardiac stem/progenitor cells) have the ability to differentiate into functional cardiac myocytes when grown with neonatal rat ventricular myocytes (NRVMs). 20 In the present study we examined whether c‐kit+ stem/progenitor cells isolated from feline bone marrow have the same capacity to differentiate into functional cardiac myocytes.
Our experiments show that when c‐kit+ stem cells from the bone marrow are cultured with NRVMs they can differentiate into cells that express cardiac contractile proteins and organize these proteins into working sarcomeres. These new myocytes have cardiac type action potentials (APs), form electrical connections with NRVMs and contract. These results strongly support the idea that c‐kit+ stem cells from the bone marrow can differentiate into functional cardiac myocytes when placed in an appropriate environment.
Methods and Materials
Bone marrow and c‐kit+ cell isolation
Crude bone marrow cells (BMCs) were dissociated from the feline femur in Ca2+‐ Mg2+ free phosphate buffered saline (PBS). Red blood cells (RBCs) were removed by incubating the BMCs in a RBC lysis buffer (155 mM NH4C1, 10 mM KHCO3, EDTA, pH 7.3) for 5 minutes on ice. The lysis buffer treated BMCs were washed and re‐suspended with PBS (pH 7.3) containing 2 mM EDTA and 0.5% IgG‐free BSA (Jackson). To isolate c‐kit+ BMSCs, the BMCs were incubated with human CD117 (c‐kit) rabbit polyclonal antibody (DAKO) and then incubated with the anti‐rabbit IgG magnetic beads at 4°C. The BMCs that bound magnetic beads were collected using the OctoMACS magnetic separation unit (Miltenyi Biotec, CA, USA). Cells isolated in this fashion are termed c‐kit+ BMSCs. BM cells that did not bind to the column are termed c‐kit+ BMCs.
Neonatal rat ventricular myocyte culture and co‐culture experiments
NRVMs were isolated and cultured by the methods previously described 21 , 22 with minor modifications. The neonatal rat hearts from 1‐day‐old Sprague‐Dawley rats were removed and atria were trimmed off. Remaining ventricles were minced and the myocytes were dissociated with a Ca2+/bicarbonate free Hanks buffer (pH 7.5) containing HEPES (10 mM), penicillin (50,000 U/L)/streptomycin (50 mg/L), gentamicin (50 mg/L), heparin (5000 USP units/mL), DNase (2 mg/mL), and trypsin (1.5 mg/mL) at 25°C with gentle mixing. The dissociated NRVMs were washed and cultured overnight in DMEM (GIBCO, Carlsbad, CA, USA) supplemented with 5% FBS (Hyclone Laboratories, Logan, UT, USA), BrdU (0.1 mM; Sigma Chemical, St. Louis, MO, USA), vitamin B12 (1.5 mM; Sigma), and antibiotics (penicillin 500,000 U/L, streptomycin 50 mg/L, gentamicin 50 mg/L; Sigma). Prior to the co‐culture, the preculture medium was replaced with co‐culture medium: DMEM (GIBCO) supplemented with 5% FBS (Hyclone), insulin (10 μg/mL; Sigma), transferrin (10 μg/mL; Sigma), vitamin B12 (1.5 mM; Sigma) and the antibiotics used in the preculture medium.
Techniques to track BMSCs in co‐culture: membrane staining with PKH26‐Red and adenoviral infection of EGFP
The isolated c‐kit+ cells were plated on a fibronectin (20 μg/mL) coated dish. The plated cells were cultured in DMEM supplemented with 10% heat inactivated FBS and transfected with adenovirus containing enhanced green fluorescence protein transcript (Ad‐EGFP) for 40 h. The transfected c‐kit+ cells were dissociated from the culture dish by mechanical force and washed with the co‐culture medium. In some experiments, isolated c‐kit+ cells were stained with PKH26‐Red dye according to the manufacturer's protocol (Sigma). For co‐culture experiments, the PKH26‐Red stained or Ad‐EGFP transfected c‐kit+, c‐kit−, and crude BM cells were directly added onto the NRVM dish and co‐cultured for specific time periods.
Immunofluorescence
The co‐cultured cells on glass coverslips were fixed in 4% paraformaldehyde and permealized with a PBS‐based permealization buffer containing 0.05% Triton X‐100 and 1% IgG free BSA (Jackson). A lower Triton X‐100 concentration (0.04%) was used to permealize PKH‐26 membrane dye stained cells to prevent loss of PKH‐26 dye from the membrane. After the permealization, the co‐cultured cells were incubated with the mouse monoclonal antibodies; sarcomeric α‐tropomyosin (Sigma); α‐actinin (Sigma); and rabbit polyclonal antibodies; connexin 43 (Millipore); Ki67 (Vector). The antibody bound molecules were detected by secondary antibodies conjugated with FITC or rhodamine X (Jackson). Nuclei were stained with DAPI. Fluorescent images were taken and analyzed with a Nikon EZ C1‐plus confocal microscope. Video images were captured using a Coolsnap cf2 digital camera (Roper Scientific).
Fluorescence recovery after photo‐bleaching (FRAP)
The gap junction mediated cell‐to‐cell communication was analyzed in the NRVM‐BMSC co‐culture. The co‐cultured cells were loaded with calcein (0.2 μg/mL calcein‐AM: CellTrace calcein red‐orange‐AM; Invitrogen) for 15 minutes at 37°C After the loading, the cells were washed three times and bathed in prewarmed (37°C) normal Tyrode solution supplemented with 1.8 mM CaCl2. Contracting, GFP expressing BMSC derived new myocytes, attached to neighboring NRVMs were located using a Nikon TE‐300 microscope. Intracellular calcein in the GFP expressing cell and immediate neighboring NRVMs was photo‐bleached using a 520–560 nm excitation light for 30 seconds. The cells were allowed to recover from the photo‐bleach for 7 minutes. As a control experiment, the FRAP protocol was repeated with 100 μM carbenoxolone, a gap junction blocker (Sigma) with a longer recovery time (15 minutes).
c‐Kit immuno‐staining
Isolated c‐kit+ BMSCs, c‐kit− BMCs, and crude BMCs were plated on fibronectin coated glass coverslips in 10% heat‐inactivated FBS DMEM for 1‐hour at room temperature (RT). The cells were then fixed with 4% paraformaldehyde for 30 minutes at RT. Prior to c‐kit staining the cells were washed with PBS and incubated in DAKO antigen retrieval buffer for 20 minutes at RT. Primary c‐kit antibody was applied at 4°C overnight. All additional primary antibodies were applied at 37°C for 1 hour in a humidified chamber. A biotinylated secondary antibody conjugated with streptavidin‐HRP was used to detect amplified c‐kit signals. c‐Kit signal was amplified using the NEN TS A Fluorescence Systems kit (NEL702 for tyramide‐tetramethyl‐rhodamine) following manufacturer recommendations. Cardiac α‐actinin was detected following the c‐kit staining using the standard indirect immunofluorescence techniques as described previously.
Electrophysiological recordings from co‐cultured cells
GFP+ BMSCs and NRVMs were bathed in Tyrodes solution containing (in mM): CaCl2, 1, glucose 10, HEPES 5, KCl 5.4, MgCL2 1.2, NaCl 150, sodium pyruvate 2, pH 7.4. GFP+ BMSC‐derived new myocytes were studied after 24–36 hours of co‐culture with NRVMs. Only those GFP+ new myocytes with visible sarcomeres and visible contractions were studied. Whole‐cell patch clamp recordings were performed at 37°C using the Axoclamp 2A amplifier (Molecular Devices, Sunnyvale, CA, USA) and pipettes (5–7 MΩ resistance) filled with a solution containing (mM): KOH 120, aspartic acid 120, KC120, Na2ATP 5, MgCl21, HEPES 10, pH 7.2. APs were recorded in the current‐clamp mode and analyzed using pClamp8 software. AP waveshape was recorded from GFP+ new myocytes in close contact with GFP− (NRVMs) cells as well as GFP+ cells that appeared to be less well coupled to NRVMs. Spontaneous APs were also recorded in isolated GFP+ NRVMs. For each cell, the AP duration at 50% repolarization (APD50) was derived from the average of 3 APs.
Statistical analysis
All data are expressed as mean ± SEM. Intergroup comparisons were made by unpaired Students t‐test. Differences with a p‐value < 0.05 were considered statistically significant.
Results
BMSC isolation
Using magnetic cell sorting, we isolated mono‐nucleated cells expressing the stem cell marker, c‐kit from the feline bone marrow. c‐Kit+ cells made up 1.6 ± 0.4% (n= 8) of total mono‐nucleated bone marrow cells (MNCs). Immunofluorescence experiments showed that BM cells isolated in this fashion had strong c‐kit staining while the crude BMCs and cells in the c‐kit isolation column out‐flow (c‐kit− cells) had low or no detectable c‐kit staining ( Figure 1 ). The majority of the c‐kit+ cells also stained positively for the hematopoietic cell/leukocyte linage marker CD45. None of these cells were detected to have cardiac myocyte proteins such as sarcomeric α‐actinin or α‐tropomyosin ( Figure 1 ). These results show that our sorting technique selectively isolates cells with abundant c‐kit expression that are not already differentiated cardiac myocytes.
Figure 1.
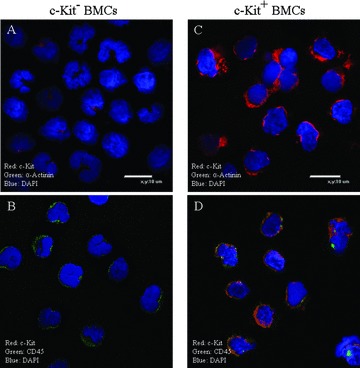
Characterization of c‐kit+ bone marrow stem cells (BMSCs): C‐kit+ and c‐kit cells (A and C) were sorted using immuno‐magnetic beads from mononucleated bone marrow cells (BMCs). Our sorting method yielded c‐kit+ cells that were 1.6 ± 0.4% (n= 10) of the total BMCs. The isolated c‐kit+ BMSCs exhibited c‐kit immunofluorescence signals (C and D), while the c‐kit cells exhibited little if any c‐kit signals (A and B). Each cell type was detected to have low CD45 signals (B and D). The cardiac myocyte marker α‐Actinin was not detected in any of the cell types (A and C).
Contractile protein expression and sarcomeric organization of bone marrow stem cell derived myocytes (BMSC‐DMs)
To investigate the potential of BMSCs to transdifferentiate into cardiac myocytes, we placed c‐kit+ BMSCs on NRVMs (NRVM‐BMSC co‐culture system). c‐Kit− and crude BMCs were also studied under these conditions. Each cell population in the co‐culture was tracked over time by labeling the cells with either PKH26 membrane dye or expressed GFP from adenoviral infection. To identify myogenic transdifferentiation, the co‐cultured cells were fixed and stained for the cardiac contractile protein, α‐tropomyosin. Twenty‐four and forty‐eight hours after co‐culture, 15.5 ± 1.8% and 41.2 ± 2.1% of c‐kit+ BMSC‐DMs were expressing cardiac contractile proteins respectively. In contrast, a significantly lower percentage of c‐kit− cells and crude BMCs expressed cardiac contractile proteins at 24 hours (4.0 ± 1.1% and 6.1 ± 1.2%, respectively) and 48 hours (12.3 ± 1.4% and 17.1 ± 1.5%, respectively) ( Figure 2 ). In order for BMSC‐DMs to contract, they must organize their contractile proteins into sarcomeres. Sarcomeric organization was detected using sarcomeric α‐actinin immunostaining. This protein is located at the Z line of each sarcomere where it helps organize and stabilize contractile proteins. Immunostaining of α‐actinin indicated that the majority of BMSC‐DMs had organized sarcomeres ( Figure 3 ). Collectively, these data indicate that some c‐kit+ BMSCs can transdifferentiate into cardiac myocytes with organized sarcomeres and the ability to contract. Some of the BMSC‐DMs were in the cell cycle, and these cells had less well organized sarcomeres. These results suggest that c‐kit+ BMSCs have the potential to transdifferentiate into myocytes and to proliferate ( Figure 4 ).
Figure 2.
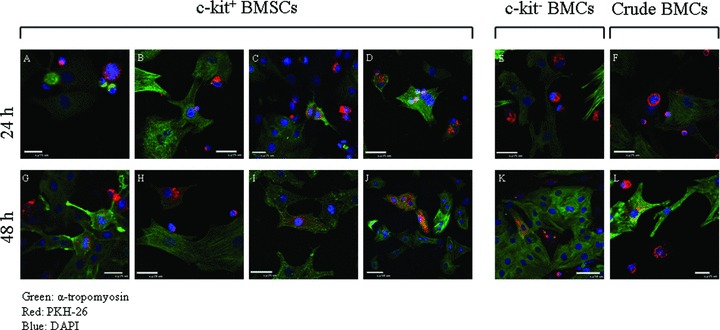
c‐Kit+‐BMSCs express cardiac contractile proteins when co‐cultured with NRVMs: c‐kit+ BMSCs, c‐kit, and crude BMCs were stained with a membrane dye PKH26 before being co‐cultured with NRVMs. The co‐culture was analyzed for the expression of cardiac contractile protein (α‐tropomyosin) at 24 h (A–F) and 48 h (G–L). During the 48 hours of co‐culturing, the c‐kit+ BMSCs expressed α‐tropomyosin. At 24 hours, the majority of the PKH26 stained BMSCs were small round shaped cells and only a few cells had α‐tropomyosin expression (A–D). Some cells were in mitosis, with (C) or without α‐tropomyosin expression (A). At 48 hours, the number of small round cells was decreased (H). The mitotic cells with α‐tropomyosin expression were still present (G) but the majority of the α‐tropomyosin expressing cells were larger (I and J). PKH26 stained c‐kit and crude BMCs were also found to express α‐tropomyosin at 48 hours (K and L); however, at 24 hours, α‐tropomyosin expression were rarely found in these cells (E and F). The percentage of c‐kit+, c‐kit−, and crude BMCs with contractile protein expression at 24 hours was 15.4 ± 1.7, 4.0 ± 1.1, and 6.1 ± 1.2%, respectively. At 48 hours, these percentages increased to 41.1 ± 2.1, 12.3 ± 1.4, and 17.1 ± 1.5%, respectively. Red: PKH26; green: α‐tropomyosin; blue: DAPI.
Figure 3.
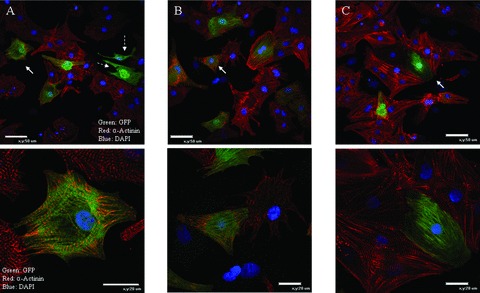
c‐Kit+‐BMSCs differentiate into cardiac myocytes with organized sarcomeres: GFP expressing c‐kit+‐BMSCs were co‐cultured for 48 hours with NRVMs and analyzed for α‐actinin immunofluorescence. 38.6 ± 1.3%(n= 4) of the GFP expressing cells were observed to have organized α‐actinin (A, B, and C). White arrows indicate the cells magnified in the bottom panels. Dotted white arrows indicate GFP expressing BMSC derived non‐myocytes that are not expressing α‐actinin (A). Green: GFP; red: α‐Actinin; blue DAPI.
Figure 4.
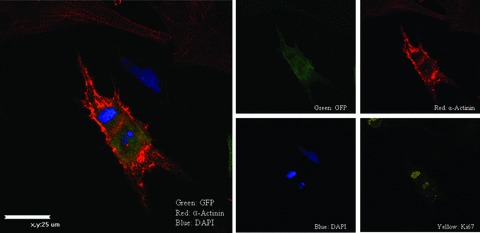
BMSC‐DMs are proliferative. Some GFP expressing BMSC‐DMs were found to be in the cell cycle.
Electrophysiological function and contraction
BMSC‐DMs exhibited spontaneous APs and contraction after 2 days of co‐culture with NRVMs ( Figure 5 and Video SI). In most instances BMSC‐DMs were beating in synchrony with NRVMs. The mean AP duration of the BMSC‐DMs was 317.8 ± 41.5 ms which was significantly longer than that of NRVMs (198.2 ± 28.2 ms). The BMSC‐DMs had less well polarized mean resting membrane potential (Em: −40.5 ± 7.9 mV) relative to the NRVMs (−62.8 ± 4.2 mV) in the same dish. The electrophysiological phenotype of the BMSC‐DMs varied with the degree of attachment to NRVMs. The BMSC‐DMs that were closely associated with NRVMs exhibited similar electrophysiological properties (AP shape, duration, and Em) to the NRVMs suggesting extensive electrical coupling via gap junctions between the two cell types. Well coupled BMSC‐DMs and NRVMs both responded to β‐adrenergic stimulation (10 nM isoproterenol) by increasing beating rate; however, the AP duration of these BMSC‐DMs did not significantly change. BMSC‐DMs that were not well coupled to NRVMs had different electrophysiological properties, with prolonged AP duration, lower peak AP, and depolarized Em. Additionally, these spatially isolated BMSC‐DMs did not respond to the β‐adrenergic stimulation ( Figure 5).
Figure 5.
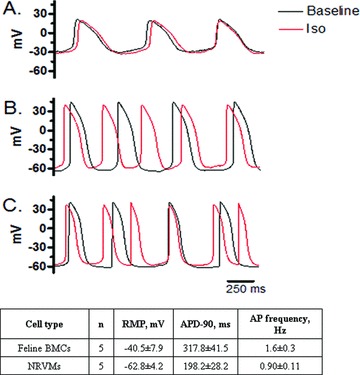
BMSC‐DMs have electrophysiological properties of cardiac myocytes: The electrical properties of the BMSC‐DMs were related to the degree of coupling with NRVMs. BMSC‐DMs with minimal contact with NRVMs (A); BMSC‐DMs attached to a NRVM (B); NRVMs with minimal contact to other cells (C). Basal electrophysiological properties of BMSC‐DMs co‐cultured with NRVMs: BMSC‐DMs exhibited spontaneous action potentials (APs), whereas some NRVMs required electrical stimulation induced AP measurement. RMP: resting membrane potential; APD‐90, action potential duration at 90% repolarization.
Communication Between BMSC‐DM and NRVM
Electrical communication between a BMSC‐DM and neighboring myocytes is critically important for a candidate cell to be considered for future cell therapy. A stem cell derived newly formed myocyte that is poorly coupled to the parent myocardium can cause arrhythmia as reported in a number of the myoblast clinical trials. 23 , 24 The existence of gap junction formation between a BMSC‐DM and a neighboring NRVM in vitro so far has been controversial. 25 , 26 , 27 To investigate the ability of the electrical communication, we first looked for the formation of gap junctions between BMSC‐DMs and neighboring NRVMs, by immunofluorescence staining for the gap junction protein connexin 43 (C×43). In our co‐culture system, the BMSC‐DMs formed gap junctions as indicated by C×43 immunofluorescence ( Figure 6 ). Our experiments showed that the greater the area of contact, the greater the C×43 signal between the BMSC‐DM and NRVMs. The BMSC‐DMs spatially isolated from a group of the NRVMs had weaker C×43 staining and the staining was not on the surface membrane ( Figure 7). In some cells with only modest contact, there was intense C×43 staining at the contact point between BMSC‐DMs and NRVMs ( Figure 7). We did not observe C×43 staining between BMSC‐DMs and non‐myocytes ( Figure 6 ).
Figure 6.
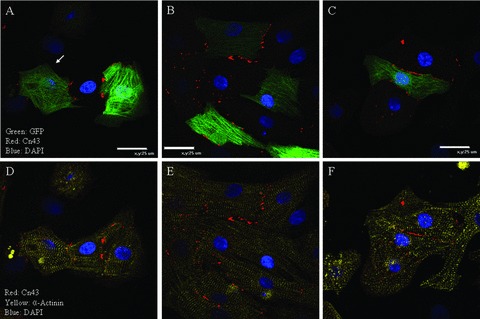
Three representative examples of BMSC‐DMs that form gap junctions with neighboring cardiac myocytes: The GFP expressing BMSCs were co‐cultured with NRVMs. At 48 hours, the co‐culture was analyzed for α‐actinin and connexin 43 (C×43) immunofluorescence. GFP (green) expressing BMSC‐DMs localized C×43 (red) at the cell border with neighboring NRVMs (A, B, and C). Cardiac myocytes were identified by α‐actinin immunofluorescence (yellow: D, E, and F). C×43 was not detected between BMSC‐DMs and non‐myocytes (white arrow: A).
Figure 7.
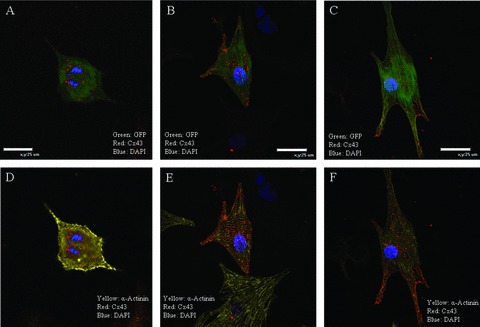
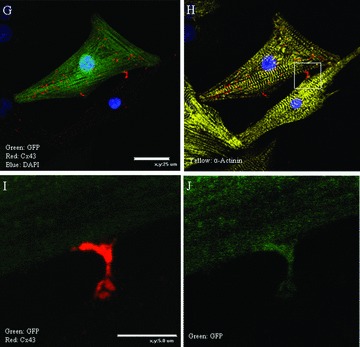
BMSC‐DMs in the cell cycle or those not making close connections with NRVMs did not have surface membrane C×43 localization; The BMSC‐DMs in the cell cycle (A and D) and the BMSC‐DMs having little (B and E) or no contact (C and F) with neighboring myocytes had punctuate, intracellular localized C×43. BMSC‐DMs with minimal contact with NRVMs had intense C×43 staining at the points of contact. Some BMSC‐DMs had small areas of contact with neighboring NRVMs (G and H) and C×43 immunofluorescence was intense at these areas (I and J).
The intracellular coupling (through gap junctions) of BMSC‐DMs and NRVMs was also explored with fluorescence recovery after photo‐bleaching (FRAP). 28 We loaded the cells with a fluorescent dye (calcein) that is small enough (less than 1 KD) to move from cell to cell through gap junctions. After being loaded with calcein, BMSC‐DMs and neighboring NRVMs were photo‐bleached. Calcein fluorescence increased in these cells over the next 7 minutes. A gap junction inhibitor, carbenoxolone blocked the FRAP ( Figure 8 ). These data indicate that there are gap junctions between BMSC‐DMs and NRVMs. These findings explain why BMSC‐DMs and NRVMs contract synchronously ( Figure 6 and Video S1).
Figure 8.
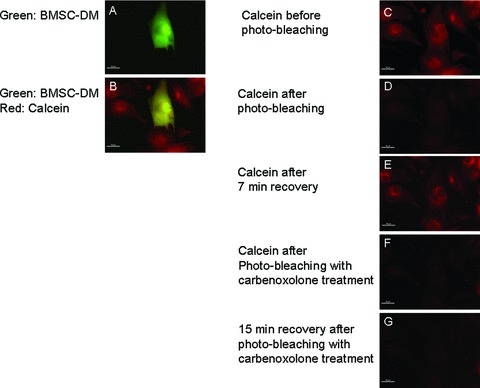
FRAP shows that BMSC‐DMs are coupled to neighboring cardiac myocytes: The GFP expressing BMSCs were co‐cultured with NRVMs. At hour 48, the co‐culture was loaded with calcein red‐orange‐AM (calcein) for 15 minutes (A, B, and C) and then photo‐bleached (D). The photobleached calcein signal recovered after 7 minutes (E). This recovery was not observed after treatment with the gap junction blocker carbenoxolone (100 μM) (G).
Discussion
The present study was designed to determine if c‐kit+ BMSCs could transdifferentiate into functional cardiac myocytes. This is a very controversial topic with evidence for 8 , 10 and against 9 , 19 the ideas. We used a BM stem cell‐cardiac myocyte co‐culture system that has been used previously to create a microenvironment sufficient to induce differentiation of many types of pluripotent cells 29 into cardiac myocytes. 20 , 25 , 26 , 28 We focused on BMCs that had the surface stem cell antigen, c‐kit, because we have shown previously that c‐kit+ cardiac progenitor cells differentiate into cardiac myocytes when co‐cultured with NRVMs. The present experiments clearly show that when c‐kit+ BMSCs are cultured with NRVMs they transdifferentiate into cardiac myocytes with cardiac contractile proteins and organized sarcomeres. Furthermore, these BMSC‐DMs were able to contract and had cardiac type action potentials. BMSC‐DMs formed functional gap junctions and contracted in rhythm with neighboring NRVMs. Importantly, BMSCs did not transdifferentiate into functional cardiac myocytes in culture without NRVMs, suggesting that NRVMs create an environment that is necessary for BMSC transdifferentiation, as previously suggested. 25 , 26 While the c‐kit+ BMSC‐DMs were coupled with the neighboring NRVMs via functional gap junctions, we did not determine whether or not cell‐cell communication between c‐kit+ BMSCs and NRVMs is necessary for myogenic transdifferentiation.
BMSC‐DMs had cardiac myocyte type electrophysiological properties. The specific electrophysiological phenotype of BMSC‐DMs varied with the degree of physical integration with the neighboring NRVMs. BMSC‐DMs that were well coupled to NRVMs exhibited electrophysiological properties similar to the NRVMs. BMSC‐DMs with minimal attachment to NRVMs had electrophysiological properties of immature myocytes, including prolonged action potential duration and depolarized resting membrane potential. The current findings are consistent with those from our previous study using human cardiac stem cell derived myocytes (hCSC‐DMs). 20 We speculate that with additional time in culture these immature new myocytes would become more tightly coupled to NRVMs and adopt a more mature electrical and mechanical phenotype.
β‐adrenergic effects also varied with the coupling of BMSC‐DMs to NRVMs with these cells responding to the β‐adrenergic stimulation with increased beating rate and shortening of the action potential duration. BMSC‐DMs that were well coupled to NRVMs also increased their beating frequency. In contrast, the BMSC‐DMs with minimum contact with NRVMs did not respond to the β‐adrenergic stimulation at all. These findings suggest that BMSC‐DMs adopt adrenergic regulation of electrophysiological and mechanical properties as they mature.
The results of the present study indicate that the BMSCs in the hematopoietic system have the capacity to transdifferentiate into new cardiac myocytes. These results are consistent with a number of studies using hearts from sex mismatched heart transplant patients that have shown that new myocytes from the host are routinely observed. 30 , 31 , 32 We showed that 1.6% of BMCs expressed the stem cell marker c‐kit+ and 41% of these cells transdifferentiated into cardiac myocytes within 2 days of co‐culture. Additionally, about 12% of the cells that were not isolated for c‐kit were also found to transdifferentiate into cardiac myocytes. These cells all expressed the surface marker CD45. We previously reported that c‐kit+ cardiac stem/progenitor cells contain a high proportion of CD45+ cells (88.3 ± 2.0% in nonfailing and 86.9 ± 7.1% in failing human hearts). Moreover, most of these c‐kit+/CD45+ cells were CD45dim/moderate relative to fte bright CD45 fluorescence intensity characteristic of mature leukocytes and many of the c‐kit−/CD45+ cells found in the human heart. 20 In the present study, we found CD45 immunofluorescence signal on the c‐kit immuno‐sorted bone marrow cells. The fluorescence intensity levels were not determined because we did not perform FACS analysis. Collectively, our results show that BMSCs have the potential to regenerate new myocytes within the damaged heart. Importantly, our studies also show that the environment in which these cells are placed is critical to whether or not they differentiate into new myocytes and couple to other myocytes. The environmental factors that best induce cardiac regeneration are an important topic for new research.
In addition, our recent study using hearts removed at transplant indicates that the failing human heart contains increased numbers of cardiac c‐kit+ cells and, as shown, some of these c‐kit+ cells also express CD45, suggesting a possible bone marrow origin. 20 These results suggest that reparative/regenerative cells are within the failing heart, but clearly these cells are unable to reverse the course of this heart failure syndrome. Future studies will likely explore novel approaches to either improve endogenous repair or to use stem cells with the ability to form new myocardium to rebuild the damaged heart.
Conclusions
Many diseases that lead to heart failure, most notably ischemic heart disease, involve the progressive death of cardiac myocytes. Cell therapy represents one option for cardiac regeneration in these individuals. Our study suggests that c‐kit+ BMSCs are capable of transdifferentiating into functional cardiac myocytes when placed in the appropriate environment. The modest improvement in ventricular function found in early clinical trials 14 maybe the result of such new myocyte formation. Much additional study is required to optimize the therapeutic potential of these cardiogenic cells.
Supporting information
Supporting info item
Acknowledgments
This study was supported by NIH grant R0133921.
References
- 1. Kajstura J, Cheng W, Reiss K, Clark WA, Sonnenblick EH, Krajewski S, Reed JC, Olivetti G, Anversa P. Apoptotic and necrotic myocyte cell deaths are independent contributing variables of infarct size in rats. Lab Invest. 1996; 74(1): 86–107. [PubMed] [Google Scholar]
- 2. Shorofsky SR, Aggarwal R, Corretti M, Baffa JM, Strum JM, Al‐Seikhan BA, Kobayashi YM, Jones LR, Wier WG, Balke CW. Cellular mechanisms of altered contractility in the hypertrophied heart: big hearts, big sparks. Cir Res. 1999; 84(4): 424–434. [DOI] [PubMed] [Google Scholar]
- 3. Bristow MR, Kantrowitz NE, Ginsburg R, Fowler MB. Beta‐adrenergic function in heart muscle disease and heart failure. J Mol Cell Cardiol. 1985; 17(Suppl 2): 41–52. [DOI] [PubMed] [Google Scholar]
- 4. Shin J, Johnson JA. beta‐Blocker pharmacogenetics in heart failure. Heart Fail Rev. 2008 April 24. (Epub ahead of print) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Ramasubbu K, Mann DL, Deswal A. Anti‐angiotensin therapy: new perspectives. Cardiol Clin. 2007; 25(4): 573–580; vi–vii. [DOI] [PubMed] [Google Scholar]
- 6. Leri A, Kajstura J, Anversa P, Frishman WH. Myocardial regeneration and stem cell repair. Curr Prob Cardiol. 2008; 33(3): 91–153. [DOI] [PubMed] [Google Scholar]
- 7. Schuldt AJ, Rosen MR, Gaudette GR, Cohen IS. Repairing damaged myocardium: evaluating cells used for cardiac regeneration. Curr Treatment Options Cardiovasc Med. 2008; 10(1): 59–72. [DOI] [PubMed] [Google Scholar]
- 8. Orlic D, Kajstura J, Chimenti S, Jakoniuk I, Anderson SM, Li B, Pickel J, McKay R, Nadal‐Ginard B, Bodine DM, Leri A, Anversa P. Bone marrow cells regenerate infarcted myocardium. Nature. 2001; 410(6829): 701–705. [DOI] [PubMed] [Google Scholar]
- 9. Balsam LB, Wagers AJ, Christensen JL, Kofidis T, Weissman IL, Robbins RC. Haematopoietic stem cells adopt mature haematopoietic fates in ischaemic myocardium. Nature. 2004; 428(6983): 668–673. [DOI] [PubMed] [Google Scholar]
- 10. Yoon YS, Wecker A, Heyd L, Park JS, Tkebuchava T, Kusano K, Hanley A, Scadova H, Qin G, Cha DH, Johnson KL, Aikawa R, Asahara T, Losordo DW. Clonally expanded novel multipotent stem cells from human bone marrow regenerate myocardium after myocardial infarction. J Clin Invest. 2005; 115(2): 326–338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Amado LC, Saliaris AP, Schuleri KH, St John M, Xie JS, Cattaneo S, Durand DJ, Fitton T, Kuang JQ, Stewart G, Lehrke S, Baumgartner WW, Martin BJ, Heldman AW, Hare JM. Cardiac repair with intramyocardial injection of allogeneic mesenchymal stem cells after myocardial infarction. Proc Nat Acad Sci USA. 2005; 102 (32): 11474–11479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Kamihata H, Matsubara H, Nishiue T, Fujiyama S, Tsutsumi Y, Ozono R, Masaki H, Mori Y, Iba O, Tateishi E, Kosaki A, Shintani S, Murohara T, Imaizumi T, Iwasaka T. Implantation of bone marrow mononuclear cells into ischemic myocardium enhances collateral perfusion and regional function via side supply of angioblasts, angiogenic ligands, and cytokines. Circulation. 2001; 104(9): 1046–1052. [DOI] [PubMed] [Google Scholar]
- 13. Kajstura J, Rota M, Whang B, Cascapera S, Hosoda T, Bearzi C, Nurzynska D, Kasahara H, Zias E, Bonafe M, Nadal‐Ginard B, Torella D, Nascimbene A, Quaini F, Urbanek K, Leri A, Anversa P. Bone marrow cells differentiate in cardiac cell lineages after infarction independently of cell fusion. Cir Res. 2005; 96(1): 127–137. [DOI] [PubMed] [Google Scholar]
- 14. Schachinger V, Erbs S, Elsasser A, Haberbosch W, Hambrecht R, Holschermann H, Yu J, Corti R, Mathey DG, Hamm CW, Suselbeck T, Assmus B, Tonn T, Dimmeler S, Zeiher AM. Intracoronary bone marrow‐derived progenitor cells in acute myocardial infarction. New Eng J Med. 2006; 355(12): 1210–1221. [DOI] [PubMed] [Google Scholar]
- 15. Assmus B, Schachinger V, Teupe C, Britten M, Lehmann R, Dobert N, Grunwald F, Aicher A, Urbich C, Martin H, Hoelzer D, Dimmeler S, Zeiher AM. Transplantation of Progenitor Cells and Regeneration Enhancement in Acute Myocardial Infarction (TOPCARE‐AMI). Circulation. 2002; 106(24): 3009–3017. [DOI] [PubMed] [Google Scholar]
- 16. Wollert KC, Meyer GP, Lotz J, Ringes‐Lichtenberg S, Lippolt P, Breidenbach C, Fichtner S, Korte T, Hornig B, Messinger D, Arseniev L, Hertenstein B, Ganser A, Drexler H. Intracoronary autologous bone‐marrow cell transfer after myocardial infarction: the BOOST randomised controlled clinical trial. Lancet. 2004; 364(9429): 141–148. [DOI] [PubMed] [Google Scholar]
- 17. Gnecchi M, He H, Noiseux N, Liang OD, Zhang L, Morello F, Mu H, Melo LG, Pratt RE, Ingwall JS, Dzau VJ. Evidence supporting paracrine hypothesis for Akt‐modified mesenchymal stem cell‐mediated cardiac protection and functional improvement. FASEBJ. 2006; 20(6): 661–669. [DOI] [PubMed] [Google Scholar]
- 18. Uemura R, Xu M, Ahmad N, Ashraf M. Bone marrow stem cells prevent left ventricular remodeling of ischemic heart through paracrine signaling. Cir Res. 2006; 98(11): 1414–1421. [DOI] [PubMed] [Google Scholar]
- 19. Murry CE, Soonpaa MH, Reinecke H, Nakajima H, Nakajima HO, Rubart M, Pasumarthi KB, Virag Jl, Bartelmez SH, Poppa V, Bradford G, Dowell JD, Williams DA, Field LJ. Haematopoietic stem cells do not transdifferentiate into cardiac myocytes in myocardial infarcts. Nature. 2004; 428(6983): 664–668. [DOI] [PubMed] [Google Scholar]
- 20. Kubo H, Jaleel N, Kumarapeli A, Berretta RM, Bratinov G, Shan X, Wang H, Houser SR, Margulies KB. Increased cardiac myocyte progenitors in failing human hearts. Circulation. 2008; 118(6): 649–657 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Simpson P, Savion S. Differentiation of rat myocytes in single cell cultures with and without proliferating nonmyocardial cells. Cross‐striations, ultrastructure, and chronotropic response to isoproterenol. Circ Res. 1982; 50(1): 101–116. [DOI] [PubMed] [Google Scholar]
- 22. Gaughan JP, Hefner CA, Houser SR. Electrophysiological properties of neonatal rat ventricular myocytes with alphal‐adrenergic‐induced hypertrophy. Am J Physiol. 1998; 275(2 Pt 2): H577–590. [DOI] [PubMed] [Google Scholar]
- 23. Menasche P, Hagege AA, Vilquin JT, Desnos M, Abergel E, Pouzet B, Bel A, Sarateanu S, Scorsin M, Schwartz K, Bruneval P, Benbunan M, Marolleau JP, Duboc D. Autologous skeletal myoblast transplantation for severe postinfarction left ventricular dysfunction. J Am Coll Cardiol. 2003; 41(7): 1078–1083. [DOI] [PubMed] [Google Scholar]
- 24. Smits PC, Van Geuns RJ, Poldermans D, Bountioukos M, Onderwater EE, Lee CH, Maat AP, Serruys PW. Catheter‐based intramyocardial injection of autologous skeletal myoblasts as a primary treatment of ischemic heart failure: clinical experience with six‐month follow‐up. J Am Coll Cardiol. 2003; 42(12): 2063–2069. [DOI] [PubMed] [Google Scholar]
- 25. Badorff C, Brandes RP, Popp R, Rupp S, Urbich C, Aicher A, Fleming I, Busse R, Zeiher AM, Dimmeler S. Transdifferentiation of blood‐derived human adult endothelial progenitor cells into functionally active cardiomyocytes. Circulation. 2003; 107(7): 1024–1032. [DOI] [PubMed] [Google Scholar]
- 26. Xu M, Wani M, Dai YS, Wang J, Yan M, Ayub A, Ashraf M. Differentiation of bone marrow stromal cells into the cardiac phenotype requires intercellular communication with myocytes. Circulation. 2004; 110(17): 2658–2665. [DOI] [PubMed] [Google Scholar]
- 27. Lagostena L, Avitabile D, De Falco E, Orlandi A, Grassi F, Lachininoto MG, Ragone G, Fucile S, Pompilio G, Eusebi F, Pesce M, Capogrossi MC. Electrophysiological properties of mouse bone marrow c‐kit+ cells co‐cultured onto neonatal cardiac myocytes. Cardiovasc Res. 2005; 66(3): 482–492. [DOI] [PubMed] [Google Scholar]
- 28. Muller‐Borer BJ, Cascio WE, Anderson PA, Snowwaert JN, Frye JR, Desai N, Esch GL, Brackham JA, Bagnell CR, Coleman WB, Grisham JW, Malouf HH. Adult‐derived liver stem cells acquire a cardiomyocyte structural and functional phenotype ex vivo. Am J Pathol. 2004; 165(1): 135–145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Lensch MW, Daheron L, Schlaeger TM. Pluripotent stem cells and their niches. Stem Cell Rev. 2006; 2(3): 185–201. [DOI] [PubMed] [Google Scholar]
- 30. Bayes‐Genis A, Salido M, Sole Ristol F, Puig M, Brossa V, Camprecios M, Corominas JM, Marinoso ML, Baro T, Vela MC, Serrano S, Padro JM, Bayes de Luna A, Cinca J. Host cell‐derived cardiomyocytes in sex‐mismatch cardiac allografts. Cardiovasc Res. 2002; 56(3): 404–410. [DOI] [PubMed] [Google Scholar]
- 31. Pfeiffer P, Muller P, Kazakov A, Kindermann I, Bohm M. Time‐dependent cardiac chimerism in gender‐mismatched heart transplantation patients. J Am College Cardiol. 2006; 48(4): 843–845. [DOI] [PubMed] [Google Scholar]
- 32. Angelini A, Castellani C, Tona F, Gambino A, Caforio AP, Feltrin G, Delia Barbera M, Valente M, Gerosa G, Thiene G. Continuous engraftment and differentiation of male recipient Y‐chromosome‐positive cardiomyocytes in donor female human heart transplants. J Heart Lung Transplant. 2007; 26(11): 1110–1118. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting info item