

Abstract
Information is limited regarding the effects of injury on neovascularization in the immature brain. We investigated effects of ischemia on cerebral cortical neovascularization after exposure of fetuses to 30 minutes of cerebral ischemia and 48- (I/R-48) or 72- (I/R-72) hours of reperfusion or sham-control treatment (Non-I/R). Immunohistochemical and morphometric analyses of cerebral cortical sections included immunostaining for glial fibrillary acidic protein and collagen type IV (Coll IV), a molecular component of the vascular basal lamina, to determine the glial-vascular network in fetal brains, and Ki67 as a proliferation marker. Cerebral cortices from I/R-48 and I/R-72 fetuses exhibited general responses to ischemia, including reactive astrocyte morphology, which was not observed in Non-I/R fetuses. Cell bodies of reactive, proliferating astrocytes along with large end-feet surrounded walls of cerebral cortical microvessels in addition to the thick Coll IV-enriched basal lamina. Morphometric analysis of Non-I/R with I/R-48 and I/R-72 groups revealed increased Coll IV density in I/R-72 cerebral cortical microvessels (p < 0.01), which also frequently displayed a sprouting appearance, characterized by growing tip cells and activated pericytes. Increases in cerebral cortical basic fibroblast growth factor were associated with neovascularization. We conclude that increased neovascularization occurs within 72 hours after ischemia in fetal cerebral cortices.
Keywords: Cerebral cortex, Fetus, Ischemia-reperfusion, Neovascularization, Ovine, Sheep
INTRODUCTION
Hypoxic-ischemic brain injury is the single most important neurologic problem occurring during the perinatal period. There is substantial evidence suggesting that a major component of brain injury is related to ischemia alone or hypoxia-ischemia (1, 2). Pathophysiological responses in the brain after hypoxia-ischemia are highly complex and involve multiple mechanisms including excitotoxicity, free radical damage, inflammation, and injury to the blood-brain barrier, all of which evolve over time and could predispose the brain to neuronal and glia injury and cell death (3, 4). There is also considerable evidence to suggest that the onset of hypoxic-ischemic brain injury in the neonate can arise before birth (5–8).
The growth of new blood vessels from a pre-existing vascular tree, known as angiogenesis, is a tightly controlled process that is regulated by angiogenic factors (9, 10). Angiogenesis is a multistep process that involves endothelial cells as well as pericytes and includes degradation of the existing vascular basement membrane with subsequent re-assembly around newly formed blood vessels (11). During early ontogenesis in the human brain, blood vessels develop according to a specific pattern of angiogenesis (12); however, developmental remodeling of the vasculature to match local metabolic tissue demands is an ongoing process, and regulated angiogenesis is a crucial component of this process (11).
In addition to the angiogenesis that occurs during normal tissue development, neovascularization by angiogenic mechanisms is one of the events by which the vascular network can be increased in various pathological situations including wound healing, arthritis, cardiovascular diseases, cancer, and cerebral ischemia. Although the vascular system of the adult brain is principally stable under normal basal conditions, endothelial cells proliferate in response to brain ischemia (13). In recent years, accumulating information suggests that the damaged brain can be surprisingly plastic with regard to the coupling of angiogenic and neurogenic mechanisms (14). After ischemia, the production of nascent blood vessels facilitates neuro-restorative processes resulting in improved recovery (13). Endothelial cells in the ischemic boundary zone proliferate within 24 hours, resulting in active neovascularization within 3 days after traumatic brain injury (15). In adult subjects, agents and manipulations that enhance neovascularization and/or neurogenesis promote functional recovery after brain injury (15). These findings may be interpreted to suggest that the manipulation of and/or changes in endogenous neural precursors, endothelial cells and endogenous and/or exogenous vascular promoting growth factors that could augment these processes might represent potential therapeutic strategies for brain injury (15). There is a paucity of information regarding the timing of the onset of neovascularization after injury the fetal brain.
Studies in neonatal rodents suggest that angiogenesis is important for recovery from hypoxic insults in the brain (16). Increased density of proliferating brain capillaries was observed as early as day 3 up to 2 weeks after hypoxia and ischemia (15, 16). Vascular endothelial growth factor (VEGF) is an important mediator of capillary proliferation; it is upregulated after hypoxic-ischemic insults and it is involved in angiogenesis, neurogenesis, and neuronal survival during the recovery process (17, 18). Disruption of the VEGF signaling pathway alters angiogenesis after recovery from hypoxic-ischemic brain injury in rodents (17, 19). In the adult brain, basic fibroblast growth factor (FGF-2, bFGF) is another growth factor that has been shown to augment angiogenesis after ischemia (20, 21). Although numerous studies have characterized the neuronal response to a variety of ischemic insults in the fetal brain (22), there is very little information on the effects of ischemia-reperfusion related injury on neovascularization in the cerebral cortex in the fetal brain (18, 23).
The ovine fetus has been used extensively to investigate many aspects of CNS development (24–28). We have previously shown that brain ischemia in the ovine fetus results in reproducible cerebral cortical and white matter lesions, increases in cerebral cortical FGF-2 levels, and increases in blood-brain barrier permeability along with concomitant alterations in the molecular composition of endothelial cell tight junctions (4, 29). In addition, a recent study has shown that by 48 hours after umbilical cord occlusion, the percentage of blood vessels expressing VEGF increased in several brain regions (18). The rationale for the current study is that, although angiogenesis is an important component in fetal brain development (11, 30) and increases have been documented after ischemia and stroke in the adult brain (31), the effects of ischemia-reperfusion-related brain injury on the potential for neovascularization are less well understood in the fetal brain. In addition, the timing of the onset of neovascularization after injury has not been well established in the fetal brain. Knowledge regarding the responses of the immature vasculature to injury could be important to the development of therapies for hypoxic-ischemic injury in the immature brain (18, 23). Therefore, the objective of the current study was to test the hypothesis that ischemia-reperfusion induces neovascularization in the cerebral cortex of the ovine fetus. Furthermore, we also sought to examine the potential relative time course for the onset of neovascularization after ischemia in the fetal brain.
MATERIALS AND METHODS
This study was conducted with the approval by the Institutional Animal Care and Use Committees of the Alpert Medical School of Brown University and Women & Infants Hospital of Rhode Island and in accordance with the National Institutes of Health Guidelines for the use of experimental animals.
Animal Preparation, Study Groups, and Experimental Design
Surgery was performed under 1–2% isoflurane anesthesia on 12 mixed breed pregnant ewes at 118 to 121 days of gestation, as described for our previous studies (29). Briefly, polyvinyl catheters were placed into a brachial artery and advanced to the thoracic aorta for blood sample withdrawal; they were then put into a femoral artery and advanced into the thoracic aorta for heart rate and blood pressure monitoring, and into a brachial vein for drug administration (29). An amniotic fluid catheter was placed for pressure monitoring to correct fetal arterial pressures. The tissue samples for this study were obtained from animals in our previous report examining the effects of ischemia on white matter lesions in the fetus (29).
After exposure of the fetal carotid arteries, the vertebral-occipital anastomoses and lingual arteries were ligated to restrict vertebral and non-cerebral blood flow, respectively (25). Two inflatable vascular occluders (In Vivo Metric, Healdsburg, CA) were placed around each carotid artery. To measure the fetal electrocorticogram, 2 pairs of screws (Small Parts, Inc., Miami Lakes, FL) were placed onto the dura with a reference electrode sewn to the scalp (25, 29, 32). The screws were connected to a recorder by shielded polyvinyl chloride insulated wires (Alpha Wire Co., Elizabeth, NJ). The electrocorticogram was used to confirm isoelectricity during the cerebral ischemia in the fetal sheep (29, 32). After recovery from surgery, at 123 to 129 days of gestation, the fetal sheep were assigned to 3 groups: 1) instrumented non-ischemic sham-control treated (Non-I/R, n = 4); 2) ischemia induced by 30 minutes of carotid occlusion (hereafter designated as “ischemia”), followed by 48-hour reperfusion (I/R-48, n = 4); and 3) 72-hour reperfusion (I/R-72, n = 4). At the conclusion of ischemia, the occluders were deflated and reperfusion continued for 48 or 72 hours (29). In the Non-I/R sham-control group, the occluders were not inflated but all other procedures were similar.
At the end of reperfusion, the ewe and fetus were killed with pentobarbital (100–200 mg/kg), a hysterotomy was performed and the fetus was withdrawn from the uterus. The brain was rapidly removed and a portion of one anterior frontal cortex was obtained, immediately frozen in liquid N2, and stored at −80°C until analysis for FGF-2. The remainder of the brain was placed in 10% formalin. Formalin-fixed whole brains were coronally cut at 1-cm intervals into 6 serial brain sections and paraffin embedded, as previously described (29). For this study, the paraffin-embedded anterior portion (i.e. section 2) of the brain containing prefrontal cortex (29) was utilized for collagen type IV (Coll IV), glial fibrillary acidic protein (GFAP), and Ki67 expression determinations by immunofluorescence confocal microscopy.
Histology and Immunofluorescence Confocal Microscopy
Immunohistochemistry and morphometry were performed on the Non-I/R, I/R-48, and I/R-72 groups. Randomly chosen sections (n = 6 per brain) from the prefrontal cerebral cortex of the Non-I/R, I/R-48, I/R-72 groups (n = 4 each) were processed for immunohistochemical and morphometric analysis as follows: Paraffin-embedded cerebral cortical samples were cut into 9-μm serial coronal sections from the anterior horn of the lateral ventricles through the head of the caudate and placed on Vectabond-treated slides (Vector Laboratories Inc., Burlingame, CA). The tissue structural preservation and microanatomy of each brain section were ascertained by routinely staining sections at 300-μm intervals with toluidine blue (n = 14 per brain). The sections were then dehydrated in acetone, clarified in xylene, and coverslipped with Entellan mounting medium (Merck, Darmstadt, Germany). Unstained sections were then selected for immunofluorescence, confocal analysis, and morphometry at intervals between toluidine-stained sections. This methodology guaranteed that only equivalent sections corresponding to the same cerebral cortical regions from each animal within the 3 groups were analyzed. Figure 1A–C shows representative toluidine blue-stained sections from the Non-I/R, I/R-48, and I/R-72 groups and illustrates that the sections obtained from each group were similar.
Figure 1.
Representative toluidine blue-stained sections from the sham-control treatment (Non-I/R) (A), ischemia/reperfusion 48 hours (I/R-48) (B), and ischemia/reperfusion 72 hours (I/R-72) (C) groups, taken at regular intervals and utilized to identify similar brain levels and cerebral cortex areas for morphometric analysis. Bars: 5 mm
Single and double immunostaining were carried out with anti-Coll IV, -GFAP, and -Ki67 antibodies. Briefly, sections were rehydrated and processed for heat-mediated antigen retrieval by microwave pre-treatment in 0.01M citrate buffer (pH 6.0) for 15 minutes (3 × 5) at 750 W. Sections were then sequentially incubated with: 1) 0.5% Triton X-100 (Merck) in phosphate-buffered saline (PBS) for 30 minutes at room temperature (RT); 2) blocking buffer ([BB]; PBS, 1% bovine serum albumin, 2% fetal calf serum; Dako Italia, Milano, Italy) for 30 minutes at RT; 3) primary antibodies, mouse IgG1 anti-GFAP (diluted 1:100 in BB; Vision Biosystem Novocastra, Newcastle upon Tyne, UK), and rabbit anti-Coll IV (diluted 1:50 in BB; Abcam, Cambridge, UK), and Ki67 (diluted 1:50 in BB; Novus Biologicals, Littleton, CO) overnight at 4°C; and 4) appropriate fluorophore-conjugated secondary antibodies, i.e. goat anti-mouse IgG Alexa 555 (diluted 1:300 in BB; Invitrogen, Eugene, OR), biotinylated goat anti-rabbit (diluted 1:300 in BB; Vector Laboratories) and streptavidin-conjugated Alexa 488 (diluted 1:400 in BB; Invitrogen) for 45 minutes at RT. After immunolabeling, the sections were post-fixed in 4% paraformaldehyde for 10 minutes and counterstained with TO-PRO3 diluted 1:10K in PBS (633; Invitrogen). The sections were washed 3 times for 5 minutes with PBS after each incubation step. Finally, the sections were coverslipped with Vectashield (Vector) and sealed with nail varnish. Negative controls were prepared by omitting the primary antibodies, pre-adsorbing the primary antibodies with an excess of antigen when available, and mismatching the secondary antibodies. The staining was examined under the Leica TCS SP5 confocal laser-scanning microscope (Leica Microsystems, Mannheim, Germany) using a sequential scan procedure. Confocal images were taken at 250–500-nm intervals through the z-axis of the sections with 20x, 40x and 63x oil lenses. Z-stacks of serial optical planes (projection images) and single optical planes were analyzed by Leica confocal software (Multicolor Package; Leica Microsystems).
Morphometry and Quantitative Analysis
Quantitative evaluation of the percentage of Coll IV-stained area as a measure of the vessel density of the cerebral cortical microvessels was performed by 2 independent observers (F.G. and M.R.), who were not aware of the group designations, by means of computer-aided morphometry on the microscopic images. An average of 15 fields per section from each brain (sections n = 6 per n = 12 brains) for a total of 360 fields in each group (Non-I/R, n = 4; I/R-48, n = 4; I/R-72, n = 4; total cerebral cortex fields, n = 1,080) were analyzed by morphometry. Confocal images were optimized by contrast enhancement functions and digital filters, segmented by an interactive thresholding modality, and the resulting binary image was processed by the measurement function of ImageJ software (NIH, Bethesda, MD). For each Coll IV immuno-labeled field, images of the immunoreactive areas of each vessel were acquired at 20x (total area 600.62 μm2) and measured on single channel and z-stacks of 15 single optical planes.
FGF-2 Assay
The protein concentration of FGF-2 was measured in frontal cerebral cortex by enzyme-linked immunosorbent assay (ELISA) as previously described (29). Briefly, frozen samples from the anterior frontal cortex were homogenized on ice in buffer consisting of 20 mM Tris-HCL, pH 7.4; 2.0 M NaCl; 1 mM EDTA; 1 mM EGTA; 0.5% Deoxycholate; 1% Igepal; proteinase inhibitor cocktail (1 mM PMSF and 1 μg/ml of each of the following: Aprotinin, Leupeptin, Pepstatin A). The samples were centrifuged at 14,000 rpm for 30 minutes at 4°C, and the total protein content of clarified supernatants was determined with a commercially available assay kit (Pierce, Rockford, IL). The FGF-2 content of the supernatants was determined by ELISA using a human FGF-2 immunoassay (Quantikine, R&D Systems, Minneapolis, MN). The assay procedures were performed according to the manufacturer’s specifications. An ELISA scanner (Titertek Multiscan Plus MKII, ICN, Costa Mesa, CA) was used to measure the optical density of total protein at 562 nm and the optical density of FGF-2 at 450 nm. Values for FGF-2 were available from the Non-I/R, n = 3, I/R-48, n = 3, and I/R-72, n = 2 groups for which the quantitative evaluation of the percentage of Coll IV-stained vessel area was also available.
Statistical Analysis
The results from each group were expressed as the percentage area (mean value ± SD). All immunohistochemical data were statistically analyzed using one-way analysis of variance (ANOVA) and the Bonferroni Multiple Comparison test (GraphPad Prism, GraphPad Software, Inc., La Jolla, CA). Correlational analysis also was used to compare the percentage of Coll IV-stained vessels to our previously reported pathological scores (29), and to compare the frontal cerebral cortex FGF-2 concentrations with the percentage of Coll IV-stained vessels within the same animals and using a least squares computerized linear regression program (StatSoft, Tulsa, OK). Results were considered significant at p values of ≤ 0.05.
RESULTS
The fetal sheep from which the cerebral cortical samples were obtained had pH, arterial blood gases, heart rates, and mean arterial blood pressures within the physiological range for our laboratory within each group (29); similar values have been reported from other laboratories (33).
Immunofluorescence Confocal Microscopy, Morphometry and Statistical Analysis
Double immunolabeling was performed with anti-GFAP as a marker of astrocytes and anti-Coll IV as a molecular component of the vascular basal lamina (VBL) to reveal the vascular network profiles in the cerebral cortices in the Non-I/R, I/R-48, and I/R-72 groups. Compared with the cerebral cortex in the Non-I/R sham control group, the cerebral cortices of the fetuses at both 48 hours and 72 hours after ischemia showed evidence of a generalized response to ischemic injury throughout the cerebral cortical areas (Fig. 2). Ischemia-reperfusion in the cerebral cortex induced reactive-type astrocyte morphology as well as a proliferative response in these cells, consistent with our previous report (29). In the Non-I/R control sections, the bodies and processes of GFAP-reactive astrocytes belonging to glia limitans abutted the pial surface and tightly enveloped the penetrating cerebral cortical microvessels (Fig. 2A). Throughout the cerebral cortical layers, GFAP-reactive, protoplasmic astrocytes showed a fine morphology and formed typical perivascular end-feet that had extensive contact with the surface of a number of small cortical microvessels that were revealed by their detectable Coll IV VBL (Fig. 2A, B).
Figure 2.

Confocal images of sheep fetal cerebral cortex double immunostained for glial fibrillary acidic protein (GFAP) and collagen type IV (Coll IV). (A, B) Control sham treatment (Non-I/R) shows healthy-appearing glio-vascular components from the outer, subpial (A), to the deeper (B) cortex layers. Both glia limitans (arrows) and perivascular astrocytes (arrowheads) tightly contact penetrating cerebral cortical microvessels and their branches revealed by Coll IV vascular basal lamina (VBL) and uniformly distributed within the cerebral cortex. (C, D) In ischemia/reperfusion 48 hours (I/R-48) fetal cerebral cortices, reactive astrogliosis involves both glia limitans (arrows) and parenchymal astrocytes, which appear hypertrophic, express high levels of GFAP, and form remarkable perivascular envelopes (arrowheads). (E, F) Apart from an evident astrogliosis, which also include the glia limitans (arrows), the main feature of ischemia/reperfusion 72 hours (I/R-72) cerebral cortices is the number of very small vessels surrounded by their Coll IV VBL and by perivascular astrocytes (arrowheads). Nuclear counterstaining TO-PRO3. Bars: 25 μm
In both the I/R-48 and I/R-72 groups, the glia limitans, parenchymal, and perivascular astrocytes, all responded to the I/R experimental conditions by hypertrophy, swelling, rapidly upregulated GFAP immunoreactivity and possibly proliferated, i.e. reactive astrogliosis. Hypertrophic astrocytes showed a large body and a high number of processes throughout all the cerebral cortical layers (Fig. 2C, D and E, F). Numerous bodies and processes extensively covered the pial surface and formed a dense network in the entire neuropil (Fig. 2C–F). Like their normal counterparts, reactive astrocytes contributed large, perivascular end-feet to microvessel walls (Fig. 3A–F), the profiles of which were revealed by the presence of a continuous, thick Coll IV-enriched VBL (Fig. 3C–F). In particular, in the I/R-72 cerebral cortex, Coll IV immunolabeling revealed a number of small, seamless-like microvessels suggestive of activated vessel proliferation by angiogenic mechanisms (11) (Figs. 2E, F 3E, F). These aspects of increased vessel density prompted a detailed analysis of the cerebral cortex vascular network to ascertain the occurrence of neo-vessel formation. Experimental animals (I/R-48 and I/R-72) were compared with Non-I/R controls applying an interactive morphometric procedure on single channel (Coll IV; green)/binary confocal images (Fig. 4A–C). To measure the percentage of Coll IV-labeled areas, ‘mean area fraction’ on randomly analyzed cortex fields (total fields, n = 1,080) were calculated. The results from each group were expressed as percentage area (mean value ± SD). The mean vessel density of 30-minute ischemia-48-hour reperfusion group did not show a significant difference vs. the mean vessel density of the instrumented Non-I/R sham-control group (I/R-48, 2.67±0.41%; Non-I/R, 2.02±1.01%). By contrast, the 30-minute ischemia-72-hour reperfusion group displayed a vessel density that was significantly higher than the controls (p < 0.01, I/R-72, 5.11±1.54%) and the I/R-48 values (p < 0.05, Fig. 5).
Figure 3.

Confocal images of sheep fetal cerebral cortex double immunostained for glial fibrillary acidic protein (GFAP) and collagen type IV (Coll IV). (A–F) Compared with sham-control treatment (Non-I/R) cerebral cortices (A, B), in both ischemia/reperfusion 48 hours (I/R-48) (C, D) and ischemia/reperfusion 72 hours (I/R-72) (E, F) groups, a diffuse astrogliosis is clearly recognizable; hypertrophic astrocytes form perivascular end-feet in contact with a thick Coll IV-enriched vascular basal lamina (VBL) (arrowheads in C–F). Nuclear counterstaining TO-PRO3. Bars: A, C, E = 25 μm; B, D, F = 10 μm
Figure 4.

Representative binary images of cerebral cortical fields submitted to morphometric analysis. Collagen type IV (Coll IV)-immunostained microvessels were segmented by an interactive thresholding function and the immunoreactive areas measured and expressed as the percentage of fractional vessel density (mean ± SD). Non-I/R, Control sham treatment; I/R-48, ischemia/reperfusion 48 hours; I/R-72, ischemia/reperfusion 72 hours. Bars: 25 μm.
Figure 5.
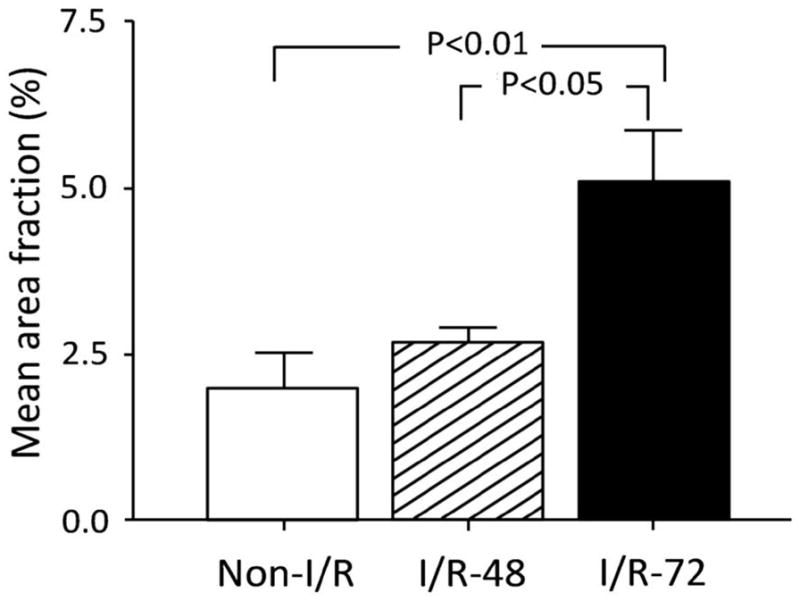
Quantification of neovascularization in randomly selected areas of sheep fetal cerebral cortex from sham-control treatment (Non-I/R), ischemia/reperfusion 48 hours (I/R-48), and ischemia/reperfusion 72 hours (I/R-72) groups. Vessel density, expressed as the percentage area (mean value ± SD) of fields (total area) segmented on anti-collagen type IV (Coll IV)-stained sections, is significantly increased when I/R-72 brains are compared with control brains (P<0.01) and with the shorter time of reperfusion (I/R-48; p < 0.05). One-way analysis of variance (ANOVA) and Bonferroni Multiple Comparison test.
These data were supported by observations carried out by single Coll IV and double Coll IV/GFAP immunostaining at a higher magnification (Figs. 6A–F, 7A–D). In these cerebral cortical fields, sprouting-type microvessels, which were already present, although rare in the I/R-48 animals (Fig. 6A, B), became a common feature of I/R-72 cerebral cortices (Fig. 6C–F). At this time, endothelial cell proliferation was recognizable as hypercellularity of the vessel wall that also appeared to be surrounded by a number of pericytes that were typically enclosed by VBL (Fig. 7A). These growing vessels were characterized by endothelial tip cells and activated pericytes forming points of vessel sprouting and always surrounded by perivascular reactive astrocytes and a Coll IV VBL (Fig. 6A–F, 7B–D) (11, 34). In the cerebral cortices of the I/R-72 group, growing microvessels consistently exhibited a high density of perivascular hypertrophic astrocytes in contact with a very thick Coll IV VBL, which characteristically ensheathed the entire associated pericytes (8A–F). Emerging vascular tubes exhibiting faint reactivity of Coll IV staining were frequent and consistently ensheathed by perivascular astrocytic end-feet (Fig. 8A, B, D, E). In areas of astrogliosis, the presence of several pairs of perivascular astrocytes suggested that ischemia-reperfusion resulted in reactive hyperplastic astrocytes (Fig. 8C).
Figure 6.

Confocal images of sheep fetal cerebral cortex double immunostained for glial fibrillary acidic protein (GFAP) and collagen type IV (Coll IV), merge (A, C, E) and single channel (B, D, F) images. (A, B) There is a rare sprouting-like microvessel in the cerebral cortex from ischemia/reperfusion 48 hours (I/R-48) group; the endothelial cell tip shows a long exploring filopodium surrounded by reactive astrocytes (arrowheads). (C–F) Sprouting-like vascular endings (arrowheads) surrounded by a number of seamless-appearing microvessels (asterisks) in ischemia/reperfusion 72 hours (I/R-72). Note that in A–F pericytes are included in the Coll IV-reactive vascular basal lamina (VBL) (arrows). Nuclear counterstaining TO-PRO3. Bars: 10 μm.
Figure 7.

Confocal images of ischemia/reperfusion 72 hours (I/R-72) cerebral cortices double immunostained for glial fibrillary acidic protein (GFAP) and collagen type IV (Coll IV) (A) and single stained for Coll IV (B, C, D). (A) Astrogliosis and activated microvessels are common at this time of cerebral cortical reperfusion; there are many endothelial cell nuclei in the wall of a growing microvessel (A, arrows). (B–D) Leading tip-like endothelial cells (arrows) are revealed by the associated Coll IV vascular basal lamina (VBL). Nuclear counterstaining TO-PRO3. Bars: A–C = 10 μm; D = 5 μm
Figure 8.

Confocal images of ischemia/reperfusion 72 hours (I/R-72) cerebral cortices double immunostained for glial fibrillary acidic protein (GFAP) and collagen type IV (Coll IV) (A–C) and on Coll IV single channel (D–F). (A, B) Activated microvessels show a thick Coll IV vascular basal lamina (VBL) with embedded pericytes (arrows), and tube-like vessel structures completely ensheathed by perivascular astrocytes (arrowheads). The single channel from these pictures (green; D, E) show pericytes located within the VLM (arrows) and vessel tubes (arrowheads) are clearly recognizable. (C, F) A pair of astrocytes (asterisk in C) extends processes to a filopodia-like protrusion of a pericyte (arrowhead in channel green, F). Bars: 10 μm.
The cell proliferation marker Ki67 was examined by immunohistochemical double staining along with GFAP and Coll IV to determine the tendency of the I/R-72 cerebral cortical astrocytes to exhibit hypertrophic/hyperplastic changes and to determine ability of the microvessels to grow by means of angiogenic mechanisms. The cerebral cortices from I/R-72 fetuses displayed numerous Ki67-reactive nuclei in both vascular and parenchymal cellular elements (Fig. 9) vs. the Non-I/R sham-control and I/R-48 groups, which showed extremely rare Ki67-immunostained nuclei (data not shown). Ki67-reactive endothelial nuclei were frequently identified in small blood vessels surrounded by a hypertrophic strongly GFAP-reactive perivascular astroglia in the Ki67/GFAP-labeled cerebral cortices of the I/R-72 group (Fig. 9A–D). Growing microvessels were observed in close relationship to a large number of Ki67-labeled proliferating astrocytes (Fig. 9A–D). Doublets of intensely stained hypertrophic, proliferating astrocytes stained with both GFAP and Ki67 were frequently observed at these sites. The findings of doublets of astrocytes strongly support the contention that there is a pool of astrocytes capable of entering the mitotic cell cycle and that proliferative events can be detected 72 hours after ischemia in the fetal brain.
Figure 9.

Confocal images of ischemia/reperfusion 72 hours (I/R-72) cerebral cortices double immunostained for glial fibrillary acidic protein (GFAP) and Ki67 (A–D) and collagen type IV (Coll IV) and Ki67 (E, F). (A) Sparse Ki67-reactive astrocytes in the outer cerebral cortical layers (arrows) and a cerebral cortical microvessel showing 2 Ki67-labeled endothelial cells (arrowheads); note the pair of astrocytes or doublets (asterisks); one is located close to the vessel wall and Ki67-reactive endothelial nuclei can be distinguished more clearly in the enlargement (C, asterisk and arrowheads, respectively). (B) A microvessel located deeper within the cerebral cortex ensheathed by Ki67-reactive perivascular astrocytes (arrows) also showing a proliferating endothelial cell (arrowhead); note a pair of Ki67-labeled perivascular cells (asterisks); the one closest to the vessel wall is shown in the enlargement (D; asterisk), along with the Ki67-reactive endothelial cell (arrowhead) and 2 proliferating astrocytes (arrows). (E, F) Cerebral cortical microvessels, whose profile is revealed by Coll IV (asterisk), show Ki67-reactive endothelial cells (arrowheads) and in (E), Ki67-labeled nuclei in the surrounding parenchyma (arrows). Bars: A, B = 25 μm; C, D = 10 μm; E, F = 7.5 μm
Double staining for Coll IV and Ki67 identified proliferating endothelial cells, confirming that cerebral cortex microvessels were frequently characterized by mitotic events in I/R-72 animals (Fig. 9E, F). Variations in the intensity of the Ki67 signal within the karyoplasm ranging from weak to intense staining also are consistent with the presence of early and late mitotic phases of the cell cycle, respectively (Fig. 9A–F) (35).
In addition, we investigated cell death in the current work based upon apoptosis using standard morphological criteria under light microscopy at 1000x magnification on Toluidine blue-stained sections. After an initial survey, cells that showed cell shrinkage and nuclear condensation or fragmentation were considered identifiable as apoptotic cells, but were only apparent in a few areas of the parenchyma in the frontal cortex. Moreover, apoptosis was not identified in the microvessels that had been exposed to ischemia and reperfusion for 48 or 72 hours.
Correlational Analysis between Neovascularization, Pathological Scores, and FGF-2 Protein Concentration in Frontal Cerebral Cortex
As previously reported, ischemia-reperfusion increases pathological injury scores of coronally sliced whole brain sections when stained with Luxol fast blue-hematoxylin and eosin (29). Ischemia for 30 minutes with reperfusion for 48 and 72 hours resulted in greater damage across 6 brain slices when compared with brain slices from the sham treated control group (29). In our former work, nuclear pyknosis, cytoplasmic reddening, and hyperchromatism were considered indicative of neuronal injury (29). Evidence of ischemic damage was also apparent in the frontal cortex, the brain region in which the percentages of Coll IV-stained vessels were increased in the current study. We did not identify a significant correlation between our previously reported pathological scores (29) and the percentage of Coll IV-stained vessels within the same animals, however (r = 0.44, n = 10, p = 0.20).
Frontal cerebral cortical concentrations of FGF-2 were available from 3 animals in the Non-I/R, 3 in the I/R-48 and 2 in the I/R-72 groups, for which the quantitative morphometric analysis of measurements of percentage of Coll IV expressed as the mean area fraction, were performed. The area fractional vessel density (%) in the frontal cerebral cortex showed a direct linear correlation with concentration of FGF-2 (FGF-2/mg total protein) in the cerebral cortex (r = 0.81, n = 8, p < 0.02, Fig. 10), suggesting that increases in FGF-2 in the cerebral cortex were associated with increases in vessels density after ischemia.
Figure 10.

The area fractional vessel density (%) of the frontal cerebral cortex plotted on the y-axis against the frontal cerebral cortical concentration of basic fibroblast growth factor (FGF-2/mg total protein on the x-axis) on the x-axis, r = 0.81, n = 8, p = 0.015.
DISCUSSION
The purpose of the current study was to determine whether ischemia-reperfusion induces neovascularization in the cerebral cortex of the ovine fetus and to establish the relative time course for the commencement of neovascularization after injury in the fetal brain. There are 3 main findings of this study. First, collagen IV-labeled reactive sprouting-type microvessels are apparent, though sparse within 48 hours after ischemia. Second, these reactive sprouting-type microvessels become a common feature by 72 hours after ischemic brain injury in the fetus, suggesting that increased neovascularization occurs within 72 hours after ischemia in the fetal cerebral cortex. Third, increases in cerebral cortical FGF-2 are associated with increases in neovascularization in the ovine cerebral cortex. Consequently, increased neovascularization represents a component of ischemic injury in the fetal brain.
The neurodevelopment of the immature ovine brain is similar to that of premature human infants with respect to completion of neurogenesis, onset of cerebral sulcation, and detection of evoked potentials (24, 36–38). Full-term pregnancy in sheep is 145 to 148 days of gestation. We examined fetal sheep at 123 to 129 days of gestation, which represents approximately 85% of the ovine gestation and, therefore, at a time in gestation that is approximately similar to late preterm and near term human infants (24). Although rodents are frequently used to study brain development and injury, the rodent brain is immature at birth (39) and almost completely agyric. In contrast, similar to non-human primate and the human brain, the sheep brain develops prenatally and is gyrencephalic. Therefore, studies of neovascularization after ischemic-reperfusion injury in the ovine fetus at 85% of gestation could have relevance to events occurring in near-term infants after brain injury (25, 29).
Neurogenesis and angiogenesis can occur in response to ischemic brain injury and could potentially be enhanced by processes that attenuate brain injury (40). In vitro evidence suggests that endothelial cells can secrete active substances that influence the neuronal survival and thereby facilitate neuroprotective effects after ischemic insults (41). The immunohistochemical and morphometric analyses in the current study demonstrated that compared to controls, the mean vessel density was significantly greater in the fetuses 72 hours after ischemia than in fetuses 48 hours after ischemia, and that there was a trend in microvessel activation also suggested by evidence of endothelial cell/pericyte activation and proliferation. These findings were confirmed by endothelial reactivity for the proliferative cellular marker Ki67 and by clearly recognizable sprouting-type microvessels. Consequently, cerebral neovascularization appears to occur within 72 hours after ischemia in the fetal cerebral cortex.
The reactive astrocytosis surrounding cerebral cortical microvessels after ischemia/reperfusion exhibited classical features of increased GFAP expression and cytoplasmic hypertrophy as well as an increase in proliferative activity. This elevated proliferative index was confirmed by the occurrence of Ki67-reactive astrocytic nuclei together with presence of Ki67 immunoreactivity in doublet astrocytes. Similar changes in the proliferation of perivascular astrocytes have recently been described using live imaging ‘in vivo’ after brain injury (42). Such studies suggest that there are specific groups of perivascular astrocytes involved in this process and that these cells may share similarities with perivascular astrocytic proliferation observed following a wound lesion in the brain (43). However, the effects of the observed early neovascularization and astrocytic proliferation upon neurogenesis in the fetal brain remain to be determined.
Although very few studies have focused on the microvascular responses of the near-term fetal brain to injury (18, 23), several previous studies have suggested that the onset of angiogenesis begins within several days after ischemic brain injury both in adults and in neonatal rodents; these findings are based on results from a variety of models including adult and neonatal stroke (14, 44), prolonged hypoxia (16) and hypoxia-ischemia (15). Angiogenesis-related genes, such as VEGF, were upregulated within minutes after focal cerebral ischemia in adult rodents, generating proteins that remain elevated in astrocytes in the ischemic area for days to weeks (14). In addition to the presence of increased VEGF, endothelial cell proliferation has been reported to occur within 12 to 24 hours after stroke and continues for a number of weeks (14). Similarly, angiogenesis related genes and proteins, including hypoxia-inducible factor-1α (HIF-1α) and VEGF, increase within hours after stroke in neonatal rodents (44). The number of HIF-1α-positive cells began to increase at 4 hours, peaked at 8 hours, and decreased at 24 hours after transiently occluding the middle cerebral artery for 1.5 hours in neonatal rats (44). Increases in VEGF mRNA and VEGF protein appear to correlate with angiogenesis in postnatally developing rat brains (16, 44). Vessel density also appeared to increase within 3 days after hypoxic-ischemic injury in neonatal rats (15). Therefore, our findings in the ovine fetus after ischemia-reperfusion combined with the previous findings in neonatal rodents suggest that the onset neovascularization can occur within 48 hours after injury both in the fetus and neonate, and that neovascularization occurs after stroke (44) and hypoxia (16) in neonatal rodents, and ischemia-reperfusion brain injury in the fetus.
Our findings are consistent with a recent report showing significant increases in the percentage of blood vessels expressing VEGF in the subventricular zone and in periventricular and subcortical white matter at 24 and 48 hours after umbilical cord occlusion in the ovine fetus (18). Umbilical cord occlusion has recently been shown to result in decreased vascular density in the caudate nucleus along with shift in the frequency of smaller to larger perimeter blood vessels in periventricular and subcortical white matter, whereas both blood vessel density and morphology remain stable up to 48 hours after ischemia in the cerebral cortex (23). These findings combined with our results, which indicate an increased rate of cerebral cortical neo-vessel formation 72 hours after ischemia, suggest that although perturbations in the microvasculature including vessel density, size and structure can be detected after both ischemia induced by carotid artery occlusion and umbilical cord occlusion in the fetal brain within 48 hours after an insult (23), features of hypoxia-induced neuro-angiogenesis appear at a later time.
A limitation of the current study is that ovine antibodies were not available to detect species-specific antigens that could reveal endothelial cell/pericyte activation and vascular sprout formation (e.g. endoglin/CD105 and proteoglycan NG2, respectively), as we previously have reported for human angiogenesis (11, 45, 46). Consequently, more selective markers to detect the onset of neovascularization could not be performed. Nonetheless, by immunostaining for Coll IV as an early component of the VBL we were able to detect vascular sprouting after ischemia in the fetal sheep brain, which appeared similar to the vascular sprouting that we have previously reported during angiogenesis in the human fetal brain. In fact, immunofluorescence data demonstrated that deposition by endothelial cells of collagen type IV in the VBL is a rather early event during angiogenesis and that discontinuities in the VBL in the tip of growing vessels may be smaller than previously thought (47). On this basis, although adequate markers were not available to localize vascular sprout formation in the ovine brain specifically, our findings are consistent with previous work documenting endothelial/pericyte mobilization in actively growing vascular sprouts (12, 34, 45), and are consistent with the similar conclusions in a recent report in which increased vascular VEGF expression was reported in several brain regions after fetal exposure to umbilical cord occlusion (18). Administration of this angiogenic factor into the adult rat brain also resulted in unanticipated proliferative effects on astroglia (48). Although endothelial cells do not represent the major source of VEGF, they do have the capacity to release this growth factor in microvessels (49), and thereby stimulate the marked proliferative responses that we observed in perivascular astrocytes. However, disruption or loss of basal lamina components has been reported to occur after ischemia resulting in damage or loss of microvascular integrity, (50–52). In contrast to these findings in adult rats, we observed increased thickness of Coll IV-stained VBL in fetal sheep after ischemia. Therefore, discrepancies between the former studies in the adult rats and our finding in fetal sheep should be considered in view of the different experimental models examined. First, there are differences in the duration of ischemia and reperfusion, the brain regions analyzed, and microvasculature organization and features. Second, there could be differences in the vascular reactivity between the fetal and adult brain, particularly when considering aged animals. Third, there could be differences in the response of immature endothelial cells and pericytes to sudden or extreme shifts in cerebral cortical microcirculatory flow. Therefore, our present results in the fetal brain after ischemia do not support the contention that there is a loss of the basal lamina components, as previously reported in the adult rat (50–52). Additional ultrastructural analysis would be required to delineate the dynamic relationships that take place between vascular cellular and non-cellular components during ischemia/reperfusion in the developing cerebral cortex.
FGF-2 has been proposed to induce angiogenesis after ischemia in the adult brain. In adult patients after ischemic stroke, FGF-2 has been detected in neurons, astrocytes, and endothelial cells (53). In ischemic areas, its expression is elevated as compared to infarcted tissue and the normal contralateral hemisphere (53). In a study of 30 patients with acute cerebral infarction, serum bFGF levels increased significantly after infarction, peaking on day 3 and persisting until day 14, and showed a positive correlation between peak bFGF levels and neurological deficit improvements (54). Similarly, FGF-2 was found to sustain blood vessels in vitro and maintain tight junction composition in neonatal mouse brain (55). The vascular architecture of the neonatal mouse brain was more complex and the vessels appeared as continuous structures as early as d 3, indicating a role for FGF-2 in angiogenesis (55). We previously reported that the concentration of FGF-2 increased in proportion to the severity of brain damage in the cerebral cortex of fetal sheep, peaking after 72 hours of reperfusion; we speculated that FGF-2 played a role in the attenuation of further injury in the fetus (29). The present study demonstrated a direct linear correlation between neovascularization as quantified by Coll IV expression and FGF-2 concentration using our previously published FGF-2 measurements (29). The correlation between area fractional vessel density (%) in the frontal cerebral cortex and FGF-2 protein concentration and Coll IV suggests that increases in FGF-2 are also associated with increased neovascularization. Our findings in the fetal brain are consistent with the previous work suggesting that FGF-2 can induce angiogenesis after ischemia in the adult brain (55).
In conclusion, neovascularization occurs within 72 hours after exposure of the fetal brain to ischemia-reperfusion. Increases in cerebral cortical FGF-2 may be involved in this process. Our findings taken together with recent work (4, 18, 23) suggest that perturbations in the microvasculature appear to represent an important component of hypoxic-ischemic injury to the fetal brain.
Acknowledgments
Supported by National Institute of General Medical Sciences of the National Institutes of Health (NIH) 1R01-HD-057100 and American Heart Association (AHA) -13POST1680015 to J.Z.
Footnotes
The authors have no duality or conflicts of interests to declare.
The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health
References
- 1.Vannucci RC. Hypoxic-ischemic encephalopathy. Am J Perinatol. 2000;17:113–20. doi: 10.1055/s-2000-9293. [DOI] [PubMed] [Google Scholar]
- 2.Volpe JJ. Neonatal encephalopathy: an inadequate term for hypoxic-ischemic encephalopathy. Ann Neurol. 2012;72:156–66. doi: 10.1002/ana.23647. [DOI] [PubMed] [Google Scholar]
- 3.Ferriero DM. Neonatal brain injury. N Engl J Med. 2004;351:1985–95. doi: 10.1056/NEJMra041996. [DOI] [PubMed] [Google Scholar]
- 4.Chen X, Threlkeld SW, Cummings EE, et al. Ischemia-reperfusion impairs blood-brain barrier function and alters tight junction protein expression in the ovine fetus. Neuroscience. 2012;226:89–100. doi: 10.1016/j.neuroscience.2012.08.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Stanley FJ. The aetiology of cerebral palsy. Early Hum Dev. 1994;36:81–8. doi: 10.1016/0378-3782(94)90035-3. [DOI] [PubMed] [Google Scholar]
- 6.Pharoah PO. The epidemiology of chronic disability in childhood. Int Rehabil Med. 1985;7:11–17. doi: 10.3109/03790798509165973. [DOI] [PubMed] [Google Scholar]
- 7.Cooke RW. Cerebral palsy in very low birthweight infants. Arch Dis Child. 1990;65:201–6. doi: 10.1136/adc.65.2.201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Leviton A, Paneth N. White matter damage in preterm newborns--an epidemiologic perspective. Early Hum Dev. 1990;24:1–22. doi: 10.1016/0378-3782(90)90002-z. [DOI] [PubMed] [Google Scholar]
- 9.Harrigan MR. Angiogenic factors in the central nervous system. Neurosurgery. 2003;53:639–60. doi: 10.1227/01.neu.0000079575.09923.59. [DOI] [PubMed] [Google Scholar]
- 10.Ballabh P, Xu H, Hu F, et al. Angiogenic inhibition reduces germinal matrix hemorrhage. Nat Med. 2007;13:477–85. doi: 10.1038/nm1558. [DOI] [PubMed] [Google Scholar]
- 11.Virgintino D, Girolamo F, Errede M, et al. An intimate interplay between precocious, migrating pericytes and endothelial cells governs human fetal brain angiogenesis. Angiogenesis. 2007;10:35–45. doi: 10.1007/s10456-006-9061-x. [DOI] [PubMed] [Google Scholar]
- 12.Risau W. Mechanisms of angiogenesis. Nature. 1997;386:671–4. doi: 10.1038/386671a0. [DOI] [PubMed] [Google Scholar]
- 13.Beck H, Plate KH. Angiogenesis after cerebral ischemia. Acta Neuropathol. 2009;117:481–96. doi: 10.1007/s00401-009-0483-6. [DOI] [PubMed] [Google Scholar]
- 14.Navaratna D, Guo S, Arai K, et al. Mechanisms and targets for angiogenic therapy after stroke. Cell Adh Migr. 2009;3:216–23. doi: 10.4161/cam.3.2.8396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Xiong Y, Mahmood A, Chopp M. Angiogenesis, neurogenesis and brain recovery of function following injury. Curr Opin Investig Drugs. 2010;11:298–308. [PMC free article] [PubMed] [Google Scholar]
- 16.Ogunshola OO, Stewart WB, Mihalcik V, et al. Neuronal VEGF expression correlates with angiogenesis in postnatal developing rat brain. Brain Res Dev Brain Res. 2000;119:139–53. doi: 10.1016/s0165-3806(99)00125-x. [DOI] [PubMed] [Google Scholar]
- 17.Shimotake J, Derugin N, Wendland M, et al. Vascular endothelial growth factor receptor-2 inhibition promotes cell death and limits endothelial cell proliferation in a neonatal rodent model of stroke. Stroke. 2010;41:343–9. doi: 10.1161/STROKEAHA.109.564229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Baburamani AA, Castillo-Melendez M, Walker DW. VEGF expression and microvascular responses to severe transient hypoxia in the fetal sheep brain. Pediatr Res. 2013;73:310–16. doi: 10.1038/pr.2012.191. [DOI] [PubMed] [Google Scholar]
- 19.Feng Y, Rhodes PG, Bhatt AJ. Neuroprotective effects of vascular endothelial growth factor following hypoxic ischemic brain injury in neonatal rats. Pediatr Res. 2008;64:370–4. doi: 10.1203/PDR.0b013e318180ebe6. [DOI] [PubMed] [Google Scholar]
- 20.Liu HM. Correlation between proto-oncogene, fibroblast growth factor and adaptive response in brain infarct. Prog Brain Res. 1995;105:239–44. doi: 10.1016/s0079-6123(08)63300-2. [DOI] [PubMed] [Google Scholar]
- 21.Lyons MK, Anderson RE, Meyer FB. Basic fibroblast growth factor promotes in vivo cerebral angiogenesis in chronic forebrain ischemia. Brain Res. 1991;558:315–20. doi: 10.1016/0006-8993(91)90784-s. [DOI] [PubMed] [Google Scholar]
- 22.Marcoux FW, Weber ML, Probert AW, Jr, et al. Hypoxic neurodegeneration in culture: calcium influx, electron microscopy, and neuroprotection with excitatory amino acid antagonists. Ann N Y Acad Sci. 1992;648:303–5. doi: 10.1111/j.1749-6632.1992.tb24563.x. [DOI] [PubMed] [Google Scholar]
- 23.Baburamani AA, Lo C, Castillo-Melendez M, et al. Morphological evaluation of the cerebral blood vessels in the late gestation fetal sheep following hypoxia in utero. Microvasc Res. 2013;85:1–9. doi: 10.1016/j.mvr.2012.09.007. [DOI] [PubMed] [Google Scholar]
- 24.Back SA, Riddle A, Hohimer AR. Role of instrumented fetal sheep preparations in defining the pathogenesis of human periventricular white-matter injury. J Child Neurol. 2006;21:582–9. doi: 10.1177/08830738060210070101. [DOI] [PubMed] [Google Scholar]
- 25.Gunn AJ, Gunn TR, de Haan HH, et al. Dramatic neuronal rescue with prolonged selective head cooling after ischemia in fetal lambs. J Clin Invest. 1997;99:248–56. doi: 10.1172/JCI119153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Riddle A, Luo NL, Manese M, et al. Spatial heterogeneity in oligodendrocyte lineage maturation and not cerebral blood flow predicts fetal ovine periventricular white matter injury. J Neurosci. 2006;26:3045–55. doi: 10.1523/JNEUROSCI.5200-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Stonestreet BS, Sadowska GB, McKnight AJ, et al. Exogenous and endogenous corticosteroids modulate blood-brain barrier development in the ovine fetus. Am J Physiol Regul Integr Comp Physiol. 2000;279:R468–77. doi: 10.1152/ajpregu.2000.279.2.R468. [DOI] [PubMed] [Google Scholar]
- 28.Stonestreet BS, Petersson KH, Sadowska GB, et al. Antenatal steroids decrease blood-brain barrier permeability in the ovine fetus. Am J Physiol. 1999;276:R283–9. doi: 10.1152/ajpregu.1999.276.2.R283. [DOI] [PubMed] [Google Scholar]
- 29.Petersson KH, Pinar H, Stopa EG, et al. White matter injury after cerebral ischemia in ovine fetuses. Pediatr Res. 2002;51:768–76. doi: 10.1203/00006450-200206000-00019. [DOI] [PubMed] [Google Scholar]
- 30.Virgintino D, Robertson D, Benagiano V, et al. Immunogold cytochemistry of the blood-brain barrier glucose transporter GLUT1 and endogenous albumin in the developing human brain. Brain Res Dev Brain Res. 2000;123:95–101. doi: 10.1016/s0165-3806(00)00086-9. [DOI] [PubMed] [Google Scholar]
- 31.Arenillas JF, Moro MA, Davalos A. The metabolic syndrome and stroke: potential treatment approaches. Stroke. 2007;38:2196–2203. doi: 10.1161/STROKEAHA.106.480004. [DOI] [PubMed] [Google Scholar]
- 32.Williams CE, Gunn AJ, Synek B, et al. Delayed seizures occurring with hypoxic-ischemic encephalopathy in the fetal sheep. Pediatr Res. 1990;27:561–5. doi: 10.1203/00006450-199006000-00004. [DOI] [PubMed] [Google Scholar]
- 33.Unno N, Wu WX, Ding XY, et al. The effects of fetal adrenalectomy at 110 days gestational age on AVP and CRH mRNA expression in the hypothalamic paraventricular nucleus of the ovine fetus. Brain Res Dev Brain Res. 1998;106:119–28. doi: 10.1016/s0165-3806(97)00203-4. [DOI] [PubMed] [Google Scholar]
- 34.Virgintino D, Ozerdem U, Girolamo F, et al. Reversal of cellular roles in angiogenesis: implications for anti-angiogenic therapy. J Vasc Res. 2008;45:129–31. doi: 10.1159/000109965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Braun N, Papadopoulos T, Muller-Hermelink HK. Cell cycle dependent distribution of the proliferation-associated Ki-67 antigen in human embryonic lung cells. Virchows Arch B Cell Pathol Incl Mol Pathol. 1988;56:25–33. doi: 10.1007/BF02889998. [DOI] [PubMed] [Google Scholar]
- 36.Barlow RM. The foetal sheep: morphogenesis of the nervous system and histochemical aspects of myelination. J Comp Neurol. 1969;135:249–62. doi: 10.1002/cne.901350302. [DOI] [PubMed] [Google Scholar]
- 37.Bernhard CG, Kolmodin GM, Meyerson BA. On the prenatal development of function and structure in the somesthetic cortex of the sheep. Prog Brain Res. 1967;26:60–77. doi: 10.1016/S0079-6123(08)61419-3. [DOI] [PubMed] [Google Scholar]
- 38.Cook CJ, Gluckman PD, Johnston BM, et al. The development of the somatosensory evoked potential in the unanaesthetized fetal sheep. J Dev Physiol. 1987;9:441–55. [PubMed] [Google Scholar]
- 39.Dobbing J, Sands J. Comparative aspects of the brain growth spurt. Early Hum Dev. 1979;3:79–83. doi: 10.1016/0378-3782(79)90022-7. [DOI] [PubMed] [Google Scholar]
- 40.Ferriero DM, Miller SP. Imaging selective vulnerability in the developing nervous system. J Anat. 2010;217:429–35. doi: 10.1111/j.1469-7580.2010.01226.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hua Q, Qing X, Li P, et al. Brain microvascular endothelial cells mediate neuroprotective effects on ischemia/reperfusion neurons. J Ethnopharmacol. 2010;129:306–13. doi: 10.1016/j.jep.2010.03.024. [DOI] [PubMed] [Google Scholar]
- 42.Bardehle S, Kruger M, Buggenthin F, et al. Live imaging of astrocyte responses to acute injury reveals selective juxtavascular proliferation. Nat Neurosci. 2013;16:580–6. doi: 10.1038/nn.3371. [DOI] [PubMed] [Google Scholar]
- 43.Cavanagh JB. The proliferation of astrocytes around a needle wound in the rat brain. J Anat. 1970;106:471–87. [PMC free article] [PubMed] [Google Scholar]
- 44.Mu D, Jiang X, Sheldon RA, et al. Regulation of hypoxia-inducible factor 1alpha and induction of vascular endothelial growth factor in a rat neonatal stroke model. Neurobiol Dis. 2003;14:524–34. doi: 10.1016/j.nbd.2003.08.020. [DOI] [PubMed] [Google Scholar]
- 45.Virgintino D, Errede M, Girolamo F, et al. Fetal blood-brain barrier P-glycoprotein contributes to brain protection during human development. J Neuropathol Exp Neurol. 2008;67:50–61. doi: 10.1097/nen.0b013e31815f65d9. [DOI] [PubMed] [Google Scholar]
- 46.Virgintino D, Rizzi M, Errede M, et al. Plasma membrane-derived microvesicles released from tip endothelial cells during vascular sprouting. Angiogenesis. 2012;15:761–9. doi: 10.1007/s10456-012-9292-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ekblo P, Klein G, Ekblom M. The basement membrane of embryonic blood vessels. In: Feinberg RN, Sherer GK, Auerbach R, editors. The Development of the Vascular System. Vol. 14. Basel: Kager; 1991. pp. 81–2. [Google Scholar]
- 48.Krum JM, Mani N, Rosenstein JM. Angiogenic and astroglial responses to vascular endothelial growth factor administration in adult rat brain. Neuroscience. 2002;110:589–604. doi: 10.1016/s0306-4522(01)00615-7. [DOI] [PubMed] [Google Scholar]
- 49.Vega-Diaz B, Herron GS, Michel S. An autocrine loop mediates expression of vascular endothelial growth factor in human dermal microvascular endothelial cells. J Invest Dermatol. 2001;116:525–30. doi: 10.1046/j.1523-1747.2001.01294.x. [DOI] [PubMed] [Google Scholar]
- 50.Hamann GF, Okada Y, Fitridge R, et al. Microvascular basal lamina antigens disappear during cerebral ischemia and reperfusion. Stroke. 1995;26:2120–6. doi: 10.1161/01.str.26.11.2120. [DOI] [PubMed] [Google Scholar]
- 51.Hamann GF, Liebetrau M, Martens H, et al. Microvascular basal lamina injury after experimental focal cerebral ischemia and reperfusion in the rat. J Cereb Blood Flow Metab. 2002;22:526–33. doi: 10.1097/00004647-200205000-00004. [DOI] [PubMed] [Google Scholar]
- 52.Vosko MR, Burggraf D, Liebetrau M, et al. Influence of the duration of ischemia and reperfusion on infarct volume and microvascular damage in mice. Neurol Res. 2006;28:200–5. doi: 10.1179/016164105X48789. [DOI] [PubMed] [Google Scholar]
- 53.Issa R, AlQteishat A, Mitsios N, et al. Expression of basic fibroblast growth factor mRNA and protein in the human brain following ischaemic stroke. Angiogenesis. 2005;8:53–62. doi: 10.1007/s10456-005-5613-8. [DOI] [PubMed] [Google Scholar]
- 54.Guo H, Huang L, Cheng M, et al. Serial measurement of serum basic fibroblast growth factor in patients with acute cerebral infarction. Neurosci Lett. 2006;393:56–9. doi: 10.1016/j.neulet.2005.09.043. [DOI] [PubMed] [Google Scholar]
- 55.Bendfeldt K, Radojevic V, Kapfhammer J, et al. Basic fibroblast growth factor modulates density of blood vessels and preserves tight junctions in organotypic cortical cultures of mice: a new in vitro model of the blood-brain barrier. J Neurosci. 2007;27:3260–7. doi: 10.1523/JNEUROSCI.4033-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]