

Abstract
Mid-gestation stage mouse embryos were cultured utilizing a serum-free culture medium prepared from commercially available stem cell media supplements in an oxygenated rolling bottle culture system. Mouse embryos at E10.5 were carefully isolated from the uterus with intact yolk sac and in a process involving precise surgical maneuver the embryos were gently exteriorized from the yolk sac while maintaining the vascular continuity of the embryo with the yolk sac. Compared to embryos prepared with intact yolk sac or with the yolk sac removed, these embryos exhibited superior survival rate and developmental progression when cultured under similar conditions. We show that these mouse embryos, when cultured in a defined medium in an atmosphere of 95% O2 / 5% CO2 in a rolling bottle culture apparatus at 37 °C for 16-40 hr, exhibit morphological growth and development comparable to the embryos developing in utero. We believe this method will be useful for investigators needing to utilize whole embryo culture to study signaling interactions important in embryonic organogenesis.
Keywords: Developmental Biology, Issue 85, mouse embryo, mid-gestation, serum-free, defined media, roller culture, organogenesis, development
Introduction
In vitro culture methods utilizing whole embryos are well suited to study signaling mechanisms involved in embryonic organogenesis that are otherwise difficult to access in utero. Whole embryos provide the tissue integrity and support appropriate for the tissue interactions that are crucial for timely occurrence of signaling mechanisms essential for different cellular processes during organogenesis. While whole embryo cultures provide a platform for a plethora of applications such as transplantation studies, genetic and tissue manipulations, bead implantation studies, toxicological studies, etc., currently utilized embryo culture systems are mainly dependent on serum for proper growth and maintenance of the embryos in the culture1-9.
Serum has been utilized as one of the major components ranging from 10-100% of the culture media6-8,10,11. However, the composition of serum is not well defined and can differ from animal to animal and each time the serum is collected. While laboratory preparation of serum is time consuming and involves stringent procedures, serum procured commercially exhibits considerable variability among different lots and raises experimental costs. Added to these, the serum may contain unknown factors such as growth factors, hormones, or other proteins, which can potentially affect the outcome of certain experiments, especially those that involve the study of signaling molecules important in tissue interactions. Studies have shown that addition of serum to culture can potentially alter the intracellular levels of certain signaling molecules such as cyclic adenosine monophosphate (cAMP) and proteins involved in mitogenic signaling and phosphoinositide 3 (PI 3)-kinase signaling pathways12-14. Contrary to these, a serum-free culture system provides the advantages of antigen free environment, abstinence from biologically active enzymes that can alter the cellular processes and enables consistency among experiments.
In the present study, we utilized a serum-free culture medium prepared from commercially available stem cell media supplements to culture mid-gestation stage whole embryos in an atmosphere of 95% O2 / 5% CO2 in a rolling bottle culture apparatus at 37 °C15,16. Mouse embryos cultured for 16 to 40 hr under these defined conditions exhibited progression in morphological development of overall embryonic body and different structures such as the heart, limbs, brain and eyes indicating appropriate levels of cellular proliferation, migration, differentiation and tissue interactions. Molecular analysis of the embryonic development in the culture for one of the complex organ systems such as the eye revealed the ocular development to be consistent with that observed in ocular tissue in embryos developing in utero (Kalaskar and Lauderdale, in preparation). Thus we show that mouse embryos cultured at mid-gestation stage, exhibit progressive growth and morphological development comparable to that observed in embryos developing in utero.
Protocol
Mouse embryo culture:
All experimental procedures were conducted in strict accordance with National Institutes of Health guidelines following protocol # A2010 07-119, which was reviewed and approved by the University of Georgia Institutional Animal Care and Use Committee, which maintains continued regulatory oversight.
1. Preparation of Culture Media
Prepare culture medium using commercially available stem cell media and supplements in the following amounts: KnockOut DMEM, KnockOut Serum Replacement (KSR) (10%), N-2 Supplement (1x), Albumin, from Bovine Serum (2%), Penicillin (50 IU/ml), Streptomycin (50 μg/ml) and Amphotericin-B (1.25 μg/ml). The culture medium prepared is similar to that previously described15 with the following changes: addition of anti-mycotics and using twice the concentration of antibiotics (for details of specific reagents and equipment, see List of Materials). Note: Antibiotic concentration as previously used (Moore-Scott, et al.15) was sufficient for embryo cultures carried up to 24 hr time period. However, when culture was continued beyond 24 hr, we experienced contamination of the culture and this was successfully controlled by doubling the antibiotic concentration.
Store the media components at 4 °C or at -20 °C as per manufacturer recommendations. KSR and N-2 Supplement should be stored in aliquots at -20 °C to avoid repeated freezing and thawing. KnockOut DMEM once opened should be utilized within 30 days as per manufacturer’s recommendation. Prior to use, warm the components in a water bath to 37 °C.
Prepare the media under sterile conditions. Disinfect the surface of all the equipment and bottles containing the media components with 70% alcohol spray before placing them in the culture hood.
The media components can be mixed either in a sterile beaker or directly in the filter system (Corning). First add the albumin powder to the KnockOut DMEM. Mix thoroughly by gently shaking until the albumin completely dissolves. Then add the N-2 Supplement, KSR and the antibiotics and mix thoroughly using a pipettor. Then filter sterilize the media using a 0.22 µm pore size filter with vacuum suction. Dispense the media into 50 ml conical tubes and store at 4 °C until used. The media once prepared should be used within 4-5 days for culture.
2. Preparation of Culture Equipment
Sterilize the glass culture bottles and rubber corks. Wrap the culture bottles in an aluminum foil and place the rubber bottle corks in a water beaker and sterilize by autoclaving.
Sterilize the culture chamber (Precision Incubator Unit) with 70% alcohol spray and warm to 37 °C before starting the culture. The culture chamber is equipped with a rolling disc at the center which holds the culture bottles. Gas flow into the chamber is regulated by a pressure gauge and the flow rate can be adjusted by observing the outflow of air bubbles through water in a glass tube at the end. This rolling bottle culture apparatus is a modified version of the original apparatus introduced by New and Cockroft17.
Connect a gas cylinder containing a gas mixture of 95% O2 / 5% CO2 to the culture chamber and adjust the gas flow to “one air bubble per second” which gives a flow rate of 50 cc/min and ensures adequate oxygen supply to the culture embryos.
3. Mouse Embryo Collection and Preparation for Culture
To obtain wild type mouse embryos, we utilized mice of C57BL/6J and CD-1 (Charles River Laboratories) genetic background strains.
Check the female mice for vaginal plugs every day morning. The noon on the day of finding the vaginal plug should be considered as E0.5 dpc (days post coitus).
Collect mouse embryos at E10.5 dpc by euthanizing the pregnant mice following standard protocols. Mice were euthanized using a CO2 inhalation device as specified by the American Veterinary Medical Association (AVMA) Guidelines for the Euthanasia of Animals (2013). To insure death, cervical dislocation was performed after exposure to CO2.
Spray 70% ethanol on the ventral abdominal surface to avoid sticking of abdominal hair to the instruments. Then open the abdominal cavity ventrally using a sharp-blunt operating scissors and a pair of 4-3/4 in microdissecting forceps to locate the uterus. Using a 4 in microdissecting forceps lift the entire uterus and separate it from the body by cutting with a light operating scissors at the uterine body and at the tips of the uterine horns.
Quickly rinse the entire uterus in 1x PBS warmed to 37 °C to remove any blood sticking to the uterus and immediately place in DMEM warmed to 37 °C in a Petri dish. Sterilize the instruments by 70% ethanol spray before further use. Note: Preheating the solutions including the PBS, DMEM and the culture medium and maintaining them at 37 °C during the entire procedure is recommended.
Under a dissecting microscope, the uterus should be segmentally dissected with a light operating scissors. This results in small openings on either side of the segmented uterus through which a pair of modified (blunted ends) microdissecting tweezers can be gently inserted to widen the opening. This permits the exposure of the placental decidua, which can then be removed by gently tearing with a pair of microdissecting tweezers to expose the parietal yolk sac (PYS) with the Reichert’s membrane. Using one edge of the tweezers gently pierce the Reichert’s membrane along with the PYS and separate from the underlying visceral yolk sac (VYS) layer to expose the embryos with intact VYS. The ectoplacental cone and the trophoectoderm derivatives can be removed either with the placental decidua or with the Reichert’s membrane. Care must be taken to avoid rupture of the VYS as the embryos pop out immediately.
The embryos with intact VYS should then be transferred to a Petri dish containing the culture medium warmed to 37 °C using a plastic transfer pipette. Note: Transfer to culture medium immediately after separation from uterus helps for better development of the embryo in culture.
A small opening should be made in the yolk sac with a pair of microdissecting tweezers avoiding major blood vessels. A sharp pair of tweezers can be used to make the opening by gently piercing in an area adjacent to the head region. Alternatively two pairs of blunt tweezers can be used to hold the yolk sac and gently tear to make a small opening. Then gently widen the opening by expanding with the tweezers to a size just enough to fit the embryonic head. The amniotic membrane which is closely wrapping the embryo should be gently held away from the body and torn using a pair of tweezers. The embryonic head and later the whole embryo should be gently exteriorized from the yolk sac while maintaining the integrity of the embryonic vasculature with that of the yolk sac15. Note: Any damage to the yolk sac vasculature can potentially affect the development of the embryo in culture. Such embryos with damaged vasculature should not be used for culture and can be used for staging the embryos which can later be discarded.
Embryonic staging criteria: Examine the embryos under the dissecting microscope and group by morphological criteria including body and head size, limb and eye morphology and stage by counting the number of somite(s) for at least two embryos from each group. Note: Usually embryos obtained from the same litter show differences in development in utero and differ in body size, morphological features and number of somites. However, we found that embryos grouped by our morphological criteria usually exhibited similar somite number (±1). For this reason, it is not required to count somites for each embryo as this would delay the time for starting the culture. Note: Do not use the embryos that were used to count the somites for culture. Count the somites after starting the culture for other embryos in order to avoid the time delay in starting the culture. The embryos that were delayed to culture after separation from the uterus usually exhibit poor development.
Transfer the embryos immediately to a Petri dish with fresh culture medium warmed to 37 °C and take it to the culture hood where the embryos should again be transferred to a Petri dish with sterile culture media before putting them into the culture bottles with medium to start the culture. Note: The multiple media transfers for the embryos before culture helps in preventing contamination as the different steps of embryo collection and dissection were performed under a dissecting microscope which was not installed in the culture hood. Note: When the litter size is large (>12 embryos), process half the embryos and start the culture or put them in a dish with culture medium and place it in a CO2 incubator at 37 °C. When embryos were left outside for longer periods (>35-40 min) we observed the heartbeat to slow down and this will affect the later development in the culture.
4. Culturing Mouse Embryos
Open the autoclaved culture bottles in the culture hood and properly label them. Transfer 3 ml of culture medium warmed to 37 °C to each of the bottles and then the mouse embryos should be transferred gently into the culture bottles using sterile plastic transfer pipettes. The culture bottles should then be capped with the rubber bottle corks which help to hold the culture bottles to the rolling disc in the culture apparatus. Note: The culture bottles with the media can be prepared and placed on the rolling disc of the culture apparatus before euthanizing the mice. This shortens the time for transfer of embryos into the culture bottles. Note: Mouse embryos can be cultured individually or as cocultures with two or three embryos in the same culture bottle.
The culture bottles should be aseptically carried to the culture apparatus at 37 °C and tightly hook them to the holes on the rolling disc with the rubber corks. Turn on the rolling disc to rotate at a constant speed of 35 rotations per minute (rpm), which enables the free floatation of the embryos in the media and also helps in free gas exchange by the embryonic tissue. The gas from the cylinder connected to the culture chamber flows through the rolling disc and the rubber corks into the culture bottles.
Culture the embryos for 16-40 hr and regularly check for the gas outflow and culture chamber temperature. Note: Mouse embryo manipulation in culture: Let the embryos get adapted to the culture conditions for at least 30 min. Then embryos that perform poorly and showing feeble heartbeat can be discarded and the remaining embryos can be utilized for further manipulation studies. Embryo manipulations such as electroporation5,9,16, microinjections18, bead implantation16, drug treatment and other procedures can be performed at this time. We successfully performed implantation of affi-gel agarose beads treated with certain signaling molecules to study their role in early eye development. The embryos after manipulations can be returned back in a fresh medium and continued in culture.
Replace the culture media totally with fresh media at specific intervals after 9-10 hr and 18-19 hr of culture when the culture was continued for 40 hr. Note: Culture media replacement may not be required if the culture is stopped by 16-18 hr.
Measures of success in culture: Embryonic survival in culture should be determined by visible heartbeat and blood circulation in the body while embryonic growth in culture should be assessed by increase in body size, somite number and morphological development of head, limbs, heart and eyes.
In utero developed embryos at E11.0 dpc (~40-41 s) and E12.0 dpc (~49-50 s) should be used as controls for comparing embryos cultured for 16-18 hr and 38-40 hr, respectively.
Embryonic development at different time points during culture and for in utero stages can be captured under dissecting microscope. Spot imaging software was utilized to capture the images and later processed using image processing software.
Representative Results
Development of mouse embryos ex utero depends on multiple factors starting from the time the uterus is isolated from the body to the time the embryos are cultured. As depicted in Figure 1, the procedure involves a series of steps including, separation of the gravid uterus from the body (Figure 1A), isolation of the embryos with intact yolk sac (Figure 1B), exteriorization of the embryos from the yolk sac (Figure 1C) and culturing the embryos in a serum-free media in an atmosphere of 95% O2 / 5% CO2 in a rolling bottle culture apparatus at 37 °C. Embryos exhibited 100% survival when transferred into culture media immediately after separation from the uterus. Under these culture conditions, mouse embryos when cultured for 16-40 hr exhibited growth and morphological development comparable to that observed in equivalent stage embryos developing in utero (Figure 2).
Embryos at E10.5 (~34-35 s) (Figure 2A) when cultured for 16-40 hr survived and advanced in development well beyond the stage of 35 s. The embryos after 16-18 hr in culture (Figures 2D and 3B; Table 1) added about 5-6 s exhibiting a growth rate of one somite for every two and a half hours in culture and were morphologically comparable to E11.0 (~40-41 s) embryos developed in utero (Figures 2B and 3A). Though the development was delayed by about an hour, these embryos in culture exhibited an overall increase in body size and a proportionate head with distinct brain vesicles. The embryos also showed developing limb buds, formation of a well chambered heart and appearance of pigmentation in the retinal pigmented epithelium. However, this type of development was observed in about 58% of the embryos cultured while 28% of the embryos exhibited an intermediate development (Figure 3C). These later embryos showed a smaller body size though they had a similar somite count compared to the in utero developed embryos. They had a relatively smaller head, although the brain vesicles were demarcated. The heart chambers were not distinct and the limb buds in some embryos exhibited dark areas at the extremities. At the other end of the spectrum, about 14% of the embryos exhibited poor development in the culture (Figure 3D). These embryos had shorter body and head sizes and showed dark areas in some parts of the body indicating degenerative changes. Though the heart was beating it was ill developed and lacked differentiation into chambers in some embryos while in others the limbs appeared dark and retarded in growth.
Embryos continued in culture for 38-40 hr (Figure 2E; Table 1) showed about 49-50 s and were comparable to E12.0 embryos (Figure 2C) developed in utero. Although delayed by 2-3 hr, these embryos in culture exhibited proportionate increase in body size and showed appropriate organogenesis and tissue differentiation as observed in the development of the pigmentation in the retinal pigmented epithelium and formation of a well demarcated snout in the head region. Though some parts at the extremities like the limb buds and the tail appeared to be under-developed, the embryos appeared to be developing comparable to the in utero embryos. However, this kind of development was observed in only 30-40% of the embryos cultured (Table 1) while the rest of them exhibited a range of developmental progression with areas of retarded growth or under-development in some parts of the body or the whole embryo.
Apart from the above differences in development, we observed a significant difference in development when the embryos were cocultured in the same culture bottle. Compared to the other embryo (Figures 4A and 4A’) in the coculture, the well-developed embryo (Figures 4B and 4B’) exhibited better growth in all aspects of the embryonic development. Major differences were observed in the overall body size and head size and sometimes in the heart and limb development. While the well-developed embryo from the coculture was morphologically comparable to the embryos developing in utero, the other embryo in the coculture has always appeared to be less developed compared to the well-developed embryo. This kind of difference in development was observed in 45% of the cocultures (N=27), while the rest of the cocultures (N=33) showed almost similar development in both the embryos in the coculture.
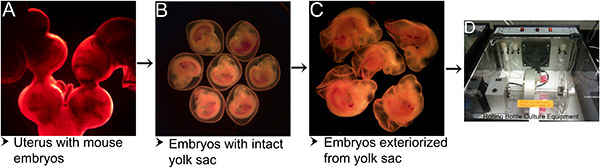 Figure 1. Steps in mouse embryo culture protocol. (A) Gravid uterus with embryos isolated from the mother mouse. (B) Embryos with intact yolk sac separated from decidua after segmental dissection of uterus. (C) Embryos exteriorized from the yolk sac. (D) Rolling bottle culture apparatus at 37 °C and supplied with 95% O2 / 5% CO2 for culturing mouse embryos. Please click here to view a larger version of this figure.
Figure 1. Steps in mouse embryo culture protocol. (A) Gravid uterus with embryos isolated from the mother mouse. (B) Embryos with intact yolk sac separated from decidua after segmental dissection of uterus. (C) Embryos exteriorized from the yolk sac. (D) Rolling bottle culture apparatus at 37 °C and supplied with 95% O2 / 5% CO2 for culturing mouse embryos. Please click here to view a larger version of this figure.
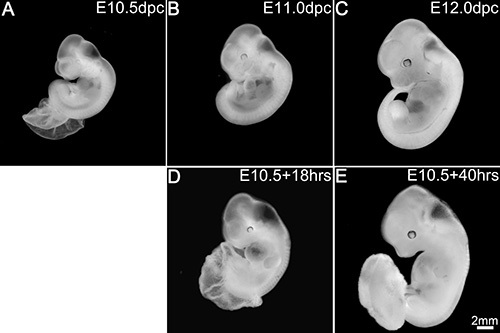 Figure 2. Embryos in culture replicate in utero development.
In utero development at E10.5 (A), E11.0 (B) and E12.0 (C). Development in culture after 18 hr (D) and 40 hr (E). Scale bar in (E) applies to all panels. Please click here to view a larger version of this figure.
Figure 2. Embryos in culture replicate in utero development.
In utero development at E10.5 (A), E11.0 (B) and E12.0 (C). Development in culture after 18 hr (D) and 40 hr (E). Scale bar in (E) applies to all panels. Please click here to view a larger version of this figure.
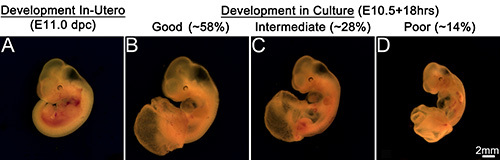 Figure 3. Embryonic development in culture. Embryo developed in utero at E11.0 dpc (A). Development in culture (B, C, D). (B) Representation of good embryonic development in culture. About 58% of total embryos cultured exhibit comparable development to in utero developed embryos. (C) Representation of intermediate development in culture. About 28% of embryos cultured show intermediate development. (D) Representation of poor embryonic development in culture. About 14% embryos show poor development in culture. Scale bar in (D) applies to all panels. Please click here to view a larger version of this figure.
Figure 3. Embryonic development in culture. Embryo developed in utero at E11.0 dpc (A). Development in culture (B, C, D). (B) Representation of good embryonic development in culture. About 58% of total embryos cultured exhibit comparable development to in utero developed embryos. (C) Representation of intermediate development in culture. About 28% of embryos cultured show intermediate development. (D) Representation of poor embryonic development in culture. About 14% embryos show poor development in culture. Scale bar in (D) applies to all panels. Please click here to view a larger version of this figure.
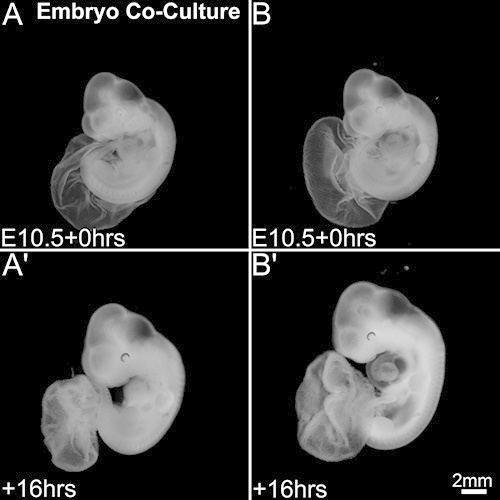 Figure 4. Developmental differences in mouse embryo coculture system. Embryos at the start of coculture (A, B). Embryonic development after 16 hr of coculture (A’, B’). One of the embryos in the coculture (A’) appears under-sized compared to the other embryo (B’) in the coculture. Scale bar in (B’) applies to all panels. Please click here to view a larger version of this figure.
Figure 4. Developmental differences in mouse embryo coculture system. Embryos at the start of coculture (A, B). Embryonic development after 16 hr of coculture (A’, B’). One of the embryos in the coculture (A’) appears under-sized compared to the other embryo (B’) in the coculture. Scale bar in (B’) applies to all panels. Please click here to view a larger version of this figure.
| Total embryos cultured for 16-18 hr | Development after Culturing for 16-18 hr | Total embryos cultured for 38-40 hr | Development after Culturing for 38-40 hr | |||
| Good | Intermediate | Poor | Good | Poor | ||
| 112 | 65 | 31 | 16 | 12 | 4 | 8 |
Table 1. Development of embryos in mouse embryo culture system. Embryos cultured for either 16-18 hr or 38-40 hr. Of the total 112 embryos cultured for 16-18 hr, 65 (58%) embryos exhibited comparable development to embryos developing in utero, 31 (28%) showed intermediate and 16 (14%) showed poor development. Of the total 12 embryos cultured for 39-40 hr, only 4 (~35%) embryos showed good development while the remaining showed poor development.
Discussion
Mid-gestation stage mouse embryos were cultured in a serum-free culture media in an atmosphere of 95% O2 / 5% CO2 in a rolling bottle culture apparatus at 37 °C. Embryo development ex utero was critically dependent on multiple factors at each step during the procedure from the time the uterus is isolated from the euthanized mice to the completion of the culture (Figure 1). The most important factor that influenced the development was the time taken to start the culture. The other critical points during the procedure that require most care include the steps such as the separation of embryos with intact yolk sac from the uterus and the exteriorization of the embryos from the yolk sac. As the rodents have discoid placenta, segmental dissection of the uterus did not damage the blood supply to the embryo even if the placental decidua is slightly damaged at the corners. In fact this left a small opening at either end through which a pair of blunt forceps can be inserted to gently tear open the uterus and the placental decidua enabling easy isolation of the embryos with intact yolk sac. However, the next step of exteriorization of the embryos from the yolk sac is critical in that any damage to the major blood vessels in the yolk sac would potentially effect the development of the embryo in the culture. Proper embryonic development was supported when embryos were exteriorized from the yolk sac maintaining the intactness of the vasculature in the yolk sac. Previous studies have indicated the opening of the yolk sac when mouse embryos beyond embryonic stage E10 were cultured1,15,18,19. Accordingly, we observed degenerative changes and the embryos could not survive the culture period when E10.5 embryos were cultured with intact yolk sac. We assume the exteriorization of the embryos would facilitate effective nutrient absorption through the tissues while maintaining the continuity in vasculature with the yolk sac. These embryos in culture exhibited effective heart beat and blood circulation in the embryonic body parts compared to the embryos cultured without the yolk sac.
Maintaining the embryos in the culture medium warmed to 37 °C right from the time they are isolated from the uterus to the time of culture would immediately provide the best suitable environment to sustain their ability for later development in the culture. This also prevents contamination due to presence of antibiotics in the medium and avoids incorporation of even minute quantities of PBS / DMEM into the culture. The defined medium we have utilized yielded superior embryonic development compared to other established defined media previously described in Wawersik et al.20,21 (data not shown). The difference in embryonic development could mainly be due to the unique components in our defined medium such as, KnockOut Serum Replacement (KSR) and N-2 Supplement. While N-2 Supplement was shown to support the growth and expression of post-mitotic neurons, KSR was shown to be a direct replacement for fetal bovine serum22,23. KSR was used in cultures to support the growth of undifferentiated pluripotent stem cells as well as human and mouse embryonic stem cells that were shown to be stable and germline competent23-25. Apart from the culture medium, the oxygen concentration in the culture bottle plays a critical role in embryonic development. Embryos could not survive the conditions of 5% CO2 in a conventional incubator. Though we have not tested other oxygen concentrations and different flow rates as previously suggested5,9, the use of 95% O2 / 5% CO2 at constant flow rate has sustained the survivability of the embryos in the culture. While rotation of culture bottles is necessary, embryos when cultured under the same conditions but with no rotation of the culture bottles resulted in nonsurvival of the embryos with no visible heartbeat. We assume that constant rotation (35 rpm) in the rolling bottle culture apparatus enabled free suspension of the embryos in the medium and as well enhanced the absorption of nutrients and oxygen through the embryonic tissues. The growth of these embryos in the culture in terms of overall body and head size, morphology, formation of a well-chambered heart, brain vesicles and ocular development was comparable to that of embryos developing in utero. In our hands, we were able to replicate the embryonic development that was observed by Moore-Scott et al.15
The embryos in the culture exhibited a range of growth and morphological development depending on multiple factors (Figure 3; Table 1). Under the defined culture conditions we used, embryos cultured for 16-18 hr showed comparable development to that observed in embryos developing in utero in about 58% of embryos, while 28% showed intermediate and 14% showed a poor development. Extension of culture to 38-40 hr further decreased the ability to show comparable development to only 30-40% of the total embryos cultured. Apart from the inherent factors that potentially influence the embryonic development ex utero, many external factors that can be managed to certain extent may play critical role when the embryos are cultured for prolonged length of time. One such potential factor that can be controlled is the time taken from the isolation of embryos from the uterus to the start of the culture. It can be interpreted that the first isolated embryos are exposed to less oxygen in the culture dish compared to the embryos isolated at the end as all the embryos go into the culture bottle and receive 95% O2 / 5% CO2 at the same time. However the oxygen available in the blood that is left in the placenta after segmental dissection may not support for extended period of time and for this reason most of the culture equipment and apparatus should be set up ready to be used before euthanizing the mouse. We usually get the embryos into the culture in 25-30 min from the time the mouse was euthanized. This short time period to start the culture would not be possible if embryos were to be processed one at a time as suggested in Zeeb, M. et al.18 When embryos are processed individually, grouping of embryos is not possible and each embryo should be counted for somite number. This procedure would take at least 5-6 min for each embryo to isolate, exteriorize from yolk sac, count somites and transfer to the culture bottle. For a litter size of 10-12 embryos, which we usually get from our CD-1 mice, the entire process would take 60-72 min for the last few embryos to get into the culture. This is pretty long period and we observed the heart beat to slow down and almost stop when embryos were exteriorized and left in PBS and delayed to culture. One other potential factor could be the handling or mishandling of the embryos during the procedure. During handling, you may induce an accidental damage to some part of the body or blood vessels in the embryo or yolk sac that is not easily detected. This can affect the development of that part of the body or sometimes the whole embryo as it was observed that even a well-developed embryo sometimes shows a kind of under-development at the extremities like the limb buds and the tail indicating insufficient blood supply. It is better to discard such embryos that appear to be compromised or damaged during the procedure. The other problem usually encountered during the procedure was, some of the embryos with intact yolk sac prop out abruptly from decidua upon segmental dissection of the uterus. This sometimes results in more blood loss at the separation point from the placenta although the vasculature with the yolk sac remains intact. This blood loss can affect the later development of the embryo due to less amount of blood retained in the embryonic body during separation.
Apart from the above factors certain conditions like, the stage of embryo at the time of starting the culture influenced the development in the culture. We successfully cultured mid-gestation mouse embryos starting with stages that ranged between 28-37 s for 16-40 hr and observed some differences in development. Embryos at 34-36 s stage exhibited better development in the culture compared to the embryos at 30-33 s stage indicating an increase in adaptability to culture conditions in embryos cultured at later stages. The other surprising fact that was observed during culture was the development of embryos when two or more embryos were cocultured (Figure 4). Coculture resulted in significant improvement in development in one of the embryos compared to the other embryo in 45% of the cocultures. Though the embryos from the same uterus look similar morphologically and in somite count, inherently no two embryos can be at the same stage of development and this could be one of the reasons for developmental difference in one of the embryos compared to the other in the coculture system.
Thus we show that mid-gestation stage mouse embryos cultured ex utero in a serum-free medium in an atmosphere of 95% O2 / 5% CO2 in a rolling bottle culture apparatus at 37 °C exhibit growth and morphological development comparable to that observed in embryos developing in utero. We believe this method of mouse embryo culture system will be useful for laboratories needing to utilize whole embryos to study signaling interactions important in early embryonic organogenesis.
Disclosures
We do not have any competing financial interests.
Acknowledgments
We would like to thank Dr. Julie Gordon and Dr. Nancy Manley for their helpful advice with the culture technique. This work has been supported by the Children’s Glaucoma Foundation and Sharon-Stewart Aniridia Research Trust.
References
- Behesti H, Holt JK, Sowden JC. The level of BMP4 signaling is critical for the regulation of distinct T-box gene expression domains and growth along the dorso-ventral axis of the optic cup. BMC Dev. Biol. 2006;6 doi: 10.1186/1471-213X-6-62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gray J, Ross ME. Neural tube closure in mouse whole embryo culture. J. Vis. Exp. 2011. [DOI] [PMC free article] [PubMed]
- Tam PP. Postimplantation mouse development: whole embryo culture and micro-manipulation. Int. J. Dev. Biol. 1998;42:895–902. [PubMed] [Google Scholar]
- Miura S, Mishina Y. Whole-embryo culture of E5.5 mouse embryos: development to the gastrulation stage. Genesis. 2003;37:38–43. doi: 10.1002/gene.10229. [DOI] [PubMed] [Google Scholar]
- Osumi N, Inoue T. Gene transfer into cultured mammalian embryos by electroporation. Methods. 2001;24:35–42. doi: 10.1006/meth.2001.1154. [DOI] [PubMed] [Google Scholar]
- Yokoo T, et al. Human mesenchymal stem cells in rodent whole-embryo culture are reprogrammed to contribute to kidney tissues. Proc. Natl. Acad. Sci. U.S.A. 2005;102:3296–3300. doi: 10.1073/pnas.0406878102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sadler TW. Culture of early somite mouse embryos during organogenesis. J. Embryol. Exp. Morphol. 1979;49:17–25. [PubMed] [Google Scholar]
- Kitchin KT, Ebron MT. Further development of rodent whole embryo culture: solvent toxicity and water insoluble compound delivery system. Toxicology. 1984;30:45–57. doi: 10.1016/0300-483x(84)90061-1. [DOI] [PubMed] [Google Scholar]
- Takahashi M, Nomura T, Osumi N. Transferring genes into cultured mammalian embryos by electroporation. Dev. Growth Different. 2008;50:485–497. doi: 10.1111/j.1440-169X.2008.01046.x. [DOI] [PubMed] [Google Scholar]
- Cuthbertson RA, Beck F. Postimplantation whole embryo culture: a new method for studying ocular development. Invest. Ophthalmol. Vis. Sci. 1990;31:1653–1656. [PubMed] [Google Scholar]
- New DA. Development of explanted rat embryos in circulating medium. J. Embryol. Exp. Morphol. 1967;17:513–525. [PubMed] [Google Scholar]
- Chung KC, Park JH, Kim CH, Ahn YS. Tumor necrosis factor-alpha and phorbol 12-myristate 13-acetate differentially modulate cytotoxic effect of nitric oxide generated by serum deprivation in neuronal PC12 cells. J. Neurochem. 1999;72:1482–1488. doi: 10.1046/j.1471-4159.1999.721482.x. [DOI] [PubMed] [Google Scholar]
- Sasaoka T, et al. Evidence for a functional role of Shc proteins in mitogenic signaling induced by insulin, insulin-like growth factor-1, and epidermal growth factor. J. Biol. Chem. 1994;269:13689–13694. [PubMed] [Google Scholar]
- Chotani MA, Mitra S, Eid AH, Han SA, Flavahan NA. Distinct cAMP signaling pathways differentially regulate alpha2C-adrenoceptor expression: role in serum induction in human arteriolar smooth muscle cells. Am. J. Physiol. Heart Circ. Physiol. 2005;288:69–76. doi: 10.1152/ajpheart.01223.2003. [DOI] [PubMed] [Google Scholar]
- Moore-Scott BA, Gordon J, Blackburn CC, Condie BG, Manley NR. New serum-free in vitro culture technique for midgestation mouse embryos. Genesis. 2003;35:164–168. doi: 10.1002/gene.10179. [DOI] [PubMed] [Google Scholar]
- Gordon J, Moore BA, Blackburn CC, Manley NR. Serum-Free Culture of Mid-gestation Mouse Embryos: A Tool for the Study of Endoderm-Derived Organs. Methods Mol. Biol. 2014;1092:183–194. doi: 10.1007/978-1-60327-292-6_12. [DOI] [PubMed] [Google Scholar]
- New DA, Cockroft DL. A rotating bottle culture method with continuous replacement of the gas phase. Experientia. 1979;35:138–140. doi: 10.1007/BF01917926. [DOI] [PubMed] [Google Scholar]
- Cockroft DL. Development in culture of rat foetuses explanted at 12.5 and 13.5 days of gestation. J. Embryol. Exp. Morphol. 1973;29:473–483. [PubMed] [Google Scholar]
- Zeeb M, et al. Pharmacological manipulation of blood and lymphatic vascularization in ex vivo-cultured mouse embryos. Nat. Protoc. 2012;7:1970–1982. doi: 10.1038/nprot.2012.120. [DOI] [PubMed] [Google Scholar]
- Wawersik S, et al. BMP7 acts in murine lens placode development. Dev. Biol. 1999;207:176–188. doi: 10.1006/dbio.1998.9153. [DOI] [PubMed] [Google Scholar]
- Thut CJ, Rountree RB, Hwa M, Kingsley DM. A large-scale in situ screen provides molecular evidence for the induction of eye anterior segment structures by the developing lens. Dev. Biol. 2001;231:63–76. doi: 10.1006/dbio.2000.0140. [DOI] [PubMed] [Google Scholar]
- Greenfield JP, et al. Estrogen lowers Alzheimer beta-amyloid generation by stimulating trans-Golgi network vesicle biogenesis. J. Biol. Chem. 2002;277:12128–12136. doi: 10.1074/jbc.M110009200. [DOI] [PubMed] [Google Scholar]
- Cheng J, Dutra A, Takesono A, Garrett-Beal L, Schwartzberg PL. Improved generation of C57BL/6J mouse embryonic stem cells in a defined serum-free media. Genesis. 2004;39:100–104. doi: 10.1002/gene.20031. [DOI] [PubMed] [Google Scholar]
- Kim JB, et al. Direct reprogramming of human neural stem cells by OCT4. Nature. 2009;461:649–643. doi: 10.1038/nature08436. [DOI] [PubMed] [Google Scholar]
- Totonchi M, et al. Feeder- and serum-free establishment and expansion of human induced pluripotent stem cells. Int. J. Dev. Biol. 2010;54:877–886. doi: 10.1387/ijdb.092903mt. [DOI] [PubMed] [Google Scholar]