

Abstract
NMDA receptors in primary afferent terminals can contribute to hyperalgesia by increasing neurotransmitter release. In rats and mice, we found that the ability of intrathecal NMDA to induce neurokinin 1 receptor (NK1R) internalization (a measure of substance P release) required a previous injection of BDNF. Selective knock-down of NMDA receptors in primary afferents decreased NMDA-induced NK1R internalization, confirming the presynaptic location of these receptors. The effect of BDNF was mediated by tropomyosin-related kinase B (trkB) receptors and not p75 neurotrophin receptors (p75NTR), because it was not produced by proBDNF and was inhibited by the trkB antagonist ANA-12 but not by the p75NTR inhibitor TAT-Pep5. These effects are probably mediated through the truncated form of the trkB receptor as there is little expression of full-length trkB in dorsal root ganglion (DRG) neurons. Src family kinase inhibitors blocked the effect of BDNF, suggesting that trkB receptors promote the activation of these NMDA receptors by Src family kinase phosphorylation. Western blots of cultured DRG neurons revealed that BDNF increased Tyr1472 phosphorylation of the NR2B subunit of the NMDA receptor, known to have a potentiating effect. Patch-clamp recordings showed that BDNF, but not proBDNF, increased NMDA receptor currents in cultured DRG neurons. NMDA-induced NK1R internalization was also enabled in a neuropathic pain model or by activating dorsal horn microglia with lipopolysaccharide. These effects were decreased by a BDNF scavenger, a trkB receptor antagonist and an Src family kinase inhibitor, indicating that BDNF released by microglia potentiates NMDA receptors in primary afferents during neuropathic pain.
Keywords: microglia, mouse, neurokinin 1 receptor, rat, substance P, trkB receptor
Introduction
NMDA receptors in the spinal dorsal horn mediate hyperalgesia in many types of chronic pain. They are present both postsynaptically in dorsal horn neurons and presynaptically in primary afferents terminals (Sato et al., 1993; Liu et al., 1994; Ma & Hargreaves, 2000; Marvizon et al., 2002; Du et al., 2003; Li et al., 2004; Nagy et al., 2004; McRoberts et al., 2007). The latter can contribute to pain transmission by increasing the release of glutamate and substance P. NMDA receptors induce substance P release in spinal cord slices (Marvizon et al., 1997; Malcangio et al., 1998; Marvizon et al., 1999; Chen et al., 2010). In vivo, however, there are conflicting reports: intrathecal NMDA evoked substance P release in one study (Liu et al., 1997) but not in another (Nazarian et al., 2008). Most of these studies measured substance P release as neurokinin 1 receptor (NK1R) internalization, a method that has higher sensitivity than immunoassay (Marvizon et al., 2003), can be used non-invasively in vivo (Mantyh et al., 1995; Allen et al., 1997; Honore et al., 1999; Adelson et al., 2009), allows the spatial location of substance P release (Abbadie et al., 1997; Hughes et al., 2007) and measures substance P release at physiologically relevant concentrations that activate NK1Rs (Trafton et al., 1999). Modulation of glutamate release by NMDA receptors has been studied through their effect on excitatory postsynaptic potentials (EPSPs) recorded in dorsal horn neurons. NMDA decreased (Bardoni et al., 2004; Zeng et al., 2006) or had no effect (Yan et al., 2013) on EPSPs in normal rats, but increased them in opiate-tolerant (Zeng et al., 2006) and neuropathic rats (Yan et al., 2013).
These findings suggest that NMDA receptors on afferent terminals are potentiated in hyperalgesic states. In many areas of the CNS, NMDA receptor function is increased by Src family kinase (SFK)-mediated tyrosine phosphorylation of their NR2B subunit. In the presence of SFK inhibitors, NMDA receptors in primary afferents, which contain the NR2B subunit (Marvizon et al., 2002; Chen et al., 2010), lose their ability to induce substance P release (Chen et al., 2010) and to evoke currents in dorsal root ganglion (DRG) neurons (Li et al., 2006). We hypothesized that brain-derived neurotrophic factor (BDNF) triggers phosphorylation and activation of NMDA receptors in primary afferent terminals, based on the following observations: (i) BDNF activates a SFK that phosphorylates NMDA receptors in cortex and hippocampus (Xu et al., 2006; Huang & McNamara, 2010); (ii) BDNF acts on presynaptic NMDA receptors to increase glutamate release (Madara & Levine, 2008); (iii) BDNF contributes to long-term potentiation (LTP) of C-fiber synapses (Zhou et al., 2008); and (iv) tropomyosin-related kinase B (trkB) receptors for BDNF are expressed by DRG neurons (Ernfors et al., 1993; Foster et al., 1994; Merighi et al., 2008a), albeit mostly as a truncated form that lacks the intracellular tyrosine kinase domain (Fenner, 2012). As neuropathic pain induces BDNF release from microglia (Coull et al., 2005; Trang et al., 2009), we further hypothesized that potentiation of these NMDA receptors occurs during neuropathic pain.
Material and methods
Animals
All animal procedures were approved by the Institutional Animal Care and Use Committee of the Veteran Affairs Greater Los Angeles Healthcare System and the Animal Research Committee of UCLA, and conform to NIH guidelines. Rats were male adult (2–4 months old) Sprague–Dawley (Harlan, Indianapolis, IND, USA). Mice were male adult C57BL/6tav (Taconic, Germantown, NY, USA). For experiments in vivo, a total of 251 rats and 27 mice were used. Additional rats were used to prepare spinal cord slices and DRG neuron cultures. Rats were given an antibiotic (enrofloxacin) and an analgesic (carprofen) twice daily for 3 days after every surgical procedure.
Mice with selective knockdown of the NR1 subunit in DRG neurons
These mice have been previously characterized (McRoberts et al., 2011). They were generated from C57BL/6tav mice with a floxed NR1 subunit gene and mice expressing Cre recombinase under the control of a portion of the peripherin promoter. The resulting Prph1-Cre_floxed-NR1 mice (Prph-fNR1) have a selective 75% knockdown of NR1 in DRG neurons. The knockdown mice used in this study were male littermates that were homozygous for the floxed NR1gene and expressed the Cre recombinase transgene (NR1−). Control littermates did not express Cre recombinase (NR1+).
Chemicals and solutions
N-[2-[[(hexahydro-2-oxo-1H-azepin-3-yl)amino]carbonyl]phenyl]-benzo[b]thiophene-2-carboxamide (ANA-12) was from Sigma; BDNF and proBDNF (Mut-mouse; BDNF precursor protein with double point mutations preventing cleavage to BDNF) were from Alomone (Jerusalem, Israel); 4-hydroxy-3,3-dimethyl-2H-benz[g]indole-2,5(3H)-dione ()BVT948, 7,8-dihydroxyflavone (7,8-DHF), (9S,10R,12R)-2,3,9,10,11,12-hexahydro-10-hydroxy-9-methyl-1-oxo-9,12-epoxy-1H-diindolo[1,2,3-fg:3′,2′,1′-kl]pyrrolo[3,4-i][1,6]benzodiazocine-10-carboxylic acid methyl ester (K252a), 3-(4-chlorophenyl) 1-(1,1-dimethylethyl)-1H-pyrazolo[3,4-d]pyrimidin-4-amine (PP2) and 1-phenyl-1H-pyrazolo[3,4-d]pyrimidin-4-amine (PP3) were from Tocris (Ellisville, MO, USA); dasatinib was from ChemieTek (Indianapolis, IN, USA); isoflurane was from Halocarbon Laboratories (River Edge, NJ, USA); neurotrophin-3 (NT-3) and H-YGRKKRRQRRR-CFFRGGFFNHNPRYC-OH, cyclic (TAT-Pep5) were from EMD Millipore (Billerica, MA, USA); recombinant chimera of human trkB receptor linked to human IgG1 (trkB-Fc) was from R&D Systems (Minneapolis, MN, USA) and other chemicals were from Sigma (St Louis, MO, USA). Stock solutions of ANA-12, BVT948, dasatinib, 7,8-DHF, K252a, minocycline, PP2, PP3 and TAT-Pep5 were prepared in DMSO.
Artificial cerebrospinal fluid (aCSF) contained (in mM) NaCl, 124; KCl, 1.9; NaHCO3, 26; KH2PO4, 1.2; MgSO4, 1.3; CaCl2 2.4; and glucose, 10; and was gassed with 95% O2 and 5% CO2. Sucrose-aCSF was the same medium with 5 mM KCl and 215 mM sucrose instead of NaCl. K+-aCSF was aCSF containing 5 mM KCl.
Intrathecal injections
Rats were implanted with chronic intrathecal catheters under isoflurane (2–4%) anesthesia (Storkson et al., 1996). After cutting the skin and muscle, a 20-gauge needle was inserted between the L5 and L6 vertebrae to puncture the dura mater. The needle was removed and the catheter (20 mm of PE-5 tube heat-fused to 150 mm of PE-10 tube) was inserted into the subarachnoid space and pushed rostrally to terminate over L5-L6. The PE-10 end of the catheter was tunneled under the skin and externalized over the head. The skin was sutured and the catheter was flushed with 10 μl saline and sealed. Rats were housed separately and used 5–7 days after surgery. The presence of motor weakness or signs of paresis was established as criterion for immediate euthanasia, but this did not occur in any of the rats. Because intrathecal NMDA produces pain (Liu et al., 1997), rats were anesthetized with isoflurane 10 min before the NMDA injection. Intrathecal injection volume was 10 μl of injectate plus 10 μl saline flush. Solutions were preloaded into a PE-10 tube and delivered within 1 min. The position of the catheter was examined post-mortem and the following exclusion criteria were used: (i) loss of the catheter; (ii) termination of the catheter inside the spinal cord; or (iii) occlusion of the catheter tip.
In mice, intrathecal injections were given using the lumbar puncture method (Hylden & Wilcox, 1980). The mouse was held by the tail and skin of the neck by one person, while another inserted the needle at a 20° angle to one side of the L5 or L6 spinous process so that it slips into the groove between the spinous and transverse processes. The needle was then moved forward to the intervertebral space as the angle of the syringe is decreased to ~ 10°. The tip of the needle was inserted 0.5 cm into the vertebral column and 2–3 μl of drug or vehicle injected.
Chronic constriction injury (CCI) of the sciatic nerve
Under isoflurane anesthesia, the sciatic nerve of the rat was exposed at the mid-thigh level proximal to the sciatic trifurcation. Four chromic gut ligatures (4/0) were loosely tied around the nerve, 1–2 mm apart (Bennett & Xie, 1988). The muscle and the skin were closed with synthetic absorbable surgical suture. Sham surgery consisted in exposing the sciatic nerve without ligation.
Measurement of mechanical allodynia
Rats moved freely in an acrylic enclosure placed on an elevated metal grid. Rats were habituated to the enclosure for periods of 30 min for 5 days. A set of calibrated plastic von Frey hairs (‘Touch-Test’, North Coast Medical, Inc., San Jose, CA, USA) of increasing size were applied to the hind paw until they bent, and the threshold force (in g) to elicit paw withdrawal was recorded.
Spinal cord slices
Coronal slices (400 μm) were prepared from the lumbar spinal cord (L2–L4) of adult rats using a vibratome (Integraslice 7550PSDS; Lafayette Instruments, Lafayette, IN, USA; Marvizon et al., 2003; Chen et al., 2010). Vibratome cutting was done in ice-cold sucrose-aCSF, after which slices were kept at 35°C in K+-aCSF for 1 h and in normal aCSF thereafter. For drug treatment (Marvizon et al., 2003; Chen et al., 2010), slices were placed on a nylon net glued to a plastic ring inserted halfway down a plastic tube containing 5 ml aCSF, which was superficially gassed with 95% O2/5% CO2 and kept at 35 °C using a metal block incubator. To change solutions, the ring and net with the slice was transferred to another tube. NMDA and D-serine (D-Ser) were applied to the slices for only 2 min to avoid excitotoxic effects. At the end of the experiment slices were incubated at 35 °C in aCSF for 10 min and then immersed in ice-cold fixative (4 % paraformaldehyde, 0.18 % picric acid).
Immunohistochemistry
Rats and mice were killed with pentobarbital (100 mg/kg) and fixed by aortic perfusion of 100 ml 0.1 M sodium phosphate (pH 7.4) containing 0.01% heparin, followed by 400 ml of ice-cold fixative. A segment of the lumbar spinal cord (L4–L6) was post-fixed, cryoprotected, frozen and sectioned at 25 μm in the coronal plane using a cryostat. Spinal cord slices were processed similarly (Marvizon et al., 2003). Sections were incubated overnight at room temperature with NK1R (1:3000) or Iba-1 (1:1000) antiserum in phosphate-buffered saline (PBS) containing 0.3 % Triton X-100, 0.001 % thimerosal and 5 % normal goat serum (Jackson ImmunoResearch Laboratories, West Grove, PA, USA). After three washes, the secondary antibody (1:2000, Alexa Fluor 488 goat anti-rabbit, Molecular Probes-Invitrogen, Eugene, OR, USA) was applied for 2 h at room temperature. Sections were coverslipped with Prolong Gold (Molecular Probes-Invitrogen).
NK1R antisera were # 94168 (CURE: Digestive Diseases Research Center, UCLA) or AB-5060 from Millipore (Billerica, MA, USA). Both antisera labeled the dorsal horn with patterns identical to those reported previously (Marvizon et al., 2003; Nazarian et al., 2008; Chen et al., 2010; Zhang et al., 2010). Antiserum # 94168 was generated in rabbits using a peptide corresponding to the C-terminus of the rat NK1R and selectively labeled cells transfected with rat NK1R (Grady et al., 1996). Iba-1 antiserum was raised in rabbit (Wako Chemicals USA, Richmond, VA, USA), and produced staining similar to that reported previously (Clark et al., 2010).
Quantification of NK1R internalization
NK1R neurons in lamina I with and without internalization were counted as reported (Marvizon et al., 2003; Chen et al., 2010) in four sections per spinal segment or slice, using a Zeiss Axio-Imager A1 (Carl Zeiss, Inc., Thornwood, NY, USA) fluorescence microscope with a 63× objective (1.40 numerical aperture). Both lamina I neurons that were strongly and those that were weakly immunoreactive to the NK1R antibody (Al Ghamdi et al., 2009) were included in the counting. The criterion for internalization was the presence of ten or more NK1R endosomes in the neuronal soma. Counting was done blinded to the treatment. Four sections per spinal segment or slice were used, counting all lamina I NK1R neurons in each section. Results were expressed as the percentage of the NK1R neurons in lamina I with NK1R internalization.
Confocal microscopy and image processing
Confocal images were acquired using a Zeiss LSM 710 confocal microscope (Carl Zeiss, Inc., Thornwood, NY, USA), with objectives of 10×, 20× and 63× oil (numerical apertures of 0.3, 0.8 and 1.4, respectively). The Alexa Fluor 488 fluorophore was excited by the 488-nm line of an argon laser and the emission window was 500–560 nm. The pinhole was 1.0 Airy unit: 37.7 μm, 31.5 μm and 50.7 μm for 10×, 20× and 63×, respectively. Images were acquired as stacks of sections of 1024 × 1024 pixels, spaced 5.89 μm, 0.85 μm and 0.38 μm for 10×, 20× and 63×, respectively (determined using the Nyquist formula).
For NK1R staining, images of the entire dorsal horn obtained with the 10× objective were used to show the location of the neurons imaged with the 63× objective. Imaris 6.1.5 software (Bitplane AG, Zurich, Switzerland) was used to crop the images into three (10× images) or four to six (63× images) optical sections. A two-dimension projection picture was imported into Adobe Photoshop 5.5 (Adobe Systems Inc., Mountain View, CA, USA), which was used to assemble the multi-panel figures.
For Iba-1 staining, images of a 425 × 425 μm area (20× objective) encompassing laminae I–II of the central dorsal horn were acquired from three histological sections per rat. Laser intensity, photomultiplier gain and offset were kept constant. Using ImageJ 1.46r (National Institutes of Health, USA), the integrated optical intensity of pixels between the thresholds of 14 and 255 was measured in one optical section from the middle of each stack.
DRG cell cultures
Primary cultures of DRG neurons from spinal levels T12–S2 were prepared as previously described (McRoberts et al., 2001; Li et al., 2004; Li et al., 2006). Neurons were plated on six-well plates coated with Matrigel (BD Biosciences, San Jose, CA, USA) for phosphorylation assays (Western blots), or on glass bottom dishes (MatTek Corp., Ashland, MA, USA) coated with Matrigel for electrophysiology. Except where noted, neurons were cultured in the presence of 200 μM ketamine for 48–72 h at 37°C in 5% CO2 incubator. The presence of ketamine or another NMDA receptor antagonist in the culture medium is necessary to preserve NMDA receptor function in cultured DRG neurons (McRoberts et al., 2001; Li et al., 2004).
Western blots
For measurement of BDNF-stimulated NR2B phosphorylation, cultured DRG neurons were incubated in culture media for 1 h in the presence or absence of 20 ng/ml BDNF, then scraped into suspension in ice-cold PBS and centrifuged at 500 g for 5 min at 4°C. The medium was aspirated and the pellet resuspended in a three-fold volume of ice cold high-SDS RIPA buffer containing 50 mM Tris-HCl, pH 7.5, 140 mM NaCl, 2 mM EDTA, 2 % SDS, 1 % NP-40 and 1% sodium deoxycholate supplemented with protease inhibitors (complete protease inhibitor cocktail; Roche Diagnostics, Mannheim, Germany) and phosphatase inhibitors (2 mM Na3VO4, 10 mM NaF and Phosphatase Inhibitor Cocktail 2; Sigma). The extract was briefly sonicated and set on ice for 10 min before centrifugation at 18,000 g for 10 min at 4°C. The supernatant was assayed for protein content using the BCA method (Thermo Scientific, Rockford, IL, USA). Approximately twenty-five micrograms of protein was electrophoresed on 3–8% NuPAGE Tris-Acetate SDS gels (Invitrogen, Dallas, TX, USA) and proteins were transferred to PVDF membranes as described previously (Li et al., 2006). Blots were blocked with 5% BSA in Tris-buffered saline containing 0.05% Tween 20 and probed with an antibody to phospho-Tyr1472-NR2B (1:1000; catalog number P1516, PhosphoSolutions). This antibody labels a band at 180–190 kDa which overlaps with that obtained with the anti-NR2B antibody used below, and was eliminated by treatment of lysates with lambda and alkaline phosphatases (Kameron Simpson, PhosphoSolutions, personal communication). Protein bands were visualized using a horseradish peroxidase-conjugated secondary antibody and an enhanced chemoluminescent detection system (GE Healthcare Biosciences, Piscataway, NJ, USA). The membranes were stripped (Restore Western Blotting Stripping Buffer; Thermo Fisher Scientific) and re-probed with well-characterized antibodies to NR2B (1:1000, catalog number sc-9057, Santa Cruz Biotechnology, Dallas, TX, USA) (Seeber et al., 2004) and monoclonal antibody 13E5 to β-actin (1:4000; catalog number 4970, Cell Signaling Technology, Danvers, MA, USA) (Gerdes et al., 2013). The relative levels of phospho-NR2B and NR2B were calculated from the ratio of the band intensities to that of β-actin.
For measurement of TrkB expression, spinal cord and DRG tissues were quickly dissected and immediately homogenized with a Teflon-glass homogenizer in ice-cold standard RIPA buffer. Standard RIPA buffer contained 0.1% SDS but was otherwise identical to that described. Cultures of DRG neurons were prepared in parallel using cage mates to the rats used for tissue harvest. After 3 days in culture, neurons were harvested and homogenized by sonication in standard RIPA buffer, identical to high SDS RIPA except that the SDS concentration was reduced to 0.1%. Homogenates were processed and electrophoresed as described above except that blots were blocked with 5% nonfat dry milk (Biorad) and probed with antibodies to TrkB (1:2000, 80E3 monoclonal antibody, catalog number 4603, Cell Signaling Technology, or 1:400 H-181 polyclonal antibody, catalog number sc-8316, Santa Cruz Biotechnology). Both antibodies recognized bands at ~130 kDa and 95 kDa corresponding to the molecular weights of TrkB.FL and TrkB.T1, respectively, in addition to one other nonspecific band which was different between the two antibodies. Protein bands were visualized, stripped and re-probed with a monoclonal antibody to β-actin. For confirmation of TrkB.FL identification, HEK293 cells were transfected with pcDNA TrkB (Addgene plasmid 24088, Cambridge, MA, USA) and an extract run along with the experimental extracts. The relative level of TrkB.FL and TrkB.T1 were calculated from the ratio of the band intensities to that of β-actin.
Patch-clamp recordings
DRG were washed in extracellular solution (in mM: NaCl, 138; KCl, 5; CaCl2, 0.5; HEPES, 10; D-glucose, 10; strychnine, 5; diethylene triamine pentaacetic acid (DTPA), 1; and tetrodotoxin, 0.25; pH 7.3) prior to obtaining a giga-Ohm seal using the following intracellular solution (in mM: CsCl, 135; MgCl2, 2; EGTA, 1; HEPES, 10; TEA, 5; Na-ATP, 2; and Na-GTP, 0.5; pH 7.25). Whole-cell currents were acquired under voltage-clamp at a gain of 1 and filtered at 2 kHz using an Axopatch 200B amplifier, 1200 Digitizer and pClamp 9.2 software (Molecular Devices, Sunnyvale, CA, USA). Currents were measured in gap-free mode at a holding potential of −70 mV, and NMDA (250 μM) and glycine (10 μM) applied by pressure ejection for 10 s once a stable basal current was reached. The data were analyzed by subtracting the mean basal current, measured over 5 s before and after the NMDA current, from the mean peak current.
Data analysis
Data were analyzed using Prism 6 (GraphPad Software, San Diego, CA, USA). All data are expressed as mean ± SEM. Statistical significance was set at 0.05. Statistical analyses consisted of t-test, one-way ANOVA followed by Holm–Sidak’s post hoc tests, or two-way ANOVA followed by Sidak’s post hoc tests. Dose–response data were fitted using non-linear regression by the dose–response function: Y = bottom + (top − bottom) / (1 + 10^(Log EC50 − Log X)). Time course data were fitted by non-linear regression to an association function: Y = Y0 + (plateau − Y0) × (1−exp(−K × x)), where K is the rate constant, Y0 is the value at time 0 and ‘plateau’ is the maximum value.
Results
BDNF increased NMDA-induced NK1R internalization in rats
To determine whether NMDA can induce substance P release and consequent NK1R internalization in vivo, anesthetized rats received intrathecal injections of saline (control) or NMDA (10 nmol). In this and later experiments, NMDA was combined with D-Ser (10 nmol) to ensure occupancy of the glycine co-agonist site of the NMDA receptor. As shown in Fig. 1A, NK1R internalization in lamina 1 neurons of spinal segments L4–L6 (see images in Fig. 2A and B) was negligible in control rats and was not increased by NMDA. However, in a large sample of rats (n = 19) we observed some variability in the effect of NMDA, with two of the 19 rats showing substantial (18% and 35%) NK1R internalization. Indeed, although a t-test yielded no significant differences between the control and the NMDA groups (P = 0.86, t25 = 0.18), there were significant differences between their variances (t-test, P = 0.0011, F18,7 = 15.8). The fact that NMDA was able to induce substance P release in a few rats suggested that there may be some factor that increases the functionality of these NMDA receptors. We previously found (Chen et al., 2010) that addition of the protein tyrosine phosphatase (PTP) inhibitor BVT948 (Liljebris et al., 2004) increased NMDA-stimulated NK1R internalization, most probably by increasing tyrosine phosphorylation of the NR2B subunit of the NMDA receptor. Indeed, an intrathecal injection of BVT948 1 h before NMDA resulted in a significant increase in NMDA-induced NK1R internalization (Fig. 1A), indicating that blocking the dephosphorylation of these NMDA receptors increases their functionality. However, the fact that the effect of BVT948 was small suggested that the main reason for their dephosphorylated state was a low activity of the SFK that phosphorylates the NR2B subunit. We reasoned that there has to be a signal that increases the activity of this SFK and hypothesized that this signal was BDNF. To test this hypothesis, we injected BDNF intrathecally (3 μg, equivalent to 111 pmol) 60 min before intrathecal NMDA (Geng et al., 2010). This resulted in a pronounced increase in NMDA-induced NK1R internalization (Fig. 1A; images in Fig. 2C). Intrathecal injection of vehicle 60 min before NMDA produced no effect (P = 0.95, n = 5).
Figure 1.

BDNF increased NMDA-induced NK1R internalization in rats and mice. (A) Rats received intrathecal saline (control), NMDA (10 nmol), or NMDA 60 min after BVT948 (10 nmol), BDNF (3 μg), proBDNF (0.3 μg), BDNF + TAT-Pep5 (1 nmol), BDNF + ANA-12 (100 nmol) or BDNF + PP2 (10 nmol). (B) Mice received intrathecal saline (control), capsaicin (100 pmol), NMDA (250 pmol) or NMDA 60 min after BDNF (75 fmol). Post hoc tests (Sidak’s): *P < 0.05, ***P < 0.001 compared to control; †P < 0.05, ††P < 0.01, †††P < 0.001 compared to NMDA. (C) Mice with NR1 subunit knockdown in DRG neurons (NR1−) or wild-type (NR1+) received intrathecal NMDA 60 min after BDNF (75 fmol). **P = 0.0078 (t-test). NMDA was always given with D-Ser (10 nmol for rats, 250 pmol for mice). Horizontal lines indicate the means.
Figure 2.

Confocal images of NK1R neurons after intrathecal BDNF and NMDA. Rats received (A) intrathecal injections of saline or (B–D) 10 nmol NMDA and D-Ser, and were fixed 10 min later for NK1R immunohistochemistry. Images were taken from the L5 spinal segment. (A) Injection of vehicle (saline) produced no NK1R internalization. (B) Injection of NMDA and D-Ser produced no NK1R internalization. (C) BDNF (3 μg) was injected intrathecally 60 min before NMDA, resulting in NK1R internalization in more than half of the neurons. (D) The SFK inhibitor PP2 (10 nmol) was coinjected with BDNF (3 μg) 60 min before NMDA, resulting in no NK1R internalization. Main panels: images taken with a 10× objective; voxel size of 830 × 830 × 5983 nm and three confocal planes. Insets: images taken with a 63× objective of the lamina I neurons indicated by the frames in the main panels; voxel size of 132 × 132 × 383 nm and three confocal planes. Adjustments in the gamma of the images in the insets were made to ensure that the staining of neurons was of similar intensity; the uncorrected intensity can be seen in the main panel images. Neurons with NK1R internalization are indicated with ‘*’ and neurons without internalization by ‘o’, with the symbol placed over the nucleus. Scale bars, 100 μm (main panels), 10 μm (insets).
This effect of BDNF can be mediated by trkB receptors or by p75 neurotrophin receptors (p75NTR receptors), which also bind BDNF. To determine which of these two receptors is involved we performed a several experiments. First, we injected intrathecally the selective agonist of p75NTR proBDNF (0.3 μg) 60 min before NMDA. We used a construct of proBDNF (Mut-mouse from Alomone) that has double point mutations to prevent its cleavage to BDNF. As shown in Fig. 1A, proBDNF failed to increase NMDA-induced NK1R internalization. Second, co-injecting BDNF with the trkB antagonist ANA-12 (100 nmol; Cazorla et al., 2011) eliminated the increase in NMDA-induced NK1R internalization produced by BDNF. Third, the p75NTR antagonist TAT-Pep5 (1 nmol) did not eliminate the increase in NMDA-induced NK1R internalization produced by BDNF.
To explore whether the effect of BDNF was mediated by phosphorylation by an SFK, we co-injected BDNF with the SFK inhibitor PP2 (10 nmol) 60 min before intrathecal NMDA. PP2 abolished the increase in NMDA-induced NK1R internalization produced by BDNF (Fig. 1A; images in Fig. 2D), bringing it to a level not significantly different from the control or the NMDA group (ANOVA of data in Fig. 1A: P < 0.0001, F4,33 = 36.6).
BDNF increased NMDA-induced NK1R internalization in mice: NMDA receptor knockdown in DRG neurons
Next, we determined whether BDNF also increases NMDA-induced NK1R internalization in mice. Anesthetized mice received intrathecal saline or NMDA (250 pmol) plus D-Ser (250 pmol). NMDA alone did not induce NK1R internalization above the levels found in saline-injected controls (Fig. 1B). Intrathecal BDNF (75 fmol) given 60 min before NMDA significantly increased NK1R internalization (Fig. 1B). Intrathecal capsaicin (100 pmol) elicited a slightly larger amount of NK1R internalization (ANOVA of data in Fig. 1B: P < 0.0001, F3,14 = 23.14).
NMDA-induced NK1R internalization is due to substance P release from primary afferents, as it was eliminated after depleting primary afferents of substance P with capsaicin (Chen et al., 2010). However, there is no direct evidence that the NMDA receptors involved are located in primary afferent terminals. To establish this we used Prph-fNR1 transgenic mice (NR1–; Fig. 1C) in which the NR1 subunit of the NMDA receptor is selectively knocked down ~75% in DRG neurons of all sizes (McRoberts et al., 2011). When these mice received intrathecal BDNF (75 fmol) and 60 min later 250 pmol NMDA (with 250 pmol D-Ser), NK1R internalization was decreased by 50% compared to their wild-type littermates (NR1+; t-test of data in Fig. 1C: P = 0.0078, n = 5, t8 = 3.526).
Time course of the effect of BDNF
To determine the time required for the onset of the effect of BNDF and its duration, we gave rats an intrathecal injection of BDNF (0.3 μg) followed by intrathecal NMDA (10 nmol), changing the interval between these two injections from 10 min to 16 h. A zero time point was obtained by giving BNDF and NMDA in a single injection. The effect of BDNF was not detected when given together with NMDA or 10 min before it, but it was fully developed by 30 min (Fig. 3A). Fitting an association function to the time points up to 4 h yielded a rate constant K = 0.047 ± 0.020 min−1 (half-time ~ 15 min). The effect of BDNF persisted up to 4 h, but it disappeared by 8 h (Fig. 3A; ANOVA of data in Fig. 3A: P < 0.0001, F7,16 = 16.22).
Figure 3.
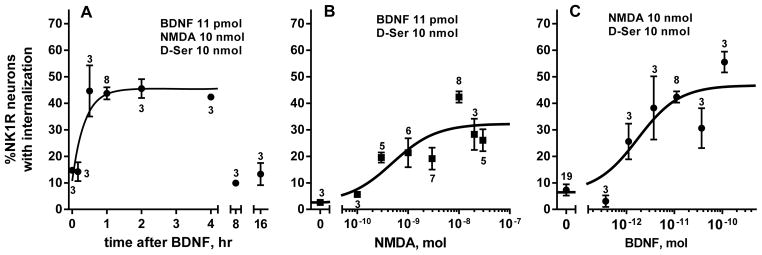
Time course and dose–responses for NMDA and BDNF in vivo. (A) Rats received intrathecal BDNF (0.3 μg) followed by NMDA (10 nmol) at the indicated times. Time 0 h: BDNF and NMDA given together. (B) Rats received intrathecal NMDA at different doses 60 min after BDNF (11 pmol, 0.3 μg). NMDA 0 nmol: BDNF without NMDA. (C) Rats received intrathecal BDNF at different doses followed 60 min later by NMDA (10 nmol). BDNF 0 nmol: NMDA without BDNF. NMDA was always given with D-Ser (10 nmol). Numbers next to data points indicate n.
A similar onset rate was obtained using spinal cord slices (Fig. 4C), which were incubated for times ranging from 15 to 90 min with 20 ng/ml BDNF and then for 2 min with 10 μM NMDA and 10 μM D-Ser. In this case fitting one-phase association function to the data yielded a rate constant K = 0.031 ± 0.026 min−1 and half-life ~ 22 min (initial value Y0 = 33 ± 3; plateau = 65 ± 8%).
Figure 4.

BDNF increased NMDA-induced NK1R internalization in spinal cord slices. (A) Rat spinal cord slices were incubated for 60 min in aCSF, BDNF (20 ng/ml), proBDNF (20 ng/ml), 7,8—DHF (1 μM) or NT-3 (100 ng/ml), and then for 2 min in aCSF or NMDA (10 μM); n = 3 except control (n = 7), NMDA (n = 12) and BDNF+NMDA (n = 15). (B) Slices were incubated for 2 min in NMDA (10 μM) after 60 min in BDNF (20 ng/ml) alone or with ANA-12 (1 μM or 10 μM), TAT-Pep5 (100 nM), K252a (30 nM), dasatinib (1 μM or 10 μM), PP2 (10 μM), PP3 (10 μM) or BVT948 (10 μM); n = 3 except control (n = 15). Post hoc tests (Sidak’s): ***P < 0.001 compared to control; †††P < 0.001 compared to NMDA. (C) Slices were incubated in BDNF (20 ng/ml) for the times indicated in the X-axis and then for 2 min in 10 μM NMDA (time 0 was NMDA alone). NMDA was always given with D-Ser (10 μM).
Dose–responses of BDNF and NMDA
To obtain a dose–response relationship for NMDA, 11 pmol (0.3 μg) BDNF was injected intrathecally followed 60 min later by different doses of NMDA (all with 10 nmol D-Ser). The dose of 0 nmol NMDA consisted of BDNF followed by D-Ser alone (in Fig. 3B) or BDNF followed by saline, both of which produced negligible amounts of NK1R internalization (3.4 ± 1.2 % and 2.6 ± 0.8 %, respectively). Hence, BDNF did not induce NK1R internalization by itself. The effect of NMDA was dose-dependent (Fig. 1E), with EC50 = 0.49 nmol (95% CI, 0.16–1.45 nmol) and a maximum effect of 32 ± 3 %.
To obtain a dose–response relationship for BDNF, different doses of BDNF were injected intrathecally followed 60 min later by 10 nmol NMDA with 10 nmol D-Ser. The dose of 0 nmol BDNF consisted of a single injection of NMDA and D-Ser. The effect of BDNF was dose-dependent (Fig. 3C), with EC50 = 1.72 pmol (95% CI, 0.55–5.40 pmol) and a maximum effect of 47 ± 4 %. The maximum effect of NMDA was somewhat smaller than the maximum effect of BDNF, probably because a non-saturating dose of BDNF was used to obtain the dose-response of NMDA.
Signaling pathways involved in the increase by BDNF of NMDA-induced NK1R internalization
The signaling pathways that mediate in the increase in NMDA-induced NK1R internalization by BDNF were studied in more detail using rat spinal cord slices, which allows the rapid generation of data using fewer animals than in vivo studies. In contrast with its lack of effect in vivo, when NMDA is applied to spinal cord slices it induces a fair amount of NK1R internalization (Marvizon et al., 1997; Marvizon et al., 1999; Chen et al., 2010). Still, the NK1R internalization induced by NMDA (10 μM with 10 μM D-Ser for 2 min) was further increased after a 60 min preincubation of the slices with the trkB receptor agonists BDNF (20 ng/ml or 0.74 nM), 7,8-DHF (1 μM; Jang et al., 2010) and NT-3 (100 ng/ml or 3.57 nM; Fig. 4A). BDNF and 7,8-DHF had no effect without NMDA (Fig. 4A). In contrast, proBDNF (20 ng/ml), an agonist of p75NTR that does not activate trkB receptors, did not increase NMDA-induced NK1R internalization (ANOVA of data in Fig. 4A: P < 0.0001, F7,41 = 57). Again, we used proBDNF with double point mutations that prevent its cleavage to BDNF.
Figure 4B shows the effect of several compounds on NMDA-induced NK1R internalization after 1 h preincubation with BDNF (20 ng/ml). All these compounds were added together with BDNF. First, NMDA-induced NK1R internalization was inhibited by the trkB receptor antagonist ANA-12 (1 μM or 10 μM; Cazorla et al., 2011) but not by the p75NTR antagonist TAT-Pep5 (Yoshizaki et al., 2008; Pearn et al., 2012), providing further support for the idea that the effect of BDNF is mediated by trkB receptors. Second, NMDA-induced NK1R internalization was also decreased by the kinase inhibitor of K252a (30 nM) which potently inhibits receptor tyrosine kinases (Huang & McNamara, 2010), and by the SFK inhibitors dasatinib (1 and 10 μM; Schittenhelm et al., 2006) and PP2 (10 μM; Hanke et al., 1996; Zhang et al., 2010), but not by the inactive PP2 analog PP3 (10 μM). Third, the PTP inhibitor BVT948 (10 μM; Liljebris et al., 2004) combined with BDNF produced a larger increase in NMDA-induced NK1R internalization than BDNF alone, suggesting that inhibiting NR2B subunit dephosphorylation by PTPs complements the increase in NMDA receptor function produced by BDNF (ANOVA of data in Fig. 4B: P < 0.0001, F9,32 = 31.6). Taken together, these results indicate that BDNF increases the function of these NMDA receptors by binding to trkB receptors and activating an SFK.
BDNF increased Tyr1472 phosphorylation of the NR2B subunit of the NMDA receptor in DRG neurons
To directly determine whether BDNF increases tyrosine phosphorylation of the NR2B subunit, 2- or 3-day-old primary cultures of DRG neurons were treated with 20 ng/ml BDNF for 1 h then harvested, and protein extracts were prepared for electrophoresis. Control cultures were left untreated. Western blots were first probed with an antibody to phospho-tyr1472-NR2B, then stripped and reprobed with antibodies to total NR2B and β-actin. As shown in Fig. 5A, there was a substantial degree of phosphorylation of NR2B in control cultures; however, BDNF increased the level of phosphorylation. Fig. 5B shows the levels of phospho-NR2B and NR2B relative to actin. BDNF had no significant effect on the expression of NR2B but did cause a 1.8 ± 0.3-fold increase in phospho-NR2B (P < 0.0185; t-test). NR2B phosphorylation after BDNF, expressed as the ratio of phospho-NR2B to total NR2B for each experimental pair, was 2.2 ± 0.4 (P < 0.001, z-test).
Figure 5.
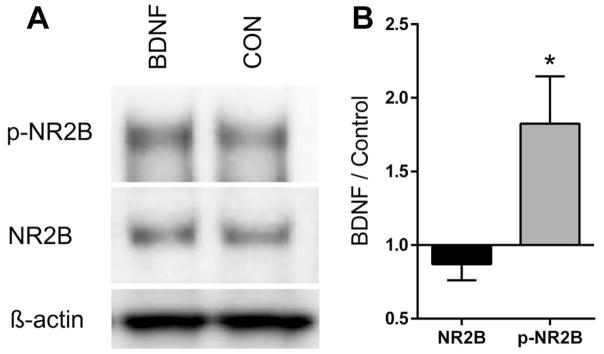
BDNF increased Tyr phosphorylation of the NR2B subunit. (A) Western blots performed on extracts of DRG neurons cultured 2–3 days in the presence of ketamine and incubated 1 h with 20 ng/ml BDNF (BDNF) or without (control; CON). The blots were probed with antibodies to (pTry1472)-NR2B (p-NR2B), NR2B and β-actin. NR2B and p-NR2B had an apparent molecular weight of 190 kDa. (B) The intensity of the bands corresponding to the p-NR2B and NR2B antibodies were normalized to actin and expressed as the ratio of BDNF versus control. *P = 0.0185 compared to NR2B, t-test, n = 6. Expressed as the ratio of p-NR2B to NR2B for each experimental pair, BDNF increased NR2B-phosphorylation an average of 2.2 ± 0.4-fold.
DRG express truncated trkB receptors
Previous studies have shown that the mRNA for full length trkB (trkB.FL) is expressed in only a small subset of DRG neurons, while a truncated form (trkB.T1) seems to be expressed in most DRG neurons and surrounding satellite cells (McMahon et al., 1994; Kashiba et al., 1995; Wright & Snider, 1995; Baxter et al., 1997). To determine whether DRG neurons cultured for 3 days expressed trkB.FL, we used an antibody that recognizes both trkB.FL and trkB.T1. We compared extracts from cultured DRG with extracts made from DRG and spinal cord tissue. As shown in Fig. 6A, trkB.FL was only detected in spinal cord extracts, which also expressed about three-fold more trkB.T1. Extracts from intact and cultured DRG contained trkB.T1, but no trkB.FL was detected (Fig. 6B). ANOVA (one-way) of the trkB.T1 data in Fig. 6B revealed significant differences (P = 0.0297, F2,6 = 6.69). Cultured and intact DRG expressed trkB.T1 at a significant lower level than the spinal cord (P < 0.05, Holm–Sidak’s post hoc test), but there were no significant differences between cultured DRG and intact DRG.
Figure 6.

Expression of trkB.FL and trkB.T1 in DRG and spinal cord. Extracts were prepared from DRG neurons cultured for 3 days and from freshly isolated DRG and spinal cord tissue and Western blots performed. (A) The image shows the results of a representative experiment. Numbers on the left are positions of the molecular weight markers in kDa. Both trkB.FL (extrapolated molecular weight 130 kDa, expected 140 kDa) and trkB.T1 (extrapolated molecular weight 95 kDa, expected 95 kDa) were detected in the spinal cord extract, but only trkB.T1 was detected in DRG tissue and cultured DRG. The band at ~70 kDa in the two tissue extracts was not seen when a different anti-trkB antibody was used and is therefore nonspecific. The last lane was an extract from HEK293 cells transfected with an expression vector for rat trkB.FL. The data were quantified by comparing the relative levels of trkB protein to β-actin. (B) The graph shows this data normalized to the level of trkB.FL in the spinal cord extract (n = 3; nd, not detected).
BDNF increased NMDA receptor currents in DRG neurons
NMDA induces an inward current in cultured DRG neurons by activating NMDA receptors (Li et al., 2004). If the increase in NMDA-induced NK1R internalization induced by BDNF is due to an increase in NMDA receptor function in primary afferents, then BDNF should also increase this NMDA current in DRG neurons. We previously found that the presence of an NMDA receptor antagonist, ketamine, in the DRG culture medium is required in order to detect NMDA receptor currents (McRoberts et al., 2001; Li et al., 2004), probably because glutamate in the medium (originating from glutamine) causes the desensitization or downregulation of the NMDA receptors. Therefore, unless stated otherwise, in the following experiments DRG neurons were cultured in the present of ketamine (250 μM), which was thoroughly washed from the media before recording.
DRG neurons from the lumbar region of adult rats were patch-clamped and held at −70 mV. When we grew DRG neurons in the absence of ketamine, addition of NMDA (250 μM with 10 μM glycine) produced negligible currents (Fig. 7A): −36 ± 9 pA (n=3) in the absence of BDNF and −26 ± 12 pA (n=4) in the presence of BDNF (20 ng/ml). In contrast, when DRG neurons were grown with 250 μM ketamine in the culture medium, NMDA produced inward currents (Fig. 7B). These NMDA currents were substantially increased when the DRG cells were preincubated for 1–2 h with 20 ng/ml BDNF (Fig. 7B).
Figure 7.

BDNF increased NMDA currents in DRG neurons. DRG neurons from adult rats were cultured for 2 or 3 days (A) without or (B–D) with ketamine (200 μM). Prior to whole-cell patch-clamp recording, cells were left untreated or were pre-treated for 1–2 h with BDNF (20 ng/ml), proBDNF (20 ng/ml) or BDNF plus ANA-12 (100 nM). Once the cells had stabilized at a holding potential of −70 mV under voltage-clamp conditions, NMDA (250 μM) and glycine (10 μM) were applied for 10 s by pressure ejection and the peak current measured. (A) Cells cultured without ketamine showed negligible currents in response to NMDA + Gly, independently of whether they were incubated with BDNF or not. (B) Cells cultured in ketamine responded to NMDA + Gly, and the currents were larger after incubation with BDNF. (C) In cells preincubated with BDNF, a first application of NMDA+Gly evoked a current that suppressed in a second application in the presence of the NMDA receptor antagonist AP-5 (50 μM) applied for 10 min. After washout of AP-5, a third application of NMDA+Gly evoked a current of the same amplitude as the first. (D) Pretreatment with BDNF (but not with proBDNF) increased the inward current induced by NMDA. The trkB antagonist ANA-12 eliminated the increase produced by BDNF. BDNF also increased the number of cells responding to NMDA (>50 pA): from 50 % in the untreated cohort (n = 25) to 95 % in the BDNF-treated cohort (n = 19). Horizontal lines indicate the mean and errors bars indicate the SEM. **P = 0.0094, Holm–Sidak’s post-test to one-way ANOVA.
The currents produced by NMDA + Gly after BDNF were mediated by NMDA receptors, because they were suppressed by the NMDA receptor antagonist AP-5 (50 μM). Fig. 7C shows the effect of three repeated applications of NMDA + Gly in the absence (first and third) or presence (second) of AP-5. The average current obtained from four cells was −157 ± 98 pA, −7 ± 2 pA and −186 ± 127 pA for the first, second (with AP-5) and third applications of NMDA + Gly, respectively. A ratio paired t-test was used to compare NMDA currents in the same cells and revealed a decrease in the NMDA current by AP-5 after the first NMDA application: P = 0.0112, t3 = 5.61.
Figure 7D shows the amplitude of the NMDA receptor currents obtained in DRG neurons untreated or preincubated for 1–2 h with BDNF (20 ng/ml), proBDNF (20 ng/ml) or BDNF plus the trkB antagonist ANA-12 (1 μM). BDNF significantly increased the currents produced by NMDA + Gly, whereas proBDNF (with double point mutations that prevent its cleavage) produced no effect and ANA-12 eliminated the increase produced by BDNF. ANOVA (one-way) revealed a significant overall effect (P = 0.0021, F3,56 = 5.542). There was also an increase in the number of cells that responded to NMDA/glycine above 50 pA, which was 50 % for untreated (n = 25) and 95 % for BDNF (n = 19, P = 0.001, Fisher’s exact test). There was no difference in the size of the cells recorded, assessed from cell capacitance (untreated, 41 ± 4 pF; BDNF, 49 ± 5 pF).
Nerve injury increased NMDA-induced NK1R internalization
It has been postulated that BDNF release occurs in the spinal cord during the onset of neuropathic pain (Coull et al., 2005; McMahon & Malcangio, 2009). If true, this would lead to an increase in NMDA-induced substance P release. To confirm this prediction, we performed unilateral CCI of the sciatic nerve (Bennett & Xie, 1988) in rats and administered NMDA (10 nmol) intrathecally at various time points thereafter. In the ipsilateral L4 spinal segment, NMDA-induced NK1R internalization was increased after CCI, peaking at 6 h and remaining elevated for 3 days (Figs 8A and 9A). Intrathecal vehicle (PBS) after CCI did not induce NK1R internalization over the same time period (Figs 8A and 9C), showing that CCI did not evoke an ongoing substance P release. Two-way ANOVA of the ipsilateral data in Fig. 8A revealed significant effects of NMDA (P < 0.0001, F1,26 = 42.7) and time (P = 0.0476, F5,26 = 2.6), but not of their interaction (P = 0.4, F5,26 = 1.0). In the contralateral L4 spinal segment, NMDA-induced NK1R internalization was negligible during the first day after CCI (Figs 8B and 9B), but it increased in days 2 (Fig. 9D) and 3 to values similar to the ipsilateral side. Two-way ANOVA of the contralateral data in Fig. 8B yielded a significant effect of NMDA (P = 0.0053, F1,26 = 9.3) but not of time (P = 0.65, F5,26 = 0.7), and no interaction (P = 0.89, F5,26 = 0.3).
Figure 8.

CCI of the sciatic nerve increased NMDA-induced NK1R internalization. Rats underwent unilateral CCI of the sciatic nerve. NK1R internalization was measured in lamina 1 neurons of segment L4. (A and B) The rats received intrathecal injections of NMDA (10 nmol) or vehicle (PBS) at the times indicated. NK1R internalization was measured in segment L4 (A) ipsilateral and (B) contralateral to CCI. (C) Comparison of the effect of intrathecal NMDA given 6 h after CCI or sham surgery. NMDA induced NK1R internalization ipsilaterally in the CCI rats but not in the sham rats. (D) CCI induced mechanical allodynia measured as responses to von Frey hairs. Data were collected from seven rats, three of which were injected with NMDA at day 7 after CCI to obtain this time point in panels A and B. (E) After CCI, rats received two intrathecal injections of vehicle (PBS), microglia inhibitors (200 nmol minocycline, 1 nmol fluorocitrate or 10 μg propentofylline), the BDNF scavenger trkB-Fc (10 μg), the trkB antagonist ANA-12 (100 nmol) or the SFK inhibitor PP2 (10 nmol). The first injection was given just after CCI and the second 3 h after CCI. All rats then received intrathecal NMDA 6 h after CCI. NMDA was always given with D-Ser (10 nmol). Numbers next to data sets indicate n. Post hoc tests (Sidak’s): *P < 0.05, **P < 0.01 ***P < 0.001 compared with vehicle (A, B and E), sham (C) or contralateral (D).
Figure 9.

Confocal images of NK1R neurons in lamina I after CCI. Rats underwent unilateral CCI of the sciatic nerve and received intrathecal (A, B and D) NMDA (10 nmol with 10 nmol D-Ser) or (C) vehicle at the times indicated. Images were taken from the L4 spinal segment. (A and B) Intrathecal NMDA 6 h after CCI resulted in (A) abundant NK1R internalization ipsilateral to CCI but (B) no NK1R internalization contralaterally. (C) Intrathecal vehicle 6 h after CCI did not result in any NK1R internalization ipsilateral to CCI. (D) Intrathecal NMDA 48 h after CCI resulted in some NK1R internalization contralateral to CCI. Main panels: images taken with a 10× objective; voxel size of 830 × 830 × 5983 nm and three confocal planes. Insets: images panels taken with a 63× objective of the lamina I neurons indicated by the frames in the main panel; voxel size of 132 × 132 × 383 nm and four to six confocal planes. Adjustments in the gamma of the images in the insets were made to ensure that the staining of neurons was of similar intensity; the uncorrected intensity can be seen in the main panel images. Neurons with NK1R internalization are indicated with ‘*’ and neurons without internalization by ‘o’. Scale bars, 100 μm (main panels), 10 μm (insets).
This increase in NMDA-induced NK1R internalization was caused by the nerve damage and not by the surgery, as sham surgery (the sciatic nerve was exposed but not constricted) resulted in little NMDA-induced NK1R internalization 6 h after the surgery (Fig. 8C). Two-way ANOVA of data in Fig. 8C yielded significant effects of sham vs. CCI (P = 0.0004, F1,8 = 35.1), ipsilateral vs. contralateral (P = 0.0003, F1,8 = 37.1) and an interaction (P = 0.001, F1,8 = 25.8).
The development of mechanical allodynia after CCI was confirmed by measuring hind-paw withdrawal to von Frey hairs. This was done in the three rats used to obtain the 7-day time point in Fig. 8A and B, and in four additional rats that underwent an identical CCI procedure, for a total of n = 7. Allodynia developed during the first 2 days after CCI, ipsilaterally to the nerve injury (Fig. 8D). Hence, NMDA-induced NK1R internalization was maximal during the time of induction of the allodynia. Two-way ANOVA (repeated-measures) showed a significant effect of time (P < 0.0001, F3,36 = 20.7) and ipsilateral vs. contralateral side (P = 0.014, F1,12 = 8.2) and an interaction (P = 0.0008, F3,36 = 6.9), and subject matching (P = 0.0004, F12,36 = 4.2).
To confirm that the increase in NMDA-induced NK1R internalization after CCI was due to BDNF release, we used intrathecal injections of the BDNF scavenger trkB-Fc (10 μg; Mannion et al., 1999) and the trkB receptor antagonist ANA-12 (100 nmol; Cazorla et al., 2011). Intrathecal injections of these compounds were given twice, one immediately after CCI and another 3 h after CCI, followed by intrathecal NMDA at 6 h after CCI injury (the peak of NMDA-induced NK1R internalization observed in Fig. 8A). Control rats were given two injections of saline at the same times. NK1R internalization induced by NMDA 6 h after CCI was abolished by the BDNF scavenger trkB-Fc (Fig. 8E), supporting the idea that the increase in the effect of NMDA produced by CCI was caused by BDNF release. The trkB receptor antagonist ANA-12 produced a less pronounced but statistically significant inhibition (Fig. 8E), indicating the involvement of trkB receptor activation. Interestingly, the SFK inhibitor PP2 (10 nmol), given intrathecally at the same times, also abolished the NMDA-induced NK1R internalization after CCI, paralleling its inhibition of the effect of intrathecal BDNF in Fig. 1A.
Microglia in the dorsal horn have been proposed as the source of BDNF release after nerve injury (Coull et al., 2005; McMahon & Malcangio, 2009). To test this idea, rats with CCI were given intrathecal injections of three inhibitors of microglia activation: minocycline (200 nmol; Tikka et al., 2001), fluorocitrate (1 nmol; Clark et al., 2007) or propentofylline (10 μg; Holdridge et al., 2007), using the same injection schedule as for trkB-Fc and ANA-12. All three microglia inhibitors produced a partial but statistically significant inhibition of NMDA-induced NK1R internalization 6 h after CCI (Fig. 8E), indicating that microglia are one of the sources of the BDNF released by nerve injury.
Two-way ANOVA of the data in Fig. 8E revealed significant effects of the compounds (P < 0.0001, F1,40 = 22.85), ipsilateral vs. contralateral side (P < 0.0001, F6,40 = 15.68) and an interaction (P = 0.0067, F6,40 = 3.53).
Microglia activation increased NMDA-induced NK1R internalization
To further test the hypothesis that microglia are the source of BDNF that increases substance P release after CCI we took the converse approach, assessing whether direct microglia activation would increase NMDA-induced NK1R internalization. To activate dorsal horn microglia we used the Gram-negative bacterial endotoxin lipopolysaccharide (LPS), a compound that induces microglia activation by binding to toll-like receptor 4 (TLR4) (Clark et al., 2010; Sorge et al., 2011; Chen et al., 2012). We gave the rats LPS (2 μg) or saline in two intrathecal injections spaced 24 h apart (Clark et al., 2010). Six hours after the second injection, we injected NMDA (10 nmol) or saline intrathecally. There was a substantial amount of NK1R internalization when NMDA was given after LPS, but not when NMDA was given after saline, when saline was given after LPS, or in the saline–saline controls (Fig. 10A). Two-way ANOVA revealed significant effects of LPS (P = 0.0307, F1,10 = 6.320) and NMDA (P < 0.0205, F1,10 = 7.559), but no interaction (P = 0.2188, F1,10 = 1.722).
Figure 10.

Microglia activation with LPS increased NMDA-induced NK1R internalization. (A) Rats (n is indicated by numbers near the bars or data points) received intrathecal LPS (2 μg) or saline twice, 24 h apart, followed 6 h later by intrathecal NMDA (10 nmol with 10 nmol D-Ser) or saline. NK1R internalization was measured in lamina I neurons of segment L4. (B) NK1R internalization induced by NMDA injected at various times after the second injection of LPS; time 0 h corresponds to two injections of LPS followed by saline. *P = 0.012, Holm-Sidak’s post hoc test compared with time 0 h. (C) The trkB receptor antagonist ANA-12 (100 nmol), the BDNF scavenger trkB-Fc (10 μg) or the SFK inhibitor PP2 (10 nmol) were added to LPS in the second injection and injected again 3 h later; this reduced NK1R internalization induced by NMDA injected 6 h after LPS (**P < 0.01, Holm–Sidak’s post hoc test). (D) Integrated optical intensity of confocal microscope images of dorsal horn microglia stained for Iba-1 after intrathecal saline or LPS (three rats per treatment, three histological sections per rat). (E and F) Representatives of the confocal images used for the measurements in panel D. Images are a square of 425 × 425 μm (20× objective) encompassing laminae I–III; white lines indicate the dorsal edge of the section.
In another experiment, we injected NMDA (10 nmol) at different times after the second injection of LPS (Fig. 10B). NMDA-induced NK1R internalization peaked 6 h after LPS and disappeared by 24 h. There was a significant effect of the time of injection of NMDA (one-way ANOVA, P = 0.0016, F5,20 = 5.907).
As in the experiment in Fig. 8E, we used intrathecal injections of the BDNF scavenger trkB-Fc (10 μg) and the trkB receptor antagonist ANA-12 (100 nmol) to confirm that the effect of LPS was mediated by BDNF release and trkB receptor activation. In addition, the SFK inhibitor PP2 (10 nmol) was used to explore the involvement of SFKs. These compounds were added to the second injection of LPS and then injected alone 3 h later. All three compounds significantly reduced the increase in NMDA-induced NK1R internalization (Fig. 10C) to levels obtained in the absence of LPS (Fig. 10A). ANOVA (one way) revealed a significant overall effect (P = 0.0065, F3,12 = 0.2864), and significant effects of the three compounds (P < 0.01) when compared with the control in a Holm–Sidak post hoc test.
To confirm that LPS did activate dorsal horn microglia, we measured the intensity of immunoreactivity for the microglia marker Iba-1 (Clark et al., 2010) in confocal images of 425 × 425 μm encompassing laminae I–II of the central dorsal horn. Iba-1 staining was appreciably higher in LPS-injected rats (Fig. 10F) than in saline-injected rats (Fig. 10E). This was confirmed quantitatively by measuring the integrated optical intensity of images obtained from three saline-injected rats and three LPS-injected rats (Fig. 10D; P = 0.0457, t4 = 2.864, unpaired t-test).
Discussion
This study shows that BDNF acting on trkB receptors potentiates NMDA receptors present in primary afferents, and that this occurs when BDNF is released in the spinal dorsal horn during neuropathic pain. This mechanism may contribute to NMDA receptor-mediated hyperalgesia associated with neuropathic pain and other forms of chronic pain.
BDNF potentiates NMDA receptor function in primary afferents
Our initial finding was that intrathecal NMDA did not induce NK1R internalization (a measure of substance P release) in the spinal cord in normal conditions, but did so after intrathecal BDNF. In spinal cord slices, NMDA receptors trigger substance P release from the central terminals of primary afferent by mediating Ca2+ entry into the terminal (Marvizon et al., 1997; Malcangio et al., 1998; Marvizon et al., 1999; Chen et al., 2010). However, there are contradictory reports on whether this occurs in vivo when NMDA is injected intrathecally to rats: whereas an initial study by Liu et al. (1997) showed that this produced extensive NK1R internalization, a later study by Nazarian et al. (2008) found that it produced absolutely no NK1R internalization. Our own results agree with those of Nazarian et al. (2008) but also show that robust NMDA-induced NK1R internalization can be brought about by a previous injection of BDNF. Moreover, we showed that the same phenomenon occurs in mice: intrathecal NMDA did not induce significant NK1R internalization in controls, but did so after intrathecal BDNF. Importantly, NMDA-induced NK1R internalization was considerably decreased in mice with selective knock-down of NMDA receptors in primary afferents, demonstrating that the NMDA receptors that induce substance P release are present in primary afferents. These findings indicate that NMDA receptors in primary afferent terminals are in an inactive state in normal conditions and can be brought into function by BDNF. One possibility is that insertion of these NMDA receptors into the membrane increases in the presence of BDNF, as this is a common mechanism for regulating the activity of glutamate receptors.
Our patch-clamp experiments in DRG neurons support the idea that BDNF potentiates NMDA receptors in primary afferents. They show that in normal conditions NMDA receptor currents are small and found only in a few DRG cells. BDNF increased both the number of cells with NMDA receptor currents and the amplitude of those currents.
The activity of NMDA receptors in primary afferent terminals can also be measured by the effect of NMDA on EPSPs recorded in dorsal horn neurons. If NMDA receptors increase glutamate release from primary afferents as they do with substance P release, this would result in a increase in the EPSP. Surprisingly, Bardoni et al. (2004) found the opposite: NMDA actually decreased EPSPs recorded in spinal cord slices from neonatal rats. Zeng et al. (2006), also working in slices from neonatal rats, found that NMDA decreased miniature excitatory postsynaptic currents (EPSCs) in naïve rats, but increased them in opiate-tolerant rats. Importantly, Yan et al. (2013) (working with adult rats) found that presynaptic NMDA receptors had no effect on EPSCs in normal rats but increased EPSCs in neuropathic rats. These findings show that NMDA receptors in primary afferents either inhibit or have no effect on glutamate release, but switch to a stimulatory effect on glutamate release during hyperalgesic conditions (opiate tolerance or neuropathic pain). This is consistent with our own results.
The effect of BDNF is mediated by trkB receptors
BDNF exerts its effects through trkB receptors or through p75NTR which, unlike trkB receptors, can also be activated by proBDNF (Greenberg et al., 2009). We found that the increase in NMDA-induced NK1R internalization produced by BDNF is mediated by trkB receptors and not by p75NTR. First, proBDNF did not increase NMDA-induced NK1R internalization, nor did it increase NMDA receptor currents in DRG neurons. Second, the p75NTR inhibitor TAT-Pep5 (Yoshizaki et al., 2008; Pearn et al., 2012) did not inhibit the increases in NMDA-induced NK1R internalization produced by BDNF. Third, the trkB antagonist ANA-12 (Cazorla et al., 2011) reversed the increase produced by BDNF in NMDA-induced NK1R internalization and NMDA receptor currents. Fourth, the trkB agonist 7,8-DHF (Jang et al., 2010) increased NMDA-induced NK1R internalization. However, full-length trkB receptors are expressed only by a small fraction of C-fibers (McMahon et al., 1994; Kashiba et al., 1995; Wright & Snider, 1995; Bergman et al., 1999). Still, many C-fibers express a truncated form of trkB receptors (Xiao et al., 2009), trkB.T1, which has been shown to have an important pronociceptive role in neuropathic and inflammatory pain (Renn et al., 2009; Wu et al., 2013). Our own data show that trkB.T1 is present in both isolated rat DRG and in cultured DRG neurons. While it is possible that full-length trkB is expressed by DRG neurons at levels below the detection limit of our Western blots, it seems likely that the effect of BDNF on NMDA receptors in primary afferents is mediated by trkB.T1.
Mechanism of BDNF potentiation of NMDA receptors
Given that the signaling mechanisms used by trkB.T1 receptors are virtually unknown, much more work will be necessary to unravel the signaling pathways by which BDNF potentiates NMDA receptors in primary afferents. Still, we present some evidence that the end result of the BDNF signal is the phosphorylation at Tyr1472 of the NR2B subunit of NMDA receptors by a SFK. The NR2B subunit is expressed by DRG neurons (Ma & Hargreaves, 2000; Marvizon et al., 2002) and forms the NMDA receptors that mediate NMDA currents in DRG neurons (Li et al., 2004; Li et al., 2006) and induce substance P release (Chen et al., 2010). SFK phosphorylation of the NR2B subunit is known to increase the conductance of NMDA receptors (Kalia et al., 2004; Xu et al., 2006). In dorsal horn neurons, phosphorylation of Tyr1472 of the NR2B subunit by Fyn contributes to neuropathic pain (Abe et al., 2005; Matsumura et al., 2010; Katano et al., 2011) and inflammatory pain (Guo et al., 2002). We have previously shown (Chen et al., 2010) that NMDA-induced substance P release is decreased by SFK inhibitors and increased by BVT948, an inhibitor of PTPs. Likewise, we show here that SFK inhibitors decrease NMDA-induced NK1R internalization stimulated by BDNF in slices and in vivo. Treating DRG neuronal cultures with BDNF increased phosphorylation of NR2B at Tyr1472, although there was a substantial amount of phosphorylated Tyr1472-NR2B in untreated cells. These observations indicate that BDNF potentiates these NMDA receptors by SFK phosphorylation of the NR2B subunit.
It has been proposed that during the onset of neuropathic pain there is BDNF release in the dorsal horn, largely from activated microglia, and that this leads to changes in nociceptive processing circuits that cause chronic pain (Coull et al., 2005; Merighi et al., 2008b; McMahon & Malcangio, 2009; Trang et al., 2009). In agreement with this idea, we found that in the CCI model of neuropathic pain there is a marked potentiation of the NMDA receptors that induce substance P release. This potentiation peaks in the first day after nerve injury, coinciding with the development of mechanical allodynia. The potentiation of the NMDA receptors produced by CCI was mediated by BDNF release, as it was abolished by the BDNF scavenger trkB-Fc and decreased by the trkB receptor antagonist ANA-12.
Surprisingly, NMDA receptors were also potentiated in the dorsal horn contralateral to CCI, albeit to a lesser extent and several days later than in the ipsilateral dorsal horn. There are previous reports of contralateral effects of unilateral nerve injury (Koltzenburg et al., 1999) and inflammation (Kelly et al., 2007). Like the effect described here, they usually have a smaller magnitude and a shorter duration. These contralateral effects could be attributed to the involvement of descending pain modulation pathways or to circuits linking the two sides of the spinal cord. It seems unlikely that they are caused by the spread of the released BDNF to the contralateral side, in view that the peak of the contralateral effect of CCI at day 2 coincides with a decline of its ipsilateral effect (Fig. 8A and B). This temporal mismatch cannot be due to a delayed effect of BDNF, which occurs in ~ 1 hr (Figs 3A and 4C).
The sources of BDNF released after CCI include microglia, because three structurally unrelated inhibitors of microglia activation decreased, but did not abolish, NMDA-induced NK1R internalization after CCI. Primary afferents also release BDNF during severe injury (Lever et al., 2001), so they could be an additional source of BDNF. The idea that BDNF originating from microglia potentiates these NMDA receptors is further confirmed by the fact that direct activation of microglia with LPS, a TLR-4 agonist, also produced an increase in NMDA-induced NK1R internalization that peaked at the same time as the increase produced by CCI. Furthermore, the effect of LPS was eliminated by trkB-Fc and ANA-12, confirming that is was due to BDNF release and trkB receptor activation.
The idea that NMDA receptors trigger neurotransmitter release from primary afferents during the induction of neuropathic pain is supported by a recent study by Yan et al. (2013). Using the spinal nerve ligation model of neuropathic pain, they found that NMDA receptors gained the ability to induce glutamate release from primary afferents, measured as EPSCs in dorsal horn neurons.
Physiological implications
This potentiation of NMDA receptors in primary afferents probably occurs in chronic pain disorders other than neuropathic pain. Thus, in a rat model of colitis there was an increase in NMDA receptor currents in DRG neurons mediated by SFKs (Li et al., 2006). Perhaps this can explain why Liu et al. (1997) found that intrathecal NMDA induced NK1R internalization: an undetected inflammation in their rats could have potentiated these NMDA receptors. Indeed, we observed NMDA-induced NK1R internalization without prior injection of BDNF in a few rats (two of 19) that did not show signs of infection or injury.
It is assumed that the glutamate that activates these NMDA receptors is released by the same primary afferent terminals where these receptors are located. However, it is also possible that these NMDA receptors are located away from the synaptic cleft and are postsynaptic to axoaxonic synapses of interneurons or descending fibers. This possibility is supported by the fact that these NMDA receptors can occasionally have inhibitory effects by coupling with slow-conductance potassium channels that limit primary afferent excitability (Bardoni et al., 2004; Pagadala et al., 2013). If the NMDA receptors were located near the release sites, Ca2+ entry through them will always induce neurotransmitter release. All this suggests that these NMDA receptors are part of a complex system of regulation of the primary afferent input.
It is likely that, once potentiated by BDNF, NMDA receptors in primary afferents contribute to the development of chronic pain. We found that mice with selective knockdown of NMDA receptors in DRG neurons have decreased pain responses in the second phase of the formalin test (McRoberts et al., 2011). However, Pagadala et al. (2013) found that knockdown of NMDA receptors in DRG neurons did not reduce pain responses to the formalin test. Still, NMDA receptors in afferent terminals mediate the long-term potentiation of primary afferent synapses produced by opioid withdrawal and tolerance (Zeng et al., 2006; Zhou et al., 2010; Heinl et al., 2011).
In conclusion, NMDA receptors in primary afferent terminals undergo a dramatic increase in their functionality by BDNF during neuropathic pain. This mechanism may contribute to the potentiation of the synapses between primary afferents and dorsal horn neurons during chronic pain.
Acknowledgments
Supported by grant R01-DA033059 to J.C.M and J.A.M. from the National Institute of Drug Abuse, grant 1I01RX000378 to J.C.M. from the Rehabilitation Research & Development Service, Department of Veterans Affairs and research funds from the Division of Digestive Diseases at the David Geffen School of Medicine, UCLA. Mutant mice were provided by the UCLA Behavioral Core by under the direction of Michael Fanselow and supported by NIH grant 1S10RR028124. This study was done under the umbrella of the following UCLA institutes: Brain Research Institute, Center for the Study of Opioid Receptors and Drugs of Abuse, CURE: Digestive Diseases Research Center and the Oppenheimer Family Center for Neurobiology of Stress.
Abbreviations
- aCSF
artificial cerebrospinal fluid
- ANA-12
N-[2-[[(hexahydro-2-oxo-1H-azepin-3-yl)amino]carbonyl]phenyl]-benzo[b]thiophene-2-carboxamide
- BDNF
brain-derived neurotrophic factor
- BVT948
4-Hydroxy-3,3-dimethyl-2H-benz[g]indole-2,5(3H)-dione
- CCI
chronic constriction injury
- D-Ser
D-serine
- EPSP
excitatory postsynaptic potential
- EPSC
excitatory postsynaptic current
- 7,8-DHF
7,8-dihydroxyflavone
- DRG
dorsal root ganglion
- K252a
9S,10R,12R)-2,3,9,10,11,12-hexahydro-10-hydroxy-9-methyl-1-oxo-9,12-epoxy-1H-diindolo[1,2,3-fg,3′,2′,1′-kl]pyrrolo[3,4-i][1,6]benzodiazocine-10-carboxylic acid methyl ester
- K+-aCSF
artificial cerebrospinal fluid with 5 mM KCl
- LPS
lipopolysaccharide
- LTP
long-term potentiation
- NK1R
neurokinin 1 receptor
- NT-3
neurotrophin-3
- p75NTR receptors
p75 neurotrophin receptors
- PBS
phosphate-buffered saline
- PP2
3-(4-chlorophenyl) 1-(1,1-dimethylethyl)-1H-pyrazolo[3,4-d]pyrimidin-4-amine
- PP3
1-phenyl-1H-pyrazolo[3,4-d]pyrimidin-4-amine
- proBDNF
BDNF precursor protein with double point mutations preventing cleavage
- PTP
protein tyrosine phosphatase
- SFK
Src family kinase
- sucrose-aCSF
aCSF with 5 mM KCl and 215 mM sucrose instead of NaCl
- TAT-Pep5
H-YGRKKRRQRRR-CFFRGGFFNHNPRYC-OH, cyclic
- TLR4
toll-like receptor 4
- trkB
tropomyosin-related kinase B
- trkB-Fc
recombinant chimera of human trkB receptor linked to human IgG1
Footnotes
The authors declare no competing financial interests.
References
- Abbadie C, Trafton J, Liu H, Mantyh PW, Basbaum AI. Inflammation increases the distribution of dorsal horn neurons that internalize the neurokinin-1 receptor in response to noxious and non-noxious stimulation. J Neurosci. 1997;17:8049–8060. doi: 10.1523/JNEUROSCI.17-20-08049.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abe T, Matsumura S, Katano T, Mabuchi T, Takagi K, Xu L, Yamamoto A, Hattori K, Yagi T, Watanabe M, Nakazawa T, Yamamoto T, Mishina M, Nakai Y, Ito S. Fyn kinase-mediated phosphorylation of NMDA receptor NR2B subunit at Tyr1472 is essential for maintenance of neuropathic pain. Eur J Neurosci. 2005;22:1445–1454. doi: 10.1111/j.1460-9568.2005.04340.x. [DOI] [PubMed] [Google Scholar]
- Adelson DW, Lao L, Zhang G, Kim W, Marvizón JC. Substance P release and neurokinin 1 receptor activation in the rat spinal cord increases with the firing frequency of C-fibers. Neuroscience. 2009;161:538–553. doi: 10.1016/j.neuroscience.2009.03.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Al Ghamdi KS, Polgár E, Todd AJ. Soma size distinguishes projection neurons from neurokinin 1 receptor-expressing interneurons in lamina I of the rat lumbar spinal dorsal horn. Neuroscience. 2009;164:1794–1804. doi: 10.1016/j.neuroscience.2009.09.071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allen BJ, Rogers SD, Ghilardi JR, Menning PM, Kuskowski MA, Basbaum AI, Simone DA, Mantyh PW. Noxious cutaneous thermal stimuli induce a graded release of endogenous substance P in the spinal cord: imaging peptide action in vivo. J Neurosci. 1997;17:5921–5927. doi: 10.1523/JNEUROSCI.17-15-05921.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bardoni R, Torsney C, Tong CK, Prandini M, MacDermott AB. Presynaptic NMDA Receptors Modulate Glutamate Release from Primary Sensory Neurons in Rat Spinal Cord Dorsal Horn. J Neurosci. 2004;24:2774–2781. doi: 10.1523/JNEUROSCI.4637-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baxter GT, Radeke MJ, Kuo RC, Makrides V, Hinkle B, Hoang R, Medina-Selby A, Coit D, Valenzuela P, Feinstein SC. Signal transduction mediated by the truncated trkB receptor isoforms, trkB.T1 and trkB.T2. J Neurosci. 1997;17:2683–2690. doi: 10.1523/JNEUROSCI.17-08-02683.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett GJ, Xie YK. A peripheral mononeuropathy in rat that produces disorders of pain sensation like those seen in man. Pain. 1988;33:87–107. doi: 10.1016/0304-3959(88)90209-6. [DOI] [PubMed] [Google Scholar]
- Bergman E, Fundin BT, Ulfhake B. Effects of aging and axotomy on the expression of neurotrophin receptors in primary sensory neurons. J Comp Neurol. 1999;410:368–386. doi: 10.1002/(sici)1096-9861(19990802)410:3<368::aid-cne2>3.0.co;2-i. [DOI] [PubMed] [Google Scholar]
- Cazorla M, Premont J, Mann A, Girard N, Kellendonk C, Rognan D. Identification of a low-molecular weight TrkB antagonist with anxiolytic and antidepressant activity in mice. J Clin Invest. 2011;121:1846–1857. doi: 10.1172/JCI43992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen W, Zhang G, Marvizon JC. NMDA receptors in primary afferents require phosphorylation by Src family kinases to induce substance P release in the rat spinal cord. Neuroscience. 2010;166:924–934. doi: 10.1016/j.neuroscience.2010.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Z, Jalabi W, Shpargel KB, Farabaugh KT, Dutta R, Yin X, Kidd GJ, Bergmann CC, Stohlman SA, Trapp BD. Lipopolysaccharide-induced microglial activation and neuroprotection against experimental brain injury is independent of hematogenous TLR4. J Neurosci. 2012;32:11706–11715. doi: 10.1523/JNEUROSCI.0730-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark AK, Gentry C, Bradbury EJ, McMahon SB, Malcangio M. Role of spinal microglia in rat models of peripheral nerve injury and inflammation. Eur J Pain. 2007;11:223–230. doi: 10.1016/j.ejpain.2006.02.003. [DOI] [PubMed] [Google Scholar]
- Clark AK, Staniland AA, Marchand F, Kaan TK, McMahon SB, Malcangio M. P2X7-dependent release of interleukin-1beta and nociception in the spinal cord following lipopolysaccharide. J Neurosci. 2010;30:573–582. doi: 10.1523/JNEUROSCI.3295-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coull JA, Beggs S, Boudreau D, Boivin D, Tsuda M, Inoue K, Gravel C, Salter MW, De Koninck Y. BDNF from microglia causes the shift in neuronal anion gradient underlying neuropathic pain. Nature. 2005;438:1017–1021. doi: 10.1038/nature04223. [DOI] [PubMed] [Google Scholar]
- Du J, Zhou S, Coggeshall RE, Carlton SM. N-methyl-D-aspartate-induced excitation and sensitization of normal and inflamed nociceptors. Neuroscience. 2003;118:547–562. doi: 10.1016/s0306-4522(03)00009-5. [DOI] [PubMed] [Google Scholar]
- Ernfors P, Rosario CM, Merlio JP, Grant G, Aldskogius H, Persson H. Expression of mRNAs for neurotrophin receptors in the dorsal root ganglion and spinal cord during development and following peripheral or central axotomy. Brain Res Mol Brain Res. 1993;17:217–226. doi: 10.1016/0169-328x(93)90005-a. [DOI] [PubMed] [Google Scholar]
- Fenner BM. Truncated TrkB: beyond a dominant negative receptor. Cytokine Growth Factor Rev. 2012;23:15–24. doi: 10.1016/j.cytogfr.2012.01.002. [DOI] [PubMed] [Google Scholar]
- Foster E, Robertson B, Fried K. trkB-like immunoreactivity in rat dorsal root ganglia following sciatic nerve injury. Brain Res. 1994;659:267–271. doi: 10.1016/0006-8993(94)90891-5. [DOI] [PubMed] [Google Scholar]
- Geng SJ, Liao FF, Dang WH, Ding X, Liu XD, Cai J, Han JS, Wan Y, Xing GG. Contribution of the spinal cord BDNF to the development of neuropathic pain by activation of the NR2B-containing NMDA receptors in rats with spinal nerve ligation. Exp Neurol. 2010;222:256–266. doi: 10.1016/j.expneurol.2010.01.003. [DOI] [PubMed] [Google Scholar]
- Gerdes MJ, Sevinsky CJ, Sood A, Adak S, Bello MO, Bordwell A, Can A, Corwin A, Dinn S, Filkins RJ, Hollman D, Kamath V, Kaanumalle S, Kenny K, Larsen M, Lazare M, Li Q, Lowes C, McCulloch CC, McDonough E, Montalto MC, Pang Z, Rittscher J, Santamaria-Pang A, Sarachan BD, Seel ML, Seppo A, Shaikh K, Sui Y, Zhang J, Ginty F. Highly multiplexed single-cell analysis of formalin-fixed, paraffin-embedded cancer tissue. Proceedings of the National Academy of Sciences. 2013;110:11982–11987. doi: 10.1073/pnas.1300136110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grady EF, Baluk P, Bohm S, Gamp PD, Wong H, Payan DG, Ansel J, Portbury AL, Furness JB, McDonald DM, Bunnett NW. Characterization of antisera specific to NK1, NK2, and NK3 neurokinin receptors and their utilization to localize receptors in the rat gastrointestinal tract. J Neurosci. 1996;16:6975–6986. doi: 10.1523/JNEUROSCI.16-21-06975.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenberg ME, Xu B, Lu B, Hempstead BL. New insights in the biology of BDNF synthesis and release: implications in CNS function. J Neurosci. 2009;29:12764–12767. doi: 10.1523/JNEUROSCI.3566-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo W, Zou S, Guan Y, Ikeda T, Tal M, Dubner R, Ren K. Tyrosine phosphorylation of the NR2B subunit of the NMDA receptor in the spinal cord during the development and maintenance of inflammatory hyperalgesia. J Neurosci. 2002;22:6208–6217. doi: 10.1523/JNEUROSCI.22-14-06208.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanke JH, Gardner JP, Dow RL, Changelian PS, Brissette WH, Weringer EJ, Pollok BA, Connelly PA. Discovery of a novel, potent, and Src family-selective tyrosine kinase inhibitor. Study of Lck- and FynT-dependent T cell activation. J Biol Chem. 1996;271:695–701. doi: 10.1074/jbc.271.2.695. [DOI] [PubMed] [Google Scholar]
- Heinl C, Drdla-Schutting R, Xanthos DN, Sandkühler J. Distinct Mechanisms Underlying Pronociceptive Effects of Opioids. J Neurosci. 2011;31:16748–16756. doi: 10.1523/JNEUROSCI.3491-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holdridge SV, Armstrong SA, Taylor AM, Cahill CM. Behavioural and morphological evidence for the involvement of glial cell activation in delta opioid receptor function: implications for the development of opioid tolerance. Mol Pain. 2007;3:7. doi: 10.1186/1744-8069-3-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Honore P, Menning PM, Rogers SD, Nichols ML, Basbaum AI, Besson JM, Mantyh PW. Spinal cord substance P receptor expression and internalization in acute, short-term, and long-term inflammatory pain states. J Neurosci. 1999;19:7670–7678. doi: 10.1523/JNEUROSCI.19-17-07670.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang YZ, McNamara JO. Mutual regulation of Src family kinases and the neurotrophin receptor TrkB. J Biol Chem. 2010;285:8207–8217. doi: 10.1074/jbc.M109.091041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes DI, Scott DT, Riddell JS, Todd AJ. Upregulation of substance P in low-threshold myelinated afferents is not required for tactile allodynia in the chronic constriction injury and spinal nerve ligation models. J Neurosci. 2007;27:2035–2044. doi: 10.1523/JNEUROSCI.5401-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hylden JL, Wilcox GL. Intrathecal morphine in mice: a new technique. Eur J Pharmacol. 1980;67:313–316. doi: 10.1016/0014-2999(80)90515-4. [DOI] [PubMed] [Google Scholar]
- Jang SW, Liu X, Yepes M, Shepherd KR, Miller GW, Liu Y, Wilson WD, Xiao G, Blanchi B, Sun YE, Ye K. A selective TrkB agonist with potent neurotrophic activities by 7,8-dihydroxyflavone. Proc Natl Acad Sci U S A. 2010;107:2687–2692. doi: 10.1073/pnas.0913572107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalia LV, Gingrich JR, Salter MW. Src in synaptic transmission and plasticity. Oncogene. 2004;23:8007–8016. doi: 10.1038/sj.onc.1208158. [DOI] [PubMed] [Google Scholar]
- Kashiba H, Noguchi K, Ueda Y, Senba E. Coexpression of trk family members and low-affinity neurotrophin receptors in rat dorsal root ganglion neurons. Brain Res Mol Brain Res. 1995;30:158–164. doi: 10.1016/0169-328x(94)00249-e. [DOI] [PubMed] [Google Scholar]
- Katano T, Nakazawa T, Nakatsuka T, Watanabe M, Yamamoto T, Ito S. Involvement of spinal phosphorylation cascade of Tyr1472-NR2B, Thr286-CaMKII, and Ser831-GluR1 in neuropathic pain. Neuropharmacology. 2011;60:609–616. doi: 10.1016/j.neuropharm.2010.12.005. [DOI] [PubMed] [Google Scholar]
- Kelly S, Dunham JP, Donaldson LF. Sensory nerves have altered function contralateral to a monoarthritis and may contribute to the symmetrical spread of inflammation. Eur J Neurosci. 2007;26:935–942. doi: 10.1111/j.1460-9568.2007.05737.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koltzenburg M, Wall PD, McMahon SB. Does the right side know what the left is doing? Trends Neurosci. 1999;22:122–127. doi: 10.1016/s0166-2236(98)01302-2. [DOI] [PubMed] [Google Scholar]
- Lever IJ, Bradbury EJ, Cunningham JR, Adelson DW, Jones MG, McMahon SB, Marvizon JC, Malcangio M. Brain-derived neurotrophic factor is released in the dorsal horn by distinctive patterns of afferent fiber stimulation. J Neurosci. 2001;21:4469–4477. doi: 10.1523/JNEUROSCI.21-12-04469.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J, McRoberts JA, Ennes HS, Trevisani M, Nicoletti P, Mittal Y, Mayer EA. Experimental colitis modulates the functional properties of NMDA receptors in dorsal root ganglia neurons. Am J Physiol Gastrointest Liver Physiol. 2006;291:G219–228. doi: 10.1152/ajpgi.00097.2006. [DOI] [PubMed] [Google Scholar]
- Li J, McRoberts JA, Nie J, Ennes HS, Mayer EA. Electrophysiological characterization of N-methyl-d-aspartate receptors in rat dorsal root ganglia neurons. Pain. 2004;109:443–452. doi: 10.1016/j.pain.2004.02.021. [DOI] [PubMed] [Google Scholar]
- Liljebris C, Baranczewski P, Bjorkstrand E, Bystrom S, Lundgren B, Tjernberg A, Warolen M, James SR. Oxidation of protein tyrosine phosphatases as a pharmaceutical mechanism of action: a study using 4-hydroxy-3,3-dimethyl-2H-benzo[g]indole-2,5(3H)-dione. J Pharmacol Exp Ther. 2004;309:711–719. doi: 10.1124/jpet.103.062745. [DOI] [PubMed] [Google Scholar]
- Liu H, Mantyh PW, Basbaum AI. NMDA-receptor regulation of substance P release from primary afferent nociceptors. Nature. 1997;386:721–724. doi: 10.1038/386721a0. [DOI] [PubMed] [Google Scholar]
- Liu H, Wang H, Sheng M, Jan LY, Jan YN, Basbaum AI. Evidence for presynaptic N-methyl-D-aspartate autoreceptors in the spinal cord dorsal horn. Proc Natl Acad Sci U S A. 1994;91:8383–8387. doi: 10.1073/pnas.91.18.8383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma QP, Hargreaves RJ. Localization of N-methyl-D-aspartate NR2B subunits on primary sensory neurons that give rise to small-caliber sciatic nerve fibers in rats. Neuroscience. 2000;101:699–707. doi: 10.1016/s0306-4522(00)00419-x. [DOI] [PubMed] [Google Scholar]
- Madara JC, Levine ES. Presynaptic and postsynaptic NMDA receptors mediate distinct effects of brain-derived neurotrophic factor on synaptic transmission. J Neurophysiol. 2008;100:3175–3184. doi: 10.1152/jn.90880.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malcangio M, Fernandes K, Tomlinson DR. NMDA receptor activation modulates evoked release of substance P from rat spinal cord. Br J Pharmacol. 1998;125:1625–1626. doi: 10.1038/sj.bjp.0702260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mannion RJ, Costigan M, Decosterd I, Amaya F, Ma QP, Holstege JC, Ji RR, Acheson A, Lindsay RM, Wilkinson GA, Woolf CJ. Neurotrophins: peripherally and centrally acting modulators of tactile stimulus-induced inflammatory pain hypersensitivity. Proc Natl Acad Sci U S A. 1999;96:9385–9390. doi: 10.1073/pnas.96.16.9385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mantyh PW, DeMaster E, Malhotra A, Ghilardi JR, Rogers SD, Mantyh CR, Liu H, Basbaum AI, Vigna SR, Maggio JE. Receptor endocytosis and dendrite reshaping in spinal neurons after somatosensory stimulation. Science. 1995;268:1629–1632. doi: 10.1126/science.7539937. [DOI] [PubMed] [Google Scholar]
- Marvizon JC, Grady EF, Stefani E, Bunnett NW, Mayer EA. Substance P release in the dorsal horn assessed by receptor internalization: NMDA receptors counteract a tonic inhibition by GABA(B) receptors. Eur J Neurosci. 1999;11:417–426. doi: 10.1046/j.1460-9568.1999.00445.x. [DOI] [PubMed] [Google Scholar]
- Marvizon JC, Martinez V, Grady EF, Bunnett NW, Mayer EA. Neurokinin 1 receptor internalization in spinal cord slices induced by dorsal root stimulation is mediated by NMDA receptors. J Neurosci. 1997;17:8129–8136. doi: 10.1523/JNEUROSCI.17-21-08129.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marvizon JC, McRoberts JA, Ennes HS, Song B, Wang X, Jinton L, Corneliussen B, Mayer EA. Two N-methyl-D-aspartate receptors in rat dorsal root ganglia with different subunit composition and localization. J Comp Neurol. 2002;446:325–341. doi: 10.1002/cne.10202. [DOI] [PubMed] [Google Scholar]
- Marvizon JC, Wang X, Matsuka Y, Neubert JK, Spigelman I. Relationship between capsaicin-evoked substance P release and neurokinin 1 receptor internalization in the rat spinal cord. Neuroscience. 2003;118:535–545. doi: 10.1016/s0306-4522(02)00977-6. [DOI] [PubMed] [Google Scholar]
- Matsumura S, Kunori S, Mabuchi T, Katano T, Nakazawa T, Abe T, Watanabe M, Yamamoto T, Okuda-Ashitaka E, Ito S. Impairment of CaMKII activation and attenuation of neuropathic pain in mice lacking NR2B phosphorylated at Tyr1472. Eur J Neurosci. 2010;32:798–810. doi: 10.1111/j.1460-9568.2010.07348.x. [DOI] [PubMed] [Google Scholar]
- McMahon SB, Armanini MP, Ling LH, Phillips HS. Expression and coexpression of Trk receptors in subpopulations of adult primary sensory neurons projecting to identified peripheral targets. Neuron. 1994;12:1161–1171. doi: 10.1016/0896-6273(94)90323-9. [DOI] [PubMed] [Google Scholar]
- McMahon SB, Malcangio M. Current challenges in glia-pain biology. Neuron. 2009;64:46–54. doi: 10.1016/j.neuron.2009.09.033. [DOI] [PubMed] [Google Scholar]
- McRoberts JA, Coutinho SV, Marvizon JC, Grady EF, Tognetto M, Sengupta JN, Ennes HS, Chaban VV, Amadesi S, Creminon C, Lanthorn T, Geppetti P, Bunnett NW, Mayer EA. Role of peripheral N-methyl-D-aspartate (NMDA) receptors in visceral nociception in rats. Gastroenterology. 2001;120:1737–1748. doi: 10.1053/gast.2001.24848. [DOI] [PubMed] [Google Scholar]
- McRoberts JA, Ennes H, Marvizon JCG, Fanselow M, Mayer EA, Vissel B. Selective knockdown of NMDA receptors in primary afferent neurons decreases pain during phase 2 of the formalin test. Neuroscience. 2011;172:474–482. doi: 10.1016/j.neuroscience.2010.10.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McRoberts JA, Li J, Ennes HS, Mayer EA. Sex-dependent differences in the activity and modulation of N-methyl-d-aspartic acid receptors in rat dorsal root ganglia neurons. Neuroscience. 2007;148:1015–1020. doi: 10.1016/j.neuroscience.2007.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merighi A, Bardoni R, Salio C, Lossi L, Ferrini F, Prandini M, Zonta M, Gustincich S, Carmignoto G. Presynaptic functional trkB receptors mediate the release of excitatory neurotransmitters from primary afferent terminals in lamina II (substantia gelatinosa) of postnatal rat spinal cord. Dev Neurobiol. 2008a;68:457–475. doi: 10.1002/dneu.20605. [DOI] [PubMed] [Google Scholar]
- Merighi A, Salio C, Ghirri A, Lossi L, Ferrini F, Betelli C, Bardoni R. BDNF as a pain modulator. Prog Neurobiol. 2008b;85:297–317. doi: 10.1016/j.pneurobio.2008.04.004. [DOI] [PubMed] [Google Scholar]
- Nagy GG, Watanabe M, Fukaya M, Todd AJ. Synaptic distribution of the NR1, NR2A and NR2B subunits of the N-methyl-d-aspartate receptor in the rat lumbar spinal cord revealed with an antigen-unmasking technique. Eur J Neurosci. 2004;20:3301–3312. doi: 10.1111/j.1460-9568.2004.03798.x. [DOI] [PubMed] [Google Scholar]
- Nazarian A, Gu G, Gracias NG, Wilkinson K, Hua XY, Vasko MR, Yaksh TL. Spinal NMDA receptors and nociception-evoked release of primary afferent substance P. Neuroscience. 2008;152:119–127. doi: 10.1016/j.neuroscience.2007.11.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pagadala P, Park CK, Bang S, Xu ZZ, Xie RG, Liu T, Han BX, Tracey WD, Wang F, Ji RR. Loss of NR1 subunit of NMDARs in primary sensory neurons leads to hyperexcitability and pain hypersensitivity: Involvement of Ca2+-activated small conductance potassium channels. The Journal of Neuroscience. 2013;33:13425–13430. doi: 10.1523/JNEUROSCI.0454-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pearn ML, Hu Y, Niesman IR, Patel HH, Drummond JC, Roth DM, Akassoglou K, Patel PM, Head BP. Propofol neurotoxicity is mediated by p75 neurotrophin receptor activation. Anesthesiology. 2012;116:352–361. doi: 10.1097/ALN.0b013e318242a48c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Renn CL, Leitch CC, Dorsey SG. In vivo evidence that truncated trkB.T1 participates in nociception. Molecular Pain. 2009;5:61. doi: 10.1186/1744-8069-5-61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato K, Kiyama H, Tae Park H, Tohyama M. AMPA, KA and NMDA receptors are expressed in the rat DRG neurones. Neuroreport. 1993;4:1263–1265. doi: 10.1097/00001756-199309000-00013. [DOI] [PubMed] [Google Scholar]
- Schittenhelm MM, Shiraga S, Schroeder A, Corbin AS, Griffith D, Lee FY, Bokemeyer C, Deininger MW, Druker BJ, Heinrich MC. Dasatinib (BMS-354825), a dual SRC/ABL kinase inhibitor, inhibits the kinase activity of wild-type, juxtamembrane, and activation loop mutant KIT isoforms associated with human malignancies. Cancer Res. 2006;66:473–481. doi: 10.1158/0008-5472.CAN-05-2050. [DOI] [PubMed] [Google Scholar]
- Seeber S, Humeny A, Herkert M, Rau T, Eschenhagen T, Becker CM. Formation of molecular complexes by N-methyl-D-aspartate receptor subunit NR2B and ryanodine receptor 2 in neonatal rat myocard. J Biol Chem. 2004;279:21062–21068. doi: 10.1074/jbc.M313009200. [DOI] [PubMed] [Google Scholar]
- Sorge RE, LaCroix-Fralish ML, Tuttle AH, Sotocinal SG, Austin JS, Ritchie J, Chanda ML, Graham AC, Topham L, Beggs S, Salter MW, Mogil JS. Spinal cord Toll-like receptor 4 mediates inflammatory and neuropathic hypersensitivity in male but not female mice. J Neurosci. 2011;31:15450–15454. doi: 10.1523/JNEUROSCI.3859-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Storkson RV, Kjorsvik A, Tjolsen A, Hole K. Lumbar catheterization of the spinal subarachnoid space in the rat. J Neurosci Methods. 1996;65:167–172. doi: 10.1016/0165-0270(95)00164-6. [DOI] [PubMed] [Google Scholar]
- Tikka T, Fiebich BL, Goldsteins G, Keinanen R, Koistinaho J. Minocycline, a tetracycline derivative, is neuroprotective against excitotoxicity by inhibiting activation and proliferation of microglia. J Neurosci. 2001;21:2580–2588. doi: 10.1523/JNEUROSCI.21-08-02580.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trafton JA, Abbadie C, Marchand S, Mantyh PW, Basbaum AI. Spinal opioid analgesia: how critical is the regulation of substance P signaling? J Neurosci. 1999;19:9642–9653. doi: 10.1523/JNEUROSCI.19-21-09642.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trang T, Beggs S, Wan X, Salter MW. P2X4-receptor-mediated synthesis and release of brain-derived neurotrophic factor in microglia is dependent on calcium and p38-mitogen-activated protein kinase activation. J Neurosci. 2009;29:3518–3528. doi: 10.1523/JNEUROSCI.5714-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright DE, Snider WD. Neurotrophin receptor mRNA expression defines distinct populations of neurons in rat dorsal root ganglia. J Comp Neurol. 1995;351:329–338. doi: 10.1002/cne.903510302. [DOI] [PubMed] [Google Scholar]
- Wu J, Renn CL, Faden AI, Dorsey SG. TrkB.T1 Contributes to Neuropathic Pain after Spinal Cord Injury through Regulation of Cell Cycle Pathways. The Journal of Neuroscience. 2013;33:12447–12463. doi: 10.1523/JNEUROSCI.0846-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao J, Wong AW, Willingham MM, Kaasinen SK, Hendry IA, Howitt J, Putz U, Barrett GL, Kilpatrick TJ, Murray SS. BDNF exerts contrasting effects on peripheral myelination of NGF-dependent and BDNF-dependent DRG neurons. J Neurosci. 2009;29:4016–4022. doi: 10.1523/JNEUROSCI.3811-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu F, Plummer MR, Len GW, Nakazawa T, Yamamoto T, Black IB, Wu K. Brain-derived neurotrophic factor rapidly increases NMDA receptor channel activity through Fyn-mediated phosphorylation. Brain Res. 2006;1121:22–34. doi: 10.1016/j.brainres.2006.08.129. [DOI] [PubMed] [Google Scholar]
- Yan X, Yan E, Gao M, Weng HR. Endogenous activation of presynaptic NMDA receptors enhances glutamate release from the primary afferents in the spinal dorsal horn in a rat model of neuropathic pain. J Physiol. 2013;591:2001–2019. doi: 10.1113/jphysiol.2012.250522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshizaki K, Yamamoto S, Yamada A, Yuasa K, Iwamoto T, Fukumoto E, Harada H, Saito M, Nakasima A, Nonaka K, Yamada Y, Fukumoto S. Neurotrophic Factor Neurotrophin-4 Regulates Ameloblastin Expression via Full-length TrkB. J Biol Chem. 2008;283:3385–3391. doi: 10.1074/jbc.M704913200. [DOI] [PubMed] [Google Scholar]
- Zeng J, Thomson LM, Aicher SA, Terman GW. Primary afferent NMDA receptors increase dorsal horn excitation and mediate opiate tolerance in neonatal rats. J Neurosci. 2006;26:12033–12042. doi: 10.1523/JNEUROSCI.2530-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang G, Chen W, Marvizon JC. Src family kinases mediate the inhibition of substance P release in the rat spinal cord by μ-opioid receptors and GABAB receptors, but not α2 adrenergic receptors. Eur J Neurosci. 2010;32:963–973. doi: 10.1111/j.1460-9568.2010.07335.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou HY, Chen SR, Chen H, Pan HL. Opioid-Induced Long-Term Potentiation in the Spinal Cord Is a Presynaptic Event. J Neurosci. 2010;30:4460–4466. doi: 10.1523/JNEUROSCI.5857-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou LJ, Zhong Y, Ren WJ, Li YY, Zhang T, Liu XG. BDNF induces late-phase LTP of C-fiber evoked field potentials in rat spinal dorsal horn. Exp Neurol. 2008;212:507–514. doi: 10.1016/j.expneurol.2008.04.034. [DOI] [PubMed] [Google Scholar]