

Abstract
Mononuclear phagocytes including monocytes and macrophages, are important defense components of innate immunity, but can be detrimental in HIV-1 infection by serving as the principal reservoirs of virus in brain and triggering a strong immune response. These viral reservoirs represent a challenge to HIV-1 eradication since they continue producing virus in tissue despite antiretroviral therapy. HIV-1 associated neurocognitive disorders (HAND) involve alterations to the blood-brain barrier and migration of activated HIV-1 infected monocytes to the brain with subsequent induced immune activation response. Our group recently showed that HIV replication in monocyte-derived macrophages is associated with increased cystatin B. This cysteine protease inhibitor also inhibits the interferon-induced antiviral response by decreasing levels of tyrosine phosphorylated STAT-1. These recent discoveries reveal novel mechanisms of HIV persistence that could be targeted by new therapeutic approaches to eliminate HIV in macrophage reservoirs. However, cystatin B has been also associated with neuroprotection. Cystatin B is an inhibitor of the cysteine protease cathepsin B, a potent neurotoxin. During HIV-1 infection cystatin B and cathepsin B are upregulated in macrophages. Reduction in cystatin/cathepsin interactions in infected macrophages leads to increased cathepsin B secretion and activity which contributes to neuronal apoptosis. Increased intracellular expression of both proteins was recently found in monocytes from Hispanic women with HAND. These findings provide new evidence for the role of cathepsin /cystatin system in the neuropathogenesis induced by HIV-infected macrophages. We summarize recent research on cystatin B and one of its substrates, cathepsin B, in HIV replication in macrophages and neuropathogenesis.
Keywords: Cathepsin B, cystatin B, cystatin C, HAND, HIV-1, macrophages
Introduction
Cystatin B is a reversible and competitive inhibitor of cysteine proteases, cathepsin L, S and B, widely distributed in most cell types and tissues [1]. Structurally, human cystatin B is a protein composed of 98 amino acid residues arranged in a single chain with a molecular mass of approximately 12 kDa. Its tertiary structure consists of five stranded beta-sheet wrapped around a five turn alpha-helix, with a carboxyl-terminal strand running on the convex side of the sheet [2]. Cystatin B has an important role as an inhibitor of a cysteine protease cathepsin B, a potent neurotoxin. Cathepsin B is a lysosomal cysteine protease with several roles in maintaining the normal metabolism of cells including the turnover of proteins in normal cells and tissues [3]. It is synthesized as a preproenzyme of 330 aminoacid residues with a molecular mass of approximately 37 kDa [3,4]. Activation of cathepsin B occurs by excision of 62 residues that produce a two-chain form of the enzyme with the excision of a dipeptide [3,4]. This protein can retain its enzymatic activity in the cytosol and in the extracellular space, in response to different stimuli [5-7].
Oxidized or reduced forms of glutathione can modify the inhibitory activity of cystatin B [8]. Reduced glutathione can react with cystatin B to form a disulfate-bond that produces two inactive forms of cystatin B: the glutathionated or the dimmer [8]. These forms are unable to insert into the binding pocket of cathepsins. Therefore, the activities of cathepsins are regulated by the intracellular redox potentials since the oxidized or reduced forms of glutathione can modify the inhibitory activity of cystatin B [8].
Mutations in the cystatin B gene cause a hereditary neurodegenerative disorder called Progressive myoclonus epilepsy of Unverricht-Lundborg type (EPM1). In this disease, cystatin B deficiency is linked to increased oxidative stress and neuronal degeneration mediated by the lysosomal protease cathepsin B [9]. Interestingly, cystatin B has a new role in HIV-1 infection that depends on its tissue and cellular localization (i.e., intracellular vs extracellular). Our group demonstrated that intracellular cystatin B induces HIV replication in blood monocyte-derived macrophages (MDM) [10,11]. These discoveries suggest that the role of cystatin B changes from that of neuroprotective cysteine protease inhibitor to a novel, detrimental role of inducing HIV replication in macrophages. Another study reported that cystatin B was significantly over-expressed in the cervicovaginal mucosa proteome of HIV-1-resistant women, suggesting a protective role of cystatin B [12]. These apparently divergent roles of cystatin B in HIV replication in different tissues deserve further study. In this review, new evidences for a role of cathepsin-cystatin system in HIV replication and the neurodegeneration induced by HIV-infected macrophages are discussed.
Macrophage derived cystatin B / cathepsin B in HIV replication
Cystatin B in Monocyte Differentiation and Inflammatory Responses
Although monocytes have been described as important HIV reservoirs, relatively few monocytes in the blood harbor HIV-1 DNA in HIV-infected individuals (<0.1%) [13]. The susceptibility of monocytes to HIV-1 replication depends on their differentiation status. Monocytes are refractory to infection and become permissive upon differentiation into macrophages [14], although their tissue localization and host factors also influence their susceptibility to infection [15,16]. Interestingly, Hashimoto showed that gene transcripts of cystatin B were significantly increased upon differentiation of monocytes (which resist HIV-1 infection) into macrophages (which are permissive for infection) [17].
Depending on their tissue localization and the inducing stimulus, macrophages are induced for polarization by a variety of factors, including cytokines and bacterial products. Through cell macrophage polarization, programmed macro-phages can respond with classical M1 (pro-inflammatory), or alternative M2 (anti-inflammatory) responses. The different-ially expressed markers of human macrophage polarization have been summarized by Cassol and others [18,19]. The classical response mediated by M1 cells is activated by IFN-γ, TNF-α, and bacterial products such as LPS [20-22] and is characterized by high levels of IL12 and low levels of IL10. The classical response activates Th2 to kill microorganisms and produces pro-inflammatory cytokines such as IL1, IL6, IL12, and TNF-α. A role of cystatin B in the classical (M1) response activated by LPS has been suggested. Treatment with LPS causes upregulation of cystatin B expression in human monocytes, whereas cystatin A is decreased and cystatin C is not affected, indicating a possible role of cystatin B in the innate immune response against bacterial infections [23,24]. The alternative response mediated by M2 cells is activated by IL4, IL13 (M2a; involved in tissue repair), immune complex (M2b; immune regulation) and IL10 (M2c; immune suppression and regulation). In contrast to the M1 response, the M2 response is characterized by low levels of IL12 and high levels of IL10 [reviewed by 13]. M2a cells activate Th2 and the type II inflammation response, and induce high levels of anti-inflammatory cytokines (such as as IL1 and IL1 receptor antagonist), whereas M2b cells produce pro-inflammatory cytokines (such as IL1, IL6, TNF-α and the anti-inflammatory IL10), and M2c cells produce IL10 and TGF-β. The expression of cystatin B and/or its role in alternative (M2) response remains to be determined.
A Novel Detrimental Role of Cystatin B as an Inducer of HIV Replication in Macrophages
Although cystatins are known as cysteine protease inhibitors, additional functions of cystatins have been found. For example, cystatin B induces TNF-α and IL10 synthesis and stimulates nitric oxide production [26,27]. These studies suggest that cystatin B could induce oxidative stress in HIV -1 infection (Fig. 1). In another recently reported novel function, cystatin B expression has been positively correlated with HIV replication in MDM. Specifically, our group reported that cystatin B is up-regulated in blood MDM compared to placental macrophages or Hofbauer cells, which are less susceptible than blood MDM to HIV-1 infection [28]. HIV-infected MDM show increased levels of both intracellular [10] and secreted cystatin B [29,30], with similar mRNA levels, suggesting that this protein is activated during HIV infection at the post-transcriptional level [31]. A direct connection of cystatin B and HIV replication was demonstrated with siRNA against cystatin B [10]. Subsequently, the signaling mechanisms for cystatin B in HIV replication were related to its interaction with STAT-1 [32]. It is known that HIV infection of macrophages activates STAT-1 [33]. Furthermore, high levels of tyrosine phosphorylated STAT-1 (STAT-1PY) have been associated with HIV-1 inhibitory activity [34]. However, another study reported the opposite: that HIV infection causes an increase in STAT-1PY at 6 days until 20 days after infection [33]. Our group demonstrated that placental macrophages, a restrictive cell for HIV replication compared to MDM [10,28,35], had higher levels of STAT-1PY, while MDM had very low levels of STAT-1PY at 12 days after infection [32]. Since STAT-1PY has been associated with HIV-1 inhibitory activity [34] and recent studies by our group showed that cystatin B decreases STAT-1PY in Vero cells [11], cystatin B may play a role in inducing/enhancing HIV replication by decreasing STAT-1PY levels.
Fig. (1).
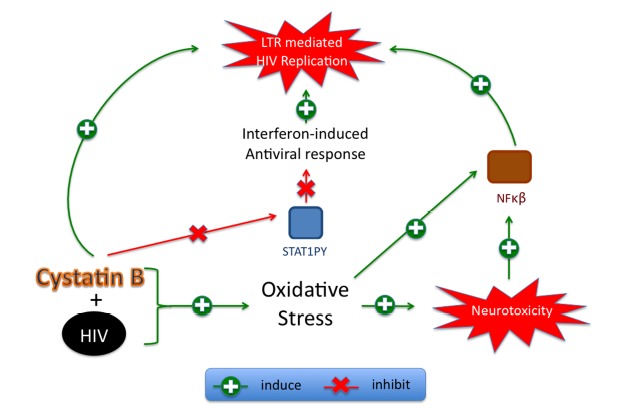
Proposed mechanism for cystatin B in HIV-1 replication and neurotoxicity induced by macrophage reservoirs. HIV replication in macrophages is associated with increased cystatin B and oxidative stress. Oxidative stress induces neurotoxicity via NF-kB. Cystatin B decreases STAT1PY and inhibits the IFN-induced antiviral genes that activate LTR-mediated HIV-1 replication.
Our group also demonstrated that cystatin B inhibited the interferon beta (IFN-β) response in Vero cells by preventing STAT-1 translocation to the nucleus and decreasing levels of STAT-1PY [11]. Whereas serine phosphorylated STAT-1 (STAT-1PS) is detrimental by inducing blood-brain barrier damage [36], STAT-1PY is beneficial since it has been associated with HIV-1 inhibitory activity mediated by CD8-T-lymphocyte antiviral factor (CAF). CAF inhibits LTR-mediated HIV replication by inducing the expression of interferon regulatory transcription factor 1 (IRF-1). This mechanism is STAT-1PY-dependent, since it requires the formation of the IRF-1/STAT-1PY complex. Activation of NF-kappa B (NF-κB) occurs in oxidative stress during HIV replication and in resistance to TNF-induced macrophage apoptosis to promote monocyte and macrophage survival [37,38]. It is postulated that HIV uses this mechanism as part of a strategy to regulate viral persistence by manipulating the apoptotic machinery. We propose that HIV-induced oxidative stress in macrophages acts in combination with cystatin B to activate NF-kB. Since cystatin B decreases STAT-1PY, this effect would release IRF-1 from the IRF-1/STAT1PY complex and allow the formation of NF-kB/IRF-1, which activates HIV-LTR induced HIV-1 replication (Fig. 1). Further studies are being conducted to elucidate the signaling pathways mediated by cystatin B.
Cathepsins and HIV Replication
Cathepsin B function is altered during HIV-1 infection of macrophages. We recently reported that during HIV-1 infection of MDM, cathepsin B no longer shows its normal cellular localization, and shows reduced interactions with its endogenous inhibitors, cystatin B and cystatin C [31]. The shuttling of cystatin B from the cytosol to the cytoplasmic membrane during HIV-infection also interferes with the formation of cystatin B/cathepsin B complexes [39]. The dysregulation of cystatin B/cathepsin B after HIV infection and its connection with HIV-1 associated neurocognitive disorders (HAND) were important in vivo findings reported recently by our group [31,40].
Cathepsin B has been associated with some aspects of HIV replication. For example, cathepsin B facilitates the release of HIV-Gag particles but also inhibits CD4 independent HIV-1 infection. A role for cathepsin B in the release of HIV-Gag particles has been demonstrated in studies with cathepsin B knockout mice and the specific cathepsin B inhibitor CA-074Me. The absence of cathepsin B leads to defects in the release of HIV-Gag particles and causes their accumulation in intracellular compartments, such as autophagosomes, in HEK293T transfected cells [41]. Cathepsin B may therefore be required for proper cleavage of HIV-Gag particles. In the endocytic pathway, the acidic late endosomes merge with the lysosomes, forming endolysosomes. If a viral particle enters a cell through the acidic late endosomes, the cathepsin B inside the lysosomes will degrade the viral particles. However, HIV entry into CD4-dependent cells occurs principally at the membrane and early endosomes escaping from late endosome; therefore inhibition of cathepsin B will not affect HIV replication. CD4-independent cell entry occurs through acidic endosomes and is inhibited by cathepsin B [42]. As reviewed [43], these findings suggest that HIV-1 may have developed an acidification-independent entry mechanism to overcome cathepsin B digestion in late endosome. Lysosomal permeabilization has also been linked with HIV replication, as diffusion of cathepsins B, L and D into the cytosol is enhanced when CD4+ lymphocytes are infected with HIV-1, and inhibited by addition of 2',3'-dideoxyinosine (didanosine or ddI), an HIV reverse transcriptase inhibitor [44].
Cystatin B and cathepsin B in neurode-generation
Cystatins B and Cathepsin B in Neurodegenerative Disorders
Cystatins comprise a large superfamily of proteins, and have protease inhibitory activity that is essential to maintain the homeostasis and physiological conditions of the cells [45,46]. Based on their inhibitory activity, cystatins are classified into three families: family I or stefins (also called cystatin A and cystatin B), family II or cystatins C, F, E/M, and finally, family or kininogens [47]. In addition to the roles discussed above, cystatin B and cathepsin B have been also implicated in neurodegenerative disorders. Cystatin B, as described above, has been associated with Progressive myoclonus epilepsy of Unverricht-Lundborg type (EPM1) [9]. In an in vitro model of EPM1, cystatin B can protect cerebellar granule neurons prepared from postnatal day 6 rat or postnatal day 5 mouse from oxidative stress-induced death and this effect is dependent on cathepsin B. Moreover, cystatin B in EPM1 mutant mice can form oligomers and protein aggregates that cause cytotoxicity [48]. A protective role for cystatin B was also proposed in another neurodegenerative disease, Alzheimer ‘s (AD), from in vitro studies with inhibition of amyloid fibril formation via regulation of Ab peptide oligomerization and aggregation [49].
Cathepsin B may also play an important role in AD [50]. For example, cathepsin B has been recently identified in secretory vesicles acting as β-secretase, and may thereby contribute to the production of neurotoxic β- amyloid (Aβ) in AD [51-54]. Consistent with this idea, the cysteine protease inhibitor E64d can reduce brain amyloid-β accumulation and improve memory deficits in AD transgenic mice [55]. Furthermore, deletion of the cathepsin B gene in AD transgenic mice improves memory deficits [56]. In contrast, other studies have suggested lysosomal modulation as a possible therapeutic approach to diseases involving protein accumulation [57,58]. For example, in AD, cathepsin B activity could be upregulated to degrade beta-amyloid aggregates [59]. It has been observed that lysosomal cathepsin B and D plays a degradative role in autophagy [60]. Both cathepsins are required to degrade dysfunctional cellular components through the lysosomal machinery [60]. In certain lysosomal storage genetic disorders, such saposin C deficiency, accumulation of autophagosomes occurs in fibroblasts due to decreased enzymatic activity of cathepsins B and D [61]. Another study suggests that dysregulation of cathepsins affects cellular homeostasis in multiple sclerosis (MS) [62], and post-mortem studies of patients with MS showed increased cathepsin B activity in brain [63]. Previous studies also described increased cathepsin B activity in peripheral blood mononuclear cells from MS patients [64]. Moreover, cathepsin B contributes to traumatic brain injury via induction of mitochondria-mediated apoptosis [65,66]. Cathepsin B is also involved in brain glioma invasiveness and migration [67]. Therefore, cathepsin-cystatin system plays important roles in the context of neurodegenerative disorders, brain injury, and cancer.
Cystatin B and cathepsin B in HIV-1 neuropathogenesis
Neurons are not susceptible to HIV-1 infection, but their dysfunction and loss are an important feature of HAND [68-70]. A hallmark of HIV neuropathology is the formation of multinucleated giant cells by the fusion of infected macrophages and microglia, and these are a common findings in patients with HIV encephalitis (HIVE) [71-73]. HIVE is the result of the severe inflammatory response ongoing in the brain parenchyma. As previously mentioned, a massive cellular activation occurs, involving perivascular macrophages, microglia and astrocytes. These activated cells orchestrate an immune defense network in which several important reactions take place. (1) Reactive oxygen species (ROS) are formed and cells undergo oxidative stress triggered by viral proteins. The antioxidant response may have a role in neuropathogenesis [74]. (2) Chemoattractant molecules such as cytokines, chemokines and viral factors disrupt the BBB and recruit more immune cells to the brain [75,76]. (3) Cytokines are secreted in order to activate the surrounding cells and trigger degranulation, which can also harm the tissue itself. (4) Cells productively infected also secrete viral proteins such as Tat, Nef and gp120. These proteins have been demonstrated to be neurotoxic, and Tat has been found inside neurons [77-79]. Tat within neurons can cause disruption of the endolysosome membrane and inhibition of autophagy, leading to neuronal damage [80]. Conditioned media from Nef-expressing astrocytes contains inflammatory cytokines that induce neuronal death [79]. Gp120 directly injected into the mouse caudate putamen induces oxidative stress and neuronal death [81]. (5) Finally, activated cells not only secrete ROS, cytokines and chemokines, but also show activation, and in many cases secretion of many enzymes and proteases, including the lysosomal cysteine protease cathepsin B [31].
The literature indicates that cells, under stress, suffer lysosomal permeabilization and leakage of proteases to the cytosol, which contribute to cell death in different conditions including cancer, liver injury and traumatic brain injury [82-85]. Cathepsin B can retain its activity once it is released into the cytosol under a range of pH from 4 to 7, and is capable of cleaving capases. In the context of HIV neuropathogenesis, cathepsin D together with other lysosomal enzymes, was found to be increased in post-mortem brains of patients with AIDS and HIVE. Moreover, this increase in lysosomal enzymes correlated with increased density of activated microglia [86], suggesting a potential connection with HIV-1 replication in the brain. Later, another group discovered that lysosomal cathepsins B and D increase and diffuse into the cytosol of HIV-infected lymphocytes, and the immunostaining intensity was correlated with the infection [44]. The authors also concluded that Nef can cause lysosomal permeabilization, leading to leaking of lysosomal contents, including cathepsin B [44]. Expression of Nef alone is also able to produce lysosomal permeabilization in CD4+ lymphocytes [44]. Although this work was done in lymphocytes, it suggests that HIV-1 infection is a potential trigger for lysosomal permeabilization, which is part of cellular pathway to apoptosis and/or necrosis. In HIV-1 neuropathogenesis, the main sources of productive viral infection in the brain are monocytes, macrophages and microglia. Lysosomal protein leakage from infected cells could be a marker of uncontrolled proteolysis and brain tissue injury. Cathepsin B, via its cysteine protease activity, is also involved in extracellular matrix modification and cellular migration [6,67,87]. Involvement of cathepsin D in cellular migration and cytoskeletal arrangements has been also reported [88]. There is also new evidence that, during HIV-1 infection in vitro, cathepsin B translocates outside of MDM lysosomes and into the extracellular space, in an active form. The molecular mechanisms underlying this translocation are not well understood. However, MDM-conditioned media (MCM), collected from HIV-1 infected MDM at 12 days post-infection, causes increased neuronal apoptosis in vitro. Moreover, this increased apoptosis can be reversed by pretreating the MCM with a monoclonal anti-cathepsin B antibody or a specific cathepsin B inhibitor, CA074 [31]. Similar results were obtained with supernatants from an HIV-infected microglial cell line (CHME-5) (unpublished results).
One hypothesis that could explain the dysregulation of cathepsin B in HIV-infected MDM is that the increase in oxidative stress induced by HIV-1 induces post-translational modifications in cathepsin B and/or cystatin B that block their interaction. We found that the two proteins no longer interact in MDM after HIV-infection [31]. The interaction between cathepsin B and cystatin C in the cytosol of MDM was also disrupted after infection. In a recent study of the plasma membrane proteome of monocytes conducted by our collaborators, HIV-infected monocytes exhibited an increased movement of cystatin B to the plasma membrane [39], supporting the idea that cathepsin-cystatin complex formation is altered during infection. We are now in the process of elucidating the link between cathepsin-associated neurotoxicity to the brain neuropathology seen after HIV-1 infection. We are also exploring the possibility that protein-protein interactions in the extracellular space are involved in triggering cathepsin B-associated neuronal apoptosis.
Cathepsin B and Cystatin B Profiles in HIV-Seropositive Women in Different Tissue Compartments and Stages of HAND
Given the associations of cystatin B with HIV replication and cathepsin B with neuronal toxicity, we recently studied these proteins and another cathepsin B inhibitor, cystatin C, as candidate biomarkers of progression to HIV-associated dementia (HAD). We found that the monocytes of Hispanic women with HAD (and being treated with cART) have higher levels of intracellular cathepsin B and cystatin B than do those of HIV-seropositive women with normal cognition [40]. Cathepsin B levels and activity were increased in the plasma of HIV-seropositive women with normal cognition or with HAD compared to healthy individuals. In our study, cystatin B was upregulated in the plasma of all HIV-seropositive women regardless their cognitive status. Interestingly, while no differences in levels or activity of cathepsin B were found in cerebrospinal fluid (CSF), cystatin B was upregulated in the CSF of HAD patients despite cART therapy. In contrast, no differences in cystatin C expression levels in CSF were observed. The higher concentrations of cystatin B in the CSF of HAD patients could be related to HIV-1 replication and oxidative stress in the brain reservoirs, and deserves further study to elucidate its potential as a drug target or as a candidate biomarker. The ratio of cathepsin B to cystatin C levels in monocytes of HIV-seropositive women who progressed to a more severe form of HAND over two consecutive visits were almost two-fold higher than in the monocytes of women whose cognitive status remained stable. Further studies are being conducted to determine the potential of cathepsin-cystatin system as candidate biomarker or therapeutic target in HAND [40].
Cystatin B/Cathepsin B as Inducer of Oxidative Stress-Mediated Neurotoxicity
HIV patients are under severe oxidative stress, which correlates with progression of disease [89]. Massive oxidative stress during HIV progression is indicated by elevated levels of two-4-hydroxynonenal (HNE) in post-mortem brain tissue and increased levels of mitochondrial toxins in the CSF [90]. It is widely believed that the principal HIV target cells in brain are the perivascular macrophages and microglia. However, although neurons are rarely infected, they are affected by the reactive oxygen species (ROS) generated by glia cells [91], and by viral proteins such as Tat and Gp120 [81]. ROS can cause oxidative damage to cellular polysaccharides, DNA, proteins, alteration of immune function, apoptosis, and modification of the redox-dependent metabolism [92]. Upon HIV infection, the binding of HIV, or gp120 alone, to the CD4 receptor induces macrophages to secrete the pro-inflammatory cytokines IL-1 and TNF-α [93], depletes intracellular glutathione (GSH) levels, and increases free radicals, causing peroxidation of lipids [94]. ROS can also stimulate viral replication through the activation of the HIV LTR via post-translational control of NF-κβ [95]. As we proposed above, HIV-induced oxidative stress, cystatin B and LTR-induced HIV replication in macrophages may all be connected with NF-κB activation (Fig. 1).
As discussed above, cystatin B has additional functions besides being an active cysteine protease inhibitor, including the induction of nitric oxide production. For example, chicken cystatin was used as treatment for experimental visceral leishmaniasis. The curative effect of cystatin was linked to upregulation of inducible nitric oxide synthase (iNOS) and mediated by the induction of IL10 and TNF-α. Induction of iNOS usually occurs in an oxidative environment when high levels of nitric oxide (NO) react with superoxide to form peroxynitrite and cause cell toxicity (Fig. 1). Recent studies from our laboratory revealed increased levels of intracellular cystatin B and cathepsin B in monocytes of patients with HAD, while previous studies showed decreased antioxidants including Cu/Zn superoxide dismutase (SOD), peroxiredoxins, thioredoxins [96], glutathione peroxidase, and catalase [97] in HAD patients. All of these antioxidants, together with reduced Cu/Zn superoxide dismutase, contribute to oxidative stress. We have also observed a down-regulation in Cu/Zn SOD expression and activity in monocytes from Hispanic women with HAD [96,97] and in in vitro assays comparing viral isolates from individuals with HAD to those without neurocognitive impairment [98]. The absence of SOD in combination with increased levels of NO leads to elevated oxidative stress that causes neurotoxicity. In summary, we propose that cystatin B contributes to macrophage-mediated HIV neurotoxicity by the induction of NO levels via induction of iNOS, and increasing pro-inflammatory cytokines associated with oxidative stress and blood-brain barrier damage.
Implications for therapy
In vitro studies by our group have linked cystatin B with oxidative stress and HIV-1 replication in macrophages, and cathepsin B to neuronal dysfunction and death after exposure to serum-free supernatants from HIV-infected MDM. These findings suggest that the cathepsin-cystatin system is a potential target to control HIV-1 replication and prevent or reduce cognitive impairment in HIV-1 patients. Cystatin B is one of the HIV Dependency Factors (HDFs), a group of human proteins that are essential for HIV replication, but are not lethal to the host cell when the gene is knock-out (KO). Cystatin B KO mice survive but serve as a model for EPM1 because they develop some symptoms typically observed in EPM1 human patients [99]. Other HDF’s that affect HIV-1 replication in monocytes/macrophages, including Alix, C/EBPβ (large), cyclin-T1, and CCR5, are druggable. Druggability is a term used in drug discovery to indicate the likelihood that a particular target can be modulated with a small-molecule drug [100]). Cystatin B might be druggable by modulation of its expression in the macrophages instead of at the systemic level using immunoliposome targeted delivery of cystatin B siRNA.
Cathepsin B inhibitors have been studied for several conditions, including cancer and neurological conditions (extensively reviewed in [101]). The cathepsin B inhibitor CA074 is currently under study by another research group for Alzheimer’s disease therapy, and it has been demonstrated that treatment with this inhibitor improves memory deficits in mouse models of AD [53-56]. Other reseach group has demonstrated that cathepsin B inhibitors prevent cell death after ischemic injury [85,102]. As both cystatin B and cathepsin B are ubiquitous, the biggest concern is that of possible side effects. But with the advancement of technology in drug delivery to the brain parenchyma, especially nanotechnology in HIV field, therapies could be targeted to specific tissues and thereby avoid side effects to other tissues [103-107]. In addition, although cathepsin B and some members of the cystatin family have specific functions in some tissues, it has been suggested that there is redundancy in the roles of these proteins [108,109], supporting the idea that partially inhibiting cathepsin B might be beneficial (reviewed in [46,110]). However, more experiments are needed to further assess the potential of cathepsin B-cystatin B system for drug targeting during HIV-1 infection, and its potential benefits to cognitive performance in HIV seropositive patients.
Conclusions
Our studies demonstrate that cystatin B plays a dual role in HIV replication and macrophage-mediated neurotoxicity: (a) induction of HIV replication by decreasing STAT-1PY (Fig. 1), and (b) loss of its normal inhibitory interaction with cathepsin B secreted from HIV-1 infected MDM in vitro, which results in increased cathepsin B neurotoxic activity, (Fig. 2). Both functions are associated with HIV-induced oxidative stress and NF-κB activation. HIV-1 infection of macrophages induces NF-kB activation and secretion of TNF-α and IL1, leading to increased ROS and the release of cathepsin B and cystatin B to the cytosol and extracellular space. Extracellular cathepsin B in the CNS can promote neuronal apoptosis. The ROS can also activate the LTR via NF-kB, which further enhance HIV replication. Intracellular cystatin/cathepsin B interaction is important in maintaining cellular homeostasis in the brain. Dysregulation of this interaction could be detrimental for brain development and is observed in other neurological conditions such as Progressive myoclonus epilepsy of EPM1, AD, MS and traumatic brain injury. HIV infection also promotes cystatin/cathepsin B dysregulation as observed in MDM infected in vitro. This dysregulation results in increased expression, secretion, and activity of cathepsin B and neuronal apoptosis (Fig. 2). The cystatin/cathepsin B system is also deregulated in plasma and CSF of HIV-seropositive women on cART, supporting a possible role of these proteins in HIV chronic infection and the development of HAND despite antiretroviral therapy. The potential of cathepsin and cystatin as candidate therapeutic agents in HIV infection and HAND deserves further investigation.
Fig. (2).
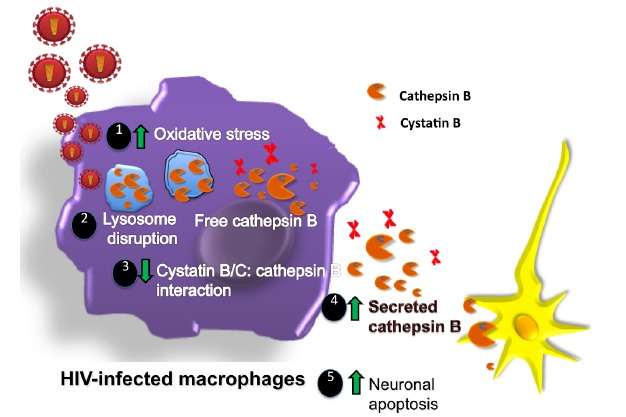
Roles of cathepsin/cystatin system in the neuropathogenesis induced by HIV-infected macrophages. (1) After HIV infection, ROS and cathepsin B are induced and macrophages undergo oxidative stress triggered by viral proteins. (2) Increased oxidative stress can also induce lysosome disruption and release of cathepsin B from lysosomes (3) with reduced cystatin/cathepsin interactions (4) leading to increased cathepsin B secretion and (5) activity that induce neuronal apoptosis.
ACKNOWLEDGEMENTS
This work was supported by grants from the National Institutes of Health R01MH083516 (LMM), U54NS043011 (LMM), NIMHHD 8G12-MD007600 (LMM) R25GM06 1838 (YC, KC), 5F32MH094210-02 (LR). Special thanks to Dr. Eillen Rodriguez-Franco for her contributions to Fig. (2) and to Dr. Edmundo Kraiselburd for careful review of the manuscript.
CONFLICT OF INTEREST
LMM has a patent application approved for cystatin B, related to this manuscript. Pat No. 8,143,231.
References
- 1.Turk V., Turk B., Guncar G., Turk D., Kos J. Lysosomal cathepsins: structure, role in antigen processing and presentation, and cancer. Adv. Enzyme Regul. 2002;42:285–303. doi: 10.1016/s0065-2571(01)00034-6. [DOI] [PubMed] [Google Scholar]
- 2.Schwabe C., Anastasi A., Crow H. Cystatin. amino acid sequence and possible secondary structure. Biochem. J. 1984;217:813–817. doi: 10.1042/bj2170813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Turk B., Turk D., Turk V. Lysosomal cysteine proteases: more than scavengers. Biochim. Biophys. Acta. 2000;1477(1-2):98–111. doi: 10.1016/s0167-4838(99)00263-0. [DOI] [PubMed] [Google Scholar]
- 4.Turk D., Podobnik M., Kuhelj R., Dolinar M., Turk V. Crystal structures of human procathepsin B at 3.2 and 3.3 Angstroms resolution reveal an interaction motif between a papain-like cysteine protease and its propeptide. FEBS Lett. 1996;384(3):211–214. doi: 10.1016/0014-5793(96)00309-2. [DOI] [PubMed] [Google Scholar]
- 5.Sakamoto M., Miyamoto K., Wu Z., Nakanishi H. Possible involvement of cathepsin B released by microglia in methylmercury-induced cerebellar pathological changes in the adult rat. Neurosci. Lett. 2008;442:292–296. doi: 10.1016/j.neulet.2008.07.019. [DOI] [PubMed] [Google Scholar]
- 6.Victor B.C., Anbalagan A., Mohamed M.M., Sloane B.F., Cavallo-Medved D. Inhibition of cathepsin B activity attenuates extracellular matrix degradation and inflammatory breast cancer invasion. Breast cancer research : BCR. BioMed Central Ltd. 2011;13(6):R115. doi: 10.1186/bcr3058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lackman R.L., Jamieson A.M., Griffith J.M., Geuze H., Cresswell P. Innate immune recognition triggers secretion of lysosomal enzymes by macrophages. Traffic. 2007;8(9):1179–1189. doi: 10.1111/j.1600-0854.2007.00600.x. [DOI] [PubMed] [Google Scholar]
- 8.Katunuma N. Posttranslational processing and modification of cathepsins and cystatins. 2010. [DOI] [PMC free article] [PubMed]
- 9.Lehtinen M.K., Tegelberg S., Schipper H., Su H., Zukor H., Manninen O., et al. Cystatin B deficiency sensitizes neurons to oxidative stress in progressive myoclonus epilepsy, EPM1. J. Neurosci. 2009;29(18):5910–5915. doi: 10.1523/JNEUROSCI.0682-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Luciano-Montalvo C., Ciborowski P., Duan F., Gendelman H.E., Meléndez L.M. Proteomic analyses associate cystatin B with restricted HIV-1 replication in placental macrophages. Placenta. 2008;29(12):1016–1023. doi: 10.1016/j.placenta.2008.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rivera-Rivera L., Perez-Laspiur J., Colón K., Meléndez L.M. Inhibition of interferon response by cystatin B: implication in HIV replication of macrophage reservoirs. J. Neurovirol. 2012;18(1):20–29. doi: 10.1007/s13365-011-0061-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Burgener A., Rahman S., Ahmad R., Lajoie J., Ramdahin S., Mesa C., et al. Comprehensive proteomic study identifies serpin and cystatin antiproteases as novel correlates of HIV-1 resistance in the cervicovaginal mucosa of female sex workers. J. Proteome Res. 2011;10(11):5139–5149. doi: 10.1021/pr200596r. [DOI] [PubMed] [Google Scholar]
- 13.Lewin S.R., Kirihara J., Sonza S., Irving L., Mills J., Crowe S.M. HIV-1 DNA and mRNA concentrations are similar in peripheral blood monocytes and alveolar macrophages in HIV-1-infected individuals. AIDS. 1998;12(7):719–727. doi: 10.1097/00002030-199807000-00008. [DOI] [PubMed] [Google Scholar]
- 14.Bergamaschi A., Pancino G. Host hindrance to HIV-1 replication in monocytes and macrophages. Retrovirology. 2010;7:31. doi: 10.1186/1742-4690-7-31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Shen R., Richter H.E., Clements R.H., Novak L., Huff K., Bimczok D., et al. Macrophages in vaginal but not intestinal mucosa are monocyte-like and permissive to human immunodeficiency virus type 1 infection. J. Virol. 2009;83(7):3258–3267. doi: 10.1128/JVI.01796-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Naif H.M., Li S., Alali M., Chang J., Mayne C., Sullivan J., et al. Definition of the stage of host cell genetic restriction of replication of human immunodeficiency virus type 1 in monocytes and monocyte-derived macrophages by using twins. J. Virol. 1999;73(6):4866–4881. doi: 10.1128/jvi.73.6.4866-4881.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hashimoto S., Suzuki T., Dong H.Y., Nagai S., Yamazaki N., Matsushima K. Serial analysis of gene expression in human monocyte-derived dendritic cells. Blood. 1999;94(3):845–852. [PubMed] [Google Scholar]
- 18.Cassol E., Cassetta L., Alfano M., Poli G. Macrophage polarization and HIV-1 infection. J. Leukoc. Biol. 2010;87(4):599–608. doi: 10.1189/jlb.1009673. [DOI] [PubMed] [Google Scholar]
- 19.Liu G., Yang H. Modulation of macrophage activation and programming in immunity. J. Cell. Physiol. 2013;228(3):502–512. doi: 10.1002/jcp.24157. [DOI] [PubMed] [Google Scholar]
- 20.Gordon S. Alternative activation of macrophages. Nat. Rev. Immunol. 2003;3(1):23–35. doi: 10.1038/nri978. [DOI] [PubMed] [Google Scholar]
- 21.Mantovani A., Sica A., Sozzani S., Allavena P., Vecchi A., Locati M. The chemokine system in diverse forms of macrophage activation and polarization. Trends Immunol. 2004;25(12):677–686. doi: 10.1016/j.it.2004.09.015. [DOI] [PubMed] [Google Scholar]
- 22.Benoit M, Desnues B, Mege J-L. Macrophage polarization in bacterial infections. 2008. [DOI] [PubMed]
- 23.Kopitar-Jerala N. The role of cystatins in cells of the immune system. FEBS Lett. 2006;580(27):6295–6301. doi: 10.1016/j.febslet.2006.10.055. [DOI] [PubMed] [Google Scholar]
- 24.Suzuki T, Hashimoto S, Toyoda N, Nagai S, Yamazaki N, Dc W, et al. Comprehensive gene expression profile of LPS-stimulated human monocytes by SAGE Comprehensive gene expression profile of LPS-stimulated human monocytes by SAGE. 2000. [PubMed]
- 25.Bol S.M., Cobos-Jiménez V. Kootstra N a, Van ’t Wout AB. HIV-1 and the macrophage. Future Virol. 2011;6(2):187–208. [Google Scholar]
- 26.Verdot L., Lalmanach G., Vercruysse V., Hartmann S., Lucius R., Hoebeke J., et al. Cystatins up-regulate nitric oxide release from interferon-gamma-activated mouse peritoneal macrophages. J. Biol. Chem. 1996;271(45):28077–28081. doi: 10.1074/jbc.271.45.28077. [DOI] [PubMed] [Google Scholar]
- 27.Verdot L, Lalmanach G, Vercruysse V, Hoebeke J, Gauthier F, Vray B. Chicken cystatin stimulates nitric oxide release from interferon-gamma-activated mouse peritoneal macrophages via cytokine synthesis. . European journal of biochemistry / FEBS . 1999. [DOI] [PubMed]
- 28.Plaud-Valentin M, Delgado R, Garcia V, Zorrilla C, Gandia J, Meléndez-Guerrero LM. HIV infection of placental macrophages: effect on the secretion of HIV stimulatory cytokines. 1999. [PubMed]
- 29.García K., García V., Pérez Laspiur J., Duan F., Meléndez L.M. Characterization of the placental macrophage secretome: implications for antiviral activity. Placenta. 2009;30(2):149–155. doi: 10.1016/j.placenta.2008.10.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ciborowski P., Kadiu I., Rozek W., Smith L., Bernhardt K., Fladseth M., et al. Investigating the human immunodeficiency virus type 1-infected monocyte-derived macrophage secretome. Virology. 2007;363(1):198–209. doi: 10.1016/j.virol.2007.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rodriguez-Franco E.J., Cantres-Rosario Y.M., Plaud-Valentin M., Romeu R., Rodríguez Y., Skolasky R., et al. Dysregulation of macrophage-secreted cathepsin B contributes to HIV-1-linked neuronal apoptosis. PLoS One. 2012;7(5):e36571. doi: 10.1371/journal.pone.0036571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Luciano-Montalvo C., Meléndez L.M. Cystatin B associates with signal transducer and activator of transcription 1 in monocyte-derived and placental macrophages. Placenta. Elsevier Ltd. 2009;30(5):464–467. doi: 10.1016/j.placenta.2009.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Magnani M., Balestra E., Fraternale A., Aquaro S., Paiardini M., Cervasi B., et al. Drug-loaded red blood cell-mediated clearance of HIV-1 macrophage reservoir by selective inhibition of STAT1 expression. J. Leukoc. Biol. 2003;74(5):764–771. doi: 10.1189/jlb.0403156. [DOI] [PubMed] [Google Scholar]
- 34.Chang T., Mosoian A., Pine R. A Soluble Factor (s) Secreted from CD8 T Lymphocytes Inhibits Human Immunodeficiency Virus Type 1 Replication through STAT1 Activation. Journalism. 2002;76(2):569–581. doi: 10.1128/JVI.76.2.569-581.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Meléndez J, García V, Sánchez E, Delgado R, Torres G, Meléndez-Guerrero LM. Is decreased HIV-1 infectivity of placental macrophages caused by high levels of beta-chemokines? Cellular and molecular biology (Noisy-le-Grand, France) 2001. [PubMed]
- 36.Chaudhuri A, Duan F, Morsey B, Persidsky Y, Kanmogne GD. HIV-1 activates proinflammatory and interferon-inducible genes in human brain microvascular endothelial cells: putative mechanisms of blood-brain barrier dysfunction. . Journal of cerebral blood flow and metabolism : official journal of the International Society of Cerebral Blood Flow and Metabolism. 2008. [DOI] [PubMed]
- 37.Asin S., Taylor J.A., Trushin S., Bren G., Paya C.V. I kappa kappa Mediates NF-kappa Beta Activation in Human Immunodeficiency Virus-Infected Cells. J. Virol. 1999;73(5):3893–3903. doi: 10.1128/jvi.73.5.3893-3903.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.McElhinny J a, MacMorran WS, Bren GD, Ten RM, Israel a, Paya C V. Regulation of I kappa B alpha and p105 in monocytes and macrophages persistently infected with human immunodeficiency virus. J. Virol. 1995;69(3):1500–1509. doi: 10.1128/jvi.69.3.1500-1509.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kadiu I, Wang T, Schlautman JD, Dubrovsky L, Ciborowski P, Bukrinsky M, et al. HIV-1 transforms the monocyte plasma membrane proteome. . Cellular immunology. Elsevier Inc . 2009. [DOI] [PMC free article] [PubMed]
- 40.Cantres-Rosario Y., Plaud-Valentín M., Gerena Y., Skolasky R.L., Wojna V., Meléndez L.M. Cathepsin B and cystatin B in HIV-seropositive women are associated with infection and HIV-1-associated neurocognitive disorders. AIDS. 2013;27(3):347–356. doi: 10.1097/QAD.0b013e32835b3e47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ha S-D., Park S., Hattlmann C.J., Barr S.D., Kim S.O. Inhibition or deficiency of cathepsin B leads defects in HIV-1 Gag pseudoparticle release in macrophages and HEK293T cells. Antiviral research. Elsevier B.V. 2012;93(1):175–184. doi: 10.1016/j.antiviral.2011.11.009. [DOI] [PubMed] [Google Scholar]
- 42.Yoshii H., Kamiyama H., Goto K., Oishi K., Katunuma N., Tanaka Y., et al. CD4-independent human immunodeficiency virus infection involves participation of endocytosis and cathepsin B. PLoS One. 2011;6(4):e19352. doi: 10.1371/journal.pone.0019352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kubo Y, Hayashi H, Matsuyama T, Sato H, Yamamoto N. Retrovirus entry by endocytosis and cathepsin proteases. . Advances in virology. 2012. [DOI] [PMC free article] [PubMed]
- 44.Laforge M., Petit F., Estaquier J., Senik A. Commitment to apoptosis in CD4(+) T lymphocytes productively infected with human immunodeficiency virus type 1 is initiated by lysosomal membrane permeabilization, itself induced by the isolated expression of the viral protein Nef. J. Virol. 2007;81(20):11426–11440. doi: 10.1128/JVI.00597-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zavasnik-Bergant T. Cystatin protease inhibitors and immune functions. Frontiers in bioscience : a journal and virtual library. 2008. [DOI] [PubMed]
- 46.Zavasnik-Bergant T., Turk B. Cysteine proteases: destruction ability versus immunomodulation capacity in immune cells. Biol. Chem. 2007;388(11):1141–1149. doi: 10.1515/BC.2007.144. [DOI] [PubMed] [Google Scholar]
- 47.Magister S., Kos J. Cystatins in immune system. J. Cancer. 2013;4(1):45–56. doi: 10.7150/jca.5044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ceru S, Layfield R, Zavasnik-Bergant T, Repnik U, Kopitar-Jerala N, Turk V, et al. Intracellular aggregation of human stefin B: confocal and electron microscopy study. . Biology of the cell / under the auspices of the European Cell Biology Organization. 2010. [DOI] [PubMed]
- 49.Skerget K., Taler-Vercic A., Bavdek A., Hodnik V., Ceru S., Tusek-Znidaric M., et al. Interaction between oligomers of stefin B and amyloid-beta in vitro and in cells. J. Biol. Chem. 2010;285(5):3201–3210. doi: 10.1074/jbc.M109.024620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hook V., Funkelstein L., Wegrzyn J., Bark S., Kindy M., Hook G. Cysteine Cathepsins in the secretory vesicle produce active peptides: Cathepsin L generates peptide neurotransmitters and cathepsin B produces beta-amyloid of Alzheimer’s disease. Biochimica et biophysica acta. Elsevier B.V. 2012;1824(1):89–104. doi: 10.1016/j.bbapap.2011.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hook V.Y., Toneff T., Aaron W., Yasothornsrikul S., Bundey R., Reisine T. Beta-amyloid peptide in regulated secretory vesicles of chromaffin cells: evidence for multiple cysteine proteolytic activities in distinct pathways for beta-secretase activity in chromaffin vesicles. J. Neurochem. 2002;81(2):237–256. doi: 10.1046/j.1471-4159.2002.00794.x. [DOI] [PubMed] [Google Scholar]
- 52.Hook V., Toneff T., Bogyo M., Greenbaum D., Medzihradszky K.F., Neveu J., et al. Inhibition of cathepsin B reduces beta-amyloid production in regulated secretory vesicles of neuronal chromaffin cells: evidence for cathepsin B as a candidate beta-secretase of Alzheimer’s disease. Biol. Chem. 2005;386(9):931–940. doi: 10.1515/BC.2005.108. [DOI] [PubMed] [Google Scholar]
- 53.Hook V.Y., Kindy M., Reinheckel T., Peters C., Hook G. Genetic cathepsin B deficiency reduces beta-amyloid in transgenic mice expressing human wild-type amyloid precursor protein. Biochem. Biophys. Res. Commun. 2009;386(2):284–288. doi: 10.1016/j.bbrc.2009.05.131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hook V., Hook G., Kindy M. Pharmacogenetic features of cathepsin B inhibitors that improve memory deficit and reduce beta-amyloid related to Alzheimer’s disease. Biol. Chem. 2010;391(8):861–872. doi: 10.1515/BC.2010.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Hook G., Hook V., Kindy M. The cysteine protease inhibitor, E64d, reduces brain amyloid-β and improves memory deficits in Alzheimer’s disease animal models by inhibiting cathepsin B, but not BACE1, β-secretase activity. Journal of Alzheimer’s disease. JAD. 2011;26(2):387–408. doi: 10.3233/JAD-2011-110101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kindy M.S., Yu J., Zhu H., El-Amouri S.S., Hook V., Hook G.R. Deletion of the cathepsin B gene improves memory deficits in a transgenic ALZHeimer’s disease mouse model expressing AßPP containing the wild-type β-secretase site sequence. Journal of Alzheimer’s disease. JAD. 2012;29(4):827–840. doi: 10.3233/JAD-2012-111604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Bahr B a Wisniewski ML, Butler D. Positive lysosomal modulation as a unique strategy to treat age-related protein accumulation diseases. Rejuvenation Res. 2012;15(2):189–197. doi: 10.1089/rej.2011.1282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Butler D., Hwang J., Estick C., Nishiyama A., Kumar S.S., Baveghems C., et al. Protective effects of positive lysosomal modulation in Alzheimer’s disease transgenic mouse models. PLoS One. 2011;6(6):e20501. doi: 10.1371/journal.pone.0020501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Wang C., Sun B., Zhou Y., Grubb A., Gan L. Cathepsin B degrades amyloid-β in mice expressing wild-type human amyloid precursor protein. J. Biol. Chem. 2012;287(47):39834–39841. doi: 10.1074/jbc.M112.371641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Oh J.E., Lee H.K. Autophagy as an innate immune modulator. Immune Netw. 2013;13(1):1–9. doi: 10.4110/in.2013.13.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Tatti M., Motta M., Di Bartolomeo S., Scarpa S., Cianfanelli V., Cecconi F., et al. Reduced cathepsins B and D cause impaired autophagic degradation that can be almost completely restored by overexpression of these two proteases in Sap C-deficient fibroblasts. Hum. Mol. Genet. 2012;21(23):5159–5173. doi: 10.1093/hmg/dds367. [DOI] [PubMed] [Google Scholar]
- 62.Haves-Zburof D., Paperna T., Gour-Lavie A., Mandel I., Glass-Marmor L., Miller A. Cathepsins and their endogenous inhibitors cystatins: expression and modulation in multiple sclerosis. J. Cell. Mol. Med. 2011;15(11):2421–2429. doi: 10.1111/j.1582-4934.2010.01229.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Bever CT, Garver DW. Increased cathepsin B activity in multiple sclerosis brain. 1995. [DOI] [PubMed]
- 64.Bever C.T., Panitch H.S., Johnson K.P. Increased cathepsin B activity in peripheral blood mononuclear cells of multiple sclerosis patients. Neurology. 1994;44(4):745–748. doi: 10.1212/wnl.44.4.745. [DOI] [PubMed] [Google Scholar]
- 65.Luo C-L., Chen X-P., Yang R., Sun Y-X., Li Q-Q., Bao H-J., et al. Cathepsin B contributes to traumatic brain injury-induced cell death through a mitochondria-mediated apoptotic pathway. J. Neurosci. Res. 2010;88(13):2847–2858. doi: 10.1002/jnr.22453. [DOI] [PubMed] [Google Scholar]
- 66.Zhang Y., Chen X., Tao L., Qin Z., Li S., Yang L., et al. Fa Yi Xue Za Zhi. 2006;22(6):404–406, 410. [Expression of cathepsin-B and -D in rat’s brain after traumatic brain injury]. [PubMed] [Google Scholar]
- 67.Rao Malla R., Gopinath S., Alapati K., Gorantla B., Gondi C.S., Rao J.S. Knockdown of cathepsin B and uPAR inhibits CD151 and α3β1 integrin-mediated cell adhesion and invasion in glioma. Mol. Carcinog. 2011;2012:1–14. doi: 10.1002/mc.21915. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 68.Spudich S.S., Ances B.M. Neurologic complications of HIV infection. Top. Antivir. Med. 2012;20(2):41–47. [PMC free article] [PubMed] [Google Scholar]
- 69.Valcour V., Chalermchai T., Sailasuta N., Marovich M., Lerdlum S., Suttichom D., et al. Central nervous system viral invasion and inflammation during acute HIV infection. J. Infect. Dis. 2012;206(2):275–282. doi: 10.1093/infdis/jis326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Spudich S., González-Scarano F. HIV-1-related central nervous system disease: current issues in pathogenesis, diagnosis, and treatment. Cold Spring Harb. Perspect. Med. 2012;2(6):a007120. doi: 10.1101/cshperspect.a007120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Koenig S., Gendelman H.E., Orenstein J.M., Dal Canto M.C., Pezeshkpour G.H., Yungbluth M., et al. Detection of AIDS virus in macrophages in brain tissue from AIDS patients with encephalopathy. Science. 1986;233(4768):1089–1093. doi: 10.1126/science.3016903. [DOI] [PubMed] [Google Scholar]
- 72.Masliah E., Achim C.L., Hansen L.A., Wiley C.A. Selective neuronal vulnerability in HIV encephalitis. J. Neuropathol. Exp. Neurol. 1992;51(6):585–593. doi: 10.1097/00005072-199211000-00003. [DOI] [PubMed] [Google Scholar]
- 73.Persidsky Y. Microglial and astrocyte chemokines regulate monocyte migration through the blood-brain barrier in human immunodeficiency virus-1 encephalitis. Am. J. Pathol. 1999;155(5):1599–1611. doi: 10.1016/S0002-9440(10)65476-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Reddy PVB, Gandhi N, Samikkannu T, Saiyed Z, Agudelo M, Yndart A, et al. HIV-1 gp120 induces antioxidant response element-mediated expression in primary astrocytes: Role in HIV associated neurocognitive disorder. . Neurochemistry international. 2011. [DOI] [PMC free article] [PubMed]
- 75.Williams K.C., Corey S., Westmoreland S.V., Pauley D., Knight H., deBakker C., et al. Perivascular macrophages are the primary cell type productively infected by simian immunodeficiency virus in the brains of macaques: implications for the neuropathogenesis of AIDS. J. Exp. Med. 2001;193(8):905–915. doi: 10.1084/jem.193.8.905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Yang B, Akhter S, Chaudhuri A, Kanmogne GD. HIV-1 gp120 induces cytokine expression, leukocyte adhesion, and transmigration across the blood-brain barrier: modulatory effects of STAT1 signaling. . Microvascular research. Elsevier Inc. 2009. [DOI] [PMC free article] [PubMed]
- 77.Agrawal L, Louboutin J-P. HIV-1 Tat neurotoxicity: a model of acute and chronic exposure, and neuroprotection by gene delivery of antioxidant enzymes. . Neurobiology of disease. Elsevier Inc . 2012. [DOI] [PubMed]
- 78.Eugenin E a D’Aversa TG, Lopez L, Calderon TM, Berman JW. MCP-1 (CCL2) protects human neurons and astrocytes from NMDA or HIV-tat-induced apoptosis. J. Neurochem. 2003;85(5):1299–1311. doi: 10.1046/j.1471-4159.2003.01775.x. [DOI] [PubMed] [Google Scholar]
- 79.Van Marle G., Henry S., Todoruk T., Sullivan A., Silva C., Rourke S.B., et al. Human immunodeficiency virus type 1 Nef protein mediates neural cell death: a neurotoxic role for IP-10. Virology. 2004;329(2):302–318. doi: 10.1016/j.virol.2004.08.024. [DOI] [PubMed] [Google Scholar]
- 80.Hui L., Chen X., Haughey N.J., Geiger J.D. Role of endolysosomes in HIV-1 Tat-induced neurotoxicity. ASN Neuro. 2012;4(4):243–252. doi: 10.1042/AN20120017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Louboutin J-P, Agrawal L. HIV-1 gp120 neurotoxicity proximally and at a distance from the point of exposure: protection by rSV40 delivery of antioxidant enzymes. . Neurobiology of disease. Elsevier Inc . 2009. [DOI] [PubMed]
- 82.Johansson A-C, Appelqvist H, Nilsson C, Kågedal K, Roberg K, Ollinger K. Regulation of apoptosis-associated lysosomal membrane permeabilization. 2010. [DOI] [PMC free article] [PubMed]
- 83.Oberle C., Huai J., Reinheckel T., Tacke M., Rassner M., Ekert P.G., et al. Lysosomal membrane permeabilization and cathepsin release is a Bax/Bak-dependent, amplifying event of apoptosis in fibroblasts and monocytes. Cell death and differentiation. Nature Publishing Group. 2010;17(7):1167–1178. doi: 10.1038/cdd.2009.214. [DOI] [PubMed] [Google Scholar]
- 84.Droga-Mazovec G., Bojic L., Petelin A., Ivanova S., Romih R., Repnik U., et al. Cysteine cathepsins trigger caspase-dependent cell death through cleavage of bid and antiapoptotic Bcl-2 homologues. J. Biol. Chem. 2008;283(27):19140–19150. doi: 10.1074/jbc.M802513200. [DOI] [PubMed] [Google Scholar]
- 85.Yamashima T., Oikawa S. The role of lysosomal rupture in neuronal death. Prog. Neurobiol. 2009;89(4):343–358. doi: 10.1016/j.pneurobio.2009.09.003. [DOI] [PubMed] [Google Scholar]
- 86.Gelman B.B. Wolf D a, Rodriguez-Wolf M, West a B, Haque a K, Cloyd M. Mononuclear phagocyte hydrolytic enzyme activity associated with cerebral HIV-1 infection. Am. J. Pathol. 1997;151(5):1437–1446. [PMC free article] [PubMed] [Google Scholar]
- 87.Murphy N. Lynch M a. Activation of the P2X7 receptor induces migration of glial cells by inducing cathepsin B degradation of tissue inhibitor of metalloproteinase 1. J. Neurochem. 2012;123(5):761–770. doi: 10.1111/jnc.12031. [DOI] [PubMed] [Google Scholar]
- 88.Koch S, Scifo E, Rokka A, Trippner P, Lindfors M, Korhonen R, et al. Cathepsin D deficiency induces cytoskeletal changes and affects cell migration pathways in the brain. . Neurobiology of disease Elsevier Inc . 2013. [DOI] [PubMed]
- 89.Suresh D.R., Annam V., Pratibha K., Prasad B.V. Total antioxidant capacity--a novel early bio-chemical marker of oxidative stress in HIV infected individuals. J. Biomed. Sci. 2009;16:61. doi: 10.1186/1423-0127-16-61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Sacktor N., Haughey N., Cutler R., Tamara A., Turchan J., Pardo C., et al. Novel markers of oxidative stress in actively progressive HIV dementia. J. Neuroimmunol. 2004;157(1-2):176–184. doi: 10.1016/j.jneuroim.2004.08.037. [DOI] [PubMed] [Google Scholar]
- 91.Viviani B., Corsini E., Binaglia M., Galli C.L., Marinovich M. Reactive oxygen species generated by glia are responsible for neuron death induced by human immunodeficiency virus-glycoprotein 120 in vitro. Neuroscience. 2001;107(1):51–58. doi: 10.1016/s0306-4522(01)00332-3. [DOI] [PubMed] [Google Scholar]
- 92.Aquaro S., Scopelliti F., Pollicita M., Perno C.F. Oxidative stress and HIV infection: target pathways for novel therapies? Future HIV Ther. 2008;2(4):327–338. [Google Scholar]
- 93.Merrill J.E., Koyanagi Y., Chen I.S. Interleukin-1 and tumor necrosis factor alpha can be induced from mononuclear phagocytes by human immunodeficiency virus type 1 binding to the CD4 receptor. J. Virol. 1989;63(10):4404–4408. doi: 10.1128/jvi.63.10.4404-4408.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Morris D, Guerra C, Donohue C, Oh H, Khurasany M, Venketaraman V. Unveiling the mechanisms for decreased glutathione in individuals with HIV infection. 2012. [DOI] [PMC free article] [PubMed]
- 95.Pyo C-W., Yang Y.L., Yoo N-K., Choi S-Y. Reactive oxygen species activate HIV long terminal repeat via post-translational control of NF-kappaB. Biochem. Biophys. Res. Commun. 2008;376(1):180–185. doi: 10.1016/j.bbrc.2008.08.114. [DOI] [PubMed] [Google Scholar]
- 96.Kraft-Terry S., Gerena Y., Wojna V., Plaud-Valentin M., Rodriguez Y., Ciborowski P., et al. Proteomic analyses of monocytes obtained from Hispanic women with HIV-associated dementia show depressed antioxidants. Proteomics Clin. Appl. 2010;4(8-9):706–714. doi: 10.1002/prca.201000010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Velázquez I., Plaud M., Wojna V., Skolasky R., Laspiur J.P., Meléndez L.M. Antioxidant enzyme dysfunction in monocytes and CSF of Hispanic women with HIV-associated cognitive impairment. J. Neuroimmunol. 2009;206(1-2):106–111. doi: 10.1016/j.jneuroim.2008.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Toro-Nieves D.M., Rodriguez Y., Plaud M., Ciborowski P., Duan F., Pérez Laspiur J., et al. Proteomic analyses of monocyte-derived macrophages infected with human immunodeficiency virus type 1 primary isolates from Hispanic women with and without cognitive impairment. J. Neurovirol. 2009;15(1):36–50. doi: 10.1080/13550280802385505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Polajnar M., Ceru S., Kopitar-Jerala N., Zerovnik E. Human stefin B normal and patho-physiological role: molecular and cellular aspects of amyloid-type aggregation of certain EPM1 mutants. Front. Mol. Neurosci. 2012;5:88. doi: 10.3389/fnmol.2012.00088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Owens J. Determining druggability. Nat. Rev. Drug Discov. 2007;6(3):187–187. [Google Scholar]
- 101.Frlan R., Gobec S. Inhibitors of cathepsin B. Curr. Med. Chem. 2006;13(19):2309–2327. doi: 10.2174/092986706777935122. [DOI] [PubMed] [Google Scholar]
- 102.Yamashima T., Kohda Y., Tsuchiya K., Ueno T., Yamashita J., Yoshioka T., et al. Inhibition of ischaemic hippocampal neuronal death in primates with cathepsin B inhibitor CA-074: a novel strategy for neuroprotection based on “calpain-cathepsin hypothesis”. Eur. J. Neurosci. 1998;10(5):1723–1733. doi: 10.1046/j.1460-9568.1998.00184.x. [DOI] [PubMed] [Google Scholar]
- 103.Sharma S, Singh A. Nanotechnology Based Targeted Drug Delivery : Current Status and Future Prospects for Drug Development 2007; 2007.
- 104.Could N., Cancer R. Nanomedicine Could Revolutionize Cancer. HIV Research; 2010. pp. 12–14. [Google Scholar]
- 105.Mamo T., Moseman E.A., Kolishetti N., Salvador-Morales C., Shi J., Kuritzkes D.R., et al. Emerging nanotechnology approaches for HIV/AIDS treatment and prevention. Nanomedicine (London, England) 2010;5(2):269–285. doi: 10.2217/nnm.10.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Kim P.S., Read S.W. Nanotechnology and HIV: potential applications for treatment and prevention. Wiley Interdiscip. Rev. Nanomed. Nanobiotechnol. 2010;2(6):693–702. doi: 10.1002/wnan.118. [DOI] [PubMed] [Google Scholar]
- 107.Ganau M., Prisco L., Pescador D., Ganau L. Challenging New Targets for CNS-HIV Infection. Front. Neurol. 2012;3:43. doi: 10.3389/fneur.2012.00043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Paperna T, Mandel I, Miller A. Cathepsins and their endogenous inhibitors cystatins : expression and modulation in multiple sclerosis 1-2. [DOI] [PMC free article] [PubMed]
- 109.Conus S., Simon H-U. Cathepsins and their involvement in immune responses. Swiss Med. Wkly. 2010;140(July):w13042. doi: 10.4414/smw.2010.13042. [DOI] [PubMed] [Google Scholar]
- 110.Reiser J, Adair B, Reinheckel T. Science in medicine Specialized roles for cysteine cathepsins in health and disease. 2010. [DOI] [PMC free article] [PubMed]