

Abstract
Here we describe the application of a new click chemistry method for fluorescent tracking of protein synthesis in individual microorganisms within environmental samples. This technique, termed bioorthogonal non-canonical amino acid tagging (BONCAT), is based on the in vivo incorporation of the non-canonical amino acid L-azidohomoalanine (AHA), a surrogate for l-methionine, followed by fluorescent labelling of AHA-containing cellular proteins by azide-alkyne click chemistry. BONCAT was evaluated with a range of phylogenetically and physiologically diverse archaeal and bacterial pure cultures and enrichments, and used to visualize translationally active cells within complex environmental samples including an oral biofilm, freshwater and anoxic sediment. We also developed combined assays that couple BONCAT with ribosomal RNA (rRNA)-targeted fluorescence in situ hybridization (FISH), enabling a direct link between taxonomic identity and translational activity. Using a methanotrophic enrichment culture incubated under different conditions, we demonstrate the potential of BONCAT-FISH to study microbial physiology in situ. A direct comparison of anabolic activity using BONCAT and stable isotope labelling by nano-scale secondary ion mass spectrometry (15NH3 assimilation) for individual cells within a sediment-sourced enrichment culture showed concordance between AHA-positive cells and 15N enrichment. BONCAT-FISH offers a fast, inexpensive and straightforward fluorescence microscopy method for studying the in situ activity of environmental microbes on a single-cell level.
Introduction
Two major overarching goals in microbial ecology are to understand the ecophysiology of microorganisms and how they react to environmental stimuli over a range of spatial and temporal scales. To achieve this, methodologies and experiments that track microbial activity in an environmental context are essential. One of the most powerful and direct approaches for deciphering structure-function relationships of microbial communities is whole-cell fluorescence microscopy. Using fluorescence in situ hybridization (FISH) techniques, the visualization of rRNA (phylogenetic identity), specific genes (genetic potential) and messenger RNA (mRNA; gene expression) has been demonstrated in individual microbial cells (DeLong et al., 1989; Amann et al., 1992; Lanoil and Giovannoni, 1997; Schönhuber et al., 1997; Pernthaler and Amann, 2004; Zwirglmaier et al., 2004; Smolina et al., 2007; Kawakami et al., 2010; Moraru et al., 2010; Stoecker et al., 2010; Yilmaz et al., 2010; Behnam et al., 2012). However, due to the limitations associated with using expression of rRNA or mRNA as metabolic tracer, and the observation that, on single-cell level, mRNA and protein expression for the same gene may be uncorrelated, detection of protein synthesis is considered to be a more reliable marker of activity (Wagner et al., 1995; Binder and Liu, 1998; Morgenroth et al., 2000; Odaa et al., 2000; Schmid et al., 2001; Bollmann et al., 2005; Foster et al., 2009; Taniguchi et al., 2010). To detect and localize target proteins involved in specific metabolisms within microbial cells, immunofluorescence analyses have been used (e.g. Lin et al., 1998; Fiencke and Bock, 2004; Wrede et al., 2013). However, this technique is not routinely applied in microbial ecology, likely due to the time and expense involved in antibody synthesis, and challenges in assessing and controlling for specificity in uncultured microorganisms. Additionally, immunofluorescence staining of proteins cannot differentiate the timing of protein synthesis. As such, the development of a general fluorescence microscopy method that can be used to study the expression of proteins in environmental microbes in situ would offer a powerful complement to the current suite of methods. In situ tracking of de novo protein synthesis in the context of the microbial community and changing ecological or physicochemical conditions would facilitate new strategies for assessing the structural and functional adaptation of microbes to environmental stimuli.
Currently, two approaches are used to monitor activity of individual cultured and uncultured microbes via the incorporation of isotopically labelled substrates. The use of stable isotopes, such as 15N-labelled ammonia (NH3) or 13C/15N-labelled amino acids can be used to identify anabolically active cells (e.g. protein synthesis) within complex samples using secondary ion mass spectrometry (SIMS) (Dekas et al., 2009; Orphan et al., 2009; Morono et al., 2011; Pernice et al., 2012) or Raman microspectroscopy (Haider et al., 2010). Additionally, radioactively labelled amino acids (e.g. 14C-leucine or 35S-methionine) have been used to track uptake into cells using micro-autoradiography (MAR) (Herndl et al., 2005; Sintes and Herndl, 2006; Teira et al., 2006). In addition, MAR-based visualization of 14CO2 assimilation by heterotrophic bacteria has been used as a general activity marker in pure cultures and environmental samples (Roslev et al., 2004; Hesselsoe et al., 2005). While these approaches offer a valuable link between anabolic activity and phylogeny for environmental microbes, they also require specialized laboratory facilities (radioactive certification in the case of MAR) or expensive instrumentation (microRaman or SIMS) which limits their general application in the field.
In addition to visualizing cellular protein and RNA expression, a complementary approach for identifying active cells is based on the incorporation of the indicator dye RedoxSensor green (RSG; Invitrogen), a nontoxic fluorescent indicator of bacterial reductase activity. While the full utility of RSG for microbial ecosystems has not yet been investigated, recent publications have successfully used this assay to detect respiratory activity in cultured aerobic microbial cells and environmental samples in near real time (Kalyuzhnaya et al., 2008; Konopka et al., 2011; Orman and Brynildsen, 2013).
Here, we explore the use of another promising approach for investigating the function and activity of environmental microorganisms based on bioorthogonal compounds coupled with click chemistry. Bioorthogonal compounds are defined as synthetic molecules that are not biologically synthesized and that do not interfere with processes within the cell. Frequently, these compounds are analogs of native biomolecules (e.g. amino acids, nucleotides, lipids, carbohydrates) that contain a functional group that is amenable for click chemistry, most commonly using an alkyne-azide reaction, which, through cycloaddition, form a triazole conjugate (i.e. click reaction; for recent reviews, see Best, 2009; Sletten and Bertozzi, 2009; Lim and Lin, 2010).
There are a series of bioorthogonal amino acids with azide or alkyne groups that have been shown to successfully compete with native amino acids and to be incorporated into the polypeptide chain during translation. However, only a subset of these non-canonical amino acids is able to exploit the substrate promiscuity of the native translational machinery without the need for genetic modification of the host cell (Ngo and Tirrell, 2011). The l-methionine (Met) surrogate L-azidohomoalanine (AHA; Fig. 1A) has been shown to be the most translationally active, with activation rates approximately 390 times lower than Met in Escherichia coli (compared with homopropargylglycine (HPG) and norleucine that have activation rates 500 and 1050 times lower than Met; Kiick et al., 2002). The rate of AHA incorporation into new proteins is regulated by methionyl-tRNA synthetase, the enzyme that catalyzes the esterification of Met with its tRNA to form a methionyl-tRNA (Kiick et al., 2002). AHA is water soluble, nontoxic and stable at physiological and most environmentally relevant conditions (with the exception of highly sulfidic, alkaline habitats; see Results section). The small difference in the molecular size of AHA compared with Met results in only minimal structural disturbance after incorporation into proteins (Kiick et al., 2002; Best, 2009). AHA has been used in a range of studies targeted to decipher translational regulation in E. coli (Kiick et al., 2002; Nessen et al., 2009; Ngo et al., 2009) as well as amphibian and mammalian cell and tissue cultures, including immune cells (Ngo et al., 2009; Howden et al., 2013), kidney cells (Dieterich et al., 2006), neurons (Dieterich et al., 2006; 2010; Yoon et al., 2012) and HeLa cells (Bagert et al., 2014). None of these studies detected a detrimental effect of AHA by microscopy. Moreover, studies that combined AHA-labelling with mass spectrometric proteomic sequencing reported that incubation with up to 1 mM AHA in the growth medium did not result in the preferential synthesis or degradation of proteins in E. coli or different human cell lines (Dieterich et al., 2006; Eichelbaum et al., 2012; Bagert et al., 2014). The properties of AHA suggest that the specificity of this assay for measuring new protein synthesis in environmental microorganisms is high. Furthermore, non-specific reactivity with azide moieties other than AHA is likely very low, as there is only one organism (a dinoflagellate) currently known to produce an azide-containing metabolite (Griffin, 1994). In contrast, click reactions using azide-modified probe molecules may have a greater potential for non-specific reactions with naturally occurring alkyne-containing biomolecules, such as polyynes (Shi Shun and Tykwinski, 2006).
Fig 1.
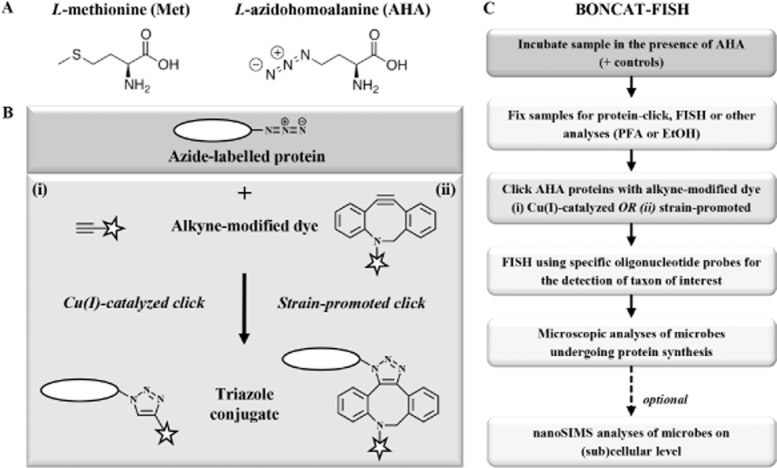
Overview of the BONCAT method for visualizing newly synthesized proteins. A. Structures of Met and its surrogate AHA, which is incorporated into newly made peptides during translation. B. The click chemistry-mediated visualization of newly produced AHA-containing proteins can be achieved via either one of two strategies: (i) in a Cu(I)-catalyzed reaction a terminal alkyne that is coupled to a fluorescence dye (star) is linked to the azide group of AHA yielding a triazole (left side); (ii) conjugation can also be achieved via strain-promoted cycloaddition, a Cu-free variation of click chemistry (right side). C. Protocol for the combinatorial labelling of AHA-containing proteins via BONCAT and rRNA-targeted FISH.
The azide moiety of AHA makes it distinguishable from the large pool of other amino acids in a cell, allowing selective click chemistry-mediated detection of proteins that have been synthesized during AHA incubation, an approach that has been termed bioorthogonal non-canonical amino acid tagging (BONCAT; Dieterich et al., 2006). The azide-alkyne reaction has a number of beneficial characteristics for use in biomolecule detection, including fast kinetics with high chemo- and regio-specificity, a single reaction product, and very high yields (Best, 2009; Lim and Lin, 2010). The assay is simple to perform under biologically relevant conditions and takes place in the presence of water or other solvents. There are two forms of azide-alkyne click reactions (Fig. 1B): (i) a Cu(I)-catalyzed cycloaddition reaction of an azide with a terminal alkyne (Huisgen, 1963; Rostovtsev et al., 2002; Torne et al., 2002); and (ii) a strain-promoted variant that makes use of a highly reactive strained cyclo-octyne system that allows the reaction to take place in the absence of a copper catalyst (Agard et al., 2004; Codelli et al., 2008). Both click reactions enable the selective conjugation of azides with alkynes within biologically and chemically complex environments (for reviews, see Carrico, 2008; Best, 2009; Sletten and Bertozzi, 2009; Jewett and Bertozzi, 2010).
Recently, an approach for the AHA-based fluorescence visualization of protein synthesis has been developed (Beatty et al., 2006; Dieterich et al., 2007). It allows microscopic detection of newly synthesized proteins that have incorporated bioorthogonal amino acids via click chemistry conjugated fluorescence dyes (Fig. 1B). Fluorescence visualization of BONCAT-labelled cells has been used to study protein synthesis in mammalian cell cultures and zebra fish (Beatty et al., 2006; 2010; Ngo et al., 2009; Dieterich et al., 2010; Hong et al., 2010; Hinz et al., 2012), as well as applied to select cultured microorganisms, i.e. E. coli (Beatty et al., 2005; Ngo et al., 2009), Pseudomonas entomophila (Chakrabarti et al., 2012), pathogenic chlamydia (Ouellette et al., 2011), and Listeria monocytogenes (Siegrist et al., 2013). These studies reveal the general applicability of BONCAT and click chemistry in traditional model organisms; however, the suitability of this method for the study of complex, natural microbial communities in situ has not been explored.
Here, we outline a protocol using BONCAT for assessing metabolic activity and protein synthesis that can be combined with existing single-cell targeted approaches in microbial ecology. BONCAT was used to visualize de novo synthesized proteins in a range of phylogenetically and physiologically diverse bacteria and archaea as well as a series of enrichments and environmental samples. Combining BONCAT and rRNA-targeted FISH, we demonstrate the ability to directly correlate translational activity of single microbial cells with their taxonomic identity by fluorescence microscopy and study the physiology of microorganisms in situ. BONCAT-FISH offers an inexpensive, simple and fast approach to study anabolically active microbes via fluorescence microscopy, which has been a core method of microbial ecology for decades. It opens a new level of inquiry to researchers interested in studying the ecophysiology and in situ activity of uncultured microbes.
Results and discussion
Cu(I)-catalyzed and strain-promoted click using fluorescent and halogenated dyes
We initially performed AHA-labelling incubations with E. coli cultures to establish a protocol for the fluorescence labelling of AHA-containing proteins in chemically fixed cells. Labelling protocols were evaluated and updated during the study (for a non-technical overview, see Fig. 1C). AHA-containing E. coli cells were used to test Cu(I)-catalyzed and strain-promoted click reactions with six alkyne-modified fluorescent dyes (spanning emission wavelengths from 524 to 571 nm; Supporting Information Fig. S1). All of these dyes are commercially available alkyne derivatives of widely used fluorophores and can be analyzed using conventional fluorescence microscopy filter sets. The removal of unincorporated dyes using the washing protocol was effective for all fluorophores except Eosin (Supporting Information Fig. S1D), a bromine-containing dye that, like the fluorinated Oregon Green 488 alkyne, can theoretically be used to detect AHA incorporation into single cells via halogen ion imaging by nano-scale secondary ion mass spectrometry (nanoSIMS; see below). The inability to remove unreacted Eosin-alkyne may be due to the inherent properties of the dye, which is commonly used as a cytoplasmic stain in histological studies.
Cu(I)-catalyzed and strain-promoted click reactions were evaluated in BONCAT-labelling of cultures of E. coli and Desulfovibrio alaskensis with two chemically similar dyes, Carboxyrhodamine 110 Alkyne and DBCO-PEG4-Carboxyrhodamine 110 (ClickChemistryTools) at equimolar concentrations (10 μM; Supporting Information Fig. S1). While signal intensities were essentially identical, much higher signal-to-noise ratios were observed for samples analyzed with the Cu(I)-catalyzed reaction as compared with the strain-promoted process (data not shown). This difference may be due to the fact that strain-promoted click chemistry can be accompanied by nonspecific reactions with free thiols (for details on thiol-blocking, see Experimental Procedures section). Decreasing the dye concentration from 10 μM to the range typically used for strain-promoted click chemistry (0.1–1 μM) substantially increased the signal-to-noise ratio for this reaction.
Click chemistry-mediated fluorescence labelling was successful with slide-immobilized biomass as well as with samples in solution, with both producing equivalent fluorescence intensities and signal-to-noise ratios (tested with dye Carboxyrhodamine 110 Alkyne and DBCO-PEG4-Carboxyrhodamine 110 for both; data not shown). To increase the ease and reproducibility of sample handling during methods optimization the majority of our testing was conducted with slide-immobilized cells.
BONCAT of physiologically and phylogenetically diverse microbes
We performed AHA incubations with several environmentally relevant pure and enrichment cultures of bacteria and archaea, representing a range of different physiologies and taxonomic affiliations, to test their ability to incorporate AHA into new proteins. Cultures included: (i) E. coli, as an example of a well-studied aerobic heterotrophic bacterium; (ii) Paracoccus denitrificans, a facultatively anaerobic denitrifying alphaproteobacterium; (iii) Desulfovibrio alaskensis, a deltaproteobacterial sulfate reducer (obligate anaerobe); (iv) Methanosarcina acetivorans, a methanogen within the Euryarchaeota (obligate anaerobe); (v) a propane-oxidizing aerobic enrichment culture in which mycobacteria (phylum Actinobacteria) were numerically dominant [∼ 95% of 4′,6-diamidino-2-phenylindole (DAPI)-stained cells]; and (vi) a micro-oxic methane-oxidizing enrichment culture, dominated by a Methylococcaceae species (∼ 97% of DAPI-stained cells; Tavormina et al., in review).
Cultures were incubated in the presence or absence of AHA (100 μM or 1 mM) and cells that incorporated AHA during the incubation were identified via click chemistry using fluorophores Carboxyrhodamine 110 Alkyne and DBCO-PEG4-Carboxyrhodamine 110 (Fig. 2). BONCAT-labelling of an E. coli culture that had been incubated in the presence of AHA (1 mM) and the protein synthesis inhibitor chloramphenicol (Camp) did not yield fluorescently labelled cells (data not shown). All incubations were performed for ≤ 1 generation of the respective culture (as assessed by OD600) to minimize excessive substitution of Met with AHA. Substantial replacement with a non-canonical amino acid such as AHA could interfere with the efficiency of the cellular machinery over time. Notably, with such short incubation times, AHA was not observed to influence the growth rate of the cultures and fluorescently labelled inclusion bodies, which can represent aggregations of misfolded proteins (Fahnert et al., 2004), were never detected within the cells. For cultures that had been incubated in the presence of AHA, generally > 97% of all DAPI-stained cells were fluorescently labelled (Fig. 2). The inability to stain the remaining cells could be due to one or a combination of the following reasons: (i) BONCAT-negative cells were not metabolically active during the incubation or did not express the respective uptake machinery (which is currently unknown for AHA); or (ii) the number of incorporated AHA molecules was insufficient to be detected via conventional fluorescence microscopy. In our parallel 16S rRNA-targeted FISH experiments (see below) essentially all cells (≥ 99%) within the individual cultures were FISH-labelled. We did not observe a difference in FISH signal intensity of BONCAT-negative as compared with positive cells. This is consistent with the finding that rRNA FISH often is not a reliable tracer of metabolic activity in microorganisms (e.g. Wagner et al., 1995; Binder and Liu, 1998; Morgenroth et al., 2000; Schmid et al., 2001; Bollmann et al., 2005). It should, however, be noted that the majority (visual estimate) of BONCAT-negative cells demonstrated weak DAPI signals, which suggests a lower DNA content as compared with intensively DAPI-stained cells (Fig. 2). Considering that fluorescently labelled oligonucleotide probes have molecular weights equal to or larger than the clickable dyes tested here, ineffective cell permeabilization is not likely to be the cause of the inability to label all cells using BONCAT.
Fig 2.

Uptake and incorporation of AHA is independent of the physiological or phylogenetic background of the target organism. Different pure and enrichment cultures were incubated in the presence or absence of AHA. BONCAT signals (green) were taken at identical exposure times for individual series (i.e. 0.1 and 1 mM AHA plus control). Note that incubation conditions were different for the individual cultures, cells have contrasting levels of background fluorescence, and that different labelling strategies were used. Together, these issues limit the value of a direct comparison of signal intensities between different cultures. DAPI-staining is shown in blue. All scale bars equal 10 μm and apply to each set of images respectively. A–D. BONCAT-labelling of four bacterial and archaeal pure cultures. E–F. BONCAT-labelling of propane- and methane-oxidizing enrichment cultures.
In addition, we performed labelling experiments using Carboxyrhodamine 110 Alkyne using E. coli and D. alaskensis cultures that had been incubated in the presence of 10 μM AHA. We observed comparable labelling efficiencies (> 97% of DAPI-staining cells; data not shown), demonstrating that under defined conditions (no Met in the medium) AHA concentrations in the range of several μM are sufficient to effectively label bacterial proteins.
We further attempted to fluorescently label AHA-containing proteins in living microorganisms, here referred to as ‘liveBONCAT’. Our preliminary results, which are summarized in the supplementary online information (Supporting Information Fig. S2), suggest that it may be possible to fluorescently label microbial cells in vivo, although the existing protocol requires further testing and optimization to enhance the percentage of cell labelling.
In gel detection of newly synthesized proteins
To test whether added AHA was directly incorporated into newly made proteins and whether this non-canonical amino acid alters the translational activity (or results in protein degradation), proteins were extracted from E. coli (3 h of incubation at 32°C, i.e. ∼ 0.67 generations), D. alaskensis (3.5 h, i.e. ∼ 0.50 generations) and the methanotroph enrichment culture (26 h of incubation, equal to ∼ 0.59 generations as judged by change of OD600) amended with AHA (0.1–1 mM). Extracted proteins were conjugated with dye DBCO-PEG4-Carboxyrhodamine 110 using strain-promoted click chemistry and analyzed by gel electrophoresis. No difference in Coomassie-stained protein bands were observed between cultures that had been incubated in the presence or in the absence of AHA (examples are shown in Fig. 3A). This is in accordance with our growth rate analyses as well as recent studies that demonstrated using quantitative proteomics that addition of AHA does not lead to protein identification artefacts in E. coli or different human cell lines (Eichelbaum et al., 2012; Bagert et al., 2014). Three independent AHA incubation experiments with E. coli, D. alaskensis and a gammaproteobacterial methanotrophic enrichment culture all demonstrated the incorporation of AHA into a diverse range of proteins (Fig. 3B). For the methanotrophic enrichment, there was a discrepancy in the apparent detection limit when comparing in gel fluorescence labelling of proteins and whole-cell detection via click chemistry. While both 100 μM and 1 mM AHA incubations yielded fluorescently labelled whole cells (Fig. 2F), fluorescent protein bands were not detected in the incubation with 100 μM AHA and only a small number of fluorescent protein bands was detected in the 1 mM AHA treatment. These results suggest that single-cell fluorescence labelling of microbial cells has a lower detection limit than in gel fluorescence. Consistent with the low molecular weight of the dye (0.88 kDa) we did not observe differences between the migrational patterns of unlabelled as compared with labelled proteins (Figs. 3A and 4B).
Fig 3.
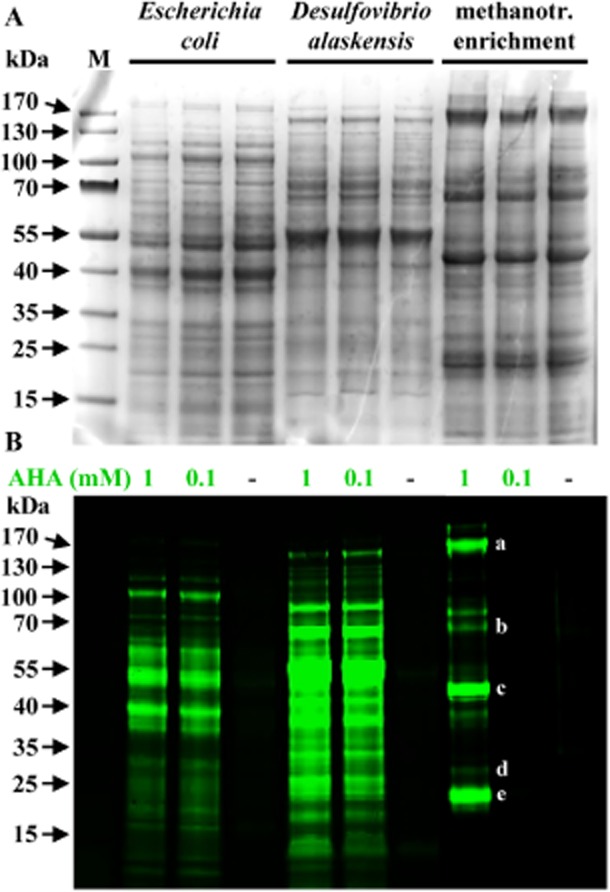
AHA does not interfere with the cellular machinery. Visualization of new proteins from cultures of E. coli, Desulfovibrio alaskensis and a methanotrophic enrichment. A. Coomassie-stained protein band patterns of cultures that had been incubated in the absence (−) or presence of AHA are indistinguishable from each other, demonstrating that AHA does not interfere with the translational machinery. B. Newly made proteins in the same gel are identified via BONCAT. Please note that the incubation time for the methanotrophic culture exposed to 100 μM AHA was too short to yield new proteins in amounts high enough to be detectable via in-gel fluorescence. At the individual cell level, AHA uptake can, however, be easily demonstrated (Fig. 2F). Some of the most intensely labelled bands were cut from the gel and analyzed via mass spectrometry. The 20 most abundant proteins from the excised bands included: (a) the two large subunits of RNA-polymerase (150.4 and 155.4 kDa); (b) a hypothetical protein (67.8 kDa) as well as two homologs of the large subunits of methanol dehydrogenase (66.6 and 68.3 kDa); (c) PmoB, 45.6 kDa; (d) PmoA (28.4 kDa) and PmoC (29.1 kDa); (e) a formaldehyde activating enzyme (17.8 kDa), superoxide dismutase (21.1 kDa), and D-arabino-3-hexulose 6-phosphate formaldehyde lyase (21.8 kDa). Letters a–e denote the bands consistent with the molecular weights of these proteins. kDa, kiloDalton; M, marker.
Fig 4.
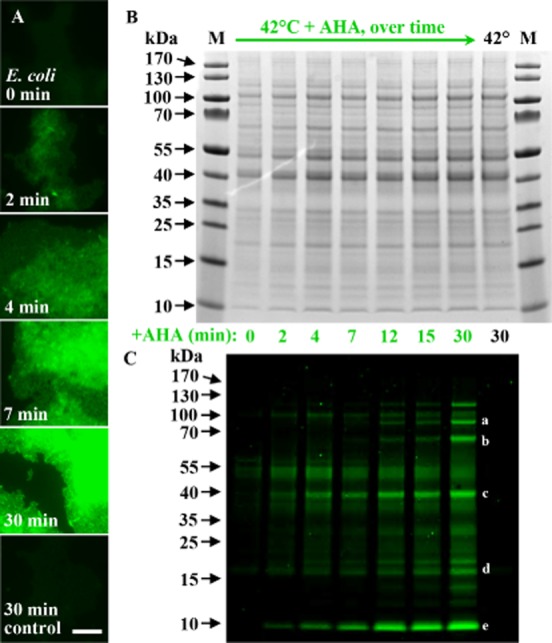
The high sensitivity of BONCAT allows detection of newly synthesized proteins after only minutes of incubation. A. Fluorescence labelling of heat-shocked (42°C) E. coli cells grown in the presence of AHA increases over time. Already after 2 min of incubation, equivalent to about 2% of E. coli's generation time under the conditions used, labelled cells can be visualized. B, C. While no differences in the relative amounts of proteins can be observed via Coomassie-staining, the fluorescent tag conjugated to newly made proteins reveals that certain proteins are preferentially amplified with time. C. Some of the most intensely fluorescently labelled bands were cut from the gel and analyzed via mass spectrometry. The obtained proteins included: (a) DNA gyrase subunits A and B as well as chaperone protein ClpB (97.0, 89.9 and 95.6 kDa respectively); (b) chaperonin GroL, a heat-inducible Lysine-tRNA ligase and chaperone DnaK (57.3, 57.8 and 69.1 kDa respectively); (c) outer membrane protein A (37.2 kDa); (d) chaperone proteins Skp (17.7 kDa) and YajL (20.8 kDa); (e) nine ribosomal proteins of low molecular mass (≤ 10 kDa) and the highest abundant protein in our dataset, major outer membrane lipoprotein Lpp (8.3 kDa). Letters a–e indicate bands consistent with the molecular weights of these proteins. Control refers to 30 min incubation at 42°C in the absence of AHA. Fluorescent signals were recorded at identical exposure times. Scale bar equals 10 μm and applies to all images. kDa, kiloDalton; M, marker.
Proteomic analysis was conducted on excised fluorescent bands from the Methylococcaceae sp. WF1 enrichment shown in Figure 3B (n = 5 fluorescently labelled bands). Consistent with the numerical dominance of Methylococcaceae in the enrichment culture, the calculated molecular weights of the most intensely labelled bands recovered from the 1 mM AHA incubation were consistent with enzymes expected to be highly expressed in aerobic methanotrophs, e.g. the subunits of particulate methane monooxygenase: PmoA (28.4 kDa), PmoB (45.6 kDa) and PmoC (29.1 kDa; Trotsenko and Murrell, 2008). The presence of PmoABC in the excised bands was additionally confirmed by mass spectrometry. Pmo subunits were among the 20 most abundant proteins in the data set, along with two homologs of the large subunit of methanol dehydrogenase (66.6 and 68.3 kDa), and the large subunits of DNA-directed RNA polymerase (150.4 and 155.4 kDa; Fig. 3B; Supporting Information Table S1A).
Although the sensitivity of these experiments is limited by the low-resolution power of one-dimensional protein gels, these results demonstrate that AHA is incorporated into a diverse range of cellular proteins and does not alter the synthesis or degradation of proteins. In contrast to Coomassie-staining, which is only able to visualize highly abundant proteins, click chemistry-based dye conjugation of AHA-containing proteins is able to distinguish highly expressed proteins from low turnover proteins with long life times.
Sensitivity of BONCAT
The sensitivity of BONCAT is influenced by several factors. Among the most important and experimentally testable are the Met content of the targeted proteins as well as the growth rate and translational activity of the cell. The average number of Met per candidate protein for the microorganisms studied here ranges from 7.0 (draft genome of Methylococcaceae sp. WF1) to 9.6 (D. alaskensis). Every protein encoded in the five genomes has at least one Met, and 10.0% (Methylococcaceae sp. WF1) to 18.9% (D. alaskensis) of all proteins have at least 15 Met in their predicted sequence (Supporting Information Fig. S3). Presumably, labelling efficiency is mostly dependent on the intracellular ratio of AHA/Met and the selectivity of the respective methionyl-tRNA synthetase. Thus, while perfect labelling efficiency is not expected, the large number of Met residues across the proteomes indicates the theoretical potential to label and detect every protein that is newly synthesized by these microorganisms.
Time course experiments with E. coli cultures were used to determine the minimum time required for detection of AHA-labelled proteins by BONCAT. Here, a heat-shock experiment with E. coli grown in the presence or absence of 1 mM AHA was performed, and then intact cells and protein extracts were analyzed after 2 to 30 min of incubation. Within 2 min of incubation, equivalent to ∼ 2% of E. coli's generation time under the experimental conditions, fluorescently labelled proteins were detected in gels as well as on the cell level (Fig. 4). Furthermore, while no changes in E. coli's total proteome were observed in Coomassie-stained protein gels, the visualization of newly made proteins via click chemistry revealed a high turnover rate for specific proteins (Fig. 4B,C). We performed proteomic analysis on several excised fluorescent bands (Fig. 4C). In total, 250 proteins were retrieved (Supporting Information Table S1B), which included several heat-shock proteins, specifically DNA gyrase subunits A and B (97.0 and 89.9 kDa), a heat-inducible Lysine-tRNA ligase (57.8 kDa), as well as chaperone proteins ClpB (95.6 kDa), GroL (57.3 kDa), DnaK (69.1 kDa), Skp (17.7 kDa), and YajL (20.8 kDa). Other highly abundant proteins contained within the excised fluorescent bands included outer membrane protein A (37.2 kDa) as well as 17 proteins of low molecular mass (≤ 10 kDa; including nine ribosomal proteins), as well as the highest abundant protein in the obtained data set, major outer membrane lipoprotein Lpp (8.3 kDa). Together, these observations demonstrate the potential of BONCAT to study rapid proteomic adaptation of fast growing microorganisms in response to environmental stimuli.
BONCAT-FISH: correlating translational activity with microbial identity
One of the most powerful characteristics of BONCAT is its potential to study the in situ activity of microbes in complex samples on the level of individual cells. To correlate translational activity with taxonomic identity, BONCAT was combined with 16S and 23S rRNA-targeted FISH. Defined mixtures of pure cultures and enrichments of bacteria and archaea, in which one of the cultures had been incubated with AHA prior to mixing with other cultures, were used to develop the BONCAT-FISH protocol (Fig. 1C). Immobilized cells from the formaldehyde (FA) fixed mixed cultures were fluorescently labelled via click chemistry and subsequently hybridized using general and species-specific oligonucleotide probes and nonsense control probes (Fig. 5A and Supporting Information Fig. S4). A comparison of mixed cultures that had been analyzed by labelling of new proteins and FISH in contrast to microbial cells stained only by protein-targeted click chemistry revealed comparable fluorescence intensities (data not shown). By combining BONCAT with FISH, we were able to detect active protein synthesis by the novel Methylococcaceae sp. WF1, which was identified using a specific FISH probe (MetI-444) within a mixture of other bacteria and archaea (Fig. 5A). The successful combination of FISH and BONCAT demonstrates the possibility to directly link species-specific identification of microbes with detection of their translational activity for individual cells using conventional fluorescence microscopy.
Fig 5.
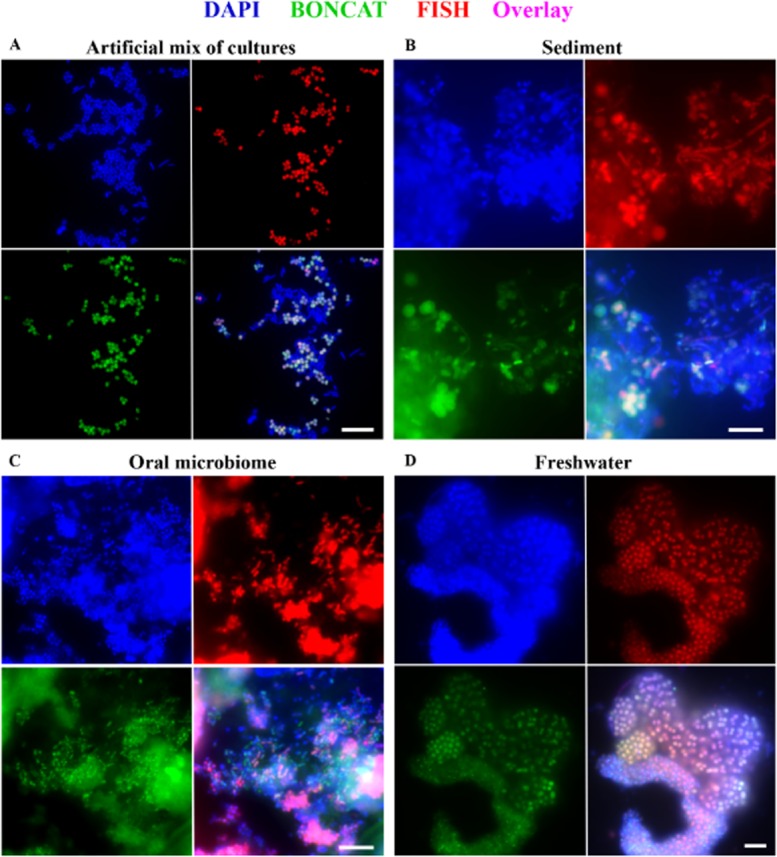
Visualization of newly synthesized proteins via BONCAT (green) in combination with rRNA-targeted FISH (red). A. An artificial mix of pure and enrichment cultures. The only microbe that had been incubated in the presence of AHA, a gamma proteobacterial methanotroph (Methylococcaceae sp. WF1), is identified via a species-specific FISH probe (MetI-444), demonstrating the feasibility of correlating translational activity with microbial identity. For details and a Cy5-probe image see Supporting Information Fig. S4. B–D. Many bacteria, identified by the general EUB338I-III FISH probe mix (B,C) or probe Gam42a (D), specific for gammaproteobacteria, are BONCAT-labelled, demonstrating in situ translational activity during time of incubation. Exposure times for click or FISH signals were identical for each sample series (i.e. AHA plus two controls), respectively. DAPI staining is shown in blue. For controls, see Supporting Information Figs. S4 and S6. All scale bars equal 10 μm.
Using BONCAT to assess the physiological potential of microorganisms in a mixed community: experiments with a methanotrophic enrichment culture
The ability to rapidly screen the anabolic activity of specific microorganisms in the environment using BONCAT-FISH offers a mechanism for assessing the activity of FISH-identified environmental microorganisms in response to substrate addition or physical manipulation in environmental samples. To demonstrate the ability to use BONCAT-FISH for discerning the physiological response of microbes under different conditions, we tested the effect of methane addition on an aerobic methanotrophic enrichment culture (WF1) originating from deep-sea sediments. This enrichment was dominated by a Methylococcaceae-related gammaproteobacterium based on FISH analyses using the methanotroph-specific probe MetI-444. Aliquots of the WF1 enrichment were incubated with 1 mM AHA in the presence or absence of methane for 26 h and then analyzed by BONCAT-FISH. FISH-identified Methylococcaceae cells showed strong BONCAT fluorescence signal (i.e. high translational activity) that increased over time in incubations with methane + oxygen, whereas a BONCAT signal was not detected in FISH-stained WF1 cells incubated without methane (Fig. 6A and Supporting Information Fig. S5). This comparative incubation experiment using BONCAT-FISH, together with recently obtained genomic data from this deep-sea Methylococcaceae strain (Tavormina et al., in review) and mass spectrometric analyses of its proteins (herein), provides clear evidence for the methanotrophic nature of this bacterium. To further test whether AHA-containing WF1 cells can be distinguished from other sediment microorganisms, we spiked an aliquot of the AHA-labelled Methylococcaeae culture (sample after 9 h of incubation) into a marine sediment sample and used Cu(I)-catalyzed click chemistry with a Carboxyrhodamine 110 Alkyne dye to screen the sample. As demonstrated in Fig. 6B, active Methylococcaceae cells can be clearly distinguished from other sediment-dwelling microbes via BONCAT. These results demonstrate the potential of using BONCAT-FISH for testing the physiology of microbes in complex environmental samples and highlight its use as a rapid and inexpensive screening tool for studying the response of cultured and uncultured microbes towards environmental stimuli.
Fig 6.
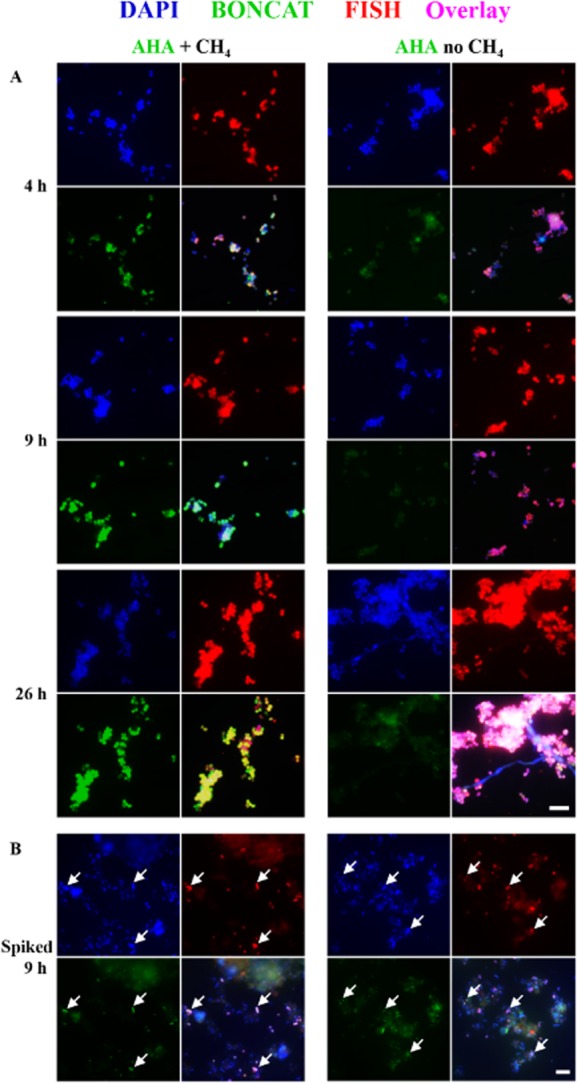
Comparative BONCAT analyses of a methanotrophic enrichment culture in the absence and presence of methane. A. Click chemistry-mediated detection of AHA incorporation (green) reveals that a gammaproteobacterium (identified by FISH probe MetI-444; red) is highly active in the presence but not in the absence of methane. B. To test whether AHA-labelled WF1 cells (examples are pointed out by arrows) would be detectable in a complex samples, an aliquot of the culture was spiked into methane seep sediment and analyzed via BONCAT. Exposure settings for recording of BONCAT signals were identical for each image set (i.e. A, B). For images taking at different settings and controls, see Supporting Information Fig. S5. Scale bars equal 10 μm and apply to all images of the respective set.
Comparison of single-cell metabolic activity proxies: BONCAT versus nanoSIMS 15N/14N analysis
Over the past 15 years, there have been a number of methods using whole-cell isotope labelling combined with FISH to measure translational activity and growth of individual microorganisms in complex environmental samples. These techniques rely on the incorporation of radiolabelled (3H, 14C or 35S) amino acids into cellular proteins using MAR or stable isotope-labelled ammonia or amino acids followed by microRaman or SIMS analyses. While these isotopic methods are gaining increasing use in microbial ecological studies, there are challenges impacting their general application by microbiologists: (i) assimilated ammonia can be used for other cellular functions in addition to translation, which may complicate data interpretation; (ii) limited access to radioactivity-certified laboratories or expensive microRaman or nanoSIMS instruments; and (iii) with SIMS and nanoSIMS analyses, considering the trade-offs between a high-sensitivity and precise measurement with relatively low sample throughput. From this perspective, BONCAT offers some advantages in accessibility and cost as compared with isotopic approaches. BONCAT uses commercially available and relatively inexpensive reagents and requires only standard molecular biological equipment (e.g. an epifluorescence microscope), offering a fast, culture-independent and cost-effective approach for direct analysis of anabolic activity in microorganisms in the environment.
To directly compare BONCAT-FISH and isotopic labelling methods by nanoSIMS, an artificial mixture of cultured microorganisms that had been grown in the presence of either AHA or AHA + 15NH4+ was prepared and then analyzed by both fluorescence microscopy and nanoSIMS. Escherichia coli cultures grown in the presence of 1.87 mM 15NH4Cl (i.e. 9% of total 14+15NH4+ pool) and 1 mM AHA were mixed with a methanotrophic enrichment culture (WF1) that had been incubated with 1 mM AHA and three unlabelled pure cultures of archaea and bacteria in roughly equal cell abundances. Cu(I)-catalyzed click chemistry using the 19F-containing dye Oregon Green 488 alkyne (Supporting Information Fig. S1A) followed by FISH was performed on the mixed culture and cells were imaged via fluorescence microscopy and by nanoSIMS to measure 15N enrichment and 19F content for each cell (Fig. 7). Using a specific FISH probe (MetI-444), we identified BONCAT-labelled cells of methanotroph WF1 in the mixed culture by epifluorescence (Fig. 7D), while E. coli cells were identified by 15N enrichment in the 15N/14N ratio images acquired by nanoSIMS (Fig. 7B). Both WF1 and E. coli exhibited strong BONCAT signals after incubation with AHA, with E. coli exhibiting slightly stronger relative fluorescence intensities per cell (Fig. 7E). All 15N-containing E. coli cells were fluorescently labelled (and vice versa, all BONCAT-positive E. coli cells had elevated 15N levels), and enriched in 19F (average 19F/12C = 0.0341; n = 35 regions of interest, ROI) as compared with non-AHA-labelled cells of different taxonomy (average 19F/12C = 0.0185; n = 15 ROI). However, WF1 methanotroph cells (average 19F/12C = 0.0177; n = 20 ROI; Fig. 7C) were indistinguishable from these control cells (t-test P-value = 0.359). This observation has two possible explanations: (i) E. coli cells had incorporated higher amounts of AHA and thus were more intensely labelled with the 19F-containing fluorescent dye. This possibility is supported by the fact that the E. coli culture was grown for ∼ 0.89 generations (1.4 h of incubation time; final OD ∼ 0.23) in the presence of AHA, while the WF1 enrichment had been exposed to AHA only for ∼ 0.59 generations (25.7 h; final OD ∼ 0.17). However, there was a smaller difference in relative fluorescence signal intensity between E. coli and methanotroph WF1 cells compared with cellular 19F enrichment by nanoSIMS (Fig. 7E versus 7C). This discrepancy may be related to the difference in detection method and subtle differences in the cellular properties of the WF1 and E. coli cells. While the epifluorescence image represents an integral of the click signal of the whole cell, the nanoSIMS-based 19F measurement is a destructive sampling method, where secondary ions are sputtered from the cell, quantified and then averaged over a Z-stack that may represent a few atomic layers to nanometers depending on the depth of pre-sputtering and analysis time. Independent of the discrepancy in fluorescence intensity and 19F-labelling, the results from these AHA incubation experiments are largely consistent with cellular 15NH3 assimilation as analyzed by nanoSIMS, demonstrating the high potential of BONCAT for studying translationally active cells by means of fluorescence microscopy.
Fig 7.
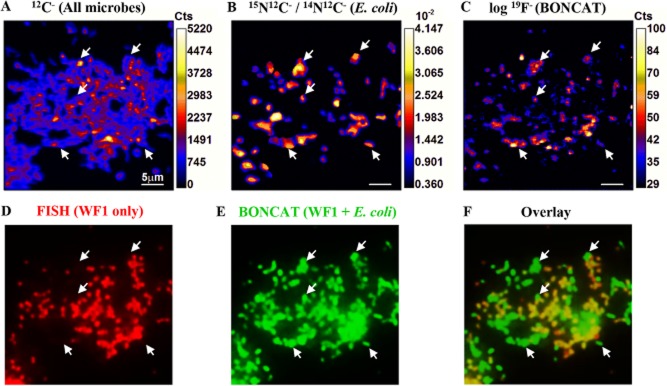
Comparison of AHA labelling of newly synthesized proteins with cellular 15N-uptake. An artificial mix of several cultures was analyzed via BONCAT, FISH and nanoSIMS. Escherichia coli had been incubated in the presence of both 15NH4Cl and AHA, while a methanotroph was exposed to AHA only. Other microbes had been grown in the absence of AHA or 15NH4Cl. A species-specific FISH probe (MetI-444) is used to localize the methanotroph WF1 (D; red), while both WF1 as well as E. coli are fluorescently labelled by a fluorine-containing alkyne-dye (E; green). While all E. coli cells (examples are pointed out by arrows) are 15N- and 19F-labelled (B, C), the 19F-signal of methanotroph cells is indistinguishable from cells that had not been incubated in the presence of AHA (C). A second halogen-containing dye (see Supporting Information Fig. S1) could not be used due to problems removing unbound dye (see main text). Scale bars equal 5 μm. Abbreviations: Cts, counts; WF1, cells of Methylococcaceae sp. WF1. A–C. Elemental and isotopic mapping of an artificial mix of cultures via nanoSIMS. D–F. Correspondent FISH and BONCAT images of the same field of view.
Visualizing active microbes in environmental samples using BONCAT-FISH
AHA incubation experiments and BONCAT-FISH were also applied to samples collected from a range of environments, including the oral microbiome (tongue scraping and saliva), pond water and anoxic sediment, to study taxonomically and physiologically diverse microorganisms within a range of sample types. These samples were selected to serve as a testing ground for the application of BONCAT-FISH with natural samples, and were incubated for several hours (4, 6 and 7.5 h for freshwater, oral and sediment samples respectively) in the presence or absence of AHA (1 mM). FA-fixed cells from each of the sample incubations were analyzed via click chemistry followed by FISH using both general (e.g. EUB338mix, Gam42a) as well as species-specific (MetI-444) oligonucleotide probes (Fig. 5B–D and Supporting Information Fig. S6).
In all samples, microbial cells with diverse morphologies were observed to be translationally active during the incubation period, which suggests a general applicability of BONCAT for microbial ecology studies. The high abundance of conspicuous aggregations of purple sulfur bacteria in the freshwater sample during time of sampling and the subsequent FISH identification of the majority of cells in dense cell clusters as gammaproteobacteria (probe Gam42a), make it likely that these BONCAT-labelled cells are members of the order Chromatiales (Fig. 5D). There were also observable differences in the anabolic activity of microorganisms between habitats. While the majority of cells that hybridized with the general bacterial probe (EUB338mix) in the oral and freshwater samples were also labelled by BONCAT (Fig. 5C and D), only a small proportion of the FISH-identified bacteria in the sediment sample were BONCAT-labelled (Fig. 5B). Unfortunately, absolute quantification of labelling efficiency was not possible because of the compact and densely packed nature of cells in all three samples. The most probable explanation for the inability to label the majority of sediment-affiliated cells is that (i) most sediment microbes did not synthesize proteins in high enough amounts for fluorescence microscopic detection during the incubation period. However, it cannot be excluded that (ii) some microbes did not take up AHA into their cells or proteins, possibly because of the absence of appropriate transporters, the high selectivity of their methionyl-tRNA-synthetases, or a high Met/AHA ratio in the cytoplasm. Arguably, the most crucial limitation of BONCAT is its dependence on an uptake mechanism. However, this is not specific to our approach, because every metabolic labelling technique is constrained by its need for an uptake system for the respective compound, be that ammonia, an amino acid, or a redox dye.
Stability of AHA
AHA is stable under all physiological and environmental conditions with the exception of high concentrations of sulfide and alkaline pH. Under such conditions, bisulfide (HS-) reduces organic azides selectively to the corresponding amines (Adachi et al., 1977). To test the extent of reduction of AHA by HS-, we performed abiotic, anoxic incubations of 1 mM AHA in the absence or presence of sulfide (H2S plus HS− equaling 1, 2 or 10 mM) and analyzed AHA and its reduction product, L-2,4-diaminobutyric acid, via nuclear magnetic resonance (NMR) spectroscopy at regular time intervals (Supporting Information Fig. S7). We found that at pH 7.0, identical to the pH of our sediment sample, after 8 days sulfide has no measurable effect on AHA (data not shown). At pH 8.0, the effect of sulfide increases with its concentration, with ≥ 10 mM being enough to reduce > 30% AHA within 40 h and concentrations ≤ 1 mM having only a minor effect (reduction of ∼ 5% AHA; Supporting Information Fig. S7). Considering that (i) typical sulfide concentrations in the sediment studied here are around the detection limit of our method (i.e. 60 μM; determined via cline assay), (ii) the pH of the sample was ≤ 7, and (iii) incubation times were substantially shorter than in our abiotic reactions (7.5 h versus 140 h), we conclude that during our freshwater and sediment incubations HS- had no significant effect on the concentration of AHA.
While not the focus of this study, some habitats inhabited by microbes with particularly slow growth rates exhibit conditions that could restrict the use of AHA over the course of long incubation times (i.e. several days to months). Examples of such systems include sulfidic deep-sea methane seeps in which inter-domain consortia, with doubling times of ∼ 3 months, catalyze the anaerobic oxidation of methane (reviewed by Knittel and Boetius, 2009). If such a system is to be studied we recommend either adding AHA at regular intervals during the incubation, or, better, using alternative surrogate amino acids (discussed below).
To test the stability of AHA at elevated temperatures, AHA (10 mM in water) was incubated at 80 ± 3°C and analyzed via NMR spectroscopy after 19.5 h of incubation. We found that at pH 5.0 and 7.1, high temperature has no measurable effect on AHA (data not shown), allowing for the study of protein turnover in (hyper)thermophilic microorganisms.
Considerations for BONCAT-FISH experiments of environmental samples
The activation rates of enzymes, specificities of transporters, etc. for bioorthogonal compounds as compared with their canonical counterparts (AHA versus Met) are likely lower. To our knowledge, the only enzyme that has been comparatively studied in this regard is the methionyl-tRNA synthetase of E. coli, where the activation rate for AHA is ∼ 0.25% of that for Met (Kiick et al., 2002). Thus, in respect to this enzyme a ‘total’ concentration of 100–1000 μM AHA in a sample translates to 0.25–2.5 μM of ‘bio-available’ amino acid. While these concentrations are still 100–1000 times higher than standing concentrations of amino acids in highly oligotrophic systems (e.g. open-ocean waters), the very low competition of in situ Met with AHA in such habitats would presumably facilitate the use of lower concentrations of AHA.
Given the complex structure, biochemistry and molecular regulation of gene expression in microbes, it is preferable to keep incubation times at a minimum to reduce the risk of system disturbance. For the environmental samples examined here, incubation times were comparable with or shorter than what is typically used in MAR- and SIMS-based experiments with incorporation of ammonia or amino acids (e.g. Ouverney and Fuhrman, 2000; Sintes and Herndl, 2006; Teira et al., 2006; Behrens et al., 2008; Finzi-Hart et al., 2009; Orphan et al., 2009; Pernice et al., 2012), and much shorter than in typical microRaman experiments (e.g. Huang et al., 2004; Huang et al., 2007; Haider et al., 2010). If longer incubation times are required, e.g. due to the study of ultra-oligotrophic systems or slow-growing microorganisms (e.g. Dekas et al., 2009; Morono et al., 2011), additional approaches that independently test for community succession or habitat disturbance should be conducted. Examples include analysis of 16S rRNA gene diversity over time, FISH, as well as geochemical and rate analyses to assess the activity of the microbes of interest.
In summary, AHA-based BONCAT-FISH is a broadly applicable technique, with the exception of samples harbouring high concentrations of sulfide in combination with high pH (e.g. sulfidic sediments) or systems characterized by high concentrations of Met. Because Met is a better substrate than AHA with respect to protein synthesis (Kiick et al., 2002), microbes living in extremely high nutrient environments, e.g. animal or human guts, will probably require modified BONCAT protocols. This idea is supported by the finding that if nutrient-rich media (Lysogeny broth, LB) were used, the high concentration of Met in these media reduced the incorporation of AHA into newly made E. coli proteins to levels below our limits of microscopic detection (data not shown).
Alternative clickable amino acid surrogates
Besides AHA, other bioorthogonal amino acids amenable to click chemistry have been described, including, most importantly, the Met surrogates homopropargylglycine (HPG) and azidonorleucine, as well as several analogs of pyrrolysine (Kiick et al., 2002; Fekner et al., 2009; Ngo and Tirrell, 2011). HPG previously has been used for the study of newly synthesized proteins in mammalian fibroblasts (Beatty et al., 2010) and neurons (Dieterich et al., 2010). Some alternatives, however, have limitations compared with the more robust and generally applicable AHA. For example, HPG has a substantially lower activation rate than AHA, while azidoalanine does not support protein synthesis in E. coli (Kiick et al., 2002). However, it has to be considered that activation rates might potentially substantially differ in phylogenetically distant and/or physiologically distinct microbes. Surrogates of unusual amino acids like pyrrolysine, for which the phylogenetic range is restricted to some groups of methanogens and deltaproteobacteria (Hao et al., 2002; Zhang and Gladyshev, 2007), may also have utility in targeted studies.
Outlook: combining BONCAT with established techniques
The successful application of BONCAT-FISH in natural ecosystems demonstrates the feasibility of correlating single-cell translational activity with phylogenetic identity in situ. In contrast to current methods used for measuring single-cell activity in microbial ecology, BONCAT enables the direct visualization of newly synthesized proteins via fluorescence microscopy, a technique commonly available in molecular biological laboratories. This method is simple, fast and comparatively high throughput using commercially available, inexpensive reagents. As azide-containing molecules are rare in the environment, the potential for nonspecific reactions is minimized. The chemistry of the click reaction is well understood, exhibits high specificity and is easily performed in the presence of a complex inorganic or organic matrix. Combined with rRNA-targeted FISH, BONCAT holds exciting prospects for the study of the spatio-temporal dynamics, ecophysiology and in situ anabolic activity of environmental microbes.
While not directly tested in this study, there are a number of promising future applications of BONCAT. For example, combining this technique with fluorescence-activated cell sorting would enable physical separation of translationally active cells, an approach analogous to the respiration response imaging method using redox sensor green (Kalyuzhnaya et al., 2008; Konopka et al., 2011). Quantification of the relationship between spatial organization and anabolic activity within structured microbial communities such as microbial mats, biofilms or consortia is also possible. Such a combination of techniques has the potential to grant us access to the genomes and physiologies of unidentified species and is independent of their respective numerical abundance in the environment.
The dynamic BONCAT fluorescent labelling of cells in response to substrate availability, demonstrated in this study with methanotrophs maintained in the presence or absence of methane, may also have utility for selective cultivation of microorganisms using liveBONCAT. Our preliminary results suggest that, with some optimization, fluorescence-labelling of living microorganisms after AHA incubation is possible. Extended to natural ecosystems, liveBONCAT could be used as a screening tool to study the reaction of uncultured microbes to the addition of potential substrates. Candidate cells that were translationally active in the presence of a compound of interest could be physically separated and subjected to culturing techniques. Such an approach could streamline compound screening and has the potential to expand the physiological and taxonomic diversity in our culture collections.
In addition to whole-cell-based detection, incorporation of azide-modified amino acids into proteins offers the possibility to physically enrich proteins that have been newly expressed during the time of incubation. By conjugating AHA-labelled proteins with alkyne-modified biotin reagents it is possible to separate those proteins on streptavidin- or neutravidin-coated affinity chromatography columns (Dieterich et al., 2006; 2007,; Szychowski et al., 2010). In theory, a single AHA residue is sufficient to separate the respective protein from the pre-existing protein fraction. This strategy has some advantages over protein stable isotope probing, which relies on the incorporation of comparatively high amounts of isotopic label for successful separation and identification of individual proteins (Jehmlich et al., 2010; von Bergen et al., 2013). Physical separation of the de novo synthesized fraction of the proteome before mass spectrometric sequencing has the potential to yield higher sequencing coverage of newly made proteins than may be observed in the bulk metaproteome.
Aside from its application to the study of protein expression, we expect click chemistry to have widespread utility in the microbial ecology field. For example, recent studies have demonstrated the successful incorporation of diverse bioorthogonal compounds into other major classes of biomolecules, including sugars (e.g. Saxon et al., 2002; Banerjee and Carrico, 2011), lipids (e.g. Kho et al., 2004; Neef and Schultz, 2009) and nucleic acids (e.g. Jao and Salic, 2008; Salic and Mitchison, 2008). We anticipate that the adaptation of these methods to complex, multi-species environmental samples will considerably expand the molecular toolbox for microbial ecologists studying uncultured and cultured microorganisms alike.
Experimental procedures
Chemicals
L-2-amino-4-azidobutanoic acid (AHA) was synthesized, purified and quality controlled according to established protocols (Link et al., 2007) with the following slight modifications: ethyl acetate washing (step 11 in the original protocol) was performed twice to remove all CuSO4; after adding the Dowex resin (Sigma Aldrich) to the chromatography column (step 14) the resin was washed with 200 ml methanol and then 400 ml water before conditioning took place as described (step 15); AHA was eluted with 1 M NH4OH until the effluent was basic (∼ 100 ml; step 18); then, the column was rinsed with an additional 100 ml of 1 M NH4OH and the effluent recycled a few times to harvest all of the amino acid (step 18). In later stages of the project, commercially available AHA (Iris Biotech) was used. Tris[(1-hydroxypropyl-1H-1,2,3-triazol-4-yl)methyl]amine (THPTA), used as ligand in copper-catalyzed click reactions, was synthesized according to a published protocol (Hong et al., 2009), but is also commercially available (e.g. Sigma). Currently, prices of AHA and THPTA offered by the above suppliers are lower than the costs associated with in-house synthesis.
Culturing
Four pure and two enrichment cultures were selected to test the BONCAT method with a range of phylogenetically and metabolically diverse microorganisms. Escherichia coli K12 was grown at either 32 or 37°C (as indicated below) at 150 r.p.m. horizontal shaking in M9 minimal medium: 0.5 g NaCl, 2.0 g glucose, 1.0 g NH4Cl, 12.8 g Na2HPO4 × 7 H2O, 3.0 g KH2PO4, 492 mg MgSO4 × 7 H2O, 11 mg CaCl2 and 100 mg thiamine per 1 L of deionized water. Incubations in LB medium were performed at 37°C and 150 r.p.m.
Paracoccus denitrificans PD1222 was grown under denitrifying conditions at 32°C at 100 r.p.m. horizontal shaking in heterotrophic medium containing 0.5 g Na-citrate × H2O, 1.0 g (NH4)2SO4, 1.8 g KNO3, 7.0 g K2HPO4, 3.0 g KH2PO4, 100 mg MgSO4, 50 mg FeSO4 × 7 H2O, 100 mg thiamine and 1.28 ml ethanol per L. 1 ml trace element solution SL-10 (http://www.dsmz.de) and 1 ml vitamin solution (see medium 141, http://www.dsmz.de) were added. Media were degassed at 100°C for 20 min before the vials were sealed with butyl rubber stoppers, the headspace flushed with N2 and the vials autoclaved.
Desulfovibrio alaskensis (formerly Desulfovibrio desulfuricans) G20 was grown in medium 383 (prepared as recommended, http://www.dsmz.de) on 20 mM lactate at 37°C with 150 r.p.m. horizontal shaking.
Methanosarcina acetivorans C2A was grown at 33°C without agitation [tubes were shaken before optical density (OD) measurements, OD600] in an artificial seawater medium that contained per litre: 20.45 g NaCl, 136 mg KH2PO4, 147 mg CaCl2 × 2 H2O, 3.05 g MgCl2 × 6 H2O, 535 mg NH4Cl, 2.52 g NaHCO3, 360 mg Na2S × 9 H2O, 1 mg resazurin, as well as 1 ml of each SL-10 trace elements and medium 141 vitamin solution (http://www.dsmz.de). Cultures were supplemented with 0.5% (v/v) of both methanol and trimethylamine and incubated under 80% H2, 20% CO2 headspace at 207 kPa (30 psi) pressure.
Methanotrophic enrichments from marine sediment and propane-oxidizing enrichments from stream sediments were grown with a modified nitrate mineral salt medium at room temperature (RT) with mild horizontal shaking (20 rpm) in the dark (Tavormina et al., in review). The headspace contained 45% N2, 30% CH4 and 25% lab air or 50% propane, 50% lab air respectively.
AHA incubations of cultures
AHA was added to log-phase cultures (assessed by optical density, OD600nm) using 100, 10 or 1 mM stock solutions yielding final concentrations of 1000, 100 or 10 μM respectively. Stock solutions were prepared in sterile filtered (0.2 μm) nanopure water and had a final pH of 7.0. AHA stock solutions for anaerobic incubations were sparged with N2 for 5 min. In addition, control incubations supplemented with water or L-Met (0.1 or 1 mM) were performed. The final volumes of all incubations for a given set of experiments were identical and ranged from 3 to 15 ml, depending on the cell density of the respective culture. All AHA and control incubations were run in duplicate. The optical densities (OD600) of cultures were regularly measured and growth rates compared. We did not observe an effect of AHA on the growth rate of any culture. After 0.2–1.0 generations (depending on growth rates and maximum cell densities of the individual cultures) incubations were stopped and cultures processed as described below.
To test on the effect of the protein synthesis inhibitor Camp on AHA incorporation in E. coli cells, we incubated an early log-phase E. coli K12 culture in the absence or presence of AHA (1 mM) and the absence or presence (290 mg l−1) of the antibiotic. After the control cells (i.e. E. coli grown with AHA but without Camp) had been grown for ∼ 0.8 generations all samples were chemically fixed (see below).
In the heat-shock experiment, AHA (1 mM final) was added to an early log-phase E. coli K12 culture and 3 ml aliquots were immediately transferred into preheated sterile glass vials. Vials were incubated for 30 min at 42°C without agitation. At the start of the experiment (after AHA addition, but before transfer to 42°C) as well as after 2, 4, 7, 12, 15 and 30 min, the entire volume of two replicate cultures was sampled. A single control without AHA was sampled after 30 min.
To compare AHA-labelling with 15NH3 uptake, a culture of E. coli K12 (grown overnight) was inoculated into M9 minimal medium and incubated for 3 h at 32°C with 150 r.p.m. horizontal shaking in the absence or presence of 1 mM AHA and 1.87 mM 15NH4Cl (Cambridge Isotope Labs; 15NH4+ constituted 9% of the total 14+15NH4+ pool).
At the end of each experiment, 1/3 of the culture volume was chemically fixed using 3% FA in 1× PBS for 60 min at RT, except for M. acetivorans, which was fixed in 3% FA in 1.5× PBS for 2.5 h at 4°C. After fixation, samples were centrifuged for 5 min at 16 100 g and the pellets washed three times with 1× PBS to remove free AHA. Pellets were resuspended in 50% EtOH in 1× PBS and stored at −20°C. The remaining 2/3 of the culture was centrifuged for 5 min at 5150 g (vol. > 4 ml) or 16 100 g (vol. < 4 ml), the supernatant (SN) discarded, and the cell pellet flash-frozen in liquid N2. Samples were stored at −80°C until further analysis.
Physiological experiments with a methanotrophic enrichment culture
To visualize the dependency of the enriched Methylococcaceae sp. WF1 on methane, we incubated the methanotrophic enrichment culture for 2.5 days in the absence of methane (energy starvation). Then, 3 ml aliquots of the culture were incubated in duplicate either with (i) 1 mM AHA and 30% methane in the headspace, (ii) 1 mM AHA without methane or (iii) 30% methane in the headspace without AHA (this last incubation was not performed in replicate). 0.5 ml aliquots were removed after 4, 9 and 26 h. Cells were pelleted and cultures fixed for 1 h in 3% FA in 1× PBS at RT. Washing steps and cell storage were performed as described for the other cultures (see above).
To test whether AHA-labelled WF1 cells can be detected in a complex sample, aliquots of the culture that been incubated under the above described conditions were spiked into a FA-fixed sample of marine methane seep top-layer sediment (not the original sample from which WF1 had been enriched from) and the sample thoroughly mixed. To separate cells from the sediment matrix, samples were sonicated on ice for 30 s with a sonicating wand (sonifier 150; Branson) at 6 W. One volume of ice-cold percoll (GE Healthcare Life Sciences) was added to the bottom of the tube and the samples centrifuged for 20 min at 16 100 g at 4°C. Afterwards, the liquid SN atop the percoll was transferred into a new tube, 1 volume 1× PBS added, and the sample centrifuged for 5 min at 16 100 g at RT. The pelleted cells were resuspended in 50% EtOH in 1× PBS and stored at −20°C.
AHA incubations of environmental samples
Three complex samples (3–15 ml each) were incubated in autoclaved glass tubes in the absence or presence (1 mM) of AHA: (i) saliva and biofilm scraped from the tongue of one of the authors; (ii) freshwater and (iii) top-layer (upper 1 cm) sediment from the ‘Lily pond’ on the Caltech campus. Oral samples were incubated for 6 h at 32°C with 100 r.p.m. horizontal shaking. We do not expect that these conditions are representative of the conditions microbes face within their human host. Rather, this experiment is intended to serve as a proof of principle demonstration and open the way for future experiments on human-associated microbes. Sediment samples (pH 7.0; 11°C) were transferred into test tubes, which were then closed with rubber stoppers and incubated in situ for 7.5 h half-submerged in water. Freshwater samples (pH 6.7; 21°C), containing aggregations of purple sulfur bacteria visible with the naked eye, were transferred into test tubes, closed with septa and incubated in the presence of AHA for 4 h. Incubations were performed during daytime, i.e. under natural light condition, and in duplicate. Aliquots of samples were fixed in 3% FA in 1× PBS for 1 h at RT. Sulfide (i.e. H2S plus HS−) concentrations in water and sediment samples were determined using the cline assay (Cline, 1969), which had a detection limit of 60 μM.
Alkyne-conjugated dyes
Several alkyne-modified dyes were tested for their suitability for protein click labelling. Dyes were purchased from the following companies: Invitrogen (Oregon Green 488 alkyne), Lumiprobe (Cy3 alkyne) and ClickChemistryTools (Carboxyrhodamine 110 Alkyne, DBCO-PEG4-Carboxyrhodamine 110, DBCO-PEG4-Tetramethylrhodamine and Eosin-alkyne).
Cu(I)-catalyzed click labelling of chemically fixed microbial cells
The azide-alkyne [3 + 2] cycloaddition (click) reaction requires a copper-catalyst, which is typically prepared with a chelating ligand, e.g. tris[(1-hydroxypropyl-1H-1,2,3-triazol-4-yl)methyl]amine (THPTA; Hong et al., 2009) or similar compounds (Besanceney-Webler et al., 2011). These molecules help to keep the metal in its Cu(I) oxidation state. Because of the instability of Cu(I) under standard lab air conditions, CuSO4 is usually added in large excess (∼ 100 μM) and in the presence of the reducing agent sodium ascorbate (∼ 5 mM). Furthermore, aminoguanidine (∼ 5 mM) is added to the reaction to inhibit protein cross-linking and precipitation (Hong et al., 2009 and references therein). Dye was used at 25 μM final concentration.
The protocol involved the following steps: fixed samples were immobilized on glass slides; dried in a hybridization oven (46°C); dehydrated and permeabilized by placing slides for 3 min each in 50, 80 and 96% ethanol; and then dried using pressurized air. Then, 1.25 μl of a 20 mM CuSO4 (in water; stored at 4°C) solution, 2.50 μl of 50 mM THPTA (in water; stored at 4°C) and 0.30 μl of alkyne dye [in dimethyl sulfoxide (DMSO) or a 1:1 DMSO : water mix] were mixed and allowed to react for 3 min at RT in the dark (i.e. dye premix). In the meantime, 12.5 μl of freshly prepared 100 mM sodium ascorbate (Sigma-Aldrich; freshly made in water; 5 mM final) and 12.5 μl of 100 mM aminoguanidine hydrochloride (Sigma-Aldrich; freshly made in water; 5 mM final) were added to 221 μl 1× PBS (pH 7.4). Then, the dye premix was added to this solution, the tube inverted once (do not mix by vortex to maintain reducing conditions), and samples were covered by 30 μl of solution. Slides were transferred into a humid chamber (water on tissue paper) and incubated in the dark at RT for 30 min. Afterwards, slides were washed three times for 3 min each in 1× PBS, treated with an increasing ethanol series (3 min each in 50, 80 and 96% ethanol) and air-dried.
In addition to on-slide-labelling, on-filter and in-solution click labelling were attempted. For on-filter click labelling, samples of pure cultures were immobilized on 0.2 μm GTBP filters (Millipore). All subsequent steps were identical to the protocol described above.
For click labelling in solution, samples were resuspended in 221 μl 1× PBS, to which solutions were added as described above. Tubes were inverted once and then incubated in the dark at RT for 30 min. Afterwards, samples were washed three times with 1x PBS, and then three times in an increasing ethanol series (50, 80 and 96%). Each washing step was followed by pelleting samples via centrifugation for 5 min at 16 100 g at RT. Finally, cells were resuspended in 50% ethanol in 1× PBS, transferred onto a glass slide, dried at 46°C, DAPI/Citifluor (Science Services) mounted, and microscopically analyzed.
Strain-promoted click labelling of chemically fixed microbes
Strain-promoted click chemistry is different from the above approach as it does not depend on the presence of a catalyst (Beatty et al., 2010; Chang et al., 2010). Instead, the reaction rate is increased by using strained dibenzocyclooctyne (DBCO) molecules to which fluorescence dyes have been conjugated via a polyethylene glycol linker (Supporting Information Fig. S1EF).
Samples were immobilized on glass slides, dried in a hybridization oven (46°C), dehydrated and permeabilized by placing slides for 3 min each in 50, 80 and 96% ethanol, and then dried using pressurized air. Slides were incubated for 1 h in 100 mM 2-chloroacetamide in Tris/HCl (pH 7.4) at 46°C in the dark to block free thiols. Then, dyes were directly added to this solution to reach final concentrations of 100 nM to 1 μM (using a 5 mM stock solution). Click reactions were carried out for 30 min at 46°C in the dark. To remove unbound dye, different washing protocols were tested, of which the following proved generally successful: after click labelling, slides were washed for 10 min at 48°C in 1× PBS (pH 7.4) and then transferred to a solution of 50% DMSO in 1x PBS (RT). Slides were incubated for 20 min and then washed three times for 3 min each in 1× PBS. Last, slides were washed/dehydrated in an increasing ethanol series (3 min each in 50, 80 and 96% ethanol) and air-dried. Slides were mounted with DAPI/Citifluor and microscopically analyzed, or, alternatively, FISH was performed. If incubation at 46/48°C is logistically not possible, reactions and washing can also be performed at RT or 37°C. However, this will result in lower signal-to-noise ratios.
FISH
To establish the BONCAT-FISH protocol, artificial mixes of pure and enrichment cultures were immobilized on glass slides. Due to the low salt concentration of the buffers used for click chemistry, FISH was always performed after click chemistry-mediated labelling of AHA-containing proteins to maintain specificity during oligonucleotide probe hybridization. This order minimizes the potential for dissociation of the hybridized probes from their target rRNAs and possible re-association with non-target rRNAs in the low stringency buffer used in click chemistry. After BONCAT had been performed and slides had been dehydrated via an increasing ethanol series (3 min in each 50, 80 and 96% ethanol), 16S and 23S rRNA-targeted FISH was carried out following established protocols. Briefly, samples were hybridized with Cy3- and Cy5-labelled oligonucleotide probes for 1.5–4 h (depending on the sample) in a humid chamber at 46°C. Formamide concentrations in the buffer were as recommended: 10–60% for probe mix EUB338 I-III (Amann et al., 1990; Daims et al., 1999) and control probe NonEUB338 (Wallner et al., 1993), 20% for probe Alf968 (Neef et al., 1999) and 35% for probes Bet42a, Gam42a (both used with unlabelled competitors probes; Manz et al., 1992) and Arch915 (Stahl and Amann, 1991). For probe MetI-444, used to detect Methylococcaceae sp. WF1 in one of our enrichment cultures, a formamide concentration of 35% was used, while 60% was originally recommended (Losekann et al., 2007). However, the sediment sample in which this probe had been recently applied (Losekann et al., 2007) is substantially more complex than our enrichment culture. Besides our target gamma proteobacterium, at the time of analysis the sample hosted up to five other bacterial species, each at abundances < 1% of the total population (based on DAPI counts). We never observed more than one FISH-positive morphotype (coccoid methanotroph WF1 cells, Fig. 4F), consistent with the in silico prediction of several mismatches to non-target rRNAs. When applied to the prestine sediment sample used in our spiking experiment, we did not observe MetI-444 (35% formamide)-positive cells. After hybridization, slides were washed for 10 min in pre-warmed washing buffer at 48°C before they were dipped into 4°C deionized water to remove salts. After slides were dried with pressurized air, they were mounted with DAPI/Citifluor and analyzed via epifluorescence microscopy. Fluorescence images were analyzed using imageJ (NIH).
nanoSIMS analysis of BONCAT-labelled cells
An artificial mix of microbes, which consisted of AHA- and 15N-labelled E. coli, the AHA-labelled methanotrophic enrichment, as well as unlabelled aliquots of P. denitrificans, D. variabilis and Methanosarcina vanielli (provided by Hiroyuki Imachi), was immobilized on silicon wafers. Cu(I)-catalyzed click labelling was performed as described above using the fluorine-containing dye Oregon Green 488 alkyne (Supporting Information Fig. S1A), followed by FISH using probe MetI-444 (see above). Biomass was microscopically imaged and analyzed via the nanoSIMS 50 L (Cameca) at Caltech's Microanalysis Center. Areas were pre-sputtered using high Cs+ beam currents before measurements were performed. Images were acquired with a 3 pA Cs+ ion primary beam, focused to a spot size of 100 nm. Analyzed regions were 30 × 30 or 40 × 40 μm in area and were rastered using a resolution of 512 × 512 pixels and a dwell time of 6.8 ms. The nanoSIMS was run in multi-collector mode with a mass resolving power of ∼ 7000 with electron multipliers positioned to detect 19F-, 12C-, 12C14N-, 12C15N-, and 31P-. Data was analyzed using the limage software (Cameca). Definitions of ROI were guided by the 12C15N- signal of E. coli, the FISH signal of methanotroph WF1 and the 12C- signal, which was used as an indicator of biomass location. Due to the physical proximity of cells to each other, it was not always possible to define ROI for individual cells. Thus, the number of cells per ROI varies between 1 and 5. Similar experiments were attempted using a Br-containing dye (Eosin) for nanoSIMS analyses. However, this dye was found to be incompatible with BONCAT due to the difficulty of removing unbound dye from the cell (Supporting Information Fig. S1D).
Extraction, labelling and purification of proteins
Frozen pellets were resuspended in 1 ml extraction buffer [1% sodium dodecyl sulfate (SDS), 50 mM Tris, 150 mM NaCl, 100 mM EDTA, 1 mM MgCl2 at pH 8.4] and boiled in a water bath for 30 min. Note that at this point, no reducing agent, such as dithiotreitol (DTT) or β-mercaptoethanol, was added because they would reduce the azide group of AHA. After cell lysis, samples were allowed to cool for 5 min at RT before they were centrifuged at 16 100 g for 5 min. The SN was transferred into a new tube and proteins were quantified using the bicinchoninic acid (BCA) protein assay according to the manufacturer's protocol (Thermo Scientific). If not immediately processed, crude protein extracts were stored at −20°C. To label AHA-containing proteins, a volume equivalent to 100–250 μg of protein was transferred into a new tube and 2-chloroacetamide was added (100 mM final concentration). Tubes were shaken for 1 h at RT in the dark before dye DBCO-PEG4-Carboxyrhodamine 110 (Supporting Information Fig. S1E) was added to a final concentration of 10 μM. Tubes were shaken for 30 min at RT in the dark before 1 mM AHA was added to stop the reaction. Proteins were extracted using a 600:150:400 μl methanol : chloroform : water mix and the pellet washed three times with 1 ml of pure methanol. All centrifugation steps were done at 16 100 g at RT. After the final wash, a volume of ∼ 200 μl was left in the tube and the sample was desiccated using a vacuum centrifuge. Extracted proteins were processed immediately or stored at 4°C for up to 3 days.
Protein gels
Pellets were resuspended in loading buffer to which 200 mM DTT had been freshly added. Proteins were denatured at 65°C for 5 min before 5–10 μg of protein were loaded and run at a constant voltage of 175 V for 45 min on NuPAGE Novex 4–12% Bis-Tris gels (Invitrogen) using MES running buffer (50 mM of 2-(N-morpholino)ethanesulfonic acid, 50 mM of Tris Base, 0.1% of SDS, 1 mM of EDTA, pH 7.3; chemicals from Sigma-Aldrich). PageRuler protein ladder (Thermo Scientific) was used as a marker of molecular weight. After electrophoresis, gels were fixed for 20 min in a 1:4:5 acetate : methanol : water mix before being washed three times in deionized water for 5 min each under horizontal shaking. Gels were scanned using a Typhoon laser scanner (GE Healthcare Life Sciences) at an excitation wavelength of 532 nm to visualize fluorescently labelled proteins. Afterwards, protein gels were stained for 1 h using GelCode Blue Stain Reagent (Thermo Scientific), washed for 1 h in deionized water under horizontal shaking, and imaged.
Extraction of labelled gel bands and protein sequencing
BONCAT-labelled proteins from a methanotrophic enrichment as well as from an E. coli culture were run on a protein gel as described above. The gel was stained for 1 h in colloidal Coomassie stain (Invitrogen) at RT and fluorescently labelled bands were excised with a scalpel, and each excised band was finely diced and transferred to an Eppendorf tube. To each gel piece 100 μl of ammonium bicarbonate (AB; filtered through a 0.2 μm filter after preparation) was added and incubated for 5 min (all steps were done at RT). Buffer was removed with a pipette and 50 μl of a 1:1(v/v) 50 mM AB/acetonitrile (ACN) was added to the gel pieces and incubated for 5 min. These de-staining steps were repeated twice to remove all Coomassie stain. Then, 25 μl of 50 mM AB and 50 μl of freshly prepared 10 mM DTT in 100 mM AB were added to the gel pieces and incubated at 50°C for 30 min. Afterwards, the solution was removed and 25 μl of 50 mM AB was added to the gel. To this mixture, 50 μl of freshly prepared 55 mM iodoacetamide in 100 mM AB was added and the gel pieces incubated in the dark for 20 min. Then, solution was removed, 100 μl of 50 mM AB was added and gel pieces incubated for 5 min. After removal of this solution, gel pieces were washed with 100 μl of ACN for 5 min. Then, 75 μl of 50 mM AB and 25 μl 6 ng μl−1 trypsin (prepared from a 100 ng μl−1 stock solution in 1 mM HCl) were added, and tubes incubated overnight at 37°C. Then, the SN was transferred into a new tube, and 100 μl of 1% formic acid, 2% ACN was added to the gel pieces. After incubation for 5 min, the solution was transferred to a vial containing SN. Gel pieces were washed with 100 μl of 1:1 (v/v) ACN : water, and after 5 min incubation the SN was again transferred. In a last washing step, 100 μl of 1% formic acid in ACN was added to the gel pieces, left for 5 min, and the SN transferred. The extracted peptides were dried using a vacuum centrifuge at 37°C, and afterwards dissolved in 0.2% formic acid. The dried peptides were resuspended in 80% ACN and separated from remaining gel pieces by purification via a C18 column. Peptides were sequenced using a liquid chromatography mass spectrometry system and raw data analyzed with the maxquant quantitative proteomics software (Cox and Mann, 2008) as recently described (Kalli et al., 2013).
Abiotic incubations of AHA
The following buffers were prepared anaerobically: 50 mM potassium phosphate pH 7.0, 100 mM potassium phosphate pH 8.0 and 100 mM carbonate pH 9.0. All buffers were prepared by mixing the appropriate amounts of KH2PO4/K2HPO4 and NaHCO3/Na2CO3 respectively. Ten volume per cent of D2O was added to all buffers for locking the NMR measurements. Each of the corresponding solutions (700 μl total) was mixed in an anaerobic chamber from a stock solution of 100 mM AHA (pH 7.0) and from a 100 mM Na2S stock solutions (adjusted to pH 7, 8 and 9 respectively). Each solution was transferred into a 5 mm NMR tube, closed with a septum and a NMR spectrum was recorded. After analysis, the NMR tubes were stored at RT in the anaerobic chamber until further measurements. For experiments at pH 9, samples were neutralized by adding 40 μl 1.0 M KH2PO4 directly into the NMR tube (pH 7.5) before measurement of the 77 h time point for easier comparison with the other spectra. An additional sample was prepared aerobically with 10 mM Na2S at pH 8.0. To test on the effect of high temperature, 10 mM AHA solutions in sterile water were adjusted to pH 5.0 and 7.1 and solutions transferred into 5 mm NMR tubes. Ten volume per cent of D2O were added, tubes closed with a septum and NMR spectra were recorded. After analysis, the tubes were incubated in a drying oven at 80 ± 3°C for 19.5 h and then again analyzed via NMR.
NMR spectroscopy
1H-NMR spectra were recorded on a Varian 400 MHz spectrometer with a broadband auto-tune OneProbe (Varian). The spectra were recorded at 25°C without spinning (90° excitation pulse, 2.6 s acquisition time) and continuous wave pre-saturation of the water signal for 2 s was employed (4.6 s repetition rate). Sixty-four scans were recorded each using identical parameters for all samples. Spectra were processed with inmr 4.1.7 software (Nucleomatica) using manual phase correction, 0.1 Hz of line broadening and automatic baseline correction. The ratio of AHA to L-2,4-diaminobutyric acid was quantified via integration of the signal RCH2N3 at 3.4 p.p.m. (AHA) and RCH2NH2 at 2.95 p.p.m. (2,4-diaminobutyric acid). For incubations in the presence of sulfide, no decomposition products other than 2,4-diaminobutyric acid could be detected. The identity of 2,4-diaminobutyric acid was verified by reducing an aliquot of AHA with tris(2-carboxyethyl)phosphine (Staudinger reduction) and comparing its NMR spectrum in pH 8.0 buffer with the sulfide incubation spectra.
Acknowledgments
We thank Katherine Dawson and Hiroyuki Imachi for advice on anaerobic culturing, Yunbin Guan for assistance with nanoSIMS analyses, John D. Bagert for discussions on click chemistry, Grayson Chadwick for calculating genomic Met contents and Jennifer Glass for helpful comments on an early version of this manuscript. We acknowledge the Caltech Proteome Exploration Laboratory (PEL) staff for analyzing mass spectrometry samples and their technical assistance with sample preparation and interpretation of results. The PEL is supported by the Beckman Institute and the Gordon & Betty Moore Foundation. Roland Hatzenpichler was supported via an O.K. Earl Postdoctoral Scholarship awarded by Caltech's Division of Geological and Planetary Sciences as well as an Erwin Schrödinger Postdoctoral Fellowship of the Austrian Science Fund (FWF), Grant No. J 3162-B20. Silvan Scheller was supported by the Swiss National Science Foundation (Grant No. PBEZP2_142903). Funding for this project was provided by the Gordon and Betty Moore Foundation through Grant No. GBMF3780 to Victoria J. Orphan, from the Department of Energy (Grant No. DE-PS02-09ER09–25) to Victoria J. Orphan and by a National Institutes of Health Grant No. NIH R01 GM062523 to David A. Tirrel.
Supporting Information
Additional Supporting Information may be found in the online version of this article at the publisher's web-site
Comparative BONCAT of newly synthesized proteins in E. coli and structures of dyes used for (A-D) Cu(I)-catalyzed and (E-F) strain-promoted click chemistry. Exposure times for cells incubated in the presence or absence of AHA are identical for each dye and were chosen to yield approximately comparable signal intensities for AHA-containing cells. Gain and offset settings of the detector were kept constant for all conditions. Note that different dyes give highly divergent signal to noise ratios when washing conditions are identical. Because of these background problems the Eosin-dye, initially believed to be a potential alternative for detection of AHA incorporation via nanoSIMS, could not be used in any of our experiments. The scale bar applies to all photos and equals 10 μm. Chemical structures were obtained from the respective company websites (see main text). Abbreviations: DBCO, Dibenzocyclooctyne; PEG, Polyethylene glycol.
Visualization of newly made proteins in living (liveBONCAT; panels A & B) and chemically fixed (C, D) AHA-labelled E. coli. Exposure times were identical for all images. Note that E. coli cells in panels (A) and (B) exhibit atypical cell morphology and that substantially lower numbers of cells are fluorescently labelled despite cells from the same culture are analyzed. The scale bar applies to all photos and equals 10 μm.
Distributions and average contents of methionine in candidate proteins encoded in the genomes of five microbes analyzed via BONCAT. Note that the genome of Methylococcaceae sp. WF1 has not been closed yet.
BONCAT-based visualization of newly synthesized proteins combined with 16S rRNA-targeted FISH in an artificial mix of microbial cultures. A methanotrophic enrichment culture that had been incubated in the presence of AHA was mixed with normally grown cultures of E. coli, P. denitrificans, Methanosarcina sp., and a propane-oxidizing enrichment culture. Consecutively, BONCAT was performed using Oregon Green 488 alkyne (green), FISH was performed using Cy3 (red) and Cy5 (turquoise) labeled oligonucleotide probes, and biomass was stained using DAPI (blue). (A) Localization of probe MetI-444 (red), specific for a group of gamma proteobacterial methanotrophs, reveals that coccoid WF1 cells from the methanotrophic enrichment culture incorporated AHA into their proteins. WF1 cells are not detected by probe Gam42a (turquoise; used with unlabeled competitor Bet42a), which is specific for many but not all gamma proteobacteria, due to a discriminating mismatch in their 23S rRNA. (B) FISH controls taken at identical exposure times for the respective channels (Cy3-labelled NonEUB338; auto-fluorescence in Cy5 channel). For BONCAT controls of the individual cultures see Fig. 2. For probe details refer to main text. The scale bars equal 10 μm.
Complimentary images to the comparative BONCAT analyses of a methanotrophic enrichment culture using different detection settings. Please note that for samples incubated for > 9 h in the presence of AHA, fluorescent signal of WF1 cells saturated the sensor (see Fig. 6). These settings were chosen in order to visualize the difference in fluorescence intensity after 4 h of incubation between WF1 cells grown in the absence and presence of methane. For images shown in this panel, exposition settings were chosen based on the much stronger signal intensities observed for later samples. All photos in this figure were taken at identical microscopic settings. Scale bar equals 10 μm and applies to all images.
BONCAT and FISH controls for microbiome and environmental samples. (A) Results for samples incubated in the absence of AHA (no BONCAT signal). (B) Tests of the specificity of the FISH protocol using a Cy3-labelled NonEUB338 probe (no FISH signal). Camera settings to record BONCAT and FISH signals were identical to those used to image samples shown in Fig. 5B–D respectively. All scale bars equal 10 μm.
Reduction of AHA in the presence of high concentrations of sulfide and high pH. (A) Results for abiotic reactions of 1 mM AHA in the absence or presence (H2S plus HS- equals 1, 2, or 10 mM) of sulfide at pH 8.0 (HPO4- buffered) and pH 9.0 (HCO3- buffered). At pH 8 and 9, AHA is slowly reduced with a reduction rate proportional to the sulfide concentration. At pH 7, the pH of the sediment sample studied here (sulfide concentration ≤ 60 μM), sulfide has no detectable effect on AHA for up to at least 8 days (data not shown). The stability of AHA itself is not influenced by pH, and sulfide has no effect on AHA at pH ≤ 7. AHA and its reduction product with sulfide, L-2,4-diaminobutyric acid (B), were analyzed via NMR spectroscopy.
Visualization of clusters of protein-synthesizing gammaproteobacterial freshwater bacteria (identified via FISH-probe Gam42a (red), which was used with its competitor). The scale bar equals 20 μm.
(A) Proteins identified via MS sequencing of gel-extracted proteins from methanotrophic enrichment culture WF1, ranked by abundance. (B) Proteins identified via MS sequencing of gel-extracted proteins from a heat-stressed E. coli K12 culture, ranked by abundance.
liveBONCAT: Attempting to visualize protein synthesis in living cells. Strain-promoted click labeling of living microbes.
References
- Adachi T, Yamada Y. Inoue I. Alternative method for selective reduction of unsaturated nucleoside azides to amines. Synthesis-Stuttgart. 1977;1977:45–46. [Google Scholar]
- Agard NJ, Prescher JA. Bertozzi CR. A strain-promoted [3 + 2] azide-alkyne cycloaddition for covalent modification of biomolecules in living systems. J Am Chem Soc. 2004;126:15046–15047. doi: 10.1021/ja044996f. [DOI] [PubMed] [Google Scholar]
- Amann RI, Binder BJ, Olson RJ, Chisholm SW, Devereux R. Stahl DA. Combination of 16S rRNA-targeted oligonucleotide probes with flow cytometry for analyzing mixed microbial populations. Appl Environ Microbiol. 1990;56:1919–1925. doi: 10.1128/aem.56.6.1919-1925.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amann RI, Zarda B, Stahl DA. Schleifer KH. Identification of individual prokaryotic cells by using enzyme-labeled, rRNA-targeted oligonucleotide probes. Appl Environ Microbiol. 1992;58:3007–3011. doi: 10.1128/aem.58.9.3007-3011.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bagert JD, Xie YJ, Sweredoski MJ, Qi Y, Hess S, Schuman EM. Tirrell DA. Quantitative, time-resolved proteomic analysis by combining bioorthogonal noncanonical amino acid tagging and pulsed stable isotope labeling by amino acids in cell culture. Mol Cell Proteomics. 2014 doi: 10.1074/mcp.M113.031914. doi: 10.1074/mcp.M113.031914 mcp.M113.031914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banerjee PS. Carrico IS. Chemoselective modification of viral proteins bearing metabolically introduced ‘clickable’ amino acids and sugars. Methods Mol Biol. 2011;751:55–66. doi: 10.1007/978-1-61779-151-2_5. [DOI] [PubMed] [Google Scholar]
- Beatty KE, Xie F, Wang Q. Tirrell DA. Selective dye-labeling of newly synthesized proteins in bacterial cells. J Am Chem Soc. 2005;127:14150–14151. doi: 10.1021/ja054643w. [DOI] [PubMed] [Google Scholar]
- Beatty KE, Liu JC, Xie F, Dieterich DC, Schuman EM, Wang Q. Tirrell DA. Fluorescence visualization of newly synthesized proteins in mammalian cells. Angew Chem Int Ed Engl. 2006;45:7364–7367. doi: 10.1002/anie.200602114. [DOI] [PubMed] [Google Scholar]
- Beatty KE, Fisk JD, Smart BP, Lu YY, Szychowski J, Hangauer MJ, et al. Live-cell imaging of cellular proteins by a strain-promoted azide-alkyne cycloaddition. Chembiochem. 2010;11:2092–2095. doi: 10.1002/cbic.201000419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Behnam F, Vilcinskas A, Wagner M. Stoecker K. A straightforward DOPE (double labeling of oligonucleotide probes)-FISH (fluorescence in situ hybridization) method for simultaneous multicolor detection of six microbial populations. Appl Environ Microbiol. 2012;78:5138–5142. doi: 10.1128/AEM.00977-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Behrens S, Losekann T, Pett-Ridge J, Weber PK, Ng WO, Stevenson BS, et al. Linking microbial phylogeny to metabolic activity at the single cell level using enhanced element labeling – catalyzed reporter deposition fluorescence in situ hybridization (EL-FISH) and NanoSIMS. Appl Environ Microbiol. 2008;74:3143–3150. doi: 10.1128/AEM.00191-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Bergen M, Jehmlich N, Taubert M, Vogt C, Bastida F, Herbst FA, et al. Insights from quantitative metaproteomics and protein-stable isotope probing into microbial ecology. ISME J. 2013;7:1877–1885. doi: 10.1038/ismej.2013.78. doi: 10.1038/ismej.2013.78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Besanceney-Webler C, Jiang H, Zheng T, Feng L, Soriano del Amo D, Wang W, et al. Increasing the efficacy of bioorthogonal click reactions for bioconjugation: a comparative study. Angew Chem Int Ed Engl. 2011;50:8051–8056. doi: 10.1002/anie.201101817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Best MD. Click chemistry and bioorthogonal reactions: unprecedented selectivity in the labeling of biological molecules. Biochemistry. 2009;48:6571–6584. doi: 10.1021/bi9007726. [DOI] [PubMed] [Google Scholar]
- Binder BJ. Liu YC. Growth rate regulation of rRNA content of a marine synechococcus (Cyanobacterium) strain. Appl Environ Microbiol. 1998;64:3346–3351. doi: 10.1128/aem.64.9.3346-3351.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bollmann A, Schmidt I, Saunders AM. Nicolaisen MH. Influence of starvation on potential ammonia-oxidizing activity and amoA mRNA levels of Nitrosospira briensis. Appl Environ Microbiol. 2005;71:1276–1282. doi: 10.1128/AEM.71.3.1276-1282.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carrico IS. Chemoselective modification of proteins: hitting the target. Chem Soc Rev. 2008;37:1423–1431. doi: 10.1039/b703364h. [DOI] [PubMed] [Google Scholar]
- Chakrabarti S, Liehl P, Buchon N. Lemaitre B. Infection-induced host translational blockage inhibits immune responses and epithelial renewal in the Drosophila gut. Cell Host Microbe. 2012;12:60–70. doi: 10.1016/j.chom.2012.06.001. [DOI] [PubMed] [Google Scholar]
- Chang PV, Prescher JA, Sletten EM, Baskin JM, Miller IA, Agard NJ, et al. Copper-free click chemistry in living animals. Proc Natl Acad Sci USA. 2010;107:1821–1826. doi: 10.1073/pnas.0911116107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cline JD. Spectrophotometric determination of hydrogen sulfide in natural waters. Limnol Oceanogr. 1969;14:454–458. [Google Scholar]
- Codelli JA, Baskin JM, Agard NJ. Bertozzi CR. Second-generation difluorinated cyclooctynes for copper-free click chemistry. J Am Chem Soc. 2008;130:11486–11493. doi: 10.1021/ja803086r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox J. Mann M. MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat Biotechnol. 2008;26:1367–1372. doi: 10.1038/nbt.1511. [DOI] [PubMed] [Google Scholar]
- Daims H, Bruhl A, Amann R, Schleifer KH. Wagner M. The domain-specific probe EUB338 is insufficient for the detection of all Bacteria: development and evaluation of a more comprehensive probe set. Syst Appl Microbiol. 1999;22:434–444. doi: 10.1016/S0723-2020(99)80053-8. [DOI] [PubMed] [Google Scholar]
- Dekas AE, Poretsky RS. Orphan VJ. Deep-sea archaea fix and share nitrogen in methane-consuming microbial consortia. Science. 2009;326:422–426. doi: 10.1126/science.1178223. [DOI] [PubMed] [Google Scholar]
- DeLong EF, Wickham GS. Pace NR. Phylogenetic stains: ribosomal RNA-based probes for the identification of single cells. Science. 1989;243:1360–1363. doi: 10.1126/science.2466341. [DOI] [PubMed] [Google Scholar]
- Dieterich DC, Link AJ, Graumann J, Tirrell DA. Schuman EM. Selective identification of newly synthesized proteins in mammalian cells using bioorthogonal noncanonical amino acid tagging (BONCAT) Proc Natl Acad Sci USA. 2006;103:9482–9487. doi: 10.1073/pnas.0601637103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dieterich DC, Lee JJ, Link AJ, Graumann J, Tirrell DA. Schuman EM. Labeling, detection and identification of newly synthesized proteomes with bioorthogonal non-canonical amino-acid tagging. Nat Protoc. 2007;2:532–540. doi: 10.1038/nprot.2007.52. [DOI] [PubMed] [Google Scholar]
- Dieterich DC, Hodas JJ, Gouzer G, Shadrin IY, Ngo JT, Triller A, et al. In situ visualization and dynamics of newly synthesized proteins in rat hippocampal neurons. Nat Neurosci. 2010;13:897–905. doi: 10.1038/nn.2580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eichelbaum K, Winter M, Berriel Diaz M, Herzig S. Krijgsveld J. Selective enrichment of newly synthesized proteins for quantitative secretome analysis. Nat Biotechnol. 2012;30:984–990. doi: 10.1038/nbt.2356. [DOI] [PubMed] [Google Scholar]
- Fahnert B, Lilie H. Neubauer P. Inclusion bodies: formation and utilisation. In: Enfors S-O, editor; Advances in Biochemical Engineering. Heidelberg, Germany: Springer; 2004. pp. 93–142. [DOI] [PubMed] [Google Scholar]
- Fekner T, Li X, Lee MM. Chan MK. A pyrrolysine analogue for protein click chemistry. Angew Chem Int Ed Engl. 2009;48:1633–1635. doi: 10.1002/anie.200805420. [DOI] [PubMed] [Google Scholar]
- Fiencke C. Bock E. Genera-specific immunofluorescence labeling of ammonia oxidizers with polyclonal antibodies recognizing both subunits of the ammonia monooxygenase. Microb Ecol. 2004;47:374–384. doi: 10.1007/s00248-003-1009-8. [DOI] [PubMed] [Google Scholar]
- Finzi-Hart JA, Pett-Ridge J, Weber PK, Popa R, Fallon SJ, Gunderson T, et al. Fixation and fate of C and N in the cyanobacterium Trichodesmium using nanometer-scale secondary ion mass spectrometry. Proc Natl Acad Sci U S A. 2009;106:6345–6350. doi: 10.1073/pnas.0810547106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foster RA, Subramaniam A. Zehr JP. Distribution and activity of diazotrophs in the Eastern Equatorial Atlantic. Environ Microbiol. 2009;11:741–750. doi: 10.1111/j.1462-2920.2008.01796.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffin RJ. The medicinal chemistry of the azido group. Prog Med Chem. 1994;31:121–232. doi: 10.1016/s0079-6468(08)70020-1. [DOI] [PubMed] [Google Scholar]
- Haider S, Wagner M, Schmid MC, Sixt BS, Christian JG, Hacker G, et al. Raman microspectroscopy reveals long-term extracellular activity of Chlamydiae. Mol Microbiol. 2010;77:687–700. doi: 10.1111/j.1365-2958.2010.07241.x. [DOI] [PubMed] [Google Scholar]
- Hao B, Gong W, Ferguson TK, James CM, Krzycki JA. Chan MK. A new UAG-encoded residue in the structure of a methanogen methyltransferase. Science. 2002;296:1462–1466. doi: 10.1126/science.1069556. [DOI] [PubMed] [Google Scholar]
- Herndl GJ, Reinthaler T, Teira E, van Aken H, Veth C, Pernthaler A. Pernthaler J. Contribution of Archaea to total prokaryotic production in the deep Atlantic Ocean. Appl Environ Microbiol. 2005;71:2303–2309. doi: 10.1128/AEM.71.5.2303-2309.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hesselsoe M, Nielsen JL, Roslev P. Nielsen PH. Isotope labeling and microautoradiography of active heterotrophic bacteria on the basis of assimilation of 14CO2. Appl Environ Microbiol. 2005;71:646–655. doi: 10.1128/AEM.71.2.646-655.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hinz FI, Dieterich DC, Tirrell DA. Schuman EM. Non-canonical amino acid labeling in vivo to visualize and affinity purify newly synthesized proteins in larval zebrafish. ACS Chem Neurosci. 2012;3:40–49. doi: 10.1021/cn2000876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong V, Presolski SI, Ma C. Finn MG. Analysis and optimization of copper-catalyzed azide-alkyne cycloaddition for bioconjugation. Angew Chem Int Ed Engl. 2009;48:9879–9883. doi: 10.1002/anie.200905087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong V, Steinmetz NF, Manchester M. Finn MG. Labeling live cells by copper-catalyzed alkyne – azide click chemistry. Bioconjug Chem. 2010;21:1912–1916. doi: 10.1021/bc100272z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howden AJ, Geoghegan V, Katsch K, Efstathiou G, Bhushan B, Boutureira O, et al. QuaNCAT: quantitating proteome dynamics in primary cells. Nat Methods. 2013;10:343–346. doi: 10.1038/nmeth.2401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang WE, Griffiths RI, Thompson IP, Bailey MJ. Whiteley AS. Raman microscopic analysis of single microbial cells. Anal Chem. 2004;76:4452–4458. doi: 10.1021/ac049753k. [DOI] [PubMed] [Google Scholar]
- Huang WE, Stoecker K, Griffiths R, Newbold L, Daims H, Whiteley AS. Wagner M. Raman-FISH: combining stable-isotope Raman spectroscopy and fluorescence in situ hybridization for the single cell analysis of identity and function. Environ Microbiol. 2007;9:1878–1889. doi: 10.1111/j.1462-2920.2007.01352.x. [DOI] [PubMed] [Google Scholar]
- Huisgen R. 1,3-dipolar cycloadditions: past and future. Angew Chem Int Ed Engl. 1963;2:565–598. [Google Scholar]
- Jao CY. Salic A. Exploring RNA transcription and turnover in vivo by using click chemistry. Proc Natl Acad Sci USA. 2008;105:15779–15784. doi: 10.1073/pnas.0808480105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jehmlich N, Schmidt F, Taubert M, Seifert J, Bastida F, von Bergen M, et al. Protein-based stable isotope probing. Nat Protoc. 2010;5:1957–1966. doi: 10.1038/nprot.2010.166. [DOI] [PubMed] [Google Scholar]
- Jewett JC. Bertozzi CR. Cu-free click cycloaddition reactions in chemical biology. Chem Soc Rev. 2010;39:1272–1279. doi: 10.1039/b901970g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalli A, Smith GT, Sweredoski MJ. Hess S. Evaluation and optimization of mass spectrometric settings during data-dependent acquisition mode: focus on LTQ-Orbitrap mass analyzers. J Proteome Res. 2013;12:3071–3086. doi: 10.1021/pr3011588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalyuzhnaya MG, Lidstrom ME. Chistoserdova L. Real-time detection of actively metabolizing microbes by redox sensing as applied to methylotroph populations in Lake Washington. ISME J. 2008;2:696–706. doi: 10.1038/ismej.2008.32. [DOI] [PubMed] [Google Scholar]
- Kawakami S, Kubota K, Imachi H, Yamaguchi T, Harada H. Ohashi A. Detection of single copy genes by two-pass tyramide signal amplification fluorescence in situ hybridization (Two-Pass TSA-FISH) with single oligonucleotide probes. Microbes Environ. 2010;25:15–21. doi: 10.1264/jsme2.me09180. [DOI] [PubMed] [Google Scholar]
- Kho Y, Kim SC, Jiang C, Barma D, Kwon SW, Cheng J, et al. A tagging-via-substrate technology for detection and proteomics of farnesylated proteins. Proc Natl Acad Sci USA. 2004;101:12479–12484. doi: 10.1073/pnas.0403413101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiick KL, Saxon E, Tirrell DA. Bertozzi CR. Incorporation of azides into recombinant proteins for chemoselective modification by the Staudinger ligation. Proc Natl Acad Sci USA. 2002;99:19–24. doi: 10.1073/pnas.012583299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knittel K. Boetius A. Anaerobic oxidation of methane: progress with an unknown process. Annu Rev Microbiol. 2009;63:311–334. doi: 10.1146/annurev.micro.61.080706.093130. [DOI] [PubMed] [Google Scholar]
- Konopka MC, Strovas TJ, Ojala DS, Chistoserdova L, Lidstrom ME. Kalyuzhnaya MG. Respiration response imaging for real-time detection of microbial function at the single-cell level. Appl Environ Microbiol. 2011;77:67–72. doi: 10.1128/AEM.01166-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lanoil BD. Giovannoni SJ. Identification of bacterial cells by chromosomal painting. Appl Environ Microbiol. 1997;63:1118–1123. doi: 10.1128/aem.63.3.1118-1123.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim RK. Lin Q. Bioorthogonal chemistry: recent progress and future directions. Chem Commun (Camb) 2010;46:1589–1600. doi: 10.1039/b925931g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin S, Henze S, Lundgren P, Bergman B. Carpenter EJ. Whole-cell immunolocalization of nitrogenase in marine diazotrophic cyanobacteria, Trichodesmium spp. Appl Environ Microbiol. 1998;64:3052–3058. doi: 10.1128/aem.64.8.3052-3058.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Link AJ, Vink MK. Tirrell DA. Preparation of the functionalizable methionine surrogate azidohomoalanine via copper-catalyzed diazo transfer. Nat Protoc. 2007;2:1879–1883. doi: 10.1038/nprot.2007.268. [DOI] [PubMed] [Google Scholar]
- Losekann T, Knittel K, Nadalig T, Fuchs B, Niemann H, Boetius A. Amann R. Diversity and abundance of aerobic and anaerobic methane oxidizers at the Haakon Mosby Mud Volcano, Barents Sea. Appl Environ Microbiol. 2007;73:3348–3362. doi: 10.1128/AEM.00016-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manz W, Amann R, Ludwig W, Wagner M. Schleifer KH. Phylogenetic oligodeoxynucleotide probes for the major subclasses of proteobacteria – problems and solutions. Syst Appl Microbiol. 1992;15:593–600. [Google Scholar]
- Moraru C, Lam P, Fuchs BM, Kuypers MM. Amann R. GeneFISH – an in situ technique for linking gene presence and cell identity in environmental microorganisms. Environ Microbiol. 2010;12:3057–3073. doi: 10.1111/j.1462-2920.2010.02281.x. [DOI] [PubMed] [Google Scholar]
- Morgenroth E, Obermayer A, Arnold E, Brühl A, Wagner M. Wilderer PA. Effect of long-term idle periods on the performance of sequencing batch reactors. Wat Sci Technol. 2000;41:105–113. [Google Scholar]
- Morono Y, Terada T, Nishizawa M, Ito M, Hillion F, Takahata N, et al. Carbon and nitrogen assimilation in deep subseafloor microbial cells. Proc Natl Acad Sci USA. 2011;108:18295–18300. doi: 10.1073/pnas.1107763108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neef A, Witzenberger R. Kampfer P. Detection of Sphingomonads and in situ identification in activated sludge using 16S rRNA-targeted oligonucleotide probes. J Ind Microbiol Biotechnol. 1999;23:261–267. doi: 10.1038/sj.jim.2900768. [DOI] [PubMed] [Google Scholar]
- Neef AB. Schultz C. Selective fluorescence labeling of lipids in living cells. Angew Chem Int Ed Engl. 2009;48:1498–1500. doi: 10.1002/anie.200805507. [DOI] [PubMed] [Google Scholar]
- Nessen MA, Kramer G, Back J, Baskin JM, Smeenk LE, de Koning LJ, et al. Selective enrichment of azide-containing peptides from complex mixtures. J Proteome Res. 2009;8:3702–3711. doi: 10.1021/pr900257z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ngo JT. Tirrell DA. Noncanonical amino acids in the interrogation of cellular protein synthesis. Acc Chem Res. 2011;44:677–685. doi: 10.1021/ar200144y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ngo JT, Champion JA, Mahdavi A, Tanrikulu IC, Beatty KE, Connor RE, et al. Cell-selective metabolic labeling of proteins. Nat Chem Biol. 2009;5:715–717. doi: 10.1038/nchembio.200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Odaa Y, Slagmana S, Meijerb WG, Forneya LJ. Gottschala JC. Influence of growth rate and starvation on fluorescent in situ hybridization of Rhodopseudomonas palustris. FEMS Microbiol Ecol. 2000;32:205–213. doi: 10.1111/j.1574-6941.2000.tb00713.x. [DOI] [PubMed] [Google Scholar]
- Orman MA. Brynildsen MP. Dormancy is not necessary or sufficient for bacterial persistence. Antimicrob Agents Chemother. 2013;57:3230–3239. doi: 10.1128/AAC.00243-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orphan VJ, Turk KA, Green AM. House CH. Patterns of 15N assimilation and growth of methanotrophic ANME-2 archaea and sulfate-reducing bacteria within structured syntrophic consortia revealed by FISH-SIMS. Environ Microbiol. 2009;11:1777–1791. doi: 10.1111/j.1462-2920.2009.01903.x. [DOI] [PubMed] [Google Scholar]
- Ouellette SP, Dorsey FC, Moshiach S, Cleveland JL. Carabeo RA. Chlamydia species-dependent differences in the growth requirement for lysosomes. PLoS ONE. 2011;6:e16783. doi: 10.1371/journal.pone.0016783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ouverney CC. Fuhrman JA. Marine planktonic archaea take up amino acids. Appl Environ Microbiol. 2000;66:4829–4833. doi: 10.1128/aem.66.11.4829-4833.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pernice M, Meibom A, Van Den Heuvel A, Kopp C, Domart-Coulon I, Hoegh-Guldberg O. Dove S. A single-cell view of ammonium assimilation in coral-dinoflagellate symbiosis. ISME J. 2012;6:1314–1324. doi: 10.1038/ismej.2011.196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pernthaler A. Amann R. Simultaneous fluorescence in situ hybridization of mRNA and rRNA in environmental bacteria. Appl Environ Microbiol. 2004;70:5426–5433. doi: 10.1128/AEM.70.9.5426-5433.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roslev P, Larsen MB, Jorgensen D. Hesselsoe M. Use of heterotrophic CO2 assimilation as a measure of metabolic activity in planktonic and sessile bacteria. J Microbiol Methods. 2004;59:381–393. doi: 10.1016/j.mimet.2004.08.002. [DOI] [PubMed] [Google Scholar]
- Rostovtsev VV, Green LG, Fokin VV. Sharpless KB. A stepwise huisgen cycloaddition process: copper(I)-catalyzed regioselective ‘ligation’ of azides and terminal alkynes. Angew Chem Int Ed Engl. 2002;41:2596–2599. doi: 10.1002/1521-3773(20020715)41:14<2596::AID-ANIE2596>3.0.CO;2-4. [DOI] [PubMed] [Google Scholar]
- Salic A. Mitchison TJ. A chemical method for fast and sensitive detection of DNA synthesis in vivo. Proc Natl Acad Sci USA. 2008;105:2415–2420. doi: 10.1073/pnas.0712168105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saxon E, Luchansky SJ, Hang HC, Yu C, Lee SC. Bertozzi CR. Investigating cellular metabolism of synthetic azidosugars with the Staudinger ligation. J Am Chem Soc. 2002;124:14893–14902. doi: 10.1021/ja027748x. [DOI] [PubMed] [Google Scholar]
- Schmid M, Schmitz-Esser S, Jetten M. Wagner M. 16S-23S rDNA intergenic spacer and 23S rDNA of anaerobic ammonium-oxidizing bacteria: implications for phylogeny and in situ detection. Environ Microbiol. 2001;3:450–459. doi: 10.1046/j.1462-2920.2001.00211.x. [DOI] [PubMed] [Google Scholar]
- Schönhuber W, Fuchs B, Juretschko S. Amann R. Improved sensitivity of whole-cell hybridization by the combination of horseradish peroxidase-labeled oligonucleotides and tyramide signal amplification. Appl Environ Microbiol. 1997;63:3268–3273. doi: 10.1128/aem.63.8.3268-3273.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi Shun AL. Tykwinski RR. Synthesis of naturally occurring polyynes. Angew Chem Int Ed Engl. 2006;45:1034–1057. doi: 10.1002/anie.200502071. [DOI] [PubMed] [Google Scholar]
- Siegrist MS, Whiteside S, Jewett JC, Aditham A, Cava F. Bertozzi CR. (D)-amino acid chemical reporters reveal peptidoglycan dynamics of an intracellular pathogen. ACS Chem Biol. 2013;8:500–505. doi: 10.1021/cb3004995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sintes E. Herndl GJ. Quantifying substrate uptake by individual cells of marine bacterioplankton by catalyzed reporter deposition fluorescence in situ hybridization combined with microautoradiography. Appl Environ Microbiol. 2006;72:7022–7028. doi: 10.1128/AEM.00763-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sletten EM. Bertozzi CR. Bioorthogonal chemistry: fishing for selectivity in a sea of functionality. Angew Chem Int Ed Engl. 2009;48:6974–6998. doi: 10.1002/anie.200900942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smolina I, Lee C. Frank-Kamenetskii M. Detection of low-copy-number genomic DNA sequences in individual bacterial cells by using peptide nucleic acid-assisted rolling-circle amplification and fluorescence in situ hybridization. Appl Environ Microbiol. 2007;73:2324–2328. doi: 10.1128/AEM.02038-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stahl DA. Amann R. Development and application of nucleic acid probes. In: Goodfellow M, editor; Stackebrandt EG, Goodfellow M, editors. Nucleic acid Techniques in Bacterial Systematics. Chichester, NY, USA: John Wiley & Sons Ltd; 1991. pp. 205–248. [Google Scholar]
- Stoecker K, Dorninger C, Daims H. Wagner M. Double labeling of oligonucleotide probes for fluorescence in situ hybridization (DOPE-FISH) improves signal intensity and increases rRNA accessibility. Appl Environ Microbiol. 2010;76:922–926. doi: 10.1128/AEM.02456-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szychowski J, Mahdavi A, Hodas JJ, Bagert JD, Ngo JT, Landgraf P, et al. Cleavable biotin probes for labeling of biomolecules via azide-alkyne cycloaddition. J Am Chem Soc. 2010;132:18351–18360. doi: 10.1021/ja1083909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taniguchi Y, Choi PJ, Li GW, Chen H, Babu M, Hearn J, et al. Quantifying E. coli proteome and transcriptome with single-molecule sensitivity in single cells. Science. 2010;329:533–538. doi: 10.1126/science.1188308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tavormina PL, Hatzenpichler R, McGlynn S, Chadwick G, Dawson K, Connon S. Orphan VJ. Methyloprofundus sedimenti gen. nov., sp. nov., an obligate methanotroph from ocean sediment belonging to the Deep Sea 1 clade of marine methanotrophs. Int J Syst Evol Microbiol. doi: 10.1099/ijs.0.062927-0. (in review) [DOI] [PubMed] [Google Scholar]
- Teira E, Van Aken H, Veth C. Herndl GJ. Archaeal uptake of enantiomeric amino acids in the meso- and bathypelagic waters of the North Atlantic. Limnol Oceanogr. 2006;51:60–69. [Google Scholar]
- Torne CW, Christensen C. Meldal M. Peptidotriazoles on solid phase: [1,2,3]-friazoles by regiospecific copper(I)-catalyzed 1,3-dipolar cycloadditions of terminal alkynes to azides. J Org Chem. 2002;67:3057–3064. doi: 10.1021/jo011148j. [DOI] [PubMed] [Google Scholar]
- Trotsenko YA. Murrell JC. Metabolic aspects of aerobic obligate methanotrophy. Adv Appl Microbiol. 2008;63:183–229. doi: 10.1016/S0065-2164(07)00005-6. [DOI] [PubMed] [Google Scholar]
- Wagner M, Rath G, Amann R, Koops HP. Schleifer KH. In situ identification of ammonia-oxidizing bacteria. Syst Appl Microbiol. 1995;18:251–264. [Google Scholar]
- Wallner G, Amann R. Beisker W. Optimizing fluorescent in situ hybridization with rRNA-targeted oligonucleotide probes for flow cytometric identification of microorganisms. Cytometry. 1993;14:136–143. doi: 10.1002/cyto.990140205. [DOI] [PubMed] [Google Scholar]
- Wrede C, Krukenberg V, Dreier A, Reitner J, Heller C. Hoppert M. Detection of metabolic key enzymes of methane turnover processes in cold seep microbial miofilms. Geomicrobiol J. 2013;30:214–227. [Google Scholar]
- Yilmaz S, Haroon MF, Rabkin BA, Tyson GW. Hugenholtz P. Fixation-free fluorescence in situ hybridization for targeted enrichment of microbial populations. ISME J. 2010;4:1352–1356. doi: 10.1038/ismej.2010.73. [DOI] [PubMed] [Google Scholar]
- Yoon BC, Jung H, Dwivedy A, O'Hare CM, Zivraj KH. Holt CE. Local translation of extranuclear lamin B promotes axon maintenance. Cell. 2012;148:752–764. doi: 10.1016/j.cell.2011.11.064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y. Gladyshev VN. High content of proteins containing 21st and 22nd amino acids, selenocysteine and pyrrolysine, in a symbiotic deltaproteobacterium of gutless worm Olavius algarvensis. Nucleic Acids Res. 2007;35:4952–4963. doi: 10.1093/nar/gkm514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zwirglmaier K, Ludwig W. Schleifer KH. Recognition of individual genes in a single bacterial cell by fluorescence in situ hybridization – RING-FISH. Mol Microbiol. 2004;51:89–96. doi: 10.1046/j.1365-2958.2003.03834.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Comparative BONCAT of newly synthesized proteins in E. coli and structures of dyes used for (A-D) Cu(I)-catalyzed and (E-F) strain-promoted click chemistry. Exposure times for cells incubated in the presence or absence of AHA are identical for each dye and were chosen to yield approximately comparable signal intensities for AHA-containing cells. Gain and offset settings of the detector were kept constant for all conditions. Note that different dyes give highly divergent signal to noise ratios when washing conditions are identical. Because of these background problems the Eosin-dye, initially believed to be a potential alternative for detection of AHA incorporation via nanoSIMS, could not be used in any of our experiments. The scale bar applies to all photos and equals 10 μm. Chemical structures were obtained from the respective company websites (see main text). Abbreviations: DBCO, Dibenzocyclooctyne; PEG, Polyethylene glycol.
Visualization of newly made proteins in living (liveBONCAT; panels A & B) and chemically fixed (C, D) AHA-labelled E. coli. Exposure times were identical for all images. Note that E. coli cells in panels (A) and (B) exhibit atypical cell morphology and that substantially lower numbers of cells are fluorescently labelled despite cells from the same culture are analyzed. The scale bar applies to all photos and equals 10 μm.
Distributions and average contents of methionine in candidate proteins encoded in the genomes of five microbes analyzed via BONCAT. Note that the genome of Methylococcaceae sp. WF1 has not been closed yet.
BONCAT-based visualization of newly synthesized proteins combined with 16S rRNA-targeted FISH in an artificial mix of microbial cultures. A methanotrophic enrichment culture that had been incubated in the presence of AHA was mixed with normally grown cultures of E. coli, P. denitrificans, Methanosarcina sp., and a propane-oxidizing enrichment culture. Consecutively, BONCAT was performed using Oregon Green 488 alkyne (green), FISH was performed using Cy3 (red) and Cy5 (turquoise) labeled oligonucleotide probes, and biomass was stained using DAPI (blue). (A) Localization of probe MetI-444 (red), specific for a group of gamma proteobacterial methanotrophs, reveals that coccoid WF1 cells from the methanotrophic enrichment culture incorporated AHA into their proteins. WF1 cells are not detected by probe Gam42a (turquoise; used with unlabeled competitor Bet42a), which is specific for many but not all gamma proteobacteria, due to a discriminating mismatch in their 23S rRNA. (B) FISH controls taken at identical exposure times for the respective channels (Cy3-labelled NonEUB338; auto-fluorescence in Cy5 channel). For BONCAT controls of the individual cultures see Fig. 2. For probe details refer to main text. The scale bars equal 10 μm.
Complimentary images to the comparative BONCAT analyses of a methanotrophic enrichment culture using different detection settings. Please note that for samples incubated for > 9 h in the presence of AHA, fluorescent signal of WF1 cells saturated the sensor (see Fig. 6). These settings were chosen in order to visualize the difference in fluorescence intensity after 4 h of incubation between WF1 cells grown in the absence and presence of methane. For images shown in this panel, exposition settings were chosen based on the much stronger signal intensities observed for later samples. All photos in this figure were taken at identical microscopic settings. Scale bar equals 10 μm and applies to all images.
BONCAT and FISH controls for microbiome and environmental samples. (A) Results for samples incubated in the absence of AHA (no BONCAT signal). (B) Tests of the specificity of the FISH protocol using a Cy3-labelled NonEUB338 probe (no FISH signal). Camera settings to record BONCAT and FISH signals were identical to those used to image samples shown in Fig. 5B–D respectively. All scale bars equal 10 μm.
Reduction of AHA in the presence of high concentrations of sulfide and high pH. (A) Results for abiotic reactions of 1 mM AHA in the absence or presence (H2S plus HS- equals 1, 2, or 10 mM) of sulfide at pH 8.0 (HPO4- buffered) and pH 9.0 (HCO3- buffered). At pH 8 and 9, AHA is slowly reduced with a reduction rate proportional to the sulfide concentration. At pH 7, the pH of the sediment sample studied here (sulfide concentration ≤ 60 μM), sulfide has no detectable effect on AHA for up to at least 8 days (data not shown). The stability of AHA itself is not influenced by pH, and sulfide has no effect on AHA at pH ≤ 7. AHA and its reduction product with sulfide, L-2,4-diaminobutyric acid (B), were analyzed via NMR spectroscopy.
Visualization of clusters of protein-synthesizing gammaproteobacterial freshwater bacteria (identified via FISH-probe Gam42a (red), which was used with its competitor). The scale bar equals 20 μm.
(A) Proteins identified via MS sequencing of gel-extracted proteins from methanotrophic enrichment culture WF1, ranked by abundance. (B) Proteins identified via MS sequencing of gel-extracted proteins from a heat-stressed E. coli K12 culture, ranked by abundance.
liveBONCAT: Attempting to visualize protein synthesis in living cells. Strain-promoted click labeling of living microbes.