

Abstract
Recent genetic studies suggest that dysfunction of ion channels and transporters may contribute to migraine pathophysiology. A migraine-associated frameshift mutation in the TWIK-related spinal cord K+ (TRESK) channel results in nonfunctional channels. Moreover, mutant TRESK subunits exert a dominant-negative effect on whole cell TRESK currents and result in hyperexcitability of small-diameter trigeminal ganglion (TG) neurons, suggesting that mutant TRESK may increase the gain of the neuronal circuit underlying migraine headache. However, the nonmigraine-associated TRESK C110R variant exhibits the same effect on TRESK currents as the mutant subunits in Xenopus oocytes, suggesting that dysfunction of TRESK is not sufficient to cause migraine. Here, we confirmed that the C110R variant formed nonfunctional channels and exerted a dominant-negative effect on TRESK currents in HEK293T cells, similar to the migraine-associated mutant TRESK. To compare the functional consequences of TRESK mutations/variants in a more physiological setting, we expressed the mutant TRESK and the C110R variant in cultured mouse TG neurons and investigated their effects on background K+ currents and neuronal excitability. Both mutant TRESK and the C110R variant reduced the endogenous TRESK currents in TG neurons, but the effect of the C110R variant was significantly smaller. Importantly, only TG neurons expressing mutant TRESK subunits, but not those expressing the C110R variant, exhibited a significant increase in excitability. Thus only the migraine-associated TRESK mutation, but not the C110R variant, reduces the endogenous TRESK currents to a degree that affects TG excitability. Our results support a potential causal relationship between the frameshift TRESK mutation and migraine susceptibility.
Keywords: migraine, TRESK K+ channel, background K+ current, trigeminal ganglion, neuronal excitability
migraine is one of the most common neurovascular disorders with a strong genetic component (Eising et al. 2013; Victor et al. 2010). One of the major symptoms of migraine, the recurring headache, is highly debilitating, poorly understood, and difficult to treat (Burstein and Jakubowski 2005; Goadsby et al. 2009; Pietrobon and Moskowitz 2013). Multiple mutations of ion channels and transporters have been associated with familial hemiplegic migraine (de Vries et al. 2009). Recent genetic studies have also implicated ion channel dysfunction in common forms of migraine (Eising et al. 2013). These discoveries offer a gateway to understanding the mechanisms underlying migraine headache.
The TWIK-related spinal cord K+ (TRESK) channel is encoded by the KCNK18 gene and is the only Ca2+-activated two-pore-domain K+ (K2P) channel (Enyedi et al. 2012; Kang et al. 2004; Sano et al. 2003). TRESK channels are abundantly expressed in almost all primary afferent neurons in trigeminal ganglion (TG) and dorsal root ganglion (Bautista et al. 2008; Dobler et al. 2007; Lafreniere et al. 2010; Sano et al. 2003; Yoo et al. 2009). Previous studies indicate that TRESK is one of the major background K+ channels in dorsal root ganglion neurons and controls neuronal excitability under both normal and chronic pain conditions (Dobler et al. 2007; Kang and Kim 2006; Liu et al. 2013; Marsh et al. 2012; Plant 2012; Tulleuda et al. 2011). A frameshift mutation in the human KCNK18 gene that leads to the truncation of TRESK protein has been linked to migraine with aura in a large pedigree (Lafreniere et al. 2010). The mutant (MT) TRESK subunits do not form functional channels. Moreover, they exert a dominant-negative effect on the currents through wild-type (WT) TRESK channels and result in hyperexcitability of small-diameter TG neurons (Lafreniere et al. 2010; Liu et al. 2013), suggesting that the frameshift mutation may increase the gain of the neuronal circuit underlying migraine headache. In support of a genotype-phenotype correlation between the TRESK function and migraine susceptibility, the missense TRESK variant A34V identified in a migraine patient but not in the control cohort exhibits the same effect on whole cell TRESK current as the frameshift mutation in Xenopus oocytes (Andres-Enguix et al. 2012). The TRESK variants R10G, S231P, and A233V have been found in both migraine and control cohorts. None of them affect the magnitude of the TRESK currents in Xenopus oocytes (Andres-Enguix et al. 2012).
An outlier is the TRESK C110R (CR) variant. It is present in both migraineurs and unaffected individuals. However, the CR variant forms a nonfunctional channel per se in Xenopus oocytes and exhibits the same dominant-negative effect on WT TRESK currents as the frameshift mutation (Andres-Enguix et al. 2012). This calls into question a direct causal relationship between TRESK dysfunction and migraine phenotype (Andres-Enguix et al. 2012; Eising et al. 2013; Lafreniere et al. 2010). On the other hand, previous works have illustrated the importance of studying migraine-associated gene mutations in neurons involved in migraine pathophysiology (Fioretti et al. 2011; Hullugundi et al. 2014; Liu et al. 2013; Tao et al. 2012; Tottene et al. 2009; van Den Maagdenberg et al. 2004). While the Xenopus oocyte system is a powerful tool for ion channel research, it may have limitations in predicting the genotype-phenotype correlation between TRESK mutations/variants and migraine susceptibility. We reason that a good starting point is to use cultured TG neurons as a platform, as it is well established that the activation and sensitization of TG neurons that project to meninges and cerebral blood vessels are crucial steps in the onset of headache attacks (Burstein 2001; Goadsby et al. 2009; Pietrobon and Moskowitz 2013; Strassman et al. 1996).
In the present study, we confirmed that the TRESK CR variant did not form functional channels in human embryonic kidney 293T (HEK293T) cells. This likely resulted from impaired channel gating, as the CR variant did not affect the expression and/or the plasma membrane trafficking of TRESK subunits. When coexpressed with WT mouse TRESK subunits, the CR variant exhibited a dominant-negative effect on the whole cell TRESK currents. Furthermore, we expressed the MT TRESK subunit and the CR variant in cultured mouse TG neurons and investigated their effects on background K+ currents and neuronal excitability. We found that both MT and CR subunits reduced the endogenous TRESK currents in TG neurons, but the effect of the CR variant was significantly smaller. Importantly, overexpression of the CR variant did not lead to hyperexcitability of TG neurons as the MT TRESK subunits (Liu et al. 2013). In one TG subpopulation, overexpression of the CR variant even increased the current threshold to elicit action potential (AP). Taken together, our results indicate a differential effect of the MT TRESK and the CR variant on TG excitability, supporting a potential causal relationship between the frameshift TRESK mutation and migraine susceptibility.
MATERIALS AND METHODS
Animals.
All procedures in this study were approved by the Animal Studies Committee at Washington University in St. Louis. The CD-1 breeders were maintained on a 12-h light/dark cycle with constant temperature (23–24°C), humidity (45–50%), as well as food and water ad libitum at the animal facility of Washington University in St. Louis.
Mouse TRESK constructs.
The migraine-associated frameshift deletion and the CR variant of human TRESK were introduced into the corresponding region of the mouse TRESK cDNA using the Stratagene QuikChange site-directed mutagenesis kit. The human TRESK CR variant contains the cysteine to arginine substitution at amino acid 110 (Andres-Enguix et al. 2012; Lafreniere et al. 2010), corresponding to the cysteine to arginine change at amino acid 121 of mouse TRESK subunit (Andres-Enguix et al. 2012). The enhanced green fluorescent protein (EGFP)- or mCherry-tagged constructs were generated by fusing TRESK CR cDNA in frame at the COOH termini of EGFP and mCherry coding region (EGFP-CR and mCherry-CR), respectively. All coding regions were downstream of the CMV promoter. All PCR-generated cDNA fragments and linker regions were completely sequenced to verify the mutation and to make sure that no additional mutations were introduced. Details of the constructs encoding EGFP- or mCherry-tagged WT and MT TRESK subunits (EGFP-WT, mCherry-WT, EGFP-MT, and mCherry-MT, respectively) were described in a previous study (Liu et al. 2013).
Cell culture, transfection, and image analysis.
HEK293T cells (ATCC) were maintained in six-well plates in DMEM with 10% FBS and were transfected with Lipofectamine 2000 (Invitrogen). One day posttransfection, cells were seeded on Matrigel-coated coverslips. Transfected cells were identified by the EGFP or mCherry fluorescence and were used 2–3 days posttransfection for patch-clamp recordings, immunostaining, or image analysis.
For each set of experiment, the EGFP and mCherry fluorescence intensity was quantified in cells that underwent a parallel experimental procedure and image analysis process. All conditions were identical, except for the DNA constructs. Fluorescent images were captured through a ×40 objective (N.A. 1.3) on a Nikon TE2000S inverted epifluorescence microscope equipped with a CoolSnap HQ2 camera (Photometrics). SimplePCI software (Hamamatsu) was used for image analysis. To measure the expression level of EGFP- or mCherry-tagged TRESK proteins, a single cell was specified as a region of interest (ROI). The intensity of EGFP or mCherry signal was determined on a pixel-by-pixel basis and was averaged for each ROI. For each image captured, the mean intensity of the cell-free regions was taken as the background level and was subtracted from the mean intensity in each ROI.
Detection of cell surface TRESK-immunoreactivity in HEK293T cells.
HEK293T cells were transfected with EGFP-WT or EGFP-CR TRESK constructs and were reseeded onto coverslips 1 day posttransfection. Two days posttransfection, cells were washed with PBS and were fixed by 4% formaldehyde for 5 min followed by PBS wash. Triton X-100 was omitted in all solutions. The coverslips were incubated in blocking buffer (PBS with 10% normal goat serum) for 1 h and were then incubated with a mouse polyclonal antibody against an extracellular domain of TRESK (1:1,000; Liu et al. 2013) in blocking buffer at 4°C overnight. Following three washes by the blocking buffer (20 min each), the coverslips were incubated with the AlexaFluor 594-conjugated goat anti-mouse secondary antibody (Invitrogen; 1:2,000) in blocking buffer for 1 h and were washed again three times in PBS. The coverslips were mounted with the Crystal Mount medium and stored at 4°C. Transfected cells were identified by the EGFP fluorescence. The fluorescent images were captured and analyzed as described above.
Protein preparation and immunodetection.
HEK293T cells were transfected with EGFP-WT, EGFP-MT, or EGFP-CR TRESK constructs and were harvested 2 days posttransfection. Cells were resuspended in Laemmli sample buffer with 10 mM dithiothreitol (Bio-Rad), Cell lysates were briefly sonicated 10 times. The insoluble material was removed by centrifugation at 20,000 g for 10 min at 4°C. The supernatants were stored at 4°C. Proteins (10 μl of each sample) were separated on 4–20% SDS-polyacrylmaide gels (SDS-PAGE; Bio-Rad). For determination of molecular weight, prestained molecular weight ladders (LamdaBio) were loaded along with protein samples. Blots were transferred to PVDF membranes (Bio-Rad) for 1 h at 350 mA. The immunodetection was performed at room temperature. The membranes were incubated in blocking buffer (Li-Cor Biosciences) for 1 h and were then incubated with a monoclonal antibody against the EGFP epitope (Clontech; 1:1,000 diluted in blocking buffer) for 1 h. Following three washes by the blocking buffer (20 min each), the membranes were incubated with the IRDye 700-conjugated anti-mouse secondary antibody (Li-Cor Biosciences; 1:20,000 diluted in blocking buffer) for 1 h and were washed again three times in PBS. Immunoblots were scanned using the Odyssey infrared imaging system (Li-Cor Biosciences).
Primary culture of neonatal mouse TG neurons, transfection, and image analysis.
TG tissues were collected from postnatal day 1 mice of either sex and were treated with 5 mg/ml trypsin for 15 min. Neurons were dissociated by triturating with fire-polished glass pipettes and were seeded on Matrigel-coated coverslips. The MEM-based culture medium contained 5% FBS, 25 ng/ml nerve growth factor, and 10 ng/ml glial cell line-derived neurotrophic factor and was replaced every 3 days. Cultured neonatal TG neurons usually grow one to three processes from soma by 2 days in vitro (DIV). Longer culture time (till 5 DIV) does not alter the number or the thickness of the processes but significantly increases the length and the branches of individual processes.
For electrophysiological experiments, TG neurons were transfected with plasmids encoding mCherry protein or mCherry-tagged TRESK subunits at 1 DIV using lipofectamine 2,000. For imaging analysis, TG neurons were transfected with plasmids encoding EGFP-tagged TRESK subunits at 1 DIV. Two days posttransfection, neurons were incubated with 10 μg/ml Alexa Fluor 594-conjugated wheat germ agglutinin (AF594-WGA; Invitrogen) for 10 min and washed with PBS for 10 min to delineate the plasma membrane (Jeng et al. 2008; Liu et al. 2013). Neurons were then fixed by 4% formaldehyde for 5 min followed by PBS wash. The coverslips were mounted with the Crystal Mount medium and stored at 4°C.
Transfected neurons were identified by the EGFP fluorescence. Both EGFP and AF594-WGA images were captured as described above. We manually traced the outer and inner boundaries of the AF594-WGA image of each neuron. The region enclosed by the outer and the inner curves represented total and cytoplasmic ROI, respectively. The region enclosed by the two curves represented the plasma membrane ROI. The area, total fluorescent intensity, and average intensity within the ROI were measured with the Simple PCI software.
Electrophysiology.
Transfected HEK293T cells were identified by the EGFP or mCherry fluorescence. Transfected neurons were identified by the mCherry fluorescence and were used between 3 and 5 DIV for patch-clamp recordings. The processes of the transfected neurons would contribute to the space-clamp error. We did not find significant differences between early (3 DIV) and late (5 DIV) recordings within individual experimental groups. Thus the space-clamp issue was not exacerbated by the prolonged culture time.
Whole cell patch-clamp recordings were performed at room temperature with a MultiClamp 700B amplifier (Molecular Devices). The recording chamber was perfused with Tyrode solution (0.5 ml/min) containing the following (in mM): 130 NaCl, 2 KCl, 2 CaCl2, 2 MgCl2, 25 HEPES, 30 glucose, pH 7.3 with NaOH, and 310 mosmol/kgH2O. The pipette solution contained the following (in mM): 130 K-gluconate, 7 KCl, 2 NaCl, 0.4 EGTA, 1 MgCl2, 4 ATP-Mg, 0.3 GTP-Na, 10 HEPES, 10 Tris-phosphocreatine, 10 U/ml creatine phosphokinase, pH 7.3 with KOH, and 290 mosmol/kgH2O. Recording pipettes had <4.5 MΩ resistance. pClamp 10 (Molecular Devices) was used to acquire and analyze data. Cell capacitance and series resistance were constantly monitored throughout the recording. Data were analyzed with the Clampfit (Molecular Devices) and Origin (OriginLab) software.
Voltage-clamp experiments.
Series resistance (<15 MΩ, average 11 ± 1 MΩ) was compensated by 80%. Current traces were not leak subtracted. Signals were filtered at 2 kHz and digitized at 20 kHz. To measure the current-voltage relationships (I–V curves) of TRESK K+ channels, HEK293T cells were held at −60 mV. Command steps from −100 to +100 mV (10-mV increments) were applied for 500 ms, and then the cell was repolarized back to −60 mV. For each cell, the peak current was normalized by the membrane capacitance (a measure of cell surface area) to reflect current density.
We also recorded background K+ currents from transfected small-diameter (15–25 μm) TG neurons between 3 and 5 DIV. The extracellular solution contained 1 μM tetrodotoxin (TTX) to inhibit TTX-sensitive Na+ currents (Bautista et al. 2008; Liu et al. 2013; Tulleuda et al. 2011). Neurons were held at −60 mV and were depolarized to −25 mV for 150 ms, and then the potential was ramped to −135 mV at 0.37 mV/ms every 10 s (Dobler et al. 2007; Liu et al. 2013; Tulleuda et al. 2011). We measured the outward currents at the end of the −25-mV depolarizing step. This minimized the transient voltage-gated K+ currents (Dobler et al. 2007). The fast TTX-resistant Na+ currents were also completely inactivated at the end of 150-ms depolarization (Liu et al. 2013). Depolarization to −25 mV only evokes very small high-voltage-activated Ca2+ currents, most of which are inactivated at the end of 150-ms depolarization (Tao et al. 2012). At −60-mV holding potential, the majority of low-voltage-activated T-type Ca2+ channels are inactivated (Perez-Reyes 2003) and thus do not contribute to the currents evoked by −25-mV depolarization. To further dissect background K+ currents through TRESK channels, we bath-applied 30 μM lamotrigine (Sigma) while evoking whole cell currents using this pulse protocol (Kang et al. 2008; Liu et al. 2013; Tulleuda et al. 2011).
Current-clamp experiments.
Series resistance (<15 MΩ) was not compensated. Signals were filtered at 10 kHz and digitized at 100 kHz. After whole cell access was established, membrane capacitance was determined with amplifier circuitry. The amplifier was then switched to current-clamp mode to measure resting membrane potential (Vrest). The input resistant (Rin) was calculated by measuring the membrane potential change in response to a 20-pA hyperpolarizing current injection from Vrest. Neurons were excluded from analysis if the Vrest was higher than −40 mV or Rin was smaller than 200 MΩ.
To test neuronal excitability, neurons were held at Vrest and were injected with 1-s depolarizing currents in 25-pA incremental steps. The rheobase was defined as the minimum amount of current required to elicit at least 1 AP. The first AP elicited using this paradigm was used to measure AP threshold (the membrane potential at which dV/dt exceeds 10 V/s), amplitude, and half-width. The amplitude of afterhyperpolarization (AHP) was measured from the single AP elicited by injecting a 1-ms depolarizing current in 200-pA incremental steps from the Vrest.
At the end of each electrophysiological recording, neurons were incubated with 3 μg/ml FITC-conjugated isolectin B4 (IB4; Sigma) for 5 min. The FITC fluorescence on soma membrane was detected after 10-min perfusion to wash off unbound IB4. The recording pipette remained attached to the neurons during IB4 staining and washing. The Vrest, Rin, capacitance, series resistance, and leak currents were not significantly altered after IB4 staining.
Statistical analysis.
All data are reported as means ± SE. The normality of each data set was assessed by χ2-test. Statistical significance was assessed by two-tailed t-test, one-way ANOVA with post hoc Bonferroni test or two-way repeated-measures (RM) ANOVA with post hoc Bonferroni test where appropriate. Differences with P < 0.05 were considered to be statistically significant.
RESULTS
Dominant-negative effect of the TRESK CR variant on whole cell K+ currents in HEK293T cells.
Our previous study shows that the migraine-associated frameshift mutation has a similar dominant-negative effect on whole cell currents through human and mouse TRESK channels (Liu et al. 2013). Here, we introduced the human TRESK CR variant into the corresponding region of the mouse TRESK cDNA and tested whether the variant has a similar effect on human and mouse TRESK channels. The variant is localized in the first pore-forming loop, a region adjacent to the selectivity filter and is also implicated in K2P channel gating (Fig. 1A) (Brohawn et al. 2012; Mathie et al. 2010; Miller and Long 2012; Piechotta et al. 2011). We tagged the WT, MT, and CR TRESK subunits with EGFP or mCherry at the NH2 terminus so as to study the effects of the CR variant on the biophysical properties as well as the expression level of TRESK channels. Previous studies indicate that the NH2-terminal EGFP or mCherry tag does not affect the properties of WT and MT TRESK channels (Guo and Cao 2014; Liu et al. 2013).
Fig. 1.

Dominant-negative effect of the TWIK-related spinal cord K+ (TRESK) C110R (CR) variant on whole cell K+ currents in HEK293T cells. A: topology of the mouse TRESK subunit. The locations of the frameshift mutation and the CR variant are labeled by × and ●, respectively. N, NH2 terminal; C, COOH terminal. B: characterization of enhanced green fluorescent protein (EGFP)-tagged wild-type (WT), CR, and mutant (MT) TRESK subunits in HEK293T cells by Western immunoblotting. The plasmid used in each transfection is indicated above. Ten microliters of lysate were loaded in each lane and were detected by an antibody against the EGFP tag. Lysate from untransfected cells is included as a control (left lane). The molecular mass markers are indicated at left. The variation in signal intensity likely results from different cell density and/or transfection efficiency between samples. C: representative current records from HEK293T cells expressing mCherry protein or various types of tagged TRESK subunits as indicated above. Transfected cells were held at −60 mV and were subject to 500-ms voltage steps from −100 to +100 mV (10-mV increments, every 10 s) and then repolarized back to −60 mV. D and E: current-voltage relationships (I–V) curves of peak TRESK current densities in HEK293T cells expressing mCherry protein or various types of TRESK subunits (the same recording protocols as in C, n = 8–14 cells in each group). F: TRESK current densities at +60 mV (the same cells as in D; one-way ANOVA with post hoc Bonferroni test; ***P < 0.001 and ^^^P < 0.001, compared with the EGFP-WT and mCherry groups, respectively; ##P < 0.01, compared with the mCherry-CR group). G: TRESK current densities at +60 mV (the same cells as in E; one-way ANOVA with post hoc Bonferroni test; ***P < 0.001, compared with the mCherry-WT group; ##P < 0.01).
First, we expressed EGFP-tagged WT, MT, and CR TRESK subunits in HEK293T cells and prepared protein lysates in the presence of reducing agent dithiothreitol (10 mM). Using an EGFP antibody to analyze the immunoblot, we detected no signal from untransfected cells (Fig. 1B). Proteins from cells expressing EGFP-WT and EGFP-CR subunits exhibited closely migrating doublet bands near the 75-kDa molecular mass marker, consistent with the predicted molecular mass of EGFP-WT and EGFP-CR monomer (73 kDa; Fig. 1B). Lysates from cells expressing EGFP-MT subunits displayed doublet bands slightly higher than the 48-kDa marker, also corresponding to the predicted size of the EGFP-MT monomer (50 kDa; Fig. 1B). The doublet bands are likely caused by the EGFP tag (Aronson et al. 2011).
Next, we recorded whole cell K+ currents through WT and CR TRESK channels in HEK293T cells. Cells expressing mCherry proteins exhibited very small background leak current densities (21 ± 2 pA/pF at +60 mV; Fig. 1, C, D, and F). Cells expressing EGFP-WT and mCherry-WT TRESK channels showed large outwardly rectifying whole cell K+ currents, with 211 ± 19 and 200 ± 25 pA/pF current density at +60 mV (Fig. 1, C–G), respectively. The I–V curves were typical of background K+ channels (Fig. 1, D and E) (Guo and Cao 2014; Kang et al. 2004; Lafreniere et al. 2010; Liu et al. 2013; Sano et al. 2003). In contrast, cells expressing EGFP-CR and mCherry-CR TRESK channels only exhibited very small outward K+ currents (32 ± 3 and 34 ± 7 pA/pF at +60 mV, respectively), similar to those of the mCherry-expressing cells (Fig. 1, C–G). Thus the CR variant resulted in a complete loss of currents through both human and mouse TRESK channels (Andres-Enguix et al. 2012).
A TRESK channel consists of two subunits. The CR variant may form dimer with WT subunits (WT-CR) and exert a dominant-negative effect on whole cell TRESK currents. To mimic the expression of TRESK from two alleles in human cells, we cotransfected HEK293T cells with plasmids encoding EGFP-WT and mCherry-CR TRESK subunits at a 1:1 molar ratio and observed a 64% reduction of the whole cell TRESK current density from 211 ± 19 pA/pF at +60 mV in the EGFP-WT group to 76 ± 7 pA/pF in the mCherry-CR + EGFFP-WT group (P < 0.001, one-way ANOVA with post hoc Bonferroni test; Fig. 1, C, D, and F). The decrease of current density was unlikely caused by the different amount of EGFP-WT plasmids used for transfection (4 μg in EGFP-WT and 2 μg in mCherry-CR + EGFFP-WT groups), as we have previously shown that varying the amount of plasmid DNA did not affect the size of WT TRESK current density (Liu et al. 2013). The CR variant-induced TRESK current reduction reached 70% when we subtracted the mean leak current density through endogenous channels (21 ± 2 pA/pF at +60 mV; Fig. 1F) from total outward current densities in individual transfected cells. This is close to the predicted 75% current reduction assuming that the WT-CR heterodimeric channel is nonfunctional. Coexpression of EGFP-WT with mCherry-MT subunits resulted in a similar magnitude of whole cell current density reduction from 211 ± 19 pA/pF at +60 mV in the EGFP-WT group to 72 ± 9 pA/pF in the mCherry-MT + EGFP-WT groups (P < 0.001; Fig. 1, C, D, and F).
To further rule out the effects of EGFP and/or mCherry tags on channel activity, we conducted the reciprocal experiment, comparing TRESK current densities in cells expressing mCherry-WT alone with cells coexpressing mCherry-WT and EGFP-CR TRESK subunits. Once again, we found that expression of the EGFP-CR TRESK subunits lead to a 64% reduction of the whole cell TRESK current density from 200 ± 25 pA/pF at +60 mV in the mCherry-WT group to 71 ± 9 pA/pF in the EGFP-CR + mCherry-WT group (P < 0.001, one-way ANOVA with post hoc Bonferroni test; Fig. 1, E and G). Taken together, we conclude that the TRESK CR variant exhibits a dominant-negative effect on whole cell currents through mouse TRESK channels, similar to the migraine-associated MT TRESK. Our results are consistent with previous studies in Xenopus oocytes using human TRESK as the backbone (Andres-Enguix et al. 2012; Lafreniere et al. 2010).
The CR variant does not affect the total expression level and/or the plasma membrane trafficking of TRESK channels in HEK293T cells.
To study the effects of the CR variant on TRESK expression and/or trafficking, we transfected HEK293T cells with EGFP- and mCherry-tagged TRESK constructs at a 1:1 ratio. Both WT and CR TRESK subunits were localized on the plasma membrane as well as in the intracellular organelles (Fig. 2A). There was a substantial overlap between the EGFP and mCherry signals in cells coexpressing EGFP-WT and mCherry-CR TRESK subunits or, reciprocally, coexpressing mCherry-WT and EGFP-CR TRESK (Fig. 2A), suggesting that WT and CR TRESK subunits may colocalize and coassemble with each other to form nonfunctional channels in HEK293T cells.
Fig. 2.

The CR variant does not affect total and plasma membrane levels of TRESK subunits in HEK293T cells. A: representative images of HEK293T cells coexpressing EGFP- and mCherry-tagged WT and CR TRESK subunits. Two micrograms of each plasmid were used in each transfection. Top and middle: EGFP- and mCherry-tagged TRESK images, respectively. Bottom: merged images. Scale bar = 10 μm. B: total EGFP fluorescence intensity is comparable in cells expressing EGFP- and mCherry-tagged WT and CR TRESK subunits (n = 50 cells in each group; one-way ANOVA). C: total mCherry fluorescence intensity is also similar in cells expressing EGFP- and mCherry-tagged WT and CR TRESK subunits (the same cells as in B, one-way ANOVA). D: representative images of HEK293T cells expressing EGFP-tagged WT and CR TRESK subunits. Cells were fixed and incubated with the TRESK antibody without Triton X-100 permeabilization. Left to right columns: EGFP fluorescence, TRESK-immunoreactivity (TRESK-ir), and the merged images. Scale bar = 5 μm. E: normalized total EGFP fluorescence intensity, surface TRESK-ir, as well as TRESK-ir vs. total EGFP intensity ratio are all comparable in cells expressing EGFP-tagged WT and CR TRESK subunits, respectively (n = 30 cells in each group; one-way ANOVA).
We went on to quantify the expression level of WT TRESK and the CR variant. The level of total EGFP-CR TRESK fluorescence intensity was comparable to that of EGFP-WT TRESK (Fig. 2B). Coexpression of mCherry-CR variant did not affect the level of total EGFP-WT TRESK fluorescence intensity (Fig. 2B). We also quantified the expression level of mCherry-WT and mCherry-CR TRESK subunits and obtained similar results (Fig. 2, A and C).
We then tested whether the CR variant affects the trafficking of TRESK subunits to the plasma membrane, using a mouse polyclonal antibody against an epitope on the first extracellular loop of the mouse TRESK protein. We have previously shown that the antibody specifically recognizes TRESK channels on the plasma membrane with a good signal-to-noise ratio (Liu et al. 2013). Here, we used this antibody to detect the level of TRESK subunits on the plasma membrane in nonpermeabilized HEK293T cells expressing EGFP-WT or EGFP-CR TRESK subunits (Fig. 2D). The fluorescence intensities of EGFP signal and TRESK-immunoreactivity (TRESK-ir) correspond to the level of total and plasma membrane TRESK subunits, respectively. The ratio of TRESK-ir intensity to EGFP intensity in individual cells indicates the efficiency of plasma membrane trafficking of the TRESK subunits. As shown in Fig. 2E, there is no difference between the WT and CR TRESK subunits in the levels of EGFP signal, surface TRESK-ir intensity, or the surface-to-total TRESK ratio, indicating that the CR variant does not affect the total expression level and/or the plasma membrane trafficking of TRESK channels in HEK293T cells.
The CR variant exhibits a weaker dominant-negative effect on the endogenous TRESK currents in small-diameter TG neurons than MT TRESK subunits.
Here, we tested whether the CR variant and the MT TRESK exhibit similar dominant-negative effects on the endogenous TRESK currents in small-diameter (15–25 μm) TG neurons. The majority of small TG neurons are primary nociceptors (Harper and Lawson 1985a,b), with a small subset representing the C-low threshold mechanorecepetors (Lawson et al. 1997; Seal et al. 2009). We overexpressed mCherry-tagged MT and CR TRESK subunits in TG neurons. To dissect currents through TRESK channels, we blocked TTX-sensitive Na+ currents and minimized other transient voltage-gated K+, Na+, and Ca2+ currents by depolarizing neurons from −60-mV holding potential to −25 mV for 150 ms. Currents measured at the end of the depolarizing step were predominantly outward K+ currents (Bautista et al. 2008; Dobler et al. 2007; Guo and Cao 2014; Liu et al. 2013; Tulleuda et al. 2011). Next, we measured the outward currents before and after bath application of 30 μM lamotrigine, which inhibits 70% of the outward current in HEK293T cells expressing mouse TRESK channels (Guo and Cao 2014). Of note, lamotrigine at this concentration did not significantly reduce the outward current in TG neurons from TRESK knockout mice (Guo Z and Cao YQ, unpublished observations), validating that TRESK channels mediate the lamotrigine-sensitive current in TG neurons under our recording conditions.
In TG neurons expressing mCherry protein, ∼30% of the outward currents were inhibited by lamotrigine (Fig. 3, A and B). The fraction of lamotrigine-sensitive currents was significantly lower in TG neurons overexpressing mCherry-CR TRESK subunits (20 ± 2%; P < 0.001, compared with the mCherry group, one-way ANOVA with post hoc Bonferroni test; Fig. 3B), indicating that the CR variant reduced the endogenous TRESK currents by 30%. On the other hand, lamotrigine only blocked 13 ± 2% of the outward currents in TG neurons overexpressing mCherry-MT TRESK subunits (P < 0.001, compared with the mCherry group; Fig. 3B), which translates to >50% reduction of the endogenous TRESK currents. We conclude that both the CR variant and the MT subunit exert a dominant-negative effect on the endogenous TRESK currents in wild-type TG neurons. However, the effect of the CR variant is significantly weaker than that of the truncated MT TRESK subunits (P < 0.05, between mCherry-CR and mCherry-MT groups; Fig. 3B). The density of the total outward K+ current at the end of the depolarizing step was highly variable between individual neurons. The mean values were not statistically different among the three groups (data not shown).
Fig. 3.
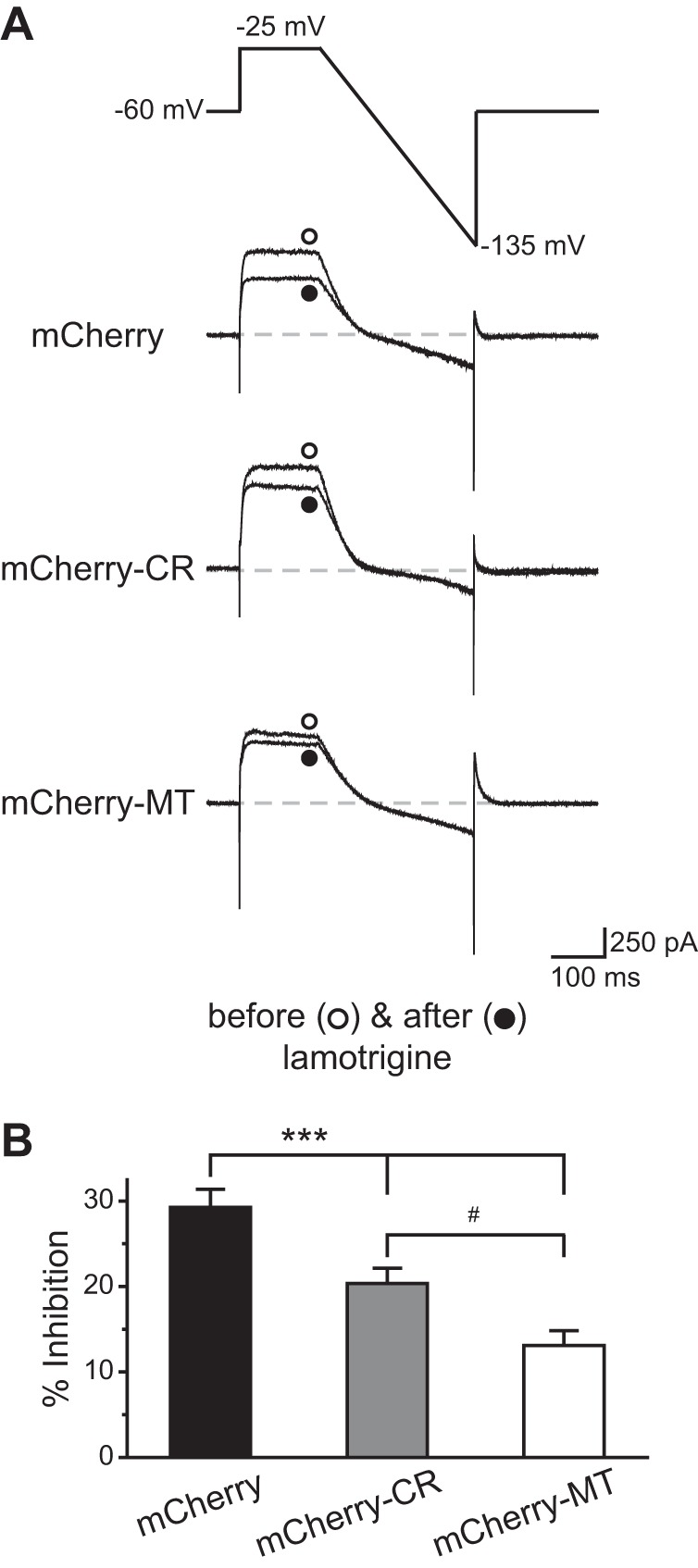
The CR variant exhibits a weaker dominant-negative effect on the endogenous TRESK currents in trigeminal ganglion (TG) neurons than MT TRESK subunits. A: voltage protocol used to minimize transient voltage-gated K+, Na+, and Ca2+ currents and the representative current traces from TG neurons expressing mCherry protein, mCherry-CR TRESK variant, and mCherry-MT TRESK subunits before and after the application of 30 μM lamotrigine, respectively. B: percentage of outward current (measured at the end of the depolarizing step) inhibited by lamotrigine (n = 12–18 neurons in each group; one-way ANOVA with post hoc Bonferroni test; ***P < 0.001, compared with the mCherry group; #P < 0.05).
The weaker dominant-negative effect of the CR variant could result from its lower expression level and/or less efficient plasma membrane trafficking than MT TRESK in TG neurons. To test this possibility, we compared total and plasma membrane expression of CR and MT TRESK subunits in TG neurons. We expressed EGFP-CR and EGFP-MT subunits in cultured TG neurons and monitored EGFP fluorescence in the soma. In both groups, the EGFP fluorescence was localized on the plasma membrane as well as aggregated intracellularly. The latter likely represented newly synthesized TRESK subunits in the intracellular organelles (Fig. 4A) (Liu et al. 2013). Quantitative analysis showed a comparable level of total somatic EGFP fluorescence intensity in TG neurons expressing EGFP-CR and EGFP-MT subunits (Fig. 4B), indicating that the expression levels of CR and MT TRESK subunits were comparable in TG neurons.
Fig. 4.

Total expression and surface expression of the TRESK CR and MT subunits are comparable in TG neurons. A: representative images of TG neurons expressing EGFP-MT and EGFP-CR TRESK subunits. Neurons were briefly stained with AF594-WGA to delineate the plasma membrane before fixation. Scale bar = 10 μm. B–D: normalized total (B), surface (C), as well as plasma membrane vs. cytosol ratio (D) of EGFP fluorescence intensity in TG neurons expressing EGFP-MT and EGFP-CR TRESK subunits, respectively (n = 30 cells in each group, same neurons in B–D). DIC, differential interference contrast; WGA, wheat germ agglutinin.
Next, we examined whether CR and MT TRESK subunits differ in trafficking to the plasma membrane. We did not use the TRESK antibody in this set of experiment as the antibody does not discriminate between the endogenous and exogenous TRESK subunits. Instead, we stained the neurons briefly with AF594-WGA to delineate the plasma membrane ROI (Fig. 4A) (Jeng et al. 2008; Liu et al. 2013). This allowed us to estimate the amount of TRESK subunits on the plasma membrane. One caveat is that, due to the resolution limit of light microscope, the ROI defined by AF594-WGA signal could also contain some signal from TRESK present in submembrane vesicles. We found that the mean EGFP intensity in the ROI was comparable between the MT and CR groups (Fig. 4C), indicating a similar steady-state plasma membrane level of MT and CR subunits. The plasma membrane vs. cytosol ratio of EGFP-MT and EGFP-CR subunits was also similar (Fig. 4D), suggesting that the efficiency of plasma membrane trafficking does not differ between the MT and CR TRESK subunits. Other mechanisms may underlie the differential dominant-negative effect of the TRESK CR variant and MT subunits in TG neurons.
Overexpression of the TRESK CR variant does not increase the excitability of small-diameter TG neurons.
Here, we used current-clamp recording to investigate whether overexpression of the mCherry-CR TRESK subunits affects the passive and active electrophysiological properties of small-diameter TG neurons. To account for the heterogeneity of TG neurons, we further divided TG neurons based on their ability to bind to fluorescently labeled IB4 at the end of each current-clamp recording. It is well documented that the small IB4-positive and IB4-negative primary afferent neurons exhibit distinct neurochemical, anatomical, and electrophysiological properties and may encode different pain modalities (Cavanaugh et al. 2009; Choi et al. 2007; Liu et al. 2013; Scherrer et al. 2009; Snider and McMahon 1998; Stucky and Lewin 1999).
In small IB4-negative TG neurons, overexpression of the TRESK CR variant did not alter the Vrest and Rin (Table 1), consistent with previous studies suggesting that there is little endogenous TRESK channel activity when membrane potential is at or below the Vrest (Dobler et al. 2007; Liu et al. 2013). To determine the rheobase (the minimum amount of current required to elicit at least 1 AP), we held the transfected neurons at Vrest and injected 1-s depolarizing currents at 25-pA incremental steps. Surprisingly, the rheobase of TG neurons expressing mCherry-CR was not statistically different from that of control neurons expressing mCherry protein (111 ± 11 and 91 ± 10 pA, respectively; P = 0.38, one-way ANOVA with post hoc Bonferroni test; Fig. 5, A and B, and Table 1), despite the fact that the CR variant significantly reduced the endogenous TRESK currents (Fig. 3B). The values of the AP threshold, amplitude, half-width, and AHP amplitude were not affected by the TRESK CR variant either (Table 1). In a parallel experiment, we confirmed the results from our previous study (Liu et al. 2013) that overexpression of the mCherry-MT TRESK subunits resulted in a significant reduction of the rheobase in small IB4-negative TG neurons (50 ± 6 pA; P < 0.05, compared with the mCherry group; Fig. 5, A and B, and Table 1).
Table 1.
Intrinsic properties of small-diameter TG neurons expressing MT TRESK subunits or CR variants
| Diameter, μm | Capacitance, pF | Rin, MΩ | Vrest, mV | Rheobase, pA | AP Threshold, mV | AP Amplitude, mV | AP Half-Width, ms | AHP Amplitude, mV | Cell Number | |
|---|---|---|---|---|---|---|---|---|---|---|
| IB4-negative neurons | ||||||||||
| mCherry | 21.3 ± 0.9 | 24.2 ± 2.2 | 628 ± 71 | −57.0 ± 1.6 | 91 ± 10 | −21.8 ± 2.0 | 108.7 ± 3.6 | 4.6 ± 0.6 | −15.4 ± 1.1 | 14 |
| mCherry-CR | 21.3 ± 0.7 | 23.5 ± 2.0 | 481 ± 31 | −54.5 ± 1.5 | 111 ± 11 | −20.5 ± 2.0 | 109.5 ± 3.5 | 3.6 ± 0.4 | −15.9 ± 1.2 | 18 |
| mCherry-MT | 18.7 ± 0.7 | 19.8 ± 1.5 | 750 ± 93† | −53.2 ± 2.0 | 50 ± 6*‡ | −24.5 ± 2.2 | 102.5 ± 4.9 | 2.9 ± 0.4 | −16.0 ± 2.1 | 9 |
| IB4-positive neurons | ||||||||||
| mCherry | 21.7 ± 0.8 | 24.5 ± 1.6 | 544 ± 64 | −55.7 ± 1.0 | 125 ± 8§ | −19.5 ± 1.8 | 109.9 ± 4.0 | 4.6 ± 0.5 | −17.1 ± 1.4 | 15 |
| mCherry-CR | 21.6 ± 0.7 | 26.5 ± 2.1 | 465 ± 34 | −57.1 ± 1.2 | 172 ± 18* | −17.4 ± 1.5 | 116.6 ± 2.6 | 4.7 ± 0.3 | −19.0 ± 1.0 | 16 |
Values are means ± SE. Input resistant (Rin) was calculated by measuring the change of membrane potential in response to a 20-pA hyperpolarizing current injection from resting membrane potential (Vrest). TG, trigeminal ganglion; MT, mutant; CR, C110R; TRESK, TWIK-related spinal cord K+; AP, action potential; AHP, afterhyperpolarization; IB4, isolectin B4.
P < 0.05, compared with the corresponding mCherry group by one-way ANOVA with post hoc Bonferroni test or two-tailed t-test.
P < 0.05,
P < 0.001, compared with the corresponding mCherry-CR group by one-way ANOVA with post hoc Bonferroni test. §P < 0.05, compared with the small IB4-negative mCherry group by two-tailed t-test.
Fig. 5.

Overexpression of the TRESK CR variant does not alter the excitability of small isolectin B4 (IB4)-negative TG neurons. A: representative traces of action potentials (APs) generated by incremental depolarizing current injections in transfected, small IB4-negative TG neurons. The values of resting membrane potential (Vrest) and the injected current are indicated. B: mean rheobase of the small IB4-negative TG neurons expressing the mCherry proteins, mCherry-CR, and mCherry-MT TRESK subunits, respectively (n = 9–18 neurons in each group; *P < 0.05, ***P < 0.001, one-way ANOVA with post hoc Bonferroni test). C: input-output plots of the transfected, small IB4-negative TG neurons (same as in B). The spike frequency was measured by injection of 1-s depolarizing current in 25-pA incremental steps in each neuron [two-way repeated-measures (RM) ANOVA and post hoc t-test with Bonferroni correction, *P < 0.05, **P < 0.01, compared with the corresponding mCherry groups; ##P < 0.01, compared with the corresponding mCherry-CR groups].
Does overexpression of the TRESK CR variant alter the spike frequency of small IB4-negative TG neurons? In control neurons expressing mCherry protein, the number of APs increased almost linearly to incremental depolarizing current injections (Fig. 5C). Overexpression of the mCherry-MT TRESK subunits significantly increased the number of APs evoked by suprathreshold current injections (P < 0.05, between mCherry and mCherry-MT groups, two-way RM ANOVA; Fig. 5C), consistent with our previous findings (Liu et al. 2013). On the contrary, overexpression of the TRESK CR variant did not alter the spike frequency of small IB4-negative TG neurons (P = 0.3, between mCherry and mCherry-CR groups, two-way RM ANOVA; Fig. 5C). Both the rheobase and spike frequency of neurons overexpressing the TRESK CR variant were significantly different from those of neurons expressing the MT TRESK subunits, respectively (Fig. 5 and Table 1). Thus unlike the migraine-associated MT TRESK, the CR variant does not affect the excitability of small IB4-negative TG neurons.
We went on to examine the effect of the TRESK CR variant on the excitability of small IB4-positive TG neurons. In agreement with previous studies on the distinct electrophysiological properties of small IB4-negative and IB4-positive primary afferent neurons (Choi et al. 2007; Fang et al. 2006; Liu et al. 2013), we found that the control small IB4-positive TG neurons had a significantly higher rheobase (P < 0.05, two-tailed t-test; Table 1) and a much flatter input-output curve relative to the control small IB4-negative TG neurons (P < 0.05, two-way RM ANOVA between mCherry groups; Figs. 5C and 6C). Overexpression of the TRESK CR variant did not alter the Vrest of small IB4-positive TG neurons (Table 1). Interestingly, the rheobase of TG neurons expressing mCherry-CR subunits was significantly higher than that of control neurons expressing mCherry protein (172 ± 18 and 125 ± 8 pA, respectively, P < 0.05, two-tailed t-test; Fig. 6, A and B, and Table 1). On the other hand, overexpression of the TRESK CR variant did not significantly alter the spike frequency of small IB4-positive TG neurons (P = 0.09, two-way RM ANOVA; Fig. 6C), even after we normalized the injected current to the rheobase of individual neurons (P = 0.09, two-way RM ANOVA; Fig. 6D) nor did it affect the AP threshold, amplitude, half-width, and AHP amplitude in this TG population (Table 1). We conclude that the TRESK CR variant decreases the excitability of small IB4-positive TG neurons mainly through increasing the current threshold for AP generation, opposite to the effects of the MT TRESK subunits in this TG subpopulation (Liu et al. 2013).
Fig. 6.

Overexpression of the TRESK CR variant increases the rheobase of small IB4-positive TG neurons. A: representative traces of APs generated by incremental depolarizing current injections in transfected, small IB4-positive TG neurons. The values of Vrest and the injected current are indicated. B: mean rheobase of small IB4-positive TG neurons expressing mCherry proteins or mCherry-CR TRESK subunits (n = 15 and 16 neurons, respectively; *P < 0.05, two-tailed t-test). C: input-output plots of the spike frequency in response to 1-s incremental depolarizing current injections in transfected, small IB4-positive TG neurons (P =0.09, two-way RM ANOVA; same neurons as in B). D: input-output plots of the spike frequency in response to 1-s depolarizing current injection from 1- to 3-fold rheobase in transfected, small IB4-positive TG neurons (P = 0.09, two-way RM ANOVA; same neurons as in B).
DISCUSSION
In this study, we investigated whether there might be a causal relationship between TRESK channel dysfunction and migraine susceptibility. We introduced the migraine-associated frameshift mutation and the CR variant into the mouse TRESK subunits and tested them in HEK293T cells. Neither MT nor CR TRESK subunits form functional channels per se. Moreover, they exert a similar dominant-negative effect on WT TRESK subunits, likely through forming heterodimeric channels with the WT TRESK subunits (Liu et al. 2013). Coexpression of either MT or CR subunits with WT TRESK reduces whole cell TRESK currents by ∼70% (Fig. 1), close to the predicted 75% current reduction assuming that both WT-CR and WT-MT heterodimeric channels are nonfunctional. The cysteine to arginine substitution in the first pore domain occurs in a region that is close to the selectivity filter and has been implicated in K2P channel gating (Mathie et al. 2010). The effect of the CR variant likely results from impaired channel gating, as the expression level and/or the plasma membrane trafficking of the CR variant is not altered in HEK293T cells (Fig. 2). Our results obtained in HEK293T cells are in complete agreement with a previous study in Xenopus oocytes (Andres-Enguix et al. 2012) indicating that the MT and CR TRESK subunits exhibited the same effects on TRESK currents in heterologous expression systems.
On the contrary, the CR variant exhibited a significantly weaker dominant-negative effect on whole cell TRESK current than the MT TRESK subunit when expressed in cultured TG neurons (Fig. 3). This is unlikely due to an insufficient expression of the CR variant in TG neurons, as the transfected TG neurons on average express twice the amount of exogenous TRESK subunits as the endogenous channels in a previous study (Guo and Cao 2014). Moreover, the total and plasma membrane levels of the CR variant are comparable to those of the MT subunits in transfected neurons, respectively (Fig. 4). On the other hand, the MT TRESK subunit is a truncated protein and the CR variant contains one amino acid change. The two proteins may exhibit different posttranslational modifications in TG neurons. One possibility is that the WT-CR heterodimer may be less stable than the WT-WT and/or WT-MT channels in TG neurons. This would account for the milder dominant-negative effect of the CR variant. Another possibility is that the WT-CR heterodimeric channels may be nonfunctional in Xenopus oocytes and HEK293T cells but may conduct small but significant K+ outflow in TG neurons. It is well established that the activity of TRESK channels can be modulated by pH, the intracellular Ca2+ level, as well as other signaling pathways (Enyedi et al. 2012). The gating of the WT-CR channels may be differentially modulated in HEK293T cells and TG neurons. Conversely, the MT TRESK subunit is truncated in the second transmembrane domain (Lafreniere et al. 2010). The WT-MT heterodimeric channels may lack a functional pore and therefore are nonfunctional in both HEK293T cells and TG neurons. All in all, more experiments are needed to understand the mechanisms underlie the differential effect of the MT and CR subunits on endogenous TRESK currents in TG neurons. Whether and how MT and CR TRESK subunits affect the size of total background K+ currents in TG neurons also merit further study.
One advantage of studying the function of MT and CR subunits in cultured TG neurons is that it allows us to directly compare their effects on TG excitability. The cell bodies of the primary afferent neurons in the neuronal circuit underlying migraine headache are predominantly localized in the TG. Activation and sensitization of these neurons are important steps in the initiation of headache attacks (Burstein 2001; Goadsby et al. 2009; Harriott and Gold 2009; Pietrobon and Moskowitz 2013; Strassman et al. 1996; Yan et al. 2013). Many triggers of migraine in human also activate TG meningeal nociceptors in rodents (Levy and Strassman 2004; Strassman et al. 1996; Zhang et al. 2010). Furthermore, mutations of the P/Q-type voltage-gated Ca2+ channel associated with familial hemiplegic migraine type-1 cause hyperexcitation of TG neurons, suggesting that migraine headache may result from abnormal membrane conductance changes of the primary afferent neurons in TG (Fioretti et al. 2011; Hullugundi et al. 2014; Tao et al. 2012). We reason that the effects of TRESK mutations/variants on TG excitability would be a better predictor of their contributions to migraine susceptibility, relative to their effects on TRESK currents in heterologous expression systems.
Consistent with our previous study (Liu et al. 2013), overexpression of MT TRESK subunits resulted in hyperexcitation of small IB4-negative TG neurons. On the contrary, overexpression of the CR variant did not alter the excitability of this TG population (Fig. 5). Previous studies indicate that there is an inverse correlation between the endogenous TRESK channel activity and primary afferent neuronal excitability (Dobler et al. 2007; Guo and Cao 2014; Lennertz et al. 2010; Liu et al. 2013; Tulleuda et al. 2011). Our results suggest that the level of endogenous TRESK activity can effectively maintain TG excitability in wild-type neurons. A moderate decrease of the TRESK current by the CR variant is well tolerated. Hyperexcitation only occurs as the result of a substantial reduction of the endogenous TRESK activity, for example, by the migraine-associated frameshift mutation. This is a critically important finding, as the functional consequence of the MT and CR TRESK subunits in small IB4-negative TG neurons is different from what was observed in oocytes or HEK293T cells. This may explain why the frameshift mutation is found exclusively in migraineurs, whereas the CR variant is seen in both migraineurs and unaffected individuals.
In small IB4-positive TG neurons, overexpression of the CR variant did not affect the spike frequency but significantly increased the current threshold to elicit AP (Fig. 6). This is opposite to the effects of the frameshift mutation, which decreases the rheobase as well as increases the spike frequency in this TG population (Liu et al. 2013). Again, the differential effects of the MT and CR TRESK subunits in small IB4-positive TG neurons implicate a causal role of MT TRESK in migraine pathogenesis. Future studies are necessary to clarify the mechanisms responsible for the unexpected effect of the CR variant in small IB4-positive TG neurons. One possibility is that other background K+ currents may overcompensate for the reduction of endogenous TRESK currents caused by the CR variant, thereby resulting in a higher rheobase.
Our results support a potential causal relationship between the frameshift TRESK mutation and migraine susceptibility. They also stress the value of cultured TG neurons as a platform for systematically investigating the contribution of ion channel mutations to migraine pathophysiology. That said, overexpressing channel subunits in cultured TG neurons from neonatal mice is a good starting point, but by no means an ideal way, to elucidate the genotype-phenotype correlations in migraine headache. Another caveat is that we have studied the functional consequences of the frameshift mutation and the CR variant in the context of mouse TRESK subunit, which shares 65% overall amino acid identity with the human TRESK. Although our results with the mouse MT and CR TRESK subunits in HEK293T cells agree completely with those of the studies using the human MT and CR TRESK subunits in Xenopus oocytes (Andres-Enguix et al. 2012; Lafreniere et al. 2010; Liu et al. 2013), it is possible that the frameshift mutation and/or the CR variant affect human and mouse TRESK channels differently in neurons. Given the limitation of the experimental system, our data should be interpreted with caution. More experiments are needed to study the functional significance of TRESK mutations/variants expressed at the endogenous level in mature neurons in the intact circuit underlying migraine headache.
In summary, our data show that the CR variant causes a smaller reduction of the endogenous TRESK currents in TG neurons than the migraine-associated frameshift mutation. Consequently, only the MT TRESK subunit, but not the CR variant, results in hyperexcitability of small-diameter TG neurons. Our results suggest a possible causal relationship between the TRESK frameshift mutation and a higher susceptibility of migraine headache.
GRANTS
This work was supported by the National Institute of Neurological Disorders and Stroke Grant R21-NS-074198 (to Y.-Q. Cao).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
AUTHOR CONTRIBUTIONS
Author contributions: Z.G. and Y.-Q.C. conception and design of research; Z.G., P.L., and F.R. performed experiments; Z.G., P.L., and F.R. analyzed data; Z.G., P.L., F.R., and Y.-Q.C. interpreted results of experiments; Z.G. and Y.-Q.C. prepared figures; Z.G. and Y.-Q.C. drafted manuscript; Z.G. and Y.-Q.C. edited and revised manuscript; Z.G., P.L., F.R., and Y.-Q.C. approved final version of manuscript.
ACKNOWLEDGMENTS
Present address of P. Liu: Dept. of Genetics, Washington Univ. School of Medicine, St. Louis, MO.
Present address of F. Ren: Dept. of Anesthesiology, Xiangya Hospital, Central South Univ., Hunan, China.
REFERENCES
- Andres-Enguix I, Shang L, Stansfeld PJ, Morahan JM, Sansom MS, Lafreniere RG, Roy B, Griffiths LR, Rouleau GA, Ebers GC, Cader ZM, Tucker SJ. Functional analysis of missense variants in the TRESK (KCNK18) K channel. Sci Rep 2: 237, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aronson DE, Costantini LM, Snapp EL. Superfolder GFP is fluorescent in oxidizing environments when targeted via the Sec translocon. Traffic 12: 543–548, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bautista DM, Sigal YM, Milstein AD, Garrison JL, Zorn JA, Tsuruda PR, Nicoll RA, Julius D. Pungent agents from Szechuan peppers excite sensory neurons by inhibiting two-pore potassium channels. Nat Neurosci 11: 772–779, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brohawn SG, del Marmol J, MacKinnon R. Crystal structure of the human K2P TRAAK, a lipid- and mechano-sensitive K+ ion channel. Science 335: 436–441, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burstein R. Deconstructing migraine headache into peripheral and central sensitization. Pain 89: 107–110, 2001 [DOI] [PubMed] [Google Scholar]
- Burstein R, Jakubowski M. Unitary hypothesis for multiple triggers of the pain and strain of migraine. J Comp Neurol 493: 9–14, 2005 [DOI] [PubMed] [Google Scholar]
- Cavanaugh DJ, Lee H, Lo L, Shields SD, Zylka MJ, Basbaum AI, Anderson DJ. Distinct subsets of unmyelinated primary sensory fibers mediate behavioral responses to noxious thermal and mechanical stimuli. Proc Natl Acad Sci USA 106: 9075–9080, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi JS, Dib-Hajj SD, Waxman SG. Differential slow inactivation and use-dependent inhibition of Nav1.8 channels contribute to distinct firing properties in IB4+ and IB4- DRG neurons. J Neurophysiol 97: 1258–1265, 2007 [DOI] [PubMed] [Google Scholar]
- de Vries B, Frants RR, Ferrari MD, van den Maagdenberg AM. Molecular genetics of migraine. Hum Genet 126: 115–132, 2009 [DOI] [PubMed] [Google Scholar]
- Dobler T, Springauf A, Tovornik S, Weber M, Schmitt A, Sedlmeier R, Wischmeyer E, Doring F. TRESK two-pore-domain K+ channels constitute a significant component of background potassium currents in murine dorsal root ganglion neurones. J Physiol 585: 867–879, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eising E, de Vries B, Ferrari MD, Terwindt GM, van den Maagdenberg AM. Pearls and pitfalls in genetic studies of migraine. Cephalalgia 33: 614–625, 2013 [DOI] [PubMed] [Google Scholar]
- Enyedi P, Braun G, Czirjak G. TRESK: the lone ranger of two-pore domain potassium channels. Mol Cell Endocrinol 353: 75–81, 2012 [DOI] [PubMed] [Google Scholar]
- Fang X, Djouhri L, McMullan S, Berry C, Waxman SG, Okuse K, Lawson SN. Intense isolectin-B4 binding in rat dorsal root ganglion neurons distinguishes C-fiber nociceptors with broad action potentials and high Nav1.9 expression. J Neurosci 26: 7281–7292, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fioretti B, Catacuzzeno L, Sforna L, Gerke-Duncan MB, van den Maagdenberg AM, Franciolini F, Connor M, Pietrobon D. Trigeminal ganglion neuron subtype-specific alterations of CaV2.1 calcium current and excitability in a Cacna1a mouse model of migraine. J Physiol 589: 5879–5895, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goadsby PJ, Charbit AR, Andreou AP, Akerman S, Holland PR. Neurobiology of migraine. Neuroscience 161: 327–341, 2009 [DOI] [PubMed] [Google Scholar]
- Guo Z, Cao YQ. Overexpression of TRESK K(+) channels reduces the excitability of trigeminal ganglion nociceptors. PLoS One 9: e87029, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harper AA, Lawson SN. Conduction velocity is related to morphological cell type in rat dorsal root ganglion neurones. J Physiol 359: 31–46, 1985a [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harper AA, Lawson SN. Electrical properties of rat dorsal root ganglion neurones with different peripheral nerve conduction velocities. J Physiol 359: 47–63, 1985b [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harriott AM, Gold MS. Electrophysiological properties of dural afferents in the absence and presence of inflammatory mediators. J Neurophysiol 101: 3126–3134, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hullugundi SK, Ansuini A, Ferrari MD, van den Maagdenberg AM, Nistri A. A hyperexcitability phenotype in mouse trigeminal sensory neurons expressing the R192Q Cacna1a missense mutation of familial hemiplegic migraine type-1. Neuroscience 266C: 244–254, 2014 [DOI] [PubMed] [Google Scholar]
- Jeng CJ, Sun MC, Chen YW, Tang CY. Dominant-negative effects of episodic ataxia type 2 mutations involve disruption of membrane trafficking of human P/Q-type Ca2+ channels. J Cell Physiol 214: 422–433, 2008 [DOI] [PubMed] [Google Scholar]
- Kang D, Kim D. TREK-2 (K2P10.1) and TRESK (K2P18.1) are major background K+ channels in dorsal root ganglion neurons. Am J Physiol Cell Physiol 291: C138–C146, 2006 [DOI] [PubMed] [Google Scholar]
- Kang D, Kim GT, Kim EJ, La JH, Lee JS, Lee ES, Park JY, Hong SG, Han J. Lamotrigine inhibits TRESK regulated by G-protein coupled receptor agonists. Biochem Biophys Res Commun 367: 609–615, 2008 [DOI] [PubMed] [Google Scholar]
- Kang D, Mariash E, Kim D. Functional expression of TRESK-2, a new member of the tandem-pore K+ channel family. J Biol Chem 279: 28063–28070, 2004 [DOI] [PubMed] [Google Scholar]
- Lafreniere RG, Cader MZ, Poulin JF, Andres-Enguix I, Simoneau M, Gupta N, Boisvert K, Lafreniere F, McLaughlan S, Dube MP, Marcinkiewicz MM, Ramagopalan S, Ansorge O, Brais B, Sequeiros J, Pereira-Monteiro JM, Griffiths LR, Tucker SJ, Ebers G, Rouleau GA. A dominant-negative mutation in the TRESK potassium channel is linked to familial migraine with aura. Nat Med 16: 1157–1160, 2010 [DOI] [PubMed] [Google Scholar]
- Lawson SN, Crepps BA, Perl ER. Relationship of substance P to afferent characteristics of dorsal root ganglion neurones in guinea-pig. J Physiol 505: 177–191, 1997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lennertz RC, Tsunozaki M, Bautista DM, Stucky CL. Physiological basis of tingling paresthesia evoked by hydroxy-alpha-sanshool. J Neurosci 30: 4353–4361, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levy D, Strassman AM. Modulation of dural nociceptor mechanosensitivity by the nitric oxide-cyclic GMP signaling cascade. J Neurophysiol 92: 766–772, 2004 [DOI] [PubMed] [Google Scholar]
- Liu P, Xiao Z, Ren F, Guo Z, Chen Z, Zhao H, Cao YQ. Functional analysis of a migraine-associated TRESK K+ channel mutation. J Neurosci 33: 12810–12824, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marsh B, Acosta C, Djouhri L, Lawson SN. Leak K(+) channel mRNAs in dorsal root ganglia: relation to inflammation and spontaneous pain behaviour. Mol Cell Neurosci 49: 375–386, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathie A, Al-Moubarak E, Veale EL. Gating of two pore domain potassium channels. J Physiol 588: 3149–3156, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller AN, Long SB. Crystal structure of the human two-pore domain potassium channel K2P1. Science 335: 432–436, 2012 [DOI] [PubMed] [Google Scholar]
- Perez-Reyes E. Molecular physiology of low-voltage-activated t-type calcium channels. Physiol Rev 83: 117–161, 2003 [DOI] [PubMed] [Google Scholar]
- Piechotta PL, Rapedius M, Stansfeld PJ, Bollepalli MK, Ehrlich G, Andres-Enguix I, Fritzenschaft H, Decher N, Sansom MS, Tucker SJ, Baukrowitz T. The pore structure and gating mechanism of K2P channels. EMBO J 30: 3607–3619, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pietrobon D, Moskowitz MA. Pathophysiology of migraine. Annu Rev Physiol 75: 365–391, 2013 [DOI] [PubMed] [Google Scholar]
- Plant LD. A role for K2P channels in the operation of somatosensory nociceptors. Front Mol Neurosci 5: 21, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sano Y, Inamura K, Miyake A, Mochizuki S, Kitada C, Yokoi H, Nozawa K, Okada H, Matsushime H, Furuichi K. A novel two-pore domain K+ channel, TRESK, is localized in the spinal cord. J Biol Chem 278: 27406–27412, 2003 [DOI] [PubMed] [Google Scholar]
- Scherrer G, Imamachi N, Cao YQ, Contet C, Mennicken F, O'Donnell D, Kieffer BL, Basbaum AI. Dissociation of the opioid receptor mechanisms that control mechanical and heat pain. Cell 137: 1148–1159, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seal RP, Wang X, Guan Y, Raja SN, Woodbury CJ, Basbaum AI, Edwards RH. Injury-induced mechanical hypersensitivity requires C-low threshold mechanoreceptors. Nature 462: 651–655, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snider WD, McMahon SB. Tackling pain at the source: new ideas about nociceptors. Neuron 20: 629–632, 1998 [DOI] [PubMed] [Google Scholar]
- Strassman AM, Raymond SA, Burstein R. Sensitization of meningeal sensory neurons and the origin of headaches. Nature 384: 560–564, 1996 [DOI] [PubMed] [Google Scholar]
- Stucky CL, Lewin GR. Isolectin B(4)-positive and -negative nociceptors are functionally distinct. J Neurosci 19: 6497–6505, 1999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tao J, Liu P, Xiao Z, Zhao H, Gerber BR, Cao YQ. Effects of familial hemiplegic migraine type 1 mutation T666M on voltage-gated calcium channel activities in trigeminal ganglion neurons. J Neurophysiol 107: 1666–1680, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tottene A, Conti R, Fabbro A, Vecchia D, Shapovalova M, Santello M, van den Maagdenberg AM, Ferrari MD, Pietrobon D. Enhanced excitatory transmission at cortical synapses as the basis for facilitated spreading depression in Ca(v)2.1 knockin migraine mice. Neuron 61: 762–773, 2009 [DOI] [PubMed] [Google Scholar]
- Tulleuda A, Cokic B, Callejo G, Saiani B, Serra J, Gasull X. TRESK channel contribution to nociceptive sensory neurons excitability: modulation by nerve injury. Mol Pain 7: 30, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Den Maagdenberg AM, Pietrobon D, Pizzorusso T, Kaja S, Broos LA, Cesetti T, Van De Ven RC, Tottene A, Van Der Kaa J, Plomp JJ, Frants RR, Ferrari MD. A cacna1a knockin migraine mouse model with increased susceptibility to cortical spreading depression. Neuron 41: 701–710, 2004 [DOI] [PubMed] [Google Scholar]
- Victor TW, Hu X, Campbell JC, Buse DC, Lipton RB. Migraine prevalence by age and sex in the United States: a life-span study. Cephalalgia 30: 1065–1072, 2010 [DOI] [PubMed] [Google Scholar]
- Yan J, Wei X, Bischoff C, Edelmayer RM, Dussor G. pH-Evoked dural afferent signaling is mediated by ASIC3 and is sensitized by mast cell mediators. Headache 53: 1250–1261, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoo S, Liu J, Sabbadini M, Au P, Xie GX, Yost CS. Regional expression of the anesthetic-activated potassium channel TRESK in the rat nervous system. Neurosci Lett 465: 79–84, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Levy D, Noseda R, Kainz V, Jakubowski M, Burstein R. Activation of meningeal nociceptors by cortical spreading depression: implications for migraine with aura. J Neurosci 30: 8807–8814, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]