

Abstract
Dopaminergic projections from the ventral tegmental area (VTA) constitute the mesolimbocortical system that underlies addiction and psychosis primarily as a result of increased dopaminergic transmission. Dopamine release is spike dependent. L-type calcium channels (LTCCs) play an important role in regulating firing activities, but the contribution of specific subtypes remains unclear. This article describes different functions of Cav1.2 and Cav1.3 subtypes in regulating firing properties with two transgenic mouse strains. For basal firing, Cav1.3-deficient (Cav1.3−/−) mice had a lower basal firing frequency. The dihydropyridine (DHP) channel blocker nifedipine reduced single-spike firing in mice expressing DHP-insensitive Cav1.2 channels (Cav1.2DHP−/− mice), confirming the significant contribution from the Cav1.3 subtype in basal firing. Moreover, the DHP channel activator (S)-(−)-Bay K8644 and the non-DHP channel activator FPL 64176 converted firing patterns from single spiking to bursting in Cav1.2DHP−/− mice. Nifedipine inhibited burst firing induced by both activators, suggesting that Cav1.3 also serves an essential role in burst firing. However, FPL 64176 also induced bursting in Cav1.3−/− mice. These results indicate that the Cav1.3 subtype is crucial to regulation of basal single-spike firing, while activation of both Cav1.2 and Cav1.3 can support burst firing of VTA neurons.
Keywords: action potential, firing patterns, calcium channel, transgenic mouse, patch-clamp recording
dopaminergic (DA) neurons in the ventral tegmental area (VTA) play an important role in cognition and reward processing (Cooper 2002; Ikemoto 2007, 2010; Tobler et al. 2005), and abnormalities in these processes are implicated in diseases such as schizophrenia and addiction (Eyles 2012; Gardner 2011; Iversen and Iversen 2007; Koob and Volkow 2010; Tritsch and Sabatini 2012). DA neurons are spontaneously active and display two major firing patterns: single-spike firing (also called pace-making spiking) and burst firing (Liu et al. 2007; Shi 2009; Zhang et al. 2005). The two firing modes are shown to be involved in different functions: pace-making spiking sets the basic tone to maintain DA levels in terminal areas and behaviorally encodes expected stimulations; however, burst firing enhances DA synaptic transmission and signals unexpected environmental stimuli (Chergui et al. 1994; Romo and Schultz 1990; Schultz 2007, 2010; Tobler et al. 2005). Therefore, it is crucial to understand the transition and regulation of the two firing modes in DA neurons.
Ca2+ influx through L-type calcium channels (LTCCs) is an important modulator of neuronal firing activities of DA neurons in the VTA. LTCCs are responsible for approximately one-third of total Ca2+ currents in DA cells (Cardozo and Bean 1995; Durante et al. 2004) and contribute preferentially to whole cell Ca2+ currents evoked by small depolarizations (Durante et al. 2004; Xu and Lipscombe 2001). They have been shown to be involved in spontaneous firing, spontaneous oscillatory potentials, and cholinergic-driven firing increases that are abolished by dihydropyridine (DHP) LTCC blockers (Mercuri et al. 1994; Nedergaard et al. 1993; Zhang et al. 2005). Also, they are involved in the regulation of burst firing. For example, the cholinergic-driven bursting and associated membrane potential oscillations are blocked by the LTCC blocker nifedipine (Zhang et al. 2005). Furthermore, direct activation of LTCCs with activators acting at different sites, (S)-(−)-Bay K8644 and FPL 64176, induces burst firing in DA neurons (Liu et al. 2007; Zhang et al. 2005).
Four subtypes of LTCCs haven been identified: Cav1.1, Cav1.2, Cav1.3, and Cav1.4; only Cav1.2 and Cav1.3 subtypes are expressed in the midbrain (Chan et al. 2007; Rajadhyaksha et al. 2004; Sinnegger-Brauns et al. 2009; Striessnig et al. 2006; Takada et al. 2001). It has been shown that Cav1.2 and Cav1.3 LTCCs are distributed throughout the entire midbrain including DA neurons (Rajadhyaksha et al. 2004; Takada et al. 2001). Compared with Cav1.2 channels, Cav1.3 has a lower activation threshold and a reduced sensitivity to DHP activators and blockers (Durante et al. 2004; Striessnig et al. 2006; Xu et al. 2001). The role of LTCCs in regulating firing activities of DA neurons has been explored; however, current pharmacological agents cannot differentiate Cav1.2 and Cav1.3 subtypes, and thus their specific contribution to functions mediated by LTCCs remains unknown. This study aimed to determine the contribution of the two LTCC subtypes to the firing activity of DA neurons in the VTA by using transgenic mouse models that carry specific defects in LTCC subtypes (Platzer et al. 2000; Sinnegger-Brauns et al. 2004, 2009; Striessnig and Koschak 2008): Cav1.3-deficient mice (Cav1.3−/−; knockout mice) as well as a knockin mouse strain (Cav1.2DHP−/−) that expresses Cav1.2 LTCCs lacking high sensitivity to DHP LTCC activators and blockers.
MATERIALS AND METHODS
All procedures involving animal handling and tissue harvesting were reviewed and approved by the Institutional Animal Care Committees at the Memorial University of Newfoundland and Shenyang Pharmaceutical University. Mice were given food and water ad libitum and housed in a room on a 12:12-h light-dark cycle with temperature maintained at 24°C.
Animals
C57BL/6J mice and transgenic strains with a C57BL/6J background were used in this study. Wild-type (WT) mice were purchased from Charles River Canada. The Cav1.2DHP−/− mice carry the T1066Y mutation within the DHP binding domain of the Cav1.2 α1-subunit that makes it insensitive to DHP modulators (Striessnig et al. 2006; Zhang et al. 2007). Cav1.2DHP−/− mice show no physiological phenotype because Cav1.2 functions normally, while Cav1.3−/− mice have abnormalities such as bradycardia, deafness, and an antidepressant-like behavior (Busquet et al. 2010; Platzer et al. 2000; Striessnig et al. 2006; Striessnig and Koschak 2008).
Congenic transgenic mice were first bred from heterozygous pairs and maintained with Cav1.2DHP−/−-Cav1.2DHP−/− breeding pairs for DHP knockin mice. Cav1.3−/− females were poor breeders, and to increase the yield of Cav1.3−/− pups a breeding group consisting of a heterozygous Cav1.3+/− female, a Cav1.3−/− female, and a Cav1.3−/− male was established. Mice were genotyped as previously described (Giordano et al. 2006, 2010; Platzer et al. 2000; Zhang et al. 2007).
Patch-Clamp Recording
Slice preparation.
Mice (2–4 mo old) were deeply anesthetized with halothane and killed by chest compression. The skull was quickly opened to expose the brain, which was cooled in situ with ice-cold, carbogenated cutting solution (composition in mM: 250 glycerol, 2.5 KCl, 1.2 NaH2PO4, 1.2 MgCl2, 2.4 CaCl2, 26 NaHCO3, and 11 glucose, pH 7.4 when bubbled with 95% O2-5% CO2) (Liu and Chen 2008; Ye et al. 2006). The brain was removed, and a block containing the midbrain was cut horizontally on a Leica vibratome (VT 1000, Nussloch, Germany). Tissue slices (300–400 μm thick) were allowed to recover in a carbogenated artificial cerebrospinal fluid (ACSF, composition in mM: 126 NaCl, 2.5 KCl, 1.2 NaH2PO4, 1.2 MgCl2, 2.4 CaCl2, 18 NaHCO3, and 11 glucose, pH 7.4 when bubbled with 95% O2-5% CO2) at 31°C for 1 h and then maintained at room temperature (22°C) until use. Slices were further trimmed to fit into a recording chamber and continuously perfused with carbogenated ACSF at a rate of 2–3 ml/min at room temperature.
Nystatin-perforated patch-clamp recording.
All recordings were made from VTA identified under a dissecting microscope (Leica MZ6). Patch electrodes were prepared from KG-33 glass micropipettes (OD 1.5 mm; Garner Glass, Claremont, CA) on a P-97 Brown-Flaming micropipette puller (Sutter Instruments, Novato, CA). Glass electrodes were filled to the tip with intracellular solution (in mM: 120 potassium acetate, 40 HEPES, 5 MgCl2, and 10 EGTA, pH adjusted to 7.35 with 0.1 N KOH) and then back-filled with the same solution containing 450 μg/ml nystatin and Pluronic F127, yielding a tip resistance of 4–8 MΩ. Gigaohm seals were made with a MultiClamp 700B (Axon Instruments, Foster City, CA) amplifier. Signals were sampled at 5 kHz and digitized by a DigiData 1320A using pCLAMP 9 software (Axon Instruments).
Selection of nystatin-perforated cell recordings in current-clamp mode was determined by the size of the action potential, since many VTA cells were spontaneously active. After adequate partitioning of nystatin into the membrane, action potentials overshot 0 mV and measured at least 50 mV. Episodic protocols were used to induce Ih and derive passive characteristics of the cell such as current-voltage relationship and input resistance. Current pulses for Ih induction were 1 s, and the intervals between pulses were 8 s to allow complete recovery of Ih channels. Cells that displayed a prominent Ih and an apparent DA-induced hyperpolarization were identified as “putative DA cells,” because the neurons recorded were not confirmed to be dopaminergic by postrecording immunohistochemistry (Liu et al. 2007; Liu and Chen 2008).
Chemicals
Reagents for extracellular and intracellular solutions were purchased from bulk distributors Fisher Scientific (Nepean, ON, Canada) and VWR International (Mississauga, ON, Canada). All other chemicals were obtained from Sigma (St. Louis, MO) and Tocris (Ellisville, MO). Chemicals were dissolved in deionized water or DMSO (0.1% final concentration) as required. Aliquots of stock solutions were kept at −30°C. Prior to application, an aliquot was diluted to working concentration and applied to the ACSF bath. DA solution was made fresh daily with an equimolar concentration of the antioxidant disodium metabisulfite.
Data Analysis
Data were analyzed off-line with Mini Analysis (Synaptosoft, Decatur, GA) and pCLAMP software. Basal firing frequencies were averaged values of at least 5-min stable baseline recording. Ih was measured as the difference in voltage or current between instantaneous and steady-state readings. Analysis of firing behavior was based on interspike intervals (ISIs) measured with the Mini Analysis program. Averaged firing frequencies were derived from those intervals. Relative densities of ISIs in 0.1- and 0.5-s bins were plotted to reveal the distribution of a given ISI series. Burst firing was defined as two spikes or more in each bursting cycle at a frequency higher than nonbursting periods and separated by a postburst hyperpolarization, which was visually tagged and quantified. Data are expressed as means and SE. Statistical comparisons of data were performed with two-tailed paired or unpaired Student's t-test and χ2-test. Values were considered significant when P < 0.05.
RESULTS
For all experiments, putative DA cells in the VTA identified according to criteria outlined in materials and methods were included.
VTA DA Cells of Cav1.2DHP−/− and WT Mice Have Similar Basic Electrophysiological Properties
The percentage of spontaneously active cells in the WT group (18 of 31, from 25 mice, 58.1%) was similar to that in Cav1.2DHP−/− mice (32 of 50, from 42 mice, 64.0%; χ2-test, P > 0.05; Fig. 1C). Both groups displayed single-spike firing with comparable basal firing frequencies (0.68 ± 0.20 Hz for WT, 0.72 ± 0.11 Hz for Cav1.2DHP−/−, unpaired t-test, P > 0.05, Fig. 1; Table 1). The resting membrane potentials (RMPs) were similar between the two groups (−52.69 ± 1.17 mV for WT vs. −51.98 ± 1.03 mV for Cav1.2DHP−/−, P > 0.05, unpaired t-test); however, there was an apparent difference in RMPs of spiking and quiescent cells in both strains. The RMPs of spiking cells [for WT, −49.87 ± 0.59 mV (n = 18, from 18 mice); for Cav1.2DHP−/−, −49.98 ± 0.88 mV (n = 32, from 32 mice)] were more depolarized than those of quiescent cells [for WT, −56.01 ± 1.68 mV (unpaired t-test, P < 0.001); for Cav1.2DHP−/−, −57.89 ± 1.70 mV (unpaired t-test, P < 0.001)]. There were no differences in RMPs of spiking cells (P > 0.05) or quiescent cells (P > 0.05) between WT and Cav1.2DHP−/− groups. In terms of spiking cells, there were no significant differences in spike amplitude (58.00 ± 2.39 mV in WT mice vs. 58.13 ± 2.09 mV in Cav1.2DHP−/− mice, unpaired t-test, P > 0.05), threshold (−38.91 ± 0.76 mV in WT mice vs. −38.87 ± 0.88 mV in Cav1.2DHP−/− mice, unpaired t-test, P > 0.05), half-width (2.97 ± 0.11 ms in WT mice vs. 3.01 ± 0.15 ms in Cav1.2DHP−/− mice, unpaired t-test, P > 0.05), or hyperpolarization afterpotentials (−13.20 ± 1.57 mV in WT mice vs. −13.57 ± 1.30 mV in Cav1.2DHP−/−, unpaired t-test, P > 0.05). In addition, the average hyperpolarization following a brief application of 50 μM DA (within 90 s) was not significantly different between WT (−9.01 ± 1.02 mV) and Cav1.2DHP−/− (−9.46 ± 1.00 mV) mice (unpaired t-test, P > 0.05). When current injections were adjusted in each cell to result in a peak hyperpolarization of about −110 mV, the Ih sag was comparable between WT (20.98 ± 1.17 mV) and Cav1.2DHP−/− (20.09 ± 1.10 mV) (unpaired t-test, P > 0.05). These results confirm that VTA neurons of Cav1.2DHP−/− mice and WT mice have indistinguishable electrophysiological properties (Table 1).
Fig. 1.
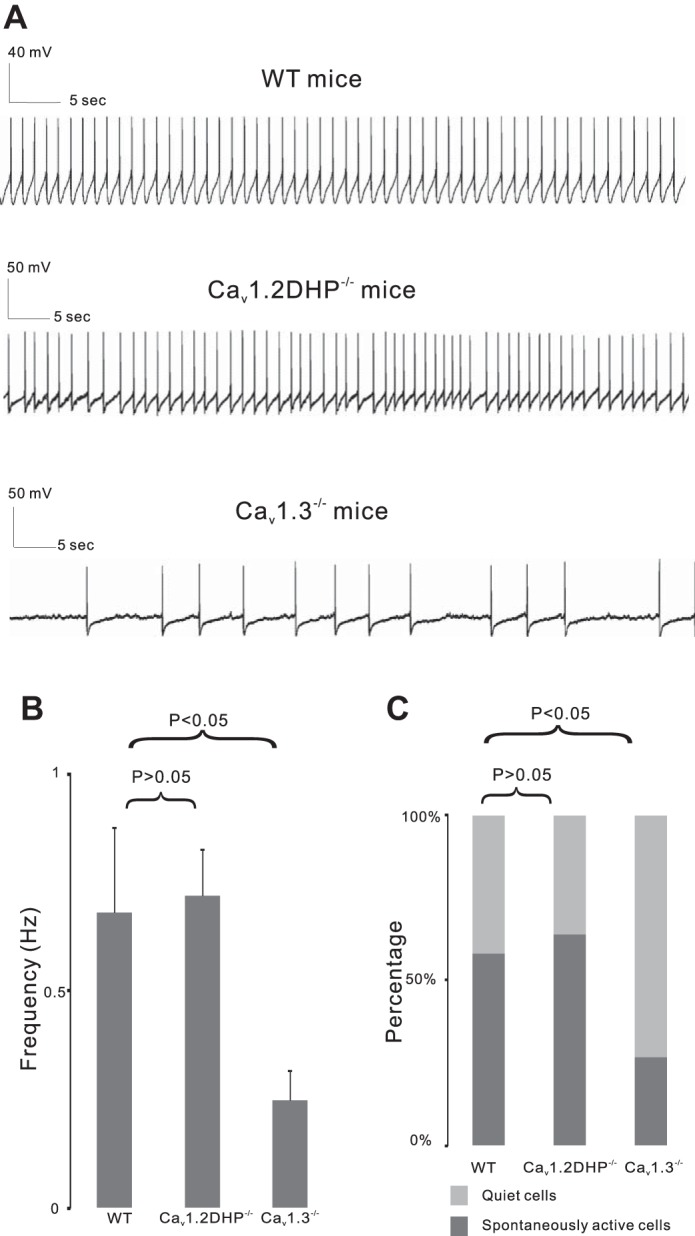
Dopaminergic cells have a lower basal firing frequency in Cav1.3−/− mice. A: current-clamp recording from representative cells showing basal firing rates in wild-type (WT) mice, mice expressing dihydropyridine (DHP)-insensitive Cav1.2 channels (Cav1.2DHP−/−), and Cav1.3-deficient (Cav1.3−/−) mice. B: basal firing frequencies in WT, Cav1.2DHP−/−, and Cav1.3−/− mice. C: % of quiet or spontaneously active cells in WT, Cav1.2DHP−/−, and Cav1.3−/− mice.
Table 1.
Basic electrophysiological properties of dopaminergic cells in wild type (WT), Cav1.2DHP−/− and Cav1.3−/− mice
| wild type (WT)1 | Cav1.2DHP−/−2 | Cav1.3−/−3 | P1,2 | P1,3 | |
|---|---|---|---|---|---|
| Number of spontaneously active cells | 18 of 31, 58.1% | 32 of 50, 64.0% | 7 of 26, 26.9% | >0.05 | <0.05 |
| Resting membrane potentials (mV) | −52.69 ± 1.17 | −51.98 ± 1.03 | −52.13 ± 1.18 | >0.05 | >0.05 |
| Basal firing frequencies (Hz) | 0.68 ± 0.20 | 0.72 ± 0.11 | 0.25 ± 0.07 | >0.05 | <0.05 |
| Spike amplitude (mV) | 58.00 ± 2.39 | 58.13 ± 2.09 | 54.62 ± 2.31 | >0.05 | >0.05 |
| Firing threshold (mV) | −38.91 ± 0.76 | −38.87 ± 0.88 | −41.03 ± 1.71 | >0.05 | >0.05 |
| Spike half-width (ms) | 2.97 ± 0.11 | 3.01 ± 0.15 | 2.97 ± 0.20 | >0.05 | >0.05 |
| Hyperpolarizing afterpotentials (mV) | −13.20 ± 1.57 | −13.57 ± 1.30 | −13.81 ± 1.51 | >0.05 | >0.05 |
| Hyperpolarization following an application of 50 μM dopamine (mV) | −9.01 ± 1.02 | −9.46 ± 1.00 | −7.96 ± 1.07 | >0.05 | >0.05 |
| Ih sag (mV) | 20.98 ± 1.17 | 20.09 ± 1.10 | 19.20 ± 1.00 | >0.05 | >0.05 |
Values are means ± SE.
DHP Channel Blocker Nifedipine Reduces Single-Spike Firing in Both WT and Cav1.2DHP−/− Mice
In WT mice with DHP-sensitive Cav1.2 and Cav1.3 channels, bath application of 5 μM nifedipine, a DHP LTCC blocker, for 10 min decreased baseline firing rate by 11.4 ± 4.1%, decreasing from 0.64 ± 0.12 Hz to 0.58 ± 0.15 Hz (n = 6, from 6 mice, paired t-test, P < 0.05) (Fig. 2). Nifedipine is lipophilic and difficult to wash out, so the firing rate continued to drop by 19.1 ± 5.5% from 0.64 ± 0.12 Hz to 0.52 ± 0.11 Hz after nifedipine was washed out for 6 min (paired t-test, P < 0.05; Fig. 2). To avoid spiking rundown, control cells without any drug application (Fig. 2) were recorded. The firing rate of control cells only decreased by 2.12 ± 1.01% from 0.58 ± 0.13 Hz to 0.57 ± 0.11 Hz after 15 min of recording (n = 6, from 6 mice, paired t-test, P > 0.05) and increased by 3.25 ± 0.98% from 0.58 ± 0.13 Hz to 0.60 ± 0.10 Hz after 21 min of recording (paired t-test, P > 0.05), demonstrating that decrease in firing rate following application of nifedipine in Fig. 2 is specific. In Cav1.2DHP−/− mice in which modulatory effects of nifedipine can only be mediated by Cav1.3, bath application of 5 μM nifedipine for 10 min reduced firing rates by 13.8 ± 6.3% from 0.61 ± 0.17 Hz to 0.52 ± 0.13 Hz (n = 6, from 6 mice, paired t-test, P < 0.05) (Fig. 2). A peak reduction of 20.7 ± 6.0% from 0.61 ± 0.17 Hz to 0.50 ± 0.15 Hz was recorded after nifedipine was washed out for 6 min (paired t-test, P < 0.05) (Fig. 2). Nifedipine at 10 μM reduced basal firing at greater percentages [20.3 ± 8.1% from 0.85 ± 0.17 Hz to 0.70 ± 0.21 Hz at 15 min (n = 5 from 5 WT mice, paired t-test, P < 0.05); 24.9 ± 7.7% from 0.77 ± 0.21 Hz to 0.63 ± 0.20 Hz at 15 min (n = 5 from 5 Cav1.2DHP−/− mice, paired t-test, P < 0.05)] (Fig. 2C). There was no significant difference in the reduction of basal firing by nifedipine between WT and Cav1.2DHP−/− mice (nifedipine 5 μM, unpaired t-test, P > 0.05; nifedipine 10 μM, unpaired t-test, P > 0.05) (Fig. 2C), indicating that the Cav1.3 subtype drives basal firing in VTA DA cells.
Fig. 2.
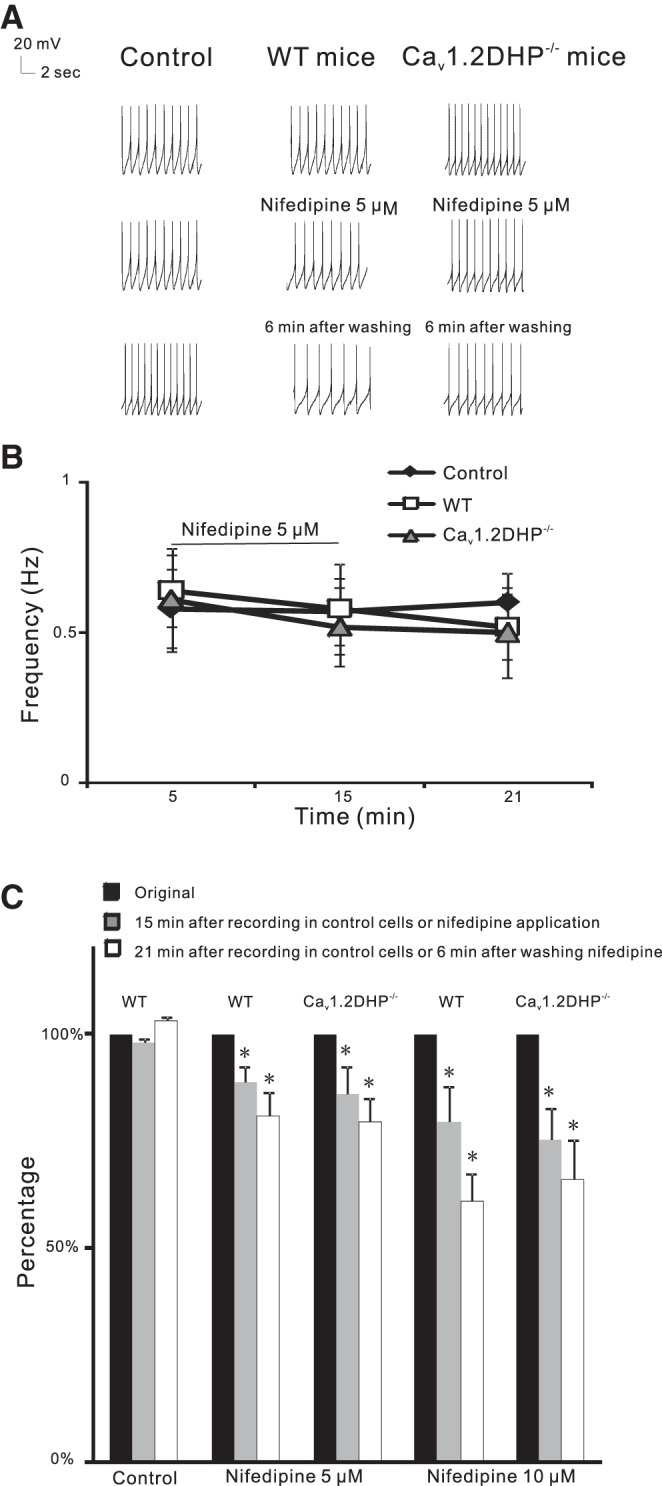
DHP site L-type calcium channel (LTCC) blocker nifedipine decreases basal firing rates in WT and Cav1.2DHP−/− mice. A: current-clamp recording from representative cells showing firing rates in control, with nifedipine (5 μM for 10 min), and after nifedipine was washed out for 6 min in control, WT, and Cav1.2DHP−/− mice. B: changes in firing frequencies after nifedipine (5 μM) in control, WT, and Cav1.2DHP−/− mice. C: % changes in firing rate reductions with or without nifedipine (5 or 10 μM for 10 min) in WT and Cav1.2DHP−/− mice. *P < 0.05.
Spontaneously Active DA Cells in Cav1.3−/− Mice Are Fewer and Have a Lower Basal Firing Frequency
To further confirm the involvement of Cav1.3 in basal firing, we studied the properties of VTA DA neurons from Cav1.3-deficient mice. Although Cav1.3−/− mice displayed an overall similarity in basic electrophysiological characteristics to WT, they exhibited a significantly lower number of spontaneously active cells and a significantly lower basal firing rate (Table 1). Seven of 26 cells from 26 mice (26.9%, χ2-test, P < 0.05 compared with that of WT group; Fig. 1C) were spontaneously active at a significantly lower basal firing frequency of 0.25 ± 0.07 Hz (unpaired t-test, P < 0.05 compared with that of WT group) (Fig. 1). Compared with WT, there were no differences in RMP (−52.13 ± 1.18 mV, unpaired t-test, P > 0.05), RMP of spiking [−47.25 ± 1.56 mV (n = 7, from 7 mice)] or quiescent [−53.82 ± 1.23 mV (n = 19, from 19 mice)] cells, spike amplitude (54.62 ± 2.31, unpaired t-test, P > 0.05), firing threshold (−41.03 ± 1.71 mV, unpaired t-test, P > 0.05), half-width (2.97 ± 0.20 ms, unpaired t-test, P > 0.05), or hyperpolarizing afterpotentials (−13.81 ± 1.51 mV, unpaired t-test, P > 0.05). Furthermore, compared with the WT group, Cav1.3−/− mice had similar average hyperpolarization following a brief application of 50 μM DA (−7.96 ± 1.07 mV, unpaired t-test, P > 0.05) and Ih sag (19.20 ± 1.00 mV, unpaired t-test, P > 0.05).
LTCC Activators Convert Single Spiking to Bursting in Mice
LTCC activators induce firing pattern conversion in rats (Liu et al. 2007; Zhang et al. 2005). We first set out to confirm that bursting could be induced by LTCC activators in mice. Bath application of 5 μM (S)-(−)-Bay K8644, a DHP LTCC activator, for 7–11 min induced burst firing in 7 of 12 treated cells from 12 WT mice regardless of their original basal firing activity being quiescent or single spiking (Table 2; Fig. 3A), the same as we reported in rats (Liu et al. 2007; Zhang et al. 2005). The lag between the start of drug application and burst firing ranged from 9 to 37 min with an average of 22 ± 4.5 min, while in control experiments (n = 6) with vehicle no bursts developed in 1 h. Bursts showed a typical action potential clustering (3–6 spikes/burst, average 4.14 ± 0.46 spikes/burst) followed by a postburst hyperpolarization. The firing frequency in the bursts (1.17 ± 0.27 Hz; Fig. 3C) was much higher than that before bursts (0.11 ± 0.08 Hz, paired t-test, P < 0.05), and the frequency of bursting cycles was 0.06 ± 0.03 Hz. The LTCC-mediated burst firing was long lasting even after prolonged washout (>30 min); however, it was readily blocked by the LTCC blocker nifedipine (10 μM for 10 min, n = 6 from 6 mice; Fig. 3A).
Table 2.
Burst properties of dopaminergic cells in wild type (WT), Cav1.2DHP−/− and Cav1.3−/− mice
| (S)-(−)-Bay K8644 |
FPL 64176 |
|||||||
|---|---|---|---|---|---|---|---|---|
| wild type (WT)1 | Cav1.2DHP−/−2 | P1,2 | wild type (WT)1 | Cav1.2DHP−/−2 | Cav1.3−/−3 | P1,2 | P1,3 | |
| Number of bursting cells after drug application | 7 of 12 | 7 of 14 | >0.05 | 11 of 16 | 7 of 14 | 10 of 22 | >0.05 | >0.05 |
| Lag between the start of drug application and bursting (min) | 9–37 | 16–40 | 11–40 | 13–42 | 14–50 | |||
| Average lag time (min) | 22 ± 4.5 | 26 ± 3.5 | >0.05 | 24 ± 3.4 | 21 ± 3.6 | 29 ± 2.5 | >0.05 | >0.05 |
| Number of long-lasting (>30 min) bursting cells | 7 of 7 | 6 of 7 | >0.05 | 11 of 11 | 6 of 7 | 6 of 10 | >0.05 | <0.05 |
| Number of inhibited cell by 10 μM nifedipine | 6 of 6 | 6 of 6 | >0.05 | 6 of 6 | 6 of 6 | 6 of 6 | >0.05 | >0.05 |
| number of spikes in bursts | 3–6 | 2–13 | 2–9 | 2–20 | 2–19 | |||
| average number of spikes | 4.14 ± 0.46 | 4.39 ± 1.18 | >0.05 | 4.28 ± 0.79 | 4.45 ± 1.12 | 5.06 ± 1.46 | >0.05 | >0.05 |
| firing frequency before bursts (Hz) | 0.11 ± 0.08 | 0.45 ± 0.16 | >0.05 | 0.75 ± 0.17 | 0.29 ± 0.12 | 0.0014 ± 0.003 | >0.05 | >0.05 |
| firing frequency in the bursts (Hz) | 1.17 ± 0.27 | 1.38 ± 0.32 | >0.05 | 2.01 ± 0.44 | 5.91 ± 4.55 | 2.26 ± 1.08 | >0.05 | >0.05 |
| bursts frequency (Hz) | 0.06 ± 0.03 | 0.10 ± 0.05 | >0.05 | 0.12 ± 0.04 | 0.20 ± 0.07 | 0.04 ± 0.01 | >0.05 | >0.05 |
| interspike interval of spikes in burst (sec) | 1.02 ± 0.19 | 0.93 ± 0.16 | >0.05 | 0.68 ± 0.17 | 0.76 ± 0.22 | 1.50 ± 0.33 | >0.05 | >0.05 |
| interburst interval (sec) | 24.86 ± 8.27 | 20.44 ± 4.68 | >0.05 | 13.86 ± 3.74 | 13.16 ± 5.49 | 32.43 ± 5.04 | >0.05 | >0.05 |
Values are means ± SE.
Fig. 3.

DHP site LTCC activator (S)-(−)-Bay K8644 converts firing pattern in WT mice. A: continuous current-clamp recording from a representative cell showing that (S)-(−)-Bay K8644 (5 μM) converted regular firing to burst firing that could be blocked by nifedipine (10 μM). B: density plot of interspike intervals (ISIs) in 0.1-s (<8 s) or 0.5-s (>8 s) bins from the same cell as in A in control conditions (241 events) and after (S)-(−)-Bay K8644 application (994 events). (S)-(−)-Bay K8644 dramatically shifted the peak to the left and gave rise to a secondary peak corresponding to the frequency of burst firing cycles. C: the firing frequency in the bursts was much higher than that before bursts (left), and the frequency of bursts was low (right), representing long pauses of firing between adjacent bursts.
Similarly, bath application of 1–4 μM FPL 64176, a benzoylpyrole site LTCC activator, for 8–24 min converted firing patterns from quiescent state or single spiking to burst firing in 11 of 16 treated cells from 16 mice (Table 2; Fig. 4). The time course was similar to that after (S)-(−)-Bay K8644, a depolarization and an increase in firing rates followed by a conversion of firing patterns 11–40 min (average: 24 ± 3.4 min) after the start of application. The burst firing was long lasting and could be reverted to single-spike firing or no firing by nifedipine (10 μM, 5–10 min, n = 6 from 6 mice; Fig. 4A). The bursts themselves had 2–9 spikes/burst (average 4.28 ± 0.79 spikes/burst). The firing frequency of spikes in the bursts (2.01 ± 0.44 Hz; Fig. 4C) was much higher than that before the bursts (0.75 ± 0.17 Hz, paired t-test, P < 0.05), and the frequency of bursting cycles was 0.12 ± 0.04 Hz.
Fig. 4.

Non-DHP site LTCC activator FPL 64176 converts firing pattern in WT mice. A: continuous current-clamp recording from a representative cell showing that FPL 64176 (2 μM) converted regular firing to burst firing that could be blocked by nifedipine (10 μM). B: density plot of ISIs in 0.1-s (<2.5 s) or 0.5-s (>2.5 s) bins from the same cell as in A in control conditions (425 events) and after FPL 64176 application (744 events). FPL 64176 dramatically shifted the peak to the left and gave rise to a secondary peak corresponding to the frequency of burst firing cycles. C: the firing frequency in the bursts was much higher than that before bursts (left), and the frequency of bursts was low (right), representing long pauses of firing between adjacent bursts.
The DHP LTCC Activator Converts Firing Patterns in Cav1.2DHP−/− Mice
To further explore which of the two channel isoforms mediates drug-induced burst firing, Cav1.2DHP−/− mice were used in which (S)-(−)-Bay K8644 selectively activates only Cav1.3 but not Cav1.2 (Sinnegger-Brauns et al. 2004). Bath application of 5 μM (S)-(−)-Bay K8644 for 8–20 min induced burst firing in 7 of 14 cells tested in 14 Cav1.2DHP−/− mice (Fig. 5), similar to WT. The lag between the start of drug application and burst firing was longer but not significantly different (range 16–40 min with the average time of 26 ± 3.5 min, unpaired t-test, P > 0.05 compared with WT group). As in WT, burst firing was long lasting in six spontaneously firing cells but only lasted for 6 min in a quiescent cell. Nifedipine (10 μM for 10 min) reverted (S)-(−)-Bay K8644-induced burst firing to single-spike firing (n = 6, from 6 mice) (Fig. 5A). The bursts had 2–13 spikes/burst with an average of 4.39 ± 1.18 spikes/burst. The intraburst firing frequency (1.38 ± 0.32 Hz; Fig. 5C) was much higher than that before the bursts (0.45 ± 0.16 Hz, paired t-test, P < 0.05), and the frequency of bursting cycles was 0.10 ± 0.05 Hz.
Fig. 5.

DHP site LTCC activator (S)-(−)-Bay K8644 converts firing pattern in Cav1.2DHP−/− mice. A: continuous current-clamp recording from a representative cell showing that (S)-(−)-Bay K8644 (5 μM) converted regular firing to burst firing that could be blocked by nifedipine (10 μM). B: density plot of ISIs in 0.1-s (<2 s) or 0.5-s (>2 s) bins from the same cell as in A in control conditions (209 events) and after (S)-(−)-Bay K8644 application (576 events). (S)-(−)-Bay K8644 dramatically shifted the peak to the left and gave rise to a secondary peak corresponding to the frequency of burst firing cycles. C: the firing frequency in the bursts was much higher than that before bursts (left), and the frequency of bursts was low (right), representing long pauses of firing between adjacent bursts.
DHP Site LTCC Blocker Inhibits Burst Firing Induced by a Non-DHP Site LTCC Activator in Cav1.2DHP−/− Mice
Similarly to WT mice, bath application of 2–4 μM FPL 64176 for 13–24 min converted firing patterns from quiescent state or single spiking to burst firing in 7 of 14 treated cells from 14 Cav1.2DHP−/− mice (Fig. 6). The responses were dose dependent: 4 μM FPL 64176 induced burst firing in three of six cells that did not respond to 2 μM. The time course was similar to that in WT mice, a depolarization and an increase in firing rates followed by a conversion of firing patterns 13–42 min (average: 21 ± 3.6 min, unpaired t-test, P > 0.05) after the start of application. The induced burst firing was long lasting except for one in the quiescent cell that lasted for only 9 min. Nifedipine (10 μM for 5–13 min, n = 6 from 6 mice; Fig. 6) inhibited the induced burst firing, being converted back into single-spike firing or into protracted and slow bursting resembling irregular firing. The bursts had 2–20 spikes/burst, (average 4.45 ± 1.12 spikes/burst). The intraburst firing frequency (5.91 ± 4.55 Hz; Fig. 6C) was much higher than that before the bursts (0.29 ± 0.12 Hz, paired t-test, P < 0.05), and the frequency of bursting cycles was 0.20 ± 0.07 Hz.
Fig. 6.

Non-DHP site LTCC activator FPL 64176 converts firing pattern in Cav1.2DHP−/− mice. A: continuous current-clamp recording from a representative cell showing that the non-DHP site LTCC activator FPL 64176 (4 μM) converted regular firing to burst firing that could be blocked by the DHP site LTCC blocker nifedipine (10 μM) in Cav1.2DHP−/− mice. B: density plot of ISIs in 0.1-s (<3 s) or 0.5-s (>3 s) bins from the same cell as in A in control conditions (108 events) and after FPL 64176 application (308 events). C: the firing frequency in the bursts was much higher than that before bursts (left), and the frequency of bursts was low (right), representing long pauses of firing between adjacent bursts.
The LTCC Activator Converts Firing Patterns in Cav1.3−/− Mice
The fact that Cav1.3 channels alone control basal firing rate does not exclude the possibility that activation of Cav1.2 channels participates in burst firing. Because LTCC activators can shift the activation voltage of Cav1.2 LTCCs to more negative voltages (Bargas et al. 1994; Fan et al. 2000) it might enhance gating of Cav1.2 at threshold voltages and thereby substitute for Cav1.3, as recently proposed for medium spiny neurons (Olson et al. 2005). To address the role of Cav1.2 we measured the response of VTA neurons in Cav1.3−/− brains. Bath application of 2–4 μM FPL 64176 for 12–32 min also converted firing patterns to burst firing in 10 of 22 treated cells from 20 mice in Cav1.3−/− mice (Fig. 7). The responses were dose dependent: 4 μM FPL 64176 induced burst firing in 6 of 11 cells that did not respond to 2 μM. The time course was a little bit different but not significantly: the lag between the start of drug application and appearance of burst firing was 14–50 min (average: 29 ± 2.5 min, unpaired t-test, P > 0.05 compared with that of WT group). The induced burst firing was not all long lasting (>30 min, Fig. 7B), and among 10 bursting cells 4 lasted only for 8, 13, 15, and 22 min (χ2-test, P < 0.05 compared with that of WT group). The bursts had 2–19 spikes/burst (average 5.06 ± 1.46 spikes/burst). The intraburst firing frequency (2.26 ± 1.08 Hz) was much higher than that before the bursts (0.0014 ± 0.0030 Hz, paired t-test, P < 0.05), and the frequency of bursting cycles was 0.04 ± 0.01 Hz. Nifedipine (10 μM for 5–21 min; Fig. 7A) reverted the induced burst firing (n = 6, from 6 mice) back to single-spike firing or no firing. These data clearly demonstrate that activation of Cav1.2 in the absence of Cav1.3 channels is sufficient to induce bursting.
Fig. 7.

LTCC activator FPL 64176 converts firing pattern in Cav1.3−/− mice. A: continuous current-clamp recording from a representative cell showing that FPL 64176 (4 μM) converted regular firing to burst firing that could be blocked by nifedipine (10 μM). B: continuous current-clamp recording from a representative cell showing that the induced burst firing by FPL 64176 in Cav1.3−/− mice was shorter in duration (<30 min).
DISCUSSION
The firing of DA cells in the VTA dictates the strength of DA transmission that is associated with normal or abnormal expression of motivation and reward processing. The LTCCs promote burst firing of these cells (Liu et al. 2007; Zhang et al. 2005), which is essential for salient stimuli (Chergui et al. 1994; Romo and Schultz 1990; Schultz 2007, 2010; Tobler et al. 2005). The novel finding of this study is that, by using two mouse models that allow the selective study of the two LTCC subtypes expressed in the brain, Cav1.2 and Cav1.3, we have uncovered their specific contributions to firing regulation. Cav1.3 LTCCs are crucial to regulate basal single-spike firing, while for burst firing both Cav1.2 and Cav1.3 LTCCs are involved.
Cav1.3 LTCCs Play an Important Role in Regulating Basal Single-Spike Firing
LTCCs are often assumed to be high voltage activated (e.g., activate between −40 and −20 mV membrane potentials) (Ertel et al. 2000), and Cav1.2 LTCCs display this typical voltage dependence (Mori et al. 1993). However, Cav1.3 LTCCs activate at relatively hyperpolarized membrane potentials (between −60 and −40 mV at physiological extracellular Ca2+ concentrations) (Xu and Lipscombe 2001), which is near the RMP of DA neurons in the VTA. The difference in activation thresholds between these two subtypes implies that they may be coupled to different molecular signaling pathways (Giordano et al. 2010; Schierberl et al. 2011; Zhang et al. 2006) so as to mediate different physiological roles in neuronal electrical tasks. For example, Cav1.3 channels are likely to be important in mediating Ca2+ influx in response to relatively small membrane depolarizations, and such properties may be important for sustaining spontaneous rhythmic firing in neurons. Consistent with this, the DHP blocker nimodipine partially suppresses spontaneous intracellular calcium oscillations and slows rhythmic firing in postnatal cerebellar Purkinje cells (Liljelund et al. 2000). More direct evidence for the involvement of Cav1.3 channels in driving rhythmic activities in excitable cells comes from our previous study of Cav1.3-deficient mice whose phenotype includes compromised sinoatrial node function (Platzer et al. 2000). In line with this, the present study shows that Cav1.3 channels mediated rhythmic firing in DA neurons in the VTA because 1) Cav1.3-deficient mice, in which there are no Cav1.3 but Cav1.2 LTCCs expressed, have significantly lower basal firing frequencies; and 2) compared with WT mice, the DHP blocker nifedipine slowed spontaneous rhythmic firing to a similar extent in Cav1.2 knockin mice, in which Cav1.2 channels were not sensitive to DHP blockers and only Cav1.3 channels were blocked by nifedipine. The essential role for Cav1.3 channels to mediate spontaneous firing of DA neurons in the substantia nigra has been shown (Chan et al. 2007); other reports showed that LTCC blocker at higher concentrations (20 μM) affects channels other than the LTCCs and has differing actions on dendritic calcium oscillations and firing (Chan et al. 2007; Guzman et al. 2009; Khaliq and Bean 2010). Our results with nifedipine at different concentrations, consistent results with LTCC activator and blocker, and reduced basal firing from Cav1.3−/− slices where no activator or blocker was present indicate that basal firing of putative DA neurons in the VTA is driven by LTCCs, as reported previously by Mercuri et al. (1994), and is not due to nifedipine's nonspecific actions on other channels.
Both Cav1.2 and Cav1.3 LTCCs Play a Role in VTA DA Burst Firing
Different from pacemaker firing, burst firing is a combination of subthreshold depolarization and action potential clustering, which is likely to be mediated by both Cav1.3 (activated at relatively hyperpolarized membrane potentials) (Xu and Lipscombe 2001) and Cav1.2 (high voltage activated) (Mori et al. 1993). It is reasonable to presume that Cav1.3 LTCCs mediate the subthreshold depolarization that raises the membrane potential to activate sodium channels initiating firing and then Cav1.2 LTCCs take over to maintain sequential firing. Our results suggest that Cav1.3 LTCCs are sufficient for bursting when both subtypes are present, while in the absence of the Cav1.3 subtype Cav1.2 subtype can drive bursting on its own.
Cav1.3 LTCCs are involved in bursting under normal conditions because 1) activating both Cav1.2 and Cav1.3 subtypes by (S)-(−)-Bay K8644 (in WT slices) induced burst firing similarly to activating Cav1.3 subtype alone (in Cav1.2DHP−/− slices); 2) Bay K-induced burst firing could be blocked by nifedipine in both WT slices (effective for both subtypes) and Cav1.2DHP−/− slices (effective only for Cav1.3); and 3) activating both subtypes with FPL 64176 induced similar burst firing that could be blocked to the same extent with nifedipine (which blocks Cav1.2 and Cav1.3 in WT slices and Cav1.3 only in Cav1.2DHP−/− slices). These results collectively demonstrate that Cav1.3 LTCCs are obligatory for burst firing, which infers that bursting would not be induced if Cav1.3 LTCCs are absent. Surprisingly, FPL 64176 also induced bursting in Cav1.3−/− mice, in which only Cav1.2 LTCCs exist. The explanations of the discrepancy between results from Cav1.2DHP−/− mice and Cav1.3−/− mice might be compensatory upregulation of Cav1.2 in these neurons in Cav1.3 knockouts. Although we cannot rule out this possibility, it appears unlikely because such upregulation has not been observed in any of the cellular systems investigated so far in these mice (Marcantoni et al. 2010; Platzer et al. 2000). A more likely explanation has recently been provided in medium spiny neurons (Olson et al. 2005). In these cells Cav1.3 stabilizes upstate potentials by means of inward current at threshold potentials allowing prolonged firing of these cells. This effect is absent in Cav1.3−/− neurons but can be rescued by treatment with (S)-(−)-Bay K8644. This can be rationalized by the fact that (S)-(−)-Bay K8644 and FPL 64176 shift activation voltage to more negative voltages, thereby promoting Cav1.2 activity at threshold voltages (Bargas et al. 1994; Fan et al. 2000). However, such Cav1.2 activation by activators might be a purely pharmacological finding, and whether it exists in the real cells remains unclear. Because of the ability of these LTCC activators to modulate channel kinetics in ways that may not occur under physiological conditions, their effects may not necessarily predict functional changes involved during physiological phenomena. FPL 64176 and (S)-(−)-Bay K8644 open LTCC channels by altering gating, prolonging opening, and increasing conductance of the channels (Fan et al. 2001; McDonough et al. 2005), probably different from physiological stimuli on LTCCs. However, it is possible that, for example, receptor-mediated protein kinase A (PKA) phosphorylation of LTCCs, which also enhances open probability and allows activation at lower voltages (Kamp and Hell 2000), can induce activity similar to pharmacological calcium channel activation.
Therefore, we believe that when both LTCC subtypes are present within the VTA Cav1.3 plays a more prominent role than Cav1.2 in regulating DA cell firing and the resulting spike-dependent DA release in its terminals. This understanding of subtype roles was further supported when we blocked the LTCC subtype Cav1.3 in the Cav1.2DHP−/− mice, as bursting was completely abolished and was returned to control levels. So given the outcomes in both WT and Cav1.2DHP−/− mice, this strongly indicates that LTCC subtype Cav1.3 has a more active and crucial role in bursting than Cav1.2.
Cav1.2 LTCCs are not necessary for burst firing under normal conditions but might aid it in some way, because our data showed that the latency to bursting (lag time) was slightly longer in Cav1.2DHP−/− slices than in WT slices, and in a few cases induced bursting did not last as long. Also, in Cav1.3−/− mice significantly fewer bursts induced by activation of LTCCs lasted as long as in WT mice. The mechanism for this might be related to contributions to the hump potential, which is already shown to be mediated by LTCCs (Zhang et al. 2005), similar to the plateau potential underlying bistable membrane behavior in motor neurons in which Cav1.3 LTCCs are involved (Carlin et al. 2000; Hsiao et al. 1998).
Cav1.3 LTCCs are Functionally Important
Burst firing of VTA DA neurons and phasic dopamine release at their terminals in the nucleus accumbens have been shown to be crucial for drug seeking behavior in rodent models of addiction (Gardner 2011; Koob and Volkow 2010). The precise molecular mechanisms involved in VTA DA neuron burst firing that underlies drug seeking behavior remain largely unknown; however, our finding in this study that Cav1.3 channels are important for the transition from spontaneous to burst firing strongly supports a role for Cav1.3 channels. This is further supported by our previous finding that Cav1.3 channels and their Ca2+-activated pathways, in VTA DA neurons, are recruited after repeated amphetamine treatment (Giordano et al. 2006; Rajadhyaksha et al. 2004) and additionally play a critical role in cocaine's long-term behavioral effects (Schierberl et al. 2011). In addition to reward seeking behavior, increased burst firing of VTA DA neurons has also been found in rodent models of depression (Cao et al. 2010; Krishnan et al. 2007; Razzoli et al. 2011). Our finding of a role for Cav1.3 channels in VTA DA neuron burst firing supports a potential role for Cav1.3-mediated burst firing in depression-related behaviors. This hypothesis is further supported by our previous finding that Cav1.3 channels modulate depression-like behaviors, as identified in the Cav1.3 knockout mice that we have used in this study (Busquet et al. 2010). Taken together, LTCCs, especially Cav1.3 subtypes, might play a significant role in central DA transmission and its related pathologies. Further experiments on the role of LTCC subtypes would have benefits for the therapies of these DA-related diseases.
GRANTS
This work was supported by the National Natural Science Foundation of China (Y. Liu, 31000483), the Scientific Research Foundation for the Returned Overseas Chinese Scholars, the State Education Ministry (Y. Liu), the National Science and Engineering Research Council of Canada (X. Chen, 202999), and the Austrian Science Fund (J. Striessnig, F44020).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
AUTHOR CONTRIBUTIONS
Author contributions: Y.L. and X.C. conception and design of research; Y.L., M.H., and A.P. performed experiments; Y.L. analyzed data; Y.L. and X.C. interpreted results of experiments; Y.L. prepared figures; Y.-d.L. drafted manuscript; Y.L., J.D., J.S., A.R., and X.C. edited and revised manuscript; Y.L., M.H., A.P., J.D., J.S., A.R., and X.C. approved final version of manuscript.
REFERENCES
- Bargas J, Howe A, Eberwine J, Cao Y, Surmeier DJ. Cellular and molecular characterization of Ca2+ currents in acutely isolated, adult rat neostriatal neurons. J Neurosci 14: 6667–6686, 1994 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Busquet P, Nguyen NK, Schmid E, Tanimoto N, Seeliger MW, Ben-Yosef T, Mizuno F, Akopian A, Striessnig J, Singewald N. CaV1.3 L-type Ca2+ channels modulate depression-like behaviour in mice independent of deaf phenotype. Int J Neuropsychopharmacol 13: 499–513, 2010 [DOI] [PubMed] [Google Scholar]
- Cao JL, Covington HE, III, Friedman AK, Wilkinson MB, Walsh JJ, Cooper DC, Nestler EJ, Han MH. Mesolimbic dopamine neurons in the brain reward circuit mediate susceptibility to social defeat and antidepressant action. J Neurosci 30: 16453–16458, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cardozo DL, Bean BP. Voltage-dependent calcium channels in rat midbrain dopamine neurons: modulation by dopamine and GABAB receptors. J Neurophysiol 74: 1137–1148, 1995 [DOI] [PubMed] [Google Scholar]
- Carlin KP, Jones KE, Jiang Z, Jordan LM, Brownstone RM. Dendritic L-type calcium currents in mouse spinal motoneurons: implications for bistability. Eur J Neurosci 12: 1635–1646, 2000 [DOI] [PubMed] [Google Scholar]
- Chan CS, Guzman JN, Ilijic E, Mercer JN, Rick C, Tkatch T, Meredith GE, Surmeier DJ. “Rejuvenation” protects neurons in mouse models of Parkinson's disease. Nature 447: 1081–1086, 2007 [DOI] [PubMed] [Google Scholar]
- Chergui K, Suaud-Chagny MF, Gonon F. Nonlinear relationship between impulse flow, dopamine release and dopamine elimination in the rat brain in vivo. Neuroscience 62: 641–645, 1994 [DOI] [PubMed] [Google Scholar]
- Cooper DC. The significance of action potential bursting in the brain reward circuit. Neurochem Int 41: 333–340, 2002 [DOI] [PubMed] [Google Scholar]
- Durante P, Cardenas CG, Whittaker JA, Kitai ST, Scroggs RS. Low-threshold L-type calcium channels in rat dopamine neurons. J Neurophysiol 91: 1450–1454, 2004 [DOI] [PubMed] [Google Scholar]
- Ertel EA, Campbell KP, Harpold MM, Hofmann F, Mori Y, Perez-Reyes E, Schwartz A, Snutch TP, Tanabe T, Birnbaumer L, Tsien RW, Catterall WA. Nomenclature of voltage-gated calcium channels. Neuron 25: 533–535, 2000 [DOI] [PubMed] [Google Scholar]
- Eyles D, Feldon J, Meyer U. Schizophrenia: do all roads lead to dopamine or is this where they start? Evidence from two epidemiologically informed developmental rodent models. Transl Psychiatry 2: e81, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan JS, Yuan Y, Palade P. Kinetic effects of FPL 64176 on L-type Ca2+ channels in cardiac myocytes. Naunyn Schmiedebergs Arch Pharmacol 361: 465–476, 2000 [DOI] [PubMed] [Google Scholar]
- Fan J, Yuan Y, Palade P. FPL-64176 modifies pore properties of L-type Ca2+ channels. Am J Physiol Cell Physiol 280: C565–C572, 2001 [DOI] [PubMed] [Google Scholar]
- Gardner EL. Addiction and brain reward and antireward pathways. Adv Psychosom Med 30: 22–60, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giordano TP, III, Satpute SS, Striessnig J, Kosofsky BE, Rajadhyaksha AM. Up-regulation of dopamine D2L mRNA levels in the ventral tegmental area and dorsal striatum of amphetamine-sensitized C57BL/6 mice: role of Cav1.3 L-type Ca2+ channels. J Neurochem 99: 1197–1206, 2006 [DOI] [PubMed] [Google Scholar]
- Giordano TP, Tropea TF, Satpute SS, Sinnegger-Brauns MJ, Striessnig J, Kosofsky BE, Rajadhyaksha AM. Molecular switch from L-type Cav1.3 to Cav1.2 Ca2+ channel signaling underlies long-term psychostimulant-induced behavioral and molecular plasticity. J Neurosci 30: 17051–17062, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guzman JN, Sanchez-Padilla J, Chan CS, Surmeier DJ. Robust pacemaking in substantia nigra dopaminergic neurons. J Neurosci 29: 11011–11019, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsiao CF, Del Negro CA, Trueblood PR, Chandler SH. Ionic basis for serotonin-induced bistable membrane properties in guinea pig trigeminal motoneurons. J Neurophysiol 79: 2847–2856, 1998 [DOI] [PubMed] [Google Scholar]
- Ikemoto S. Dopamine reward circuitry: two projection systems from the ventral midbrain to the nucleus accumbens-olfactory tubercle complex. Brain Res Rev 56: 27–78, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikemoto S. Brain reward circuitry beyond the mesolimbic dopamine system: a neurobiological theory. Neurosci Biobehav Rev 35: 129–150, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iversen SD, Iversen LL. Dopamine: 50 years in perspective. Trends Neurosci 30: 188–193, 2007 [DOI] [PubMed] [Google Scholar]
- Kamp TJ, Hell JW. Regulation of cardiac L-type calcium channels by protein kinase A and protein kinase C. Circ Res 87: 1095–1102, 2000 [DOI] [PubMed] [Google Scholar]
- Khaliq ZM, Bean BP. Pacemaking in dopaminergic ventral tegmental area neurons: depolarizing drive from background and voltage-dependent sodium conductances. J Neurosci 30: 7401–7413, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koob GF, Volkow ND. Neurocircuitry of addiction. Neuropsychopharmacology 35: 217–238, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krishnan V, Han MH, Graham DL, Berton O, Renthal W, Russo SJ, Laplant Q, Graham A, Lutter M, Lagace DC, Ghose S, Reister R, Tannous P, Green TA, Neve RL, Chakravarty S, Kumar A, Eisch AJ, Self DW, Lee FS, Tamminga CA, Cooper DC, Gershenfeld HK, Nestler EJ. Molecular adaptations underlying susceptibility and resistance to social defeat in brain reward regions. Cell 131: 391–404, 2007 [DOI] [PubMed] [Google Scholar]
- Liljelund P, Netzeband JG, Gruol DL. L-type calcium channels mediate calcium oscillations in early postnatal Purkinje neurons. J Neurosci 20: 7394–7403, 2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Chen X. Cholinergic excitation of dopaminergic cells depends on sequential activation of protein kinase C and the L-type calcium channel in ventral tegmental area slices. Brain Res 1245: 41–51, 2008 [DOI] [PubMed] [Google Scholar]
- Liu Y, Dore J, Chen X. Calcium influx through L-type channels generates protein kinase M to induce burst firing of dopamine cells in the rat ventral tegmental area. J Biol Chem 282: 8594–8603, 2007 [DOI] [PubMed] [Google Scholar]
- Marcantoni A, Vandael DH, Mahapatra S, Carabelli V, Sinnegger-Brauns MJ, Striessnig J, Carbone E. Loss of Cav1.3 channels reveals the critical role of L-type and BK channel coupling in pacemaking mouse adrenal chromaffin cells. J Neurosci 30: 491–504, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDonough SI, Mori Y, Bean BP. FPL 64176 modification of CaV1.2 L-type calcium channels: dissociation of effects on ionic current and gating current. Biophys J 88: 211–223, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mercuri NB, Bonci A, Calabresi P, Stratta F, Stefani A, Bernardi G. Effects of dihydropyridine calcium antagonists on rat midbrain dopaminergic neurones. Br J Pharmacol 113: 831–838, 1994 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mori Y, Niidome T, Fujita Y, Mynlieff M, Dirksen RT, Beam KG, Iwabe N, Miyata T, Furutama D, Furuichi T. Molecular diversity of voltage-dependent calcium channel. Ann NY Acad Sci 707: 87–108, 1993 [DOI] [PubMed] [Google Scholar]
- Nedergaard S, Flatman JA, Engberg I. Nifedipine- and omega-conotoxin-sensitive Ca2+ conductances in guinea-pig substantia nigra pars compacta neurones. J Physiol 466: 727–747, 1993 [PMC free article] [PubMed] [Google Scholar]
- Olson PA, Tkatch T, Hernandez-Lopez S, Ulrich S, Ilijic E, Mugnaini E, Zhang H, Bezprozvanny I, Surmeier DJ. G-protein-coupled receptor modulation of striatal CaV1.3 L-type Ca2+ channels is dependent on a Shank-binding domain. J Neurosci 25: 1050–1062, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Platzer J, Engel J, Schrott-Fischer A, Stephan K, Bova S, Chen H, Zheng H, Striessnig J. Congenital deafness and sinoatrial node dysfunction in mice lacking class D L-type Ca2+ channels. Cell 102: 89–97, 2000 [DOI] [PubMed] [Google Scholar]
- Rajadhyaksha A, Husson I, Satpute SS, Kuppenbender KD, Ren JQ, Guerriero RM, Standaert DG, Kosofsky BE. L-type Ca2+ channels mediate adaptation of extracellular signal-regulated kinase 1/2 phosphorylation in the ventral tegmental area after chronic amphetamine treatment. J Neurosci 24: 7464–7476, 2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Razzoli M, Andreoli M, Michielin F, Quarta D, Sokal DM. Increased phasic activity of VTA dopamine neurons in mice 3 weeks after repeated social defeat. Behav Brain Res 218: 253–257, 2011 [DOI] [PubMed] [Google Scholar]
- Romo R, Schultz W. Dopamine neurons of the monkey midbrain: contingencies of responses to active touch during self-initiated arm movements. J Neurophysiol 63: 592–606, 1990 [DOI] [PubMed] [Google Scholar]
- Schierberl K, Hao J, Tropea TF, Ra S, Giordano TP, Xu Q, Garraway SM, Hofmann F, Moosmang S, Striessnig J, Inturrisi CE, Rajadhyaksha AM. Cav1.2 L-type Ca2+ channels mediate cocaine-induced GluA1 trafficking in the nucleus accumbens, a long-term adaptation dependent on ventral tegmental area Cav1.3 channels. J Neurosci 31: 13562–13575, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schultz W. Multiple dopamine functions at different time courses. Annu Rev Neurosci 30: 259–288, 2007 [DOI] [PubMed] [Google Scholar]
- Schultz W. Dopamine signals for reward value and risk: basic and recent data. Behav Brain Funct 6: 24, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi WX. Electrophysiological characteristics of dopamine neurons: a 35-year update. J Neural Transm Suppl 2009: 103–119, 2009 [DOI] [PubMed] [Google Scholar]
- Sinnegger-Brauns MJ, Hetzenauer A, Huber IG, Renstrom E, Wietzorrek G, Berjukov S, Cavalli M, Walter D, Koschak A, Waldschutz R, Hering S, Bova S, Rorsman P, Pongs O, Singewald N, Striessnig JJ. Isoform-specific regulation of mood behavior and pancreatic beta cell and cardiovascular function by L-type Ca2+ channels. J Clin Invest 113: 1430–1439, 2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sinnegger-Brauns MJ, Huber IG, Koschak A, Wild C, Obermair GJ, Einzinger U, Hoda JC, Sartori SB, Striessnig J. Expression and 1,4-dihydropyridine-binding properties of brain L-type calcium channel isoforms. Mol Pharmacol 75: 407–414, 2009 [DOI] [PubMed] [Google Scholar]
- Striessnig J, Koschak A. Exploring the function and pharmacotherapeutic potential of voltage-gated Ca2+ channels with gene knockout models. Channels (Austin) 2: 233–251, 2008 [DOI] [PubMed] [Google Scholar]
- Striessnig J, Koschak A, Sinnegger-Brauns MJ, Hetzenauer A, Nguyen NK, Busquet P, Pelster G, Singewald N. Role of voltage-gated L-type Ca2+ channel isoforms for brain function. Biochem Soc Trans 34: 903–909, 2006 [DOI] [PubMed] [Google Scholar]
- Takada M, Kang Y, Imanishi M. Immunohistochemical localization of voltage-gated calcium channels in substantia nigra dopamine neurons. Eur J Neurosci 13: 757–762, 2001 [DOI] [PubMed] [Google Scholar]
- Tobler PN, Fiorillo CD, Schultz W. Adaptive coding of reward value by dopamine neurons. Science 307: 1642–1645, 2005 [DOI] [PubMed] [Google Scholar]
- Tritsch NX, Sabatini BL. Dopaminergic modulation of synaptic transmission in cortex and striatum. Neuron 76: 33–50, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu W, Lipscombe D. Neuronal CaV1.3alpha1 L-type channels activate at relatively hyperpolarized membrane potentials and are incompletely inhibited by dihydropyridines. J Neurosci 21: 5944–5951, 2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye JH, Zhang J, Xiao C, Kong JQ. Patch-clamp studies in the CNS illustrate a simple new method for obtaining viable neurons in rat brain slices: glycerol replacement of NaCl protects CNS neurons. J Neurosci Methods 158: 251–259, 2006 [DOI] [PubMed] [Google Scholar]
- Zhang H, Fu Y, Altier C, Platzer J, Surmeier DJ, Bezprozvanny I. Ca1.2 and CaV1.3 neuronal L-type calcium channels: differential targeting and signaling to pCREB. Eur J Neurosci 23: 2297–2310, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Berra-Romani R, Sinnegger-Brauns MJ, Striessnig J, Blaustein MP, Matteson DR. Role of Cav1.2 L-type Ca2+ channels in vascular tone: effects of nifedipine and Mg2+. Am J Physiol Heart Circ Physiol 292: H415–H425, 2007 [DOI] [PubMed] [Google Scholar]
- Zhang L, Liu Y, Chen X. Carbachol induces burst firing of dopamine cells in the ventral tegmental area by promoting calcium entry through L-type channels in the rat. J Physiol 568: 469–481, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]