

Abstract
Fasting and hypoglycemia elicit powerful gastrointestinal contractions. Whereas the relationship between utilizable nutrient and gastric motility is well recognized, the explanation of this phenomenon has remained incomplete. A relatively recent controversial report suggested that astrocytes in the dorsal hindbrain may be the principal detectors of glucoprivic stimuli. Our own studies also show that a subset of astrocytes in the solitary nucleus (NST) is activated by low glucose. It is very likely that information about glucopenia may directly impact gastric control because the hindbrain is also the location of the vago-vagal reflex circuitry regulating gastric motility. Our in vivo single unit neurophysiological recordings in intact rats show fourth ventricular application of 2-deoxyglucose (2-DG) inhibits NST neurons and activates dorsal motor nucleus (DMN) neurons involved in the gastric accommodation reflex. Additionally, as shown in earlier studies, either systemic insulin or central 2-DG causes an increase in gastric motility. These effects on motility were blocked by fourth ventricle pretreatment with the astrocyte inactivator fluorocitrate. Fluorocitrate administered alone has no effect on gastric-NST or -DMN neuron responsiveness, or on gastric motility. These results suggest that glucoprivation-induced increases in gastric motility are dependent on intact hindbrain astrocytes.
Keywords: astrocytes, dorsal motor nucleus, fluorocitrate, solitary nucleus, vago-vagal reflex
Introduction
Nutrient levels, feeding behavior, and gastric motility have been linked in a correlational sense for >100 years. The relationship was heralded by William Beaumont's (1833) observation of the digestive processes of Alexis St. Martin. Next, Pavlov (1910) observed that increases in gastric motility and secretion were related to periods of food deprivation as well as the anticipation of eating. This led to Pavlov's (1910) proposal of a “cephalic phase” of digestion whereby stimuli, both internal (e.g., glucoprivation) and external (e.g., cues related to the expectation of feeding), were integrated by the brain to augment digestion in advance of feeding. Cannon and Washburn (1912) connected food deprivation and the sensation of gastric contractions with the urge to eat. Classic studies from the 1920s through the 1990s (Bulatao and Carlson, 1924; Richter, 1941; Novin et al., 1973; Cato et al., 1990) established that glucoprivation is a strong signal for the initiation of feeding and perhaps the strongest known stimulant of gastric motility.
The nucleus of the solitary tract (NST) is the most likely site for the convergence of glucodetection and the regulation of gastric motility. Several reports provide evidence for the NST as a gluco-sensory structure that maintains the connections necessary for regulating nutrient homeostasis and digestion (Mizuno and Oomura, 1984; Adachi et al., 1995; Yettefti et al., 1995; Dallaporta et al., 1999, 2000; Rogers and Hermann, 2012). Additionally, the NST is the recipient of substantial vagal afferent projections from the gut. The NST integrates this vagal input and controls gastric motility and secretion via short axon projections to the subjacent dorsal motor nucleus of the vagus (DMN; Fig. 1).
Figure 1.
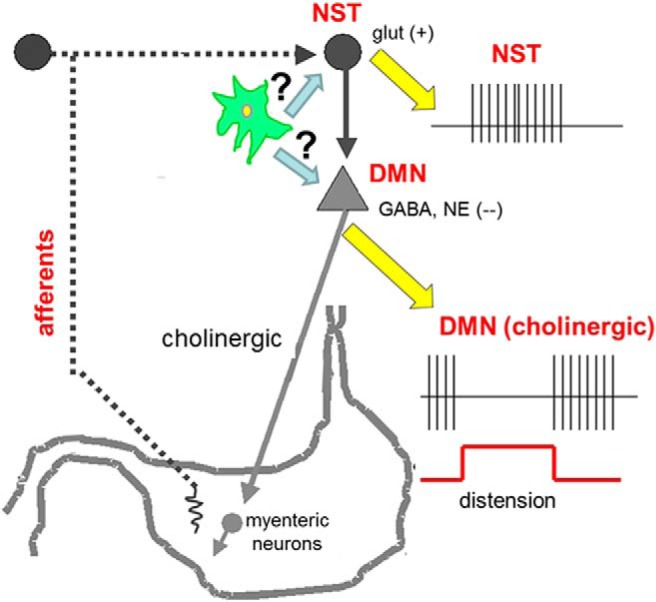
Schematic diagram of primary components involved in gastric accommodation reflex responsible for controlling gastric tone and motility (adapted from Rogers and Hermann, 2012). Vagal afferent sensors are activated when the antrum is distended with a small balloon. Vagal afferents release glutamate onto second-order NST neurons. In turn, NST neurons inhibit tonically active gastric vagal motor neurons that control tone and motility. The central hypothesis of these studies is that glucose-sensitive astrocytes in the NST can modulate gastric vago-vagal control in response to cytoglucopenia. Given the high density of astrocytes in the NST versus the DMN (Hermann et al., 2009), it is highly likely that low glucose challenges are transduced by glia in the NST. Still, we cannot rule out the possibility of direct astrocyte-DMN communications.
Until recently, there would have been nearly unanimous agreement that the cells responsible for sensing, signaling, and transmitting data concerning glucose availability are neurons. However, a provocative paper by Marty et al. (2005) has thrown the primacy of central hypoglycemia detection into question in favor of the astrocyte. Specifically, transgenic mice with the type II glucose transporter (GLUT2) knocked out (KO) demonstrated an inability to respond to a hypoglycemic challenge by activating glucose counter-regulatory pathways. GLUT2 is a critical component of both the pancreatic β cell and some neuronal glucodetection mechanisms (Marty et al., 2007). The selective re-expression of GLUT2 in astrocytes, but not neurons, rescued the normal hypoglycemia defense mechanism in these global GLUT2-KO mice (Marty et al., 2005), thus suggesting that astrocytes form an obligatory component of CNS glucodetection mechanisms.
To address this question, we applied the techniques of single-unit extracellular recordings from identified gastric vago-vagal reflex control neurons in the hindbrain and strain gauge recordings of gastric motility in the intact anesthetized rat. Our results suggest that intact astrocyte function in the dorsal hindbrain is essential to the development of the hypermotility evoked by central cytoglucopenia.
Materials and Methods
All experimental procedures were conducted under the approval of the Pennington Biomedical Research Center's Institutional Animal Care and Use Committee and were performed according to the guidelines set forth by the National Institutes of Health. Long–Evans rats of either sex (body weight between 300 and 500 g) obtained from the Pennington Biomedical Research breeding colony were used in these studies. Animals were housed in a temperature-controlled room under 12 h light/dark cycle and provided water and food ad libitum.
In vivo single unit extracellular recordings from identified gastric-NST and -DMN neurons
Surgical preparation.
Methods for recording from identified gastric-NST and -DMN vago-vagal reflex neurons were previously described in detail (Rogers and McCann, 1989; McCann and Rogers, 1990, 1992; Viard et al., 2012). Animals were not food deprived before the experiment; however, to facilitate the insertion of the gastric stimulating balloon in the rat (described later in this section), these animals were maintained on a nutritionally complete liquid diet of Ensure (Abbott Laboratories) for a minimum of 24 h before the experiment. This liquid diet was so readily accepted by most rats, we frequently needed to refill the bottles a second time during the day to insure that no animal was food deprived before 5:00 P.M. Rats (N = 91) were deeply anesthetized with thiobutabarbital (Inactin, Sigma-Aldrich; 150 mg/kg, i.p.). This long-term anesthesia is preferred due to its minimal interference with autonomic reflexes (Buelke-Sam et al., 1978). Using aseptic technique, a cannula was secured in the trachea to insure a clear airway. A laparotomy was done to expose the stomach and the proximal part of the duodenum. A small incision in the proximal duodenum was made to remove gastric contents via lavage through the pylorus. Once the stomach was empty, a small gastric balloon was inserted through the duodenal incision, past the pylorus, and into the antrum. The gastric balloon was constructed from the small finger of a surgical glove and attached to a piece of silastic tubing (0.065 inch outer diameter) which was exteriorized through the duodenal incision and secured via purse-string ligature. The abdominal muscle wall and skin were closed with the tubing from the balloon exiting via the incision. The tubing was connected to a pressure transducer (Isotec, Harvard Instruments) to monitor gastric pressure. Instrumented rats were secured in a stereotaxic frame. A midline incision was made in the scalp and the cervical musculature was retracted. The foramen magnum was opened; removal of the dura and arachnoid membranes exposed the caudal portion of the floor of the fourth ventricle.
Extracellular electrophysiological recordings.
A single glass micropipette (tip diameter = 1 μm), filled with 2 m NaCl plus 1% pontamine sky blue for iontophoretic marking of recording sites, was used in the identification and recording of activity of gastric-NST or -DMN neurons, as described previously (McCann et al., 1992; Viard et al., 2012). Extracellular signals from the micropipette were amplified (5000×; WPI DAM 50 Differential Amplifier) and bandpass filtered (300–3000 Hz; Warner Instruments LPF 202A) before being displayed on an oscilloscope and stored for later analysis on an AM Systems LabChart 7 PC-based waveform analysis system (AD Instruments).
Gastric-DMN neurons typically exhibit a characteristic, spontaneous, pacemaker activity; tonic basal DMN firing rate (FR) of ∼2–3 spikes/s. Moderate distension (0.1–1.0 ml) of the stomach via the antral balloon causes a sharp and stimulus-dependent reduction in spontaneous DMN-FR that is time-locked to the period of antral balloon distension (McCann and Rogers, 1990, 1992, 1994; Viard et al., 2012).
The gastric-NST neurons located just dorsal to the DMN were identified by their transition from quiescence to activation during a 10 s antral balloon distension of 1 ml. This 1 ml stimulus challenge was chosen as it produces a response midway between resting activity and maximal activation firing rates (for the NST) or inhibition (for the DMN; McCann and Rogers, 1990, 1992, 1994; Viard et al., 2012). Examples of gastric-NST or -DMN responses to gastric distension are shown in Figure 2.
Figure 2.

Effects of 4V 2DG glucoprivation and FC on single identified gastric reflex control neurons in the hindbrain. Left panels, Single-unit NST neuron responses to gastric balloon distension (1 ml). Note that 2DG reduces the excitation of the NST neuron responding to vagal afferent distension information. Although the astrocytic metabolic blocker (FC) alone has no effect on NST neuron responsiveness, FC blunts the 2DG effect. Right panels, These DMN neurons are reflexly inhibited by NST neurons as can be readily observed from their response to the same gastric distension signal. 2DG greatly accelerates basal DMN firing and obstructs the firing rate inhibition normally caused by gastric distension. Again, FC has no effect of and by itself on DMN firing; however, the 2DG effect on DMN responsiveness is blocked by FC. A, Control; B, 4V 2DG alone; C, 4V FC + 2DG; D, 4V FC alone. Insets in A (in both left and right panels), Multiple extracellular action potentials of the neuron being monitored are superimposed on an expanded time scale to show the unitary nature of the recordings. Compression of the spike train records for the purpose of evaluating 10 s intervals causes an apparent distortion of spike shape and amplitude. All spike train records analyzed for this study were confirmed to be from single units.
Experimental design: single-unit extracellular recordings from gastric-DMN and -NST neurons.
Once the instrumented animal was placed in the stereotaxic frame and all surgical preparations for neurophysiological recordings of neurons in the hindbrain were complete, the recording micropipette was directed toward either the NST or DMN nuclei via an hydraulic microdrive (∼300 μm anterior and lateral to calamus; ∼200–600 μm below brainstem surface). All animals were monitored for normal gastric-NST or -DMN responses to 1.0 ml antral distension. Animals were randomly assigned to one of five experimental groups: (1) 2-deoxy-d-glucose (2DG; Sigma-Aldrich) alone (3 mg in 10 μl = 18 μmol; Granneman and Friedman, 1983). Application of 2DG (competitive blocker of glucose utilization) to the floor of the 4V was used to mimic a hypoglycemic condition by interfering with hindbrain intracellular glucose availability (Ritter et al., 1978, 1998). (2) Fluorocitrate (FC; Sigma-Aldrich) alone (1 or 5 nmole in 5 μl; as per Hassel et al., 1992; Swanson and Graham, 1994; Martín et al., 2007; Erlichman and Leiter, 2010) intraventricular or intrathecal injection studies). Astrocyte function and signaling was interrupted by the use of FC, a reversible metabolic inhibitor that selectively affects glia (Hassel et al., 1992; Erlichman et al., 1998; Erlichman and Leiter, 2010). (3) FC (1 or 5 nmole) followed by 2DG to determine whether neuronal responses are influenced by astrocytic activity.
In each case, the experimental agent was applied to the floor of the fourth ventricle (4V) using a Hamilton syringe. Sixty minutes later, gastric-NST or -DMN neurons were again identified according to their responses to the 1.0 ml antral distension; 10 s duration. (In the case of the FC+2DG combination, the FC was applied 4V first. Thirty minutes later, the 2DG was applied 4V and the usual 60 min exposure preceded neurophysiological recordings).
At the end of the neurophysiological recording session, the location of the last recorded gastric neuron (either NST or DMN) could be marked by applying 2 uA positive direct current for 1 min to the pontamine-filled recording micropipette using a stimulus isolation unit (WPI). Locations of the recording sites were then validated histologically as previously described (Viard et al., 2012).
Data analysis.
Recordings were made from both gastric-NST and -DMN neurons, which form the afferent and efferent portion of the gastric-accommodation reflex (GAR), respectively. Vagal afferent activation of the normally quiescent NST neurons provokes a reflex suppression of tonic DMN activity (McCann and Rogers, 1992; Rogers and Hermann, 2012; Viard et al., 2012). Basal FR was defined as the number of action potential spikes occurring during the 10 s interval immediately before evoking GAR response (i.e., 1 ml gastric distension held for 10 s). The number of action potentials that occurred during the 10 s gastric stimulation (GAR) was compared with the preceding basal FR. Modulation of normal gastric-NST or -DMN responses to consistent stimulation (i.e., 1 ml antral distension) during glucoprivation was evaluated in the presence of 2DG. Contribution of astrocyte activity to neuronal activity during glucoprivation was evaluated in the presence of 2DG preceded by FC. The effect of astrocyte input on NST or DMN activity, in general, was also evaluated in the presence of FC alone (Fig. 3).
Figure 3.

Averaged (mean ± SEM) basal and evoked firing rates of identified gastric-NST and -DMN neurons responding to 1 ml balloon distension of the antrum (GAR). Top row, Normally quiescent gastric-NST neurons are readily activated by 1 ml balloon distension of the antrum (McCann et al., 1992; Rogers et al., 2013). This response is not significantly altered by 4V pretreatment with the glial metabolic blocker, FC alone, at either dose level. In contrast, 4V 2DG substantially reduces NST responsiveness to the same stimulus. 4V FC pretreatment eliminates the inhibitory effect of 2DG on NST responsiveness to antral distension (N = 136; Dunnett's post hoc, *p < 0.05). Bottom row, Activity of gastric-DMN neurons is essentially the mirror image of the NST with antral distension causing a reduction in spontaneous DMN firing. As seen with the gastric-NST, FC alone does not affect this relationship. However, 4V 2DG causes a substantial increase in basal DMN activity. Although antral distension still causes a proportional reduction in DMN firing rate, the evoked response to the distension-GAR by the DMN neurons is significantly elevated. Pretreatment with 4V FC eliminates these 2DG effects (N = 178; Dunnett's post hoc; *p < 0.05, firing rate during GAR; #p < 0.05, basal firing rate).
Statistical comparisons of either basal FR or evoked GAR responses were made across all experimental groups of NST (or DMN) neurons by using overall one-way ANOVA. Dunnett's post hoc test of differences between individual groups and the control condition were conducted. P < 0.05 was considered statistically significant.
Gastric motility: effects of glucoprivic stimuli in the hindbrain
Surgical preparations.
Rats were prepared for gastric strain gauge recording of motility patterns as previously described in detail (Rogers and Hermann, 1987; Hermann and Rogers, 1995; Chen et al., 1997; Hermann et al., 2002, 2006). Briefly, rats (N = 35) that had not been food deprived were anesthetized with thiobutabarbital, as described in Surgical preparation, above. A tracheal cannula was placed to maintain a clear airway. After a midline laparotomy, the gastric antrum was exposed and a miniature strain gauge (RB Products) was sutured to the exterior of the antrum. Strain gauge leads were exteriorized through the laparotomy incision and the abdomen sutured closed, in layers. Rats were then placed in a stereotaxic frame and the floor of the 4V was exposed, as described in Extracellular recording, above.
Before surgical placement of the strain gauge in the animal, strain gauge outputs were calibrated in units of grams by suspending weights from the gauges. In this way, we could label the y-axis of the sample motility plots in convenient units of mass. Strain gauge leads were connected to a Wheatstone bridge amplifier whose output was analyzed by a PC-driven AD Instruments Powerlab 8/30.
Experimental design.
Two studies were performed to evaluate the relationship between intact hindbrain astrocyte functioning and increases in gastric motility triggered by glucoprivic or low glucose stimuli. Both studies used the same surgical and animal preparations and instrumentation. Gastric motility and tone were monitored continuously. In the first set of experiments, all stimuli where administered directly onto the floor of the 4V. Animals were randomly assigned to one of four groups: (1) Control = Saline, 10 μl; (2) Glucoprivic condition = 2DG, 3 mg in 10 μl; (3) Astrocytic metabolic blocker = FC, either 1 or 5 nmole in 10 μl; or (4) FC followed by 2DG = FC + 2DG, in this case, FC preceded the 4V application of 2DG by 60 min.
In the second study, either saline or FC was applied to the 4V as described above. One hour after the 4V pretreatment, insulin (10 units/kg, s.c.) was administered systemically. As above, gastric tone and motility were monitored continuously.
To verify that vago-vagal innervation of the stomach had not been disrupted during the surgical instrumentation session or as a consequence of exposure to FC, thyrotropin releasing hormone (TRH; 0.2 nmole in 2 μl) was applied 4V at the end of the experiment. TRH causes a vagally mediated increase in gastric motility via direct action on neurons in the NST and DMN (Rogers and Hermann, 1987; McCann et al., 1988; Rogers and McCann, 1989;Taché et al., 1990). Thus, if FC causes any generalized effects to damage or disrupt hindbrain neuronal circuits regulating motility, then we would expect FC pretreatment to also cause a significant disruption in TRH-induced increases in motility relative to the saline pretreated control group.
Data analysis.
Gastric activity was quantified across 10 min epochs by motility indexing (MI) according to a formulation put forward by Ormsbee and Bass (1976). Contraction data are graded according to the relationship:
where MI is the final index per unit of time selected (e.g., 10 min); nA(1–2) is the number of contractions in the amplitude range from just-detectable to twice just-detectable. nA(2–4) is the number of contractions 2–4 times just detectable, etc. In these experiments, the 10 min MI for the period immediately before either 4V or SC drug injection served as baseline activity; motility indices were also calculated at 30, 60, and 120 min after the treatment for comparison.
The size of the MI between preparations can be influenced by numerous factors, such as the specific location of attachment of the gastric strain gauge, the tension across the points of attachment, the amount of residual food, etc.; therefore, each animal served as its own control. That is, the MI under basal conditions was considered “100%” for each individual animal; any changes in MI were reflected against this 100% background. Examples of raw gastric motility are shown in Figure 4; quantitated motility indices across the four time periods are shown in Figure 5. One-way ANOVA was done on the 60 min MI across the experimental groups. Dunnett's post hoc test of differences between individual groups and the control condition were conducted. P < 0.05 was considered statistically significant.
Figure 4.

Glucoprivic stimuli evoke increases in gastric motility that can be blocked by fourth ventricular application of FC in the anesthetized rat. Plots on the left are gastric strain gauge recordings taken before any experimental manipulation. Plots on the right represent strain gauge recordings 1 h after the application of a stimulus. A, 4V 2DG causes a significant increase in gastric motility. B, FC, an astrocyte-specific metabolic inhibitor, applied to the 4V before 2DG (4V FC+2DG) blocks the 2DG effect. C, FC applied to the 4V (4V FC) alone has no effect on basal gastric motility pattern. D, Subcutaneous insulin produces a dramatic increase in gastric motility (Insulin, s.c.). E, 4V FC largely blocks the effects of insulin to increase gastric motility (4V FC + Insulin s.c.). These data suggest that astrocytes in the hindbrain detect glucoprivic conditions and can activate vagal pro-motility circuits.
Figure 5.

Plots of averaged (mean ± SEM) gastric motility indices over time following glucoprivic challenges. A, At time “0 min” either saline, FC, or 2DG was applied to the floor of the 4V. Statistical analyses were performed on the “60 min” data (Fig. 6A, circled). Compared with saline control group, 4V 2DG produced a significant increase in MI within 60 min of delivery of 2DG. 4V FC (either 1 or 5 nmole) alone had no effect on motility. However, pretreatment with either dose of 4V FC blocked the 4V 2DG-induced increase in motility. B, At time “0 min,” the instrumented animal received a subcutaneous injection of insulin (10 unit/kg). One hour before the systemic insulin injection, the animal had received either saline or FC via 4V. Within 60 min of systemic injection, insulin evoked large increases in gastric motility. Pretreatment with 1 nmole 4V-FC reduced but did not block this insulin-induced increase in motility. However, the 5 nmole dose of 4V-FC completely blocked the insulin-induced hypermotility.
Results
In vivo single-unit extracellular recordings from identified gastric neurons
NST recording studies
A total of 136 gastric-NST neurons were identified by antral balloon distension. NST neurons involved in the GAR are typically quiescent, producing <2 spikes every 10 s (Fig. 2; McCann and Rogers, 1992; Viard et al., 2012). These basal firing rates of gastric-NST neurons were not changed relative to control under any of the conditions tested (p > 0.05).
Under control conditions, antral distension (1 ml) evoked an increased firing rate for GAR-NST neurons averaging 10.4 ± 1.4 spikes/10 s (average ± SEM; Fig. 3). 2DG pretreatment caused a substantial reduction in reflex-induced spike production (1.8 ± 0.4 spikes/10 s). Neither 1 nmole nor 5 nmole FC treatment, alone, had any effects on either the basal NST firing rate or the GAR-induced firing rate compared with control condition (13.7 ± 2.0 and 10.1 ± 1.8, respectively). However, 4V pretreatment with either dose of FC blocked the effect of 2DG to reduce NST-reflex responsiveness (12.3 ± 2.8 and 9.1 ± 1.6, respectively), such that the NST response to gastric distension was not different from control conditions [Fig. 3; ANOVA of GAR-NST firing rate in response to 1 ml antral distension F(5,130) = 6.409, p < 0.0001; Dunnett's post-test *p < 0.05; Note that, statistical analyses were performed on the male only data (N = 75), as well as the male plus female data (N = 91), no significant differences were observed between those datasets (data not shown). Therefore, all reported statistical analyses included data from both males and females].
DMN recording studies
Gastric-DMN neurons (N = 178) were identified by antral balloon distension. As previously reported (McCann and Rogers, 1992; Viard et al., 2012), these neurons characteristically maintain a steady basal firing rate that is markedly inhibited in response to gastric distension. Under control conditions, gastric-DMN neurons presented an average basal firing rate of 23.7 ± 2.2 spikes/10 s (Fig. 3). After pretreatment with 4V 2DG, basal firing of gastric-DMN neurons increased significantly to 43.6 ± 5.7 spikes/10 s (F(5,172) = 5.408, p = 0.0001; Dunnett's post-test # p < 0.05). Pretreatment with FC, alone (either 1 or 5 nmole dose), had no effect on basal DMN firing (21.7 ± 2.9 and 23.2 ± 3.0 spikes/10 s, respectively). However, pretreatment with FC (either 1 or 5 nmole) blocked the 2DG effect to increase the basal firing rate of gastric-DMN neurons firing (25.0 ± 2.7 and 20.4 ± 2.2 spikes/10 s, respectively).
Antral distension reduced the firing rate of these identified gastric-DMN neurons. Under control conditions, 1 ml distension suppressed the basal firing rate to 11.4 ± 1.3 spikes/10 s (Fig. 3B). Pretreatment with 4V 2DG yielded a significantly different reflex-induced firing rate of 22.0 ± 5.1 spikes/10 s (F(5,172) = 4.025, p = 0.0018; Dunnet's post-test *p < 0.05). Whereas pretreatment with either 1 or 5 nmole FC alone had no effect on reflex-induced firing rate (7.4 ± 1.5 and 8.3 ± 2.0 spikes/10 s, respectively), pretreatment with either dose of FC before 2DG exposure suppressed 2DG's effect on reflex-induced DMN firing rate. In other words, pretreatment with FC before 2DG challenge yielded essentially the same gastric-DMN firing rate during antral distension as seen under control conditions (9.8 ± 2.8 and 7.5 ± 1.6 spikes/10 s, respectively).
Although 2DG affected both the basal and reflex-induced change in firing rate of gastric-DMN neurons, the amount of suppression in firing rate was proportional to that seen in the control group. That is, basal firing rates of gastric-DMN neurons were suppressed ∼50% in response to antral distension (Fig. 3).
Motility studies
Central glucoprivic challenge: 4V 2DG
Compared with saline control group, 4V 2DG produced a significant increase in MI within 60 min of delivery of 2DG (Figs. 5A, 6A). This effect on motility is similar to that previously reported by Cato et al. (1990) following systemic glucoprivation with 2DG. Fourth ventricular FC, alone, had no effect on motility but pretreatment with 4V FC (either 1 or 5 nmoles) blocked the 4V 2DG-induced increase in MI (ANOVA F(4,16) = 6.26, p = 0.0031; Dunnett's post hoc comparisons; *p ≤ 0.05). 4V application of TRH 3 h after other 4V challenges caused a large increase in MI that was not different across the saline, FC, or FC + 2DG groups (Fig. 6C); thus vago-vagal neurocircuitry involved in gastric function was not compromised by surgical preparations or exposure to FC. Note that TRH was not applied to the “2DG alone” cases because gastric motility was still quite elevated at this time and there was no concern about integrity of vago-vagal circuits.
Figure 6.

Statistical comparisons of motility indices. A, 2DG applied to the 4V significantly increases gastric motility compared with control (saline 4V). Although 4V FC alone has no effect on gastric motility, either dose of 4V FC (1 or 5 nmole) blocks the pro-motility effects of 4V 2DG (N = 20; Dunnett's post hoc comparisons; *p ≤ 0.05). B, Subcutaneous insulin (10 units/kg) produces a dramatic increase in motility index. Although 1 nmole 4V FC effects to reduce this insulin effect are not significant, 5 nmole FC was able to block the increase in gastric motility at the 60 min time point (N = 15; Dunnett's post hoc comparisons; *p ≤ 0.05). C, 4V application of TRH 3 h after other 4V challenges caused a large increase in MI that was not different across the saline, FC, or FC + 2DG groups. This final challenge was administered to verify that the vago-vagal neurocircuitry involved in gastric function was not compromised by surgical preparations or exposure to FC. Note that TRH was not applied to the 2DG alone cases because gastric motility was still elevated at this time and there was no concern about integrity of vago-vagal circuit.
Systemic low glucose challenge: subcutaneous insulin
Subcutaneous injection of insulin (10 unit/kg) evoked large increase in gastric motility (Figs. 5B, 6B) similar to that reported by Bulatao and Carlson (1924) in the dog and Cato et al. (1990) in the rat. Pretreatment with 4V-FC (1 nmole) reduced but did not eliminate this insulin-induced increase in motility. However, the 5 nmole dose of 4V-FC was able to suppress the insulin-induced increase in motility (ANOVA F(2,12) = 4.24, p = 0.0406; Dunnett's post hoc comparisons; *p < 0.05).
Discussion
Our results show that a localized glucoprivic challenge (4V-2DG) reduces the responsiveness of NST neurons to antral distension. Perhaps as a result, DMN neurons belonging to this gastric control reflex also show a marked increase in basal firing under the local effects of 2DG. This connection is expected as a result of the strong inhibitory input from the NST to the DMN; thus, removing NST input to DMN GAR neurons will disinhibit them (McCann and Rogers, 1992; Rogers and Hermann, 2012). Though FC, alone, had no effects on either NST or DMN basal or GAR activity, FC blocked the effects of 2DG on NST and DMN behavior (Figs. 2, 3). Our observations of 2DG effects on gastric-NST and -DMN neurons are readily explained by the basic vago-vagal relationship of these neurons as described above. However, these studies have not ruled out the unusual possibility that gluco-sensitive astrocytes may be having direct effects on DMN neurons as well.
These in vivo electrophysiological results were mirrored in the results of our in vivo motility studies using strain gauges to monitor gastric activity of the anesthetized preparation. 4V-2DG caused a significant increase in basal motility that would be the result of increased cholinergic vagal-motor (DMN) input to the gastric enteric plexus (Schemann and Grundy, 1992). This gastrointestinal effect of dorsal hindbrain cellular glucoprivation was blocked by pretreatment with the glial specific inhibitor, FC. As we saw in our in vivo electrophysiological studies, 4V-FC alone had no effect on motility. Together, these results suggest that the hypoglycemic or local glucoprivic effects to increase gastric motility involve astrocytic modulation of the synapses between vagal afferents, the NST, and the DMN.
The NST and dorsal hindbrain have long been associated with the central detection of the glucose availability and the control of the glycemic state (Bernard, 1855). Systemic administration of 2DG evokes increased c-fos expression in the NST of rats (Ritter et al., 1998; Young et al., 2000). Injection of a similar glucoprivic agent (5-thio-d-glucose) directly into the NST also drives counter regulatory responses, such as increased food intake (Ritter et al., 2000) and increases in serum glucagon and stress hormone levels (Andrew et al., 2007).
At first glance, the c-fos data (Ritter et al., 1998; Young et al., 2000) seems at odds with our present results. That is, our neurophysiological data on identified gastric-NST neurons shows that 2DG clearly inhibits the NST-neurons involved in the GAR and inhibition of neuronal activity does not cause c-fos expression (Hoffman et al., 1993). However, these earlier c-fos observations (Ritter et al., 1998; Young et al., 2000) were made before the realization that astrocytes can express c-fos when activated (Edling et al., 2007) and that they are active participants in autonomic signaling (Hermann and Rogers, 2009). Indeed, our recent work shows that low glucose levels or 2DG can activate significant calcium accumulation in astrocytes (McDougal et al., 2013) and large increases in cytoplasmic calcium triggers c-Fos gene transcription in astrocytes (Edling et al., 2007). Additionally, Young et al. (2000) did note that the brain c-fos response to 2DG was 70% blocked by a gliotoxin, methionine sulfoximine, suggesting that astrocytes may have a role in gluco-sensing. Thus, in all likelihood, many of the c-fos-positive cells in the NST responding to 2DG in those earlier studies were probably astrocytes. We obtained a similar result in our studies of the effects of the cytokine tumor necrosis factor-α (TNFα) in the NST. Specifically, TNFα activated significant numbers of cells in the NST; ∼29% were phenotypically identified to be tyrosine-hydroxylase-positive (i.e., presumably noradrenergic) neurons, ∼10% were nitrergic neurons, and 54% of the c-fos-activated cells in the NST were phenotypically identified to be astrocytes (Hermann and Rogers, 2009).
In our neurophysiological experiments, this astrocyte activation apparently provoked an inhibition of gastric-NST neurons and an excitation of gastric-DMN neurons. This activity is further reflected in increased gastric motility elicited by hindbrain glucoprivation (2DG) or systemic insulin injection.
Marty et al. (2005) showed that transgenic mice with a global GLUT2-deletion were unable to elicit the counter-regulation response of induced glucagon release following hypoglycemic conditions of glucoprivation. Selective re-expression of GLUT2 (an obligatory component of many glucodetection schemes) (Thorens, 2001) in astrocytes, but not neurons, rescued the normal counter regulation response in these knock-out mice (Marty et al., 2005). The results from our present studies suggest that astrocytes in the NST may also regulate vago-vagal reflex control of gastric motility in response to hypoglycemic challenge.
Previous work (Kahlert and Reiser, 2000; Arnold, 2005) and our own data (McDougal et al., 2013) suggest that glucoprivic stimuli can cause an increase in calcium flux in astrocytes, though the mechanism explaining this effect is not clear. Evidence in cultured systems suggests that astrocytes are highly dependent on glycolysis for ATP production (Kahlert and Reiser, 2000); thus, removal of glucose or blocking glucose utilization may transiently starve astrocytes of glucose for ATP production. This effect could be quite rapid in astrocytes expressing the bidirectional GLUT2 transporter. With impaired glycolysis and the resulting drop in ATP, the calcium-ATPase pump in the endoplasmic reticulum (ER) of astrocytes fails and ER calcium is released to the cytoplasm (Kahlert and Reiser, 2000; Arnold, 2005). More cellular physiological studies will be required to determine whether this mechanism applies to gluco-sensitive NST-astrocytes.
Cytoplasmic calcium is a trigger for “gliotransmission” (Araque et al., 1999; Zhang and Haydon, 2005; Verkhratsky et al., 2012). ATP and/or glutamate are commonly released from astrocytes by gliotransmission. ATP, hydrolyzed to adenosine, can act as a potent inhibitory purinergic agonist and could be responsible for inhibiting NST neurons (Pascual et al., 2005; Fellin et al., 2006). Further imaging and electrophysiological studies of astrocyte-NST neuron interactions will be required to determine whether ATP is released by astrocytes activated by cytoglucopenia.
For now, we posit the hypothesis that: (1) astrocytes in the NST are necessary for the normal detection of glucoprivic stimuli; (2) low-glucose-activated NST astrocytes release an inhibitory gliotransmitter, perhaps adenosine, to inhibit NST neurons involved in the inhibitory control of excitatory gastric-DMN neurons; and (3) inhibition of NST neurons disinhibits spontaneously active gastric DMN neurons, causing their increase in firing and the resultant increase in motility.
Footnotes
This work was supported by NIH Grants NS52142, DK56373, HD47643, and NS60664. A preliminary report of some of this work was presented at the 1st Joint ISAN/AAS meeting in Buzios, Brazil (2011). The symposium review appeared in Autonomic Neuroscience: Basic and Clinical (2013, 175:61–69).
The authors declare no competing financial interests.
References
- Adachi A, Kobashi M, Funahashi M. Glucose-responsive neurons in the brainstem. Obes Res. 1995;3:735S–740S. doi: 10.1002/j.1550-8528.1995.tb00493.x. [DOI] [PubMed] [Google Scholar]
- Andrew SF, Dinh TT, Ritter S. Localized glucoprivation of hindbrain sites elicits corticosterone and glucagon secretion. Am J Physiol Regul Integr Comp Physiol. 2007;292:R1792–R1798. doi: 10.1152/ajpregu.00777.2006. [DOI] [PubMed] [Google Scholar]
- Araque A, Parpura V, Sanzgiri RP, Haydon PG. Tripartite synapses: glia, the unacknowledged partner. Trends Neurosci. 1999;22:208–215. doi: 10.1016/S0166-2236(98)01349-6. [DOI] [PubMed] [Google Scholar]
- Arnold S. Estrogen suppresses the impact of glucose deprivation on astrocytic calcium levels and signaling independently of the nuclear estrogen receptor. Neurobiol Dis. 2005;20:82–92. doi: 10.1016/j.nbd.2005.02.002. [DOI] [PubMed] [Google Scholar]
- Beaumont W. Experiments and observations on the gastric juice. Plattsburgh: F.P. Allen; 1833. [DOI] [PubMed] [Google Scholar]
- Bernard C. Lecons de physiologie experimentale appliques á la medecine. Paris: Bailliere; 1855. pp. 296–313. [Google Scholar]
- Buelke-Sam J, Holson JF, Bazare JJ, Young JF. Comparative stability of physiological parameters during sustained anesthesia in rats. Lab Anim Sci. 1978;28:157–162. [PubMed] [Google Scholar]
- Bulatao E, Carlson A. Contributions to the physiology of the stomach: influence of experimental changes in blood sugar level on gastric hunger contractions. Am J Physiol. 1924;69:107–115. [Google Scholar]
- Cannon WB, Washburn AL. An explanation of hunger. Am J Physiol. 1912;29:441–454. [Google Scholar]
- Cato RK, Flanagan LM, Verbalis JG, Stricker EM. Effects of glucoprivation on gastric motility and pituitary oxytocin secretion in rats. Am J Physiol. 1990;259:R447–R452. doi: 10.1152/ajpregu.1990.259.3.R447. [DOI] [PubMed] [Google Scholar]
- Chen CH, Stephens RL, Jr, Rogers RC. PYY and NPY: control of gastric motility via action on Y1 and Y2 receptors in the DVC. Neurogastroenterol Motil. 1997;9:109–116. doi: 10.1046/j.1365-2982.1997.d01-26.x. [DOI] [PubMed] [Google Scholar]
- Dallaporta M, Himmi T, Perrin J, Orsini JC. Solitary tract nucleus sensitivity to moderate changes in glucose level. Neuroreport. 1999;10:2657–2660. doi: 10.1097/00001756-199908200-00040. [DOI] [PubMed] [Google Scholar]
- Dallaporta M, Perrin J, Orsini JC. Involvement of adenosine triphosphate-sensitive K+ channels in glucose-sensing in the rat solitary tract nucleus. Neurosci Lett. 2000;278:77–80. doi: 10.1016/S0304-3940(99)00898-8. [DOI] [PubMed] [Google Scholar]
- Edling Y, Ingelman-Sundberg M, Simi A. Glutamate activates c-fos in glial cells via a novel mechanism involving the glutamate receptor subtype mGlu5 and the transcriptional repressor DREAM. Glia. 2007;55:328–340. doi: 10.1002/glia.20464. [DOI] [PubMed] [Google Scholar]
- Erlichman JS, Leiter JC. Glia modulation of the extracellular milieu as a factor in central CO2 chemosensitivity and respiratory control. J Appl Physiol. 2010;108:1803–1811. doi: 10.1152/japplphysiol.01321.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erlichman JS, Li A, Nattie EE. Ventilatory effects of glial dysfunction in a rat brain stem chemoreceptor region. J Appl Physiol. 1998;85:1599–1604. doi: 10.1152/jappl.1998.85.5.1599. [DOI] [PubMed] [Google Scholar]
- Fellin T, Sul JY, D'Ascenzo M, Takano H, Pascual O, Haydon PG. Bidirectional astrocyte-neuron communication: the many roles of glutamate and ATP. Novartis Found Symp. 2006;276:208–217. doi: 10.1002/9780470032244.ch16. [DOI] [PubMed] [Google Scholar]
- Granneman J, Friedman MI. Feeding after recovery from 2-deoxyglucose injection: cerebral and peripheral factors. Am J Physiol. 1983;244:R383–R388. doi: 10.1152/ajpregu.1983.244.3.R383. [DOI] [PubMed] [Google Scholar]
- Hassel B, Paulsen RE, Johnsen A, Fonnum F. Selective inhibition of glial cell metabolism in vivo by fluorocitrate. Brain Res. 1992;576:120–124. doi: 10.1016/0006-8993(92)90616-H. [DOI] [PubMed] [Google Scholar]
- Hermann G, Rogers RC. Tumor necrosis factor-alpha in the dorsal vagal complex suppresses gastric motility. Neuroimmunomodulation. 1995;2:74–81. doi: 10.1159/000096874. [DOI] [PubMed] [Google Scholar]
- Hermann GE, Rogers RC. TNF activates astrocytes and catecholaminergic neurons in the solitary nucleus: implications for autonomic control. Brain Res. 2009;1273:72–82. doi: 10.1016/j.brainres.2009.03.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hermann GE, Tovar CA, Rogers RC. LPS-induced suppression of gastric motility relieved by TNFR:Fc construct in dorsal vagal complex. Am J Physiol Gastrointest Liver Physiol. 2002;283:G634–G639. doi: 10.1152/ajpgi.00412.2001. [DOI] [PubMed] [Google Scholar]
- Hermann GE, Travagli RA, Rogers RC. Esophageal-gastric relaxation reflex in rat: dual control of peripheral nitrergic and cholinergic transmission. Am J Physiol Regul Integr Comp Physiol. 2006;290:R1570–R1576. doi: 10.1152/ajpregu.00717.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hermann GE, Van Meter MJ, Rood JC, Rogers RC. Proteinase-activated receptors in the nucleus of the solitary tract: evidence for glial-neural interactions in autonomic control of the stomach. J Neurosci. 2009;29:9292–9300. doi: 10.1523/JNEUROSCI.6063-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffman GE, Smith MS, Verbalis JG. c-Fos and related immediate early gene products as markers of activity in neuroendocrine systems. Front Neuroendocrinol. 1993;14:173–213. doi: 10.1006/frne.1993.1006. [DOI] [PubMed] [Google Scholar]
- Kahlert S, Reiser G. Requirement of glycolytic and mitochondrial energy supply for loading of Ca(2+) stores and InsP(3)-mediated Ca(2+) signaling in rat hippocampus astrocytes. J Neurosci Res. 2000;61:409–420. doi: 10.1002/1097-4547(20000815)61:4<409::AID-JNR7>3.0.CO;2-M. [DOI] [PubMed] [Google Scholar]
- Martín ED, Fernández M, Perea G, Pascual O, Haydon PG, Araque A, Ceña V. Adenosine released by astrocytes contributes to hypoxia-induced modulation of synaptic transmission. Glia. 2007;55:36–45. doi: 10.1002/glia.20431. [DOI] [PubMed] [Google Scholar]
- Marty N, Dallaporta M, Foretz M, Emery M, Tarussio D, Bady I, Binnert C, Beermann F, Thorens B. Regulation of glucagon secretion by glucose transporter type 2 (glut2) and astrocyte-dependent glucose sensors. J Clin Invest. 2005;115:3545–3553. doi: 10.1172/JCI26309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marty N, Dallaporta M, Thorens B. Brain glucose sensing, counterregulation, and energy homeostasis. Physiology. 2007;22:241–251. doi: 10.1152/physiol.00010.2007. [DOI] [PubMed] [Google Scholar]
- McCann MJ, Rogers RC. Oxytocin excites gastric-related neurones in rat dorsal vagal complex. J Physiol. 1990;428:95–108. doi: 10.1113/jphysiol.1990.sp018202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCann MJ, Rogers RC. Impact of antral mechanoreceptor activation on the vago-vagal reflex in the rat: functional zonation of responses. J Physiol. 1992;453:401–411. doi: 10.1113/jphysiol.1992.sp019235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCann MJ, Rogers RC. Functional and chemical neuroanatomy of a gastric vago-vagal reflex. In: Taché Y, Wingate DL, Burks TF, editors. Innervation of the gut: pathophysiological implications. Boca Raton, FL: CRC; 1994. pp. 81–92. [Google Scholar]
- McCann MJ, Hermann GE, Rogers RC. Dorsal medullary serotonin and gastric motility: enhancement of effects by thyrotropin-releasing hormone. J Auton Nerv Syst. 1988;25:35–40. doi: 10.1016/0165-1838(88)90005-7. [DOI] [PubMed] [Google Scholar]
- McDougal DH, Hermann GE, Rogers RC. Astrocytes in the nucleus of the solitary tract are activated by low glucose or glucoprivation: evidence for glial involvement in glucose homeostasis. Front Neurosci. 2013;7:249. doi: 10.3389/fnins.2013.00249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizuno Y, Oomura Y. Glucose responding neurons in the nucleus tractus solitarius of the rat: in vitro study. Brain Res. 1984;307:109–116. doi: 10.1016/0006-8993(84)90466-9. [DOI] [PubMed] [Google Scholar]
- Novin D, VanderWeele DA, Rezek M. Infusion of 2-deoxy-d-glucose into the hepatic-portal system causes eating: evidence for peripheral glucoreceptors. Science. 1973;181:858–860. doi: 10.1126/science.181.4102.858. [DOI] [PubMed] [Google Scholar]
- Ormsbee HS, 3rd, Bass P. Gastroduodenal motor gradients in the dog after pyloroplasty. Am J Physiol. 1976;230:389–397. doi: 10.1152/ajplegacy.1976.230.2.389. [DOI] [PubMed] [Google Scholar]
- Pascual O, Casper KB, Kubera C, Zhang J, Revilla-Sanchez R, Sul JY, Takano H, Moss SJ, McCarthy K, Haydon PG. Astrocytic purinergic signaling coordinates synaptic networks. Science. 2005;310:113–116. doi: 10.1126/science.1116916. [DOI] [PubMed] [Google Scholar]
- Pavlov IP. The work of the digestive glands. Ed 2. Philadelphia: Charles Griffin; 1910. The centrifugal (efferent) nerves to the gastric glands and of the pancreas; pp. 48–59. [Google Scholar]
- Richter CP. Increased dextrose appetite of normal rats treated with insulin. Am J Physiol. 1941;135:781–787. [Google Scholar]
- Ritter RC, Roelke M, Neville M. Glucoprivic feeding behavior in absence of other signs of glucoprivation. Am J Physiol. 1978;234:E617–E621. doi: 10.1152/ajpendo.1978.234.6.E617. [DOI] [PubMed] [Google Scholar]
- Ritter S, Llewellyn-Smith I, Dinh TT. Subgroups of hindbrain catecholamine neurons are selectively activated by 2-deoxy-d-glucose induced metabolic challenge. Brain Res. 1998;805:41–54. doi: 10.1016/S0006-8993(98)00655-6. [DOI] [PubMed] [Google Scholar]
- Ritter S, Dinh TT, Zhang Y. Localization of hindbrain glucoreceptive sites controlling food intake and blood glucose. Brain Res. 2000;856:37–47. doi: 10.1016/S0006-8993(99)02327-6. [DOI] [PubMed] [Google Scholar]
- Rogers RC, Hermann GE. Oxytocin, oxytocin antagonist, TRH, and hypothalamic paraventricular nucleus stimulation effects on gastric motility. Peptides. 1987;8:505–513. doi: 10.1016/0196-9781(87)90017-9. [DOI] [PubMed] [Google Scholar]
- Rogers RC, McCann MJ. Effects of TRH on the activity of gastric inflation-related neurons in the solitary nucleus in the rat. Neurosci Lett. 1989;104:71–76. doi: 10.1016/0304-3940(89)90331-5. [DOI] [PubMed] [Google Scholar]
- Rogers RC, Viard E, Hermann GE. CXCL12 sensitizes vago-vagal reflex neurons in the dorsal medulla. Brain Res. 2013;1492:46–52. doi: 10.1016/j.brainres.2012.11.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogers RC, Hermann GE. Brainstem control of gastric function. In: Johnson LR, editor. Physiology of the gastrointestinal tract. Ed 4. New York: Elsevier; 2012. pp. 861–892. [Google Scholar]
- Schemann M, Grundy D. Electrophysiological identification of vagally innervated enteric neurons in guinea pig stomach. Am J Physiol. 1992;263:G709–G718. doi: 10.1152/ajpgi.1992.263.5.G709. [DOI] [PubMed] [Google Scholar]
- Swanson RA, Graham SH. Fluorocitrate and fluoroacetate effects on astrocyte metabolism in vitro. Brain Res. 1994;664:94–100. doi: 10.1016/0006-8993(94)91958-5. [DOI] [PubMed] [Google Scholar]
- Taché Y, Garrick T, Raybould H. Central nervous system action of peptides to influence gastrointestinal motor function. Gastroenterology. 1990;98:517–528. doi: 10.1016/0016-5085(90)90849-v. [DOI] [PubMed] [Google Scholar]
- Thorens B. GLUT2 in pancreatic and extra-pancreatic gluco-detection (review) Mol Membr Biol. 2001;18:265–273. doi: 10.1080/09687680110100995. [DOI] [PubMed] [Google Scholar]
- Verkhratsky A, Rodríguez JJ, Parpura V. Calcium signalling in astroglia. Mol Cell Endocrinol. 2012;353:45–56. doi: 10.1016/j.mce.2011.08.039. [DOI] [PubMed] [Google Scholar]
- Viard E, Rogers RC, Hermann GE. Systemic cholecystokinin amplifies vago-vagal reflex responses recorded in vagal motor neurones. J Physiol. 2012;590:631–646. doi: 10.1113/jphysiol.2011.224477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yettefti K, Orsini JC, el Ouazzani T, Himmi T, Boyer A, Perrin J. Sensitivity of nucleus tractus solitarius neurons to induced moderate hyperglycemia, with special reference to catecholaminergic regions. J Auton Nerv Syst. 1995;51:191–197. doi: 10.1016/0165-1838(94)00130-C. [DOI] [PubMed] [Google Scholar]
- Young JK, Baker JH, Montes MI. The brain response to 2-deoxy glucose is blocked by a glial drug. Pharmacol Biochem Behav. 2000;67:233–239. doi: 10.1016/S0091-3057(00)00315-4. [DOI] [PubMed] [Google Scholar]
- Zhang Q, Haydon PG. Roles for gliotransmission in the nervous system. J Neural Transm. 2005;112:121–125. doi: 10.1007/s00702-004-0119-x. [DOI] [PubMed] [Google Scholar]