

Abstract
Chronic pain caused by insults to the CNS (central neuropathic pain) is widely assumed to be maintained exclusively by central mechanisms. However, chronic hyperexcitablility occurs in primary nociceptors after spinal cord injury (SCI), suggesting that SCI pain also depends upon continuing activity of peripheral sensory neurons. The present study in rats (Rattus norvegicus) found persistent upregulation after SCI of protein, but not mRNA, for a voltage-gated Na+ channel, Nav1.8, that is expressed almost exclusively in primary afferent neurons. Selectively knocking down Nav1.8 after SCI suppressed spontaneous activity in dissociated dorsal root ganglion neurons, reversed hypersensitivity of hindlimb withdrawal reflexes, and reduced ongoing pain assessed by a conditioned place preference test. These results show that activity in primary afferent neurons contributes to ongoing SCI pain.
Keywords: Chronic pain, dorsal root ganglion, Nav1.8, neuropathic pain, nociceptor, spinal contusion
Introduction
After peripheral injury or inflammation, central sensitization in the spinal cord promotes allodynia, hyperalgesia, and spontaneous pain, but this central sensitization often requires continuing sensory activity (Baron et al., 2013). In contrast, pain resulting from injury or inflammation within the CNS (central neuropathic pain) is usually assumed to be maintained by central alterations (Finnerup, 2013). However, recent findings suggest that persistent hyperexcitability and spontaneous activity (SA) in primary sensory neurons might promote spinal cord injury (SCI) pain. First, SA occurs in peripheral terminals (Carlton et al., 2009) and in cell bodies of nociceptors in vivo and after dissociation from dorsal root ganglia (DRG; Bedi et al., 2010) long after SCI. Second, reduction of TRPV1 function reverses behavioral hypersensitivity after SCI (Wu et al., 2013). TRPV1 is expressed most abundantly in primary nociceptors (Cavanaugh et al., 2011), suggesting that activity in primary sensory neurons might drive reflex hypersensitivity after SCI. Furthermore, the aversive quality of pain (Baastrup et al., 2010; Navratilova et al., 2013) might also be driven by primary afferent activity after SCI.
A strong test of the hypothesis that activity in primary afferent neurons helps to maintain SCI pain (Walters, 2012) is enabled by the selective expression of a voltage-gated Na+ channel, Nav1.8, in somatic sensory neurons. Nav1.8 is absent in central neurons (Akopian et al., 1999; Shields et al., 2012) and is important for SA in primary afferent neurons after peripheral insults (Roza et al., 2003; Jarvis et al., 2007). Here, we report that knockdown of Nav1.8 channels after SCI reduces SA in primary afferent neurons, reverses reflex hypersensitivity, and ameliorates a pain-like aversive state.
Materials and Methods
Procedures.
All procedures complied with guidelines of the International Association for the Study of Pain and were approved by the institutional animal care and use committee. Male rats (200–350 g) were maintained under a 12 h reversed light/dark cycle and tested during the dark phase. Additional methodological details are available (Bedi et al., 2010; Wu et al., 2013).
SCI procedures.
Contusion injury occurred at vertebral segment T10 (Bedi et al., 2010). Rats were deeply anesthetized with ketamine (80 mg/kg), xylazine (10 mg/kg), and acepromazine (0.75 mg/kg) before laminectomy at T10, followed by a spinal impact using an Infinite Horizon impactor (150 kdyne, 1 s dwell time). Sham-operated (“sham”) animals received identical procedures except for spinal impact. Animals accepted for study exhibited Basso, Beattie, and Bresnahan (BBB) hindlimb motor scores of 0 1 d after SCI (Basso et al., 1995). All showed partial locomotor recovery by the end of testing, with extensive movement of all joints in the hindlimbs (BBB score ≥7).
Antisense oligodeoxynucleotide (ODN) knockdown of Nav1.8.
Previous studies identified an antisense oligodeoxynucleotide (ASO) sequence that is taken up in vivo by DRG neurons after intrathecal delivery and reduces expression of Nav1.8 protein (Porreca et al., 1999; Lai et al., 2002). This sequence, 5′-TCC-TCT-GTG-CTT-GGT-TCT-GGC-CT-3′, and a mismatched oligodeoxynucleotide (MMO) sequence, 5′-TCC-TTC-GTG-CTG-TGT-TCG-TGC-CT-3′, were purchased from Sigma-Aldrich. Approximately 1 month after SCI, rats were anesthetized with isoflurane and a chronic intrathecal catheter was inserted at the atlantooccipital joint terminating at the lumbar enlargement. Animals showing additional impairment after catheterization (altered body posture or forelimb function) were killed. Intrathecal injections (45 μg of ODN in 5 μl of saline, followed by a 10 μl of saline flush) were given 1–2 months after injury, twice daily for 3 d, and then once daily for 2 d.
Western blot analysis of Nav protein expression.
At the end of ODN injection, animals were deeply anesthetized and bilateral L4 and L5 ganglia were removed and immediately homogenized in RIPA buffer (Teknova) with protease inhibitors. Lysate protein concentrations were determined by bicinchoninic acid assay (Pierce). Equal amounts of total protein (30 μg) were resolved by SDS-PAGE (4–20% Tris-HCl; Bio-Rad) after 1:1 dilution with Laemmli buffer, transferred to a PVDF membrane, blocked with 10% nonfat dry milk, and incubated overnight at 4°C with antibodies against Nav1.8 (catalog #AB9274; Millipore), Nav1.6 (catalog #ASC-009; Alomone Labs), Nav1.7 (catalog #ASC-008; Alomone Labs), or Nav1.9 (catalog #AB-9222; Millipore), and β-actin (catalog #ab6276; Abcam). Secondary HRP anti-rabbit or anti-mouse IgGs were incubated for 1 h at 22°C. Blots were developed using an enhanced chemiluminescence substrate (Pierce). Optical densities were normalized to β-actin.
RT-PCR analysis of Nav1.8 mRNA expression in DRG neurons and spinal cord.
Total RNA was extracted from homogenized DRG or spinal cord with on-column DNase digestion (E.Z.N.A. Total RNA Kit I) and cDNA was synthesized by MMLV reverse transcriptase (Invitrogen) using random primer. Rat Nav1.8 primers were TCCCGGGGAAGGCTACATTA (forward) and TAATGTTGGCCCGGTCACTC (reverse; Hu et al., 2013); rat GAPDH primers were CCCCCAATGTATCCGTTGTG (forward) and TAGCCCAGGATGCCCTTTAGT (reverse; Piller et al., 2013). mRNA abundance was determined by real-time PCR (LightCycler 480; Roche) with SYBR Green Master Mix (Sigma). Preincubation at 95°C for 3 min was followed by 45 amplification cycles (95°C for 30 s, 57°C for 30 s, and 72°C for 30 s) and fluorescence collection at 60°C. Gene expression was normalized to Gapdh and expressed as fold change of sham control averaged over three replicates from each of three animals in each group.
Dissociation and culture of DRG neurons.
Selected DRG neurons (L5, L4, T12, T11, T9, T8) were minced and incubated for 40 min at 34°C with trypsin (0.4 mg/ml) and collagenase D (1.6 mg/ml). DRG fragments were triturated and the neurons were plated without serum or growth factors onto dishes coated with poly-l-lysine and kept overnight in DMEM under 5% CO2, 95% humidity at 37°C.
Recording from dissociated DRG neurons.
Whole-cell patch recordings of SA were made at ∼23°C from small neurons 18–26 h after dissociation, as described previously (Bedi et al., 2010). Tetrodotoxin (TTX)-resistant currents were measured in solution containing the following (in mm): 130 NaCl, 4 KCl, 2 CaCl2, 1 MgCl2, 0.1 CdCl2, 10 TEA-Cl, 10 HEPES, and 5 glucose, and a pipette solution containing the following (in mm): 100 CsCl, 30 CsF, 8 NaCl, 1 CaCl2, 1 MgCl2, 0.4 Na2GTP, 4 MgATP, 10 EGTA, and 10 HEPES. Current–voltage relationships were determined with 250 nm TTX and a holding potential of −60 mV to isolate Nav1.8 channels (Cummins et al., 1999); 200 ms command potentials were delivered from −100 to 50 mV in 10 mV increments at 5 s intervals, with each command following a 100 ms conditioning prepulse to −120 mV.
Reflex hypersensitivity tests.
Tests were conducted by investigators blinded to treatment before injury and just before and at the end of ODN treatment (Bedi et al., 2010; Wu et al., 2013). Animals were habituated to test chambers on day 1. On days 2 and 3, heat and mechanical test stimuli were given for habituation to test procedures; data were collected from tests on days 4 and 5. Hindlimb heat hypersensitivity was tested by the Hargreaves radiant heat method. Mechanical hypersensitivity was tested with calibrated von Frey filaments delivered to the glabrous surface of hindpaws.
Conditioned place preference test for ongoing pain.
We modified conditioned place preference (CPP) procedures used in peripheral pain models (King et al., 2009). A commercial CPP box (Med Associates) with automated data collection had three chambers with equal levels of dim illumination: black and white end chambers and a connecting gray chamber. On day 1 (∼2 months after injury), each rat was permitted to explore the gray and black (but not white) chambers. Conditioning trials occurred on days 2–4. Each morning, 0.5 ml of vehicle (saline) was injected intraperitoneally 10 min before confinement in the black chamber for 60 min. Three hours later, the same rat was injected with retigabine (10 mg/kg in 0.5 ml of saline; Yang et al., 2013) 10 min before confinement in the white chamber. On day 5, each rat was placed in the gray chamber with unrestricted access to all chambers for 15 min. Data are presented as the difference in time spent in the retigabine-paired (white) chamber minus time in the vehicle-paired (black) chamber during the drug-free day 5 test. The total number of crossings into all three chambers provided objective criteria to exclude SCI animals exhibiting excessive locomotor impairment (<21 crossings; two SCI + MMO and two SCI + ASO animals) or insufficient spinal injury (>250 crossings; one SCI + MMO and one SCI + ASO animal). For comparison, sham animals exhibited 190 ± 26 total crossings during the test.
Statistical analysis.
Data are presented as means ± SEM. Comparisons were made with Student's t tests or one- or two-way ANOVA, followed by Bonferroni's post hoc tests. SA incidence was compared using Fisher's exact tests.
Results
Nav1.8 antisense treatment selectively reduces Nav1.8 expression in DRG neurons after upregulation by SCI
We investigated whether SCI had any effect on Nav1.8 expression in L4 and L5 DRG. These DRG are sufficiently far from the vertebral T10 contusion site that few C-fiber neurons should be injured directly by the T10 injury (Bedi et al., 2010), but neurons in these DRG may be exposed to inflammatory signals disseminated after SCI (McKay and McLachlan, 2004; Alexander and Popovich, 2009), which might alter Nav1.8 expression (Yu et al., 2011). Processing for Western blot analysis began immediately after excision of L4 and L5 DRG 1 month after injury (Fig. 1A). The amount of Nav1.8 protein differed among DRG from naive, sham, and SCI animals (F(2,11) = 8.83; p = 0.005), with the levels being significantly higher in ganglia from SCI than naive or sham animals (p < 0.01 and p < 0.05, respectively). No difference was found between SCI and sham groups in Nav1.8 mRNA expression in lumbar DRG, nor was any evidence found for Nav1.8 mRNA expression in lumbar spinal cord in either group (Fig. 1B). Using a Nav1.8 ASO sequence (Lai et al., 2002), we compared the expression of four different Nav proteins after Nav1.8 ASO treatment to the corresponding expression after Nav1.8 MMO treatment (Fig. 1C). No significant differences were found in expression of Nav1.6, Nav1.7, or Nav1.9 channels in SCI animals after lumbar Nav1.8 ASO treatment, whereas Nav1.8 protein expression was significantly lower after Nav1.8 ASO treatment than after MMO treatment (p = 0.002).
Figure 1.
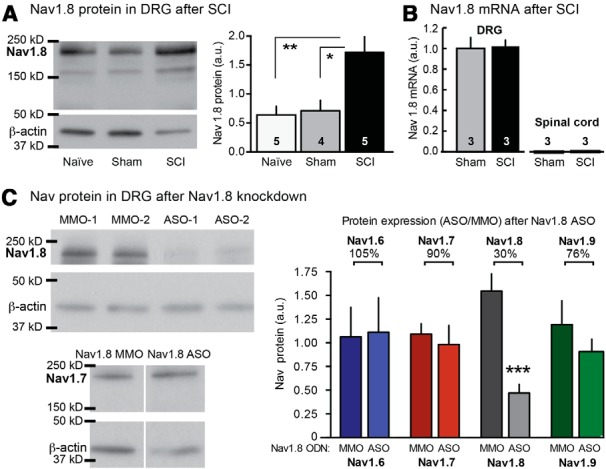
Effects of SCI and Nav1.8 antisense treatment on Nav channel expression. A, SCI increases Nav1.8 protein in L4 and L5 DRG 1 month after injury. Left, Sample Western blots. Right, Increase in Nav1.8 protein after SCI (normalized to β-actin). Number of animals tested (four DRG per animal) is indicated in each bar. B, Nav1.8 mRNA expression is detectable in DRG, but not spinal cord, and is not altered by SCI. C, Nav1.8 ASO treatment selectively knocks down Nav1.8 protein expression. Left, Sample Western blot (two animals each for Nav1.8 and one each for Nav1.7). Right, Normalized expression of four Nav channels. Percentages indicate mean relative expression for each channel protein after Nav1.8 ASO treatment compared with Nav1.8 MMO treatment.
Nav1.8 antisense treatment reduces TTX-resistant Na+ current and spontaneous activity in DRG neurons after SCI
Nav1.8 channels mediate a distinctive Na+ current that is resistant to TTX and has an unusually depolarized voltage dependence of fast inactivation (Akopian et al., 1996; Dib-Hajj et al., 1997). We assessed the degree of reduction of Nav1.8-mediated, TTX-resistant Na+ current by antisense knockdown in small DRG neurons from SCI animals. In the presence of 250 nm TTX, a whole-cell voltage-clamp pulse protocol that inactivates the other TTX-resistant Nav channel in DRG neurons, Nav1.9, was used (Cummins et al., 1999). Depolarizing steps from a −60 mV holding potential produced inward currents with relatively slow activation and inactivation kinetics (Fig. 2A, left), with a threshold level of ∼−30 mV and a peak at ∼0 mV (Fig. 2A, middle), suggestive of an Nav1.8-mediated current. The peak amplitude of evoked current was lower in DRG neurons after in vivo Nav1.8 ASO injections (Fig. 2A, right) than after MMO injections when tested ∼20 h (p < 0.0001, unpaired t test), but not ∼40 h, after the last injection, consistent with known rates of turnover of Nav channels (Porreca et al., 1999).
Figure 2.

In vivo Nav1.8 antisense treatment reduces TTX-resistant Na+ current and SA in small primary afferent neurons tested in vitro after SCI. A, Reduction of TTX-resistant current 18–24 h after Nav1.8 ASO injection. Depolarizing steps evoked smaller currents compared with MMO treatment (left and middle). Peak TTX-resistant current was significantly reduced ∼20 h, but not ∼40 h, after the last ASO injection (right). B, ASO treatment decreased the incidence of SA (example in left) 3 d and 1–2 months after SCI (right). Significant suppression from lumbar Nav1.8 ASO injection occurred in lumbar, but not thoracic (T8, T9, T11, T12), DRG neurons.
An important prediction was that knockdown of Nav1.8 expression would reduce SCI-induced SA. SCI quadrupled the incidence of SA in small DRG neurons (Fig. 2B, left) dissociated from L4 and L5 ganglia 3 d after injury compared with neurons dissociated from naive animals (p < 0.0001; Fig. 2B, right), and this increase was abolished by Nav1.8 ASO injections at the lumbar level (p = 0.003 vs SCI). Similarly, SCI increased the incidence of SA 1–3 months after SCI, and this increase was abolished by lumbar SCI + ASO treatment (p = 0.006). SA incidence was significantly greater in lumbar DRG neurons after SCI or SCI + MMO treatment than in lumbar DRG neurons after lumbar SCI + ASO treatment (p = 0.003). No significant effects on SCI-induced increases in SA were found in DRG neurons dissociated from thoracic ganglia (Fig. 2B, right) after Nav1.8 ASO treatment at the lumbar level.
Nav1.8 antisense treatment reverses hindlimb hyperreflexia and ongoing pain after SCI
Persistent sensitization of hindlimb withdrawal responses evoked by mechanical and heat test stimuli was found 1–3 months after SCI, similar to earlier comparisons of SCI, sham, and naive groups (Bedi et al., 2010), and this sensitization was reversed by Nav1.8 ASO treatment. For mechanical sensitivity (Fig. 3A), significant effects were found for test sequence (pre-SCI, post-SCI, post-ODN; F(2,84) = 23.9; p < 0.0001), ODN treatment (F(1,57) = 4.5; p = 0.039), and their interaction (F(2,57) = 4.82; p = 0.0012). Post hoc comparison revealed higher post-ODN withdrawal threshold in ASO-treated than MMO-treated rats (p = 0.0009). Twenty-eight of 29 animals showed mechanical hypersensitivity after SCI (before ODN treatment) and 72% also showed heat hypersensitivity. In these animals, significant effects were found for test sequence (F(2,57) = 14.2; p < 0.0001), ODN treatment (F(1,9) = 11.03; p = 0.009), and their interaction (F(2,57) = 4.82; p = 0.011). Post hoc comparison revealed higher post-ODN withdrawal latency in ASO-treated than in MMO-treated rats (p = 0.0019).
Figure 3.

In vivo Nav1.8 ASO treatment reverses behavioral indications of hyperreflexia and pain 1–3 months after SCI. A, Nav1.8 ASO treatment reversed the reduction in mechanical threshold for hindlimb withdrawal. B, Nav1.8 ASO treatment reversed the reduction in latency for withdrawal to heat. C, Intraperitoneal injections of retigabine supported CPP after SCI (left), which was prevented in SCI animals by Nav1.8 ASO treatment (right). CPP score (left axis) quantifies relative preference (indicated by arrows on right) for the white (retigabine paired) and black (vehicle paired) chambers after conditioning.
To investigate ongoing pain, we used operant conditioning of place preference to a chamber paired with retigabine injection. Retigabine opens KCNQ K+ channels, reducing neuronal excitability and, in other models, behavioral hypersensitivity (Brown and Passmore, 2009). Importantly, we found recently that retigabine suppresses SA in small DRG neurons and reverses hyperreflexia after SCI (Yang et al., 2013). One day after the 3 d differential conditioning procedure, sham animals preferred the vehicle-paired black chamber (as do naive animals, unpublished observations), whereas SCI animals showed relative preference for the white, retigabine-paired chamber (p = 0.003, unpaired t test; Fig. 3C, left). Preference for the white chamber in SCI, but not sham, animals indicates that retigabine is only rewarding when an SCI-induced aversive state is present. To test whether knockdown of Nav1.8 reduces the conditioned shift in preference toward the white chamber in SCI animals, we tested animals after MMO or ASO treatment and found that, compared with the SCI + MMO animals, SCI + ASO animals significantly preferred the black chamber (p = 0.045; Fig. 3C, right). The absence in SCI + ASO animals of a shift in preference away from the innately preferred, vehicle-paired black chamber suggests that Nav1.8 function is necessary to maintain ongoing pain after SCI.
Discussion
Finding that persistent pain induced by SCI requires Nav1.8, which is expressed almost exclusively in primary afferent neurons (Akopian et al., 1999; Shields et al., 2012), indicates that Nav1.8-expressing sensory neurons play a major role in driving SCI pain and perhaps associated central neuropathic alterations (Finnerup, 2013). Our results confirm that intrathecal application of a Nav1.8 ASO sequence reduces expression of Nav1.8 protein in DRG and functional activity of Nav1.8 channels (Porreca et al., 1999; Lai et al., 2002; Gold et al., 2003; Yu et al., 2011). We also demonstrate that this knockdown is highly specific; Nav1.8 ASO treatment did not significantly reduce DRG expression of related Na+ channels: Nav1.6 and Nav1. 7 (see also Porreca et al., 1999) and Nav 1.9. Reversal of reflex hypersensitivity by Nav1.8 ASO treatment has also been shown in peripheral injury and inflammation models (Yoshimura et al., 2001; Villarreal et al., 2005; Joshi et al., 2006; Morgan and Gebhart, 2008; Miao et al., 2010).
Our hypothesis that SA in primary afferent neurons drives chronic pain after SCI (Bedi et al., 2010; Walters, 2012) is supported by three findings. First, Nav1.8 mRNA was expressed in DRG, but not the spinal cord, even after SCI. Second, Nav1.8 knockdown eliminated the increase in SA in DRG neurons and hyperreflexia after SCI, suggesting that electrical activity in DRG neurons maintains hyperreflexia. This probably explains the strong correlation between the incidence of SA in dissociated DRG neurons and the degree of reflex hypersensitivity (Bedi et al., 2010). Third, an intervention that selectively reduces activity in primary sensory neurons (Nav1.8 knockdown) blocks a CPP measure of ongoing pain that captures its aversive quality (Navratilova et al., 2013). Because retigabine's effects may involve central as well as peripheral mechanisms (Brown and Passmore, 2009), it was important that the drug was only present during conditioning, not testing. Suppression by retigabine of ongoing pain during conditioning rather than long-lasting central actions or intrinsic reward value was indicated by the lack of retigabine-dependent CPP in sham, naive (unpublished observations), or Nav1.8 SCI + ASO animals. The finding of retigabine-dependent CPP after SCI adds to operant evidence for SCI-induced aversive states in rodents (Baastrup et al., 2010; Davoody et al., 2011; Lau et al., 2012; Vierck et al., 2013).
In many nociceptors, Nav1.8 channels are responsible for the upstroke of the action potential (Renganathan et al., 2001), which could explain suppression of SA by knockdown of Nav1.8. Upregulation of Nav1.8 after SCI is likely to contribute to generalized nociceptor hyperexcitability and might contribute to increased SA (Choi and Waxman, 2011), although multiple electrophysiological alterations probably promote nociceptor SA after SCI (Bedi et al., 2010; Wu et al., 2013). Regardless of the complexity of SA mechanisms, the requirement for Nav1.8 suggests that interventions preferentially targeting nociceptive primary afferent neurons, such as antagonists of Nav1.8 (Jarvis et al., 2007) or TRPV1 (Wu et al., 2013), may relieve some forms of central neuropathic pain.
Footnotes
This work was supported by grants from the United States Army Medical Research Acquisition Activity–United States Department of Defense, Mission Connect/TIRR Foundation, and the Craig H. Neilsen Foundation. We thank Carmen Dessauer, Raymond Grill, and Hongzhen Hu for helpful suggestions.
The authors declare no competing financial interests.
References
- Akopian AN, Sivilotti L, Wood JN. A tetrodotoxin-resistant voltage-gated sodium channel expressed by sensory neurons. Nature. 1996;379:257–262. doi: 10.1038/379257a0. [DOI] [PubMed] [Google Scholar]
- Akopian AN, Souslova V, England S, Okuse K, Ogata N, Ure J, Smith A, Kerr BJ, McMahon SB, Boyce S, Hill R, Stanfa LC, Dickenson AH, Wood JN. The tetrodotoxin-resistant sodium channel SNS has a specialized function in pain pathways. Nat Neurosci. 1999;2:541–548. doi: 10.1038/9195. [DOI] [PubMed] [Google Scholar]
- Alexander JK, Popovich PG. Neuroinflammation in spinal cord injury: therapeutic targets for neuroprotection and regeneration. Prog Brain Res. 2009;175:125–137. doi: 10.1016/S0079-6123(09)17508-8. [DOI] [PubMed] [Google Scholar]
- Baastrup C, Maersk-Moller CC, Nyengaard JR, Jensen TS, Finnerup NB. Spinal-, brainstem- and cerebrally mediated responses at- and below-level of a spinal cord contusion in rats: evaluation of pain-like behavior. Pain. 2010;151:670–679. doi: 10.1016/j.pain.2010.08.024. [DOI] [PubMed] [Google Scholar]
- Baron R, Hans G, Dickenson AH. Peripheral input and its importance for central sensitization. Ann Neurol. 2013;74:630–636. doi: 10.1002/ana.24017. [DOI] [PubMed] [Google Scholar]
- Basso DM, Beattie MS, Bresnahan JC. A sensitive and reliable locomotor rating scale for open field testing in rats. J Neurotrauma. 1995;12:1–21. doi: 10.1089/neu.1995.12.1. [DOI] [PubMed] [Google Scholar]
- Bedi SS, Yang Q, Crook RJ, Du J, Wu Z, Fishman HM, Grill RJ, Carlton SM, Walters ET. Chronic spontaneous activity generated in the somata of primary nociceptors is associated with pain-related behavior after spinal cord injury. J Neurosci. 2010;30:14870–14882. doi: 10.1523/JNEUROSCI.2428-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown DA, Passmore GM. Neural KCNQ (Kv7) channels. Br J Pharmacol. 2009;156:1185–1195. doi: 10.1111/j.1476-5381.2009.00111.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlton SM, Du J, Tan HY, Nesic O, Hargett GL, Bopp AC, Yamani A, Lin Q, Willis WD, Hulsebosch CE. Peripheral and central sensitization in remote spinal cord regions contribute to central neuropathic pain after spinal cord injury. Pain. 2009;147:265–276. doi: 10.1016/j.pain.2009.09.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cavanaugh DJ, Chesler AT, Jackson AC, Sigal YM, Yamanaka H, Grant R, O'Donnell D, Nicoll RA, Shah NM, Julius D, Basbaum AI. Trpv1 reporter mice reveal highly restricted brain distribution and functional expression in arteriolar smooth muscle cells. J Neurosci. 2011;31:5067–5077. doi: 10.1523/JNEUROSCI.6451-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi JS, Waxman SG. Physiological interactions between Na(v)1.7 and Na(v)1.8 sodium channels: a computer simulation study. J Neurophysiol. 2011;106:3173–3184. doi: 10.1152/jn.00100.2011. [DOI] [PubMed] [Google Scholar]
- Cummins TR, Dib-Hajj SD, Black JA, Akopian AN, Wood JN, Waxman SG. A novel persistent tetrodotoxin-resistant sodium current in SNS-null and wild-type small primary sensory neurons. J Neurosci. 1999;19:RC43. doi: 10.1523/JNEUROSCI.19-24-j0001.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davoody L, Quiton RL, Lucas JM, Ji Y, Keller A, Masri R. Conditioned place preference reveals tonic pain in an animal model of central pain. J Pain. 2011;12:868–874. doi: 10.1016/j.jpain.2011.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dib-Hajj SD, Ishikawa K, Cummins TR, Waxman SG. Insertion of a SNS-specific tetrapeptide in S3–S4 linker of D4 accelerates recovery from inactivation of skeletal muscle voltage-gated Na channel mu1 in HEK293 cells. FEBS Lett. 1997;416:11–14. doi: 10.1016/S0014-5793(97)01154-X. [DOI] [PubMed] [Google Scholar]
- Finnerup NB. Pain in patients with spinal cord injury. Pain. 2013;154:S71–S76. doi: 10.1016/j.pain.2012.12.007. [DOI] [PubMed] [Google Scholar]
- Gold MS, Weinreich D, Kim CS, Wang R, Treanor J, Porreca F, Lai J. Redistribution of Na(V)1.8 in uninjured axons enables neuropathic pain. J Neurosci. 2003;23:158–166. doi: 10.1523/JNEUROSCI.23-01-00158.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu S, Xiao Y, Zhu L, Li L, Hu CY, Jiang X, Xu GY. Neonatal maternal deprivation sensitizes voltage-gated sodium channel currents in colon-specific dorsal root ganglion neurons in rats. Am J Physiol Gastrointest Liver Physiol. 2013;304:G311–G321. doi: 10.1152/ajpgi.00338.2012. [DOI] [PubMed] [Google Scholar]
- Jarvis MF, Honore P, Shieh CC, Chapman M, Joshi S, Zhang XF, Kort M, Carroll W, Marron B, Atkinson R, Thomas J, Liu D, Krambis M, Liu Y, McGaraughty S, Chu K, Roeloffs R, Zhong C, Mikusa JP, Hernandez G, et al. A-803467, a potent and selective Nav1.8 sodium channel blocker, attenuates neuropathic and inflammatory pain in the rat. Proc Natl Acad Sci U S A. 2007;104:8520–8525. doi: 10.1073/pnas.0611364104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joshi SK, Mikusa JP, Hernandez G, Baker S, Shieh CC, Neelands T, Zhang XF, Niforatos W, Kage K, Han P, Krafte D, Faltynek C, Sullivan JP, Jarvis MF, Honore P. Involvement of the TTX-resistant sodium channel Nav 1.8 in inflammatory and neuropathic, but not post-operative, pain states. Pain. 2006;123:75–82. doi: 10.1016/j.pain.2006.02.011. [DOI] [PubMed] [Google Scholar]
- King T, Vera-Portocarrero L, Gutierrez T, Vanderah TW, Dussor G, Lai J, Fields HL, Porreca F. Unmasking the tonic-aversive state in neuropathic pain. Nat Neurosci. 2009;12:1364–1366. doi: 10.1038/nn.2407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai J, Gold MS, Kim CS, Bian D, Ossipov MH, Hunter JC, Porreca F. Inhibition of neuropathic pain by decreased expression of the tetrodotoxin-resistant sodium channel, NaV1.8. Pain. 2002;95:143–152. doi: 10.1016/S0304-3959(01)00391-8. [DOI] [PubMed] [Google Scholar]
- Lau D, Harte SE, Morrow TJ, Wang S, Mata M, Fink DJ. Herpes simplex virus vector-mediated expression of interleukin-10 reduces below-level central neuropathic pain after spinal cord injury. Neurorehabil Neural Repair. 2012;26:889–897. doi: 10.1177/1545968312445637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKay SM, McLachlan EM. Inflammation of rat dorsal root ganglia below a mid-thoracic spinal transection. Neuroreport. 2004;15:1783–1786. doi: 10.1097/01.wnr.0000135700.52904.77. [DOI] [PubMed] [Google Scholar]
- Miao XR, Gao XF, Wu JX, Lu ZJ, Huang ZX, Li XQ, He C, Yu WF. Bilateral downregulation of Nav1.8 in dorsal root ganglia of rats with bone cancer pain induced by inoculation with Walker 256 breast tumor cells. BMC Cancer. 2010;10:216. doi: 10.1186/1471-2407-10-216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgan JR, Gebhart GF. Characterization of a model of chronic orofacial hyperalgesia in the rat: contribution of NA(V) 1.8. J Pain. 2008;9:522–531. doi: 10.1016/j.jpain.2008.01.326. [DOI] [PubMed] [Google Scholar]
- Navratilova E, Xie JY, King T, Porreca F. Evaluation of reward from pain relief. Ann N Y Acad Sci. 2013;1282:1–11. doi: 10.1111/nyas.12095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piller N, Decosterd I, Suter MR. Reverse transcription quantitative real-time polymerase chain reaction reference genes in the spared nerve injury model of neuropathic pain: validation and literature search. BMC Res Notes. 2013;6:266. doi: 10.1186/1756-0500-6-266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porreca F, Lai J, Bian D, Wegert S, Ossipov MH, Eglen RM, Kassotakis L, Novakovic S, Rabert DK, Sangameswaran L, Hunter JC. A comparison of the potential role of the tetrodotoxin-insensitive sodium channels, PN3/SNS and NaN/SNS2, in rat models of chronic pain. Proc Natl Acad Sci U S A. 1999;96:7640–7644. doi: 10.1073/pnas.96.14.7640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Renganathan M, Cummins TR, Waxman SG. Contribution of Na(v)1.8 sodium channels to action potential electrogenesis in DRG neurons. J Neurophysiol. 2001;86:629–640. doi: 10.1152/jn.2001.86.2.629. [DOI] [PubMed] [Google Scholar]
- Roza C, Laird JM, Souslova V, Wood JN, Cervero F. The tetrodotoxin-resistant Na+ channel Nav1.8 is essential for the expression of spontaneous activity in damaged sensory axons of mice. J Physiol. 2003;550:921–926. doi: 10.1113/jphysiol.2003.046110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shields SD, Ahn HS, Yang Y, Han C, Seal RP, Wood JN, Waxman SG, Dib-Hajj SD. Nav1.8 expression is not restricted to nociceptors in mouse peripheral nervous system. Pain. 2012;153:2017–2030. doi: 10.1016/j.pain.2012.04.022. [DOI] [PubMed] [Google Scholar]
- Vierck CJ, Cannon RL, Acosta-Rua AJ. Evaluation of lateral spinal hemisection as a preclinical model of spinal cord injury pain. Exp Brain Res. 2013;228:305–312. doi: 10.1007/s00221-013-3563-8. [DOI] [PubMed] [Google Scholar]
- Villarreal CF, Sachs D, Cunha FQ, Parada CA, Ferreira SH. The role of Na(V)1.8 sodium channel in the maintenance of chronic inflammatory hypernociception. Neurosci Lett. 2005;386:72–77. doi: 10.1016/j.neulet.2005.04.060. [DOI] [PubMed] [Google Scholar]
- Walters ET. Nociceptors as chronic drivers of pain and hyperreflexia after spinal cord injury: an adaptive-maladaptive hyperfunctional state hypothesis. Front Physiol. 2012;3:309. doi: 10.3389/fphys.2012.00309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Z, Yang Q, Crook RJ, O'Neil RG, Walters ET. TRPV1 channels make major contributions to behavioral hypersensitivity and spontaneous activity in nociceptors after spinal cord injury. Pain. 2013;154:2130–2141. doi: 10.1016/j.pain.2013.06.040. [DOI] [PubMed] [Google Scholar]
- Yang Q, Wu Z, Crook RJ, Du J, Carlton SM, Walters ET. Opening of KCNQ/Kv7 channels blocks both spontaneous activity in small DRG neurons and signs of chronic pain after spinal cord injury. Journal of Pain. 2013;14:S54. [Google Scholar]
- Yoshimura N, Seki S, Novakovic SD, Tzoumaka E, Erickson VL, Erickson KA, Chancellor MB, de Groat WC. The involvement of the tetrodotoxin-resistant sodium channel Na(v)1.8 (PN3/SNS) in a rat model of visceral pain. J Neurosci. 2001;21:8690–8696. doi: 10.1523/JNEUROSCI.21-21-08690.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu YQ, Zhao F, Guan SM, Chen J. Antisense-mediated knockdown of Na(V)1.8, but not Na(V)1.9, generates inhibitory effects on complete Freund's adjuvant-induced inflammatory pain in rat. PLoS One. 2011;6:e19865. doi: 10.1371/journal.pone.0019865. [DOI] [PMC free article] [PubMed] [Google Scholar]