

Abstract
We determined the complete mitochondrial (mt) genomes of three evaniomorph species, Ceraphron sp. (Ceraphronoidea), Gasteruption sp. (Evanioidea), and Orthogonalys pulchella (Trigonalyoidea) as well as the nearly complete mt genome from another evaniomorph species, Megalyra sp. (Megalyroidea). Each of them possesses dramatic gene rearrangements, including protein-coding or rRNA genes. Gene inversions were identified in all of these mt genomes; for example, the two rRNA genes have inverted and moved into the nad2-cox1 junction in the Megalyra sp. mt genome. In addition, we found two copies of a 10-bp complementary repeat at the beginning of rrnS and at the end of trnL2 in the Gasteruption sp. mt genome, consistent with recombination as the possible mechanism for gene inversion and long-range movement. Although each of the genomes contains a number of repeats of varying size, there was no consistent association of the size or number of repeats with the extent or type of gene rearrangement. The breakpoint distance analysis showed the Evaniomorpha has an intermediate rate of gene rearrangement. Sequence-based phylogenetic analyses of 13 protein-coding and 2 rRNA genes in 22 hymenopteran taxa recovered a paraphyletic Evaniomorpha with the Aculeata nested within it. Within the Evaniomorpha, our analyses confirmed the Trigonalyoidea + Megalyroidea as the sister group to the Aculeata and recovered a novel clade, Ceraphronoidea + Evanioidea. In contrast to previous hymenopteran phylogenetic studies, the internal relationships of the Evaniomorpha were highly supported and robust to the variation of alignment approach and phylogenetic inference approach.
Keywords: mitochondrial genome, Evaniomorpha, Aculeata, phylogeny, gene rearrangement
Introduction
Knowledge of the forces that shape the organization of the animal mitochondrial (mt) genome has suffered from a lack of suitable model systems. This is because the rate of gene rearrangement in most animal mt genomes is generally extremely slow; for example, most vertebrates have identically arranged mt genomes (Boore 1999). The ideal model system would provide multiple gene rearrangements for comparison (to identify common themes), but in a lineage that is not rearranging so frequently that hidden and convergent rearrangements obscure the interpretation of evolutionary events. In the insects, a number of higher level lineages have been identified with accelerated rates of mt gene rearrangement—the Hymenoptera (Dowton and Austin 1999) and the hemipteroid orders (Shao et al. 2001) are two examples. Within these rapidly rearranging lineages, taxonomic sampling at lower levels may be all that is required to identify good model groups; that is, ones in which the evolutionary trajectory of genome reorganization is straightforward to interpret. In this study, we investigated whether one group of the Hymenoptera (the Evaniomorpha) might provide such a model system.
The Hymenoptera (sawflies, wasps, ants, and bees) is one of the most important components of insect diversity. As the third largest insect order, it is estimated to contain more than 140,000 extant species, placed into two traditional suborders (Symphyta and Apocrita) (Huber 2009). The Apocrita has long been considered as a natural group and comprises more than 90% of the Hymenoptera (Sharkey 2007; Huber 2009). Many apocritan species play valuable roles in biological control, ecosystem, and production of commercial products (LaSalle and Gauld 1993).
Traditionally, the Apocrita is subdivided into two groups, the Aculeata (stinging wasps) and the Parasitica (parasitoid wasps), with the Aculeata now widely accepted as being derived from within the Parasitica (Sharkey 2007). The reconstruction of a robust phylogeny for the Apocrita has long been of great interest to hymenopteran systematists (reviewed by Sharkey [2007]). In 1988, Rasnitsyn proposed a fully resolved phylogenetic hypothesis of higher level hymenopteran relationships based on morphological and fossil evidence (Rasnitsyn 1988). Despite the use of noncladistic methodology, Rasnitsyn’s research remains influential and sets a stage for current research of hymenopteran phylogeny. In his hypothesis, the Apocrita was divided into four lineages, the Ichneumonomorpha, the Vespomorpha (Aculeata), the Proctotrupomorpha, and the Evaniomorpha (fig. 1). The Ichneumonomorpha and Aculeata have long been recovered as natural groups (Dowton et al. 1997; Dowton and Austin 2001; Castro and Dowton 2006; Rasnitsyn and Zhang 2010; Vilhelmsen et al. 2010; Heraty et al. 2011; Sharkey et al. 2011; Klopfstein et al. 2013), and the monophyly of the Proctotrupomorpha has been supported with several comprehensive studies (Castro and Dowton 2006; Heraty et al. 2011; Sharkey et al. 2011; Klopfstein et al. 2013). However, the monophyly of the Evaniomorpha (including the Ceraphronoidea, Evanioidea, Megalyroidea, Stephanoidea, and Trigonalyoidea) remains unclear from both morphological and molecular analyses. In morphological analyses, a subsequent numerical cladistic analysis of Rasnitsyn’s (1988) data failed to retrieve the Evaniomorpha (Ronquist et al. 1999). Gibson (1999) suspected that the mesocoxal articulatory structure, which supported the monophyly of the Evaniomorpha according to Rasnitsyn (1988), was a retained symplesiomorphy rather than a synapomorphy (Gibson 1999). Moreover, Rasnitsyn himself proposed that the Evaniomorpha was not monophyletic and divided it into three lineages (Rasnitsyn and Zhang 2010). Most molecular analyses recovered the Evaniomorpha as paraphyletic, with the Aculeata nested within the Evaniomorpha. Further, the sister group of the Aculeata varied among different analyses (Castro and Dowton 2006; Heraty et al. 2011; Sharkey et al. 2011; Klopfstein et al. 2013). Within the Evaniomorpha, there is little consensus on the phylogenetic relationships among the superfamilies due to a lack of reliable morphological characters and the limited number of molecular markers.
Fig. 1.—
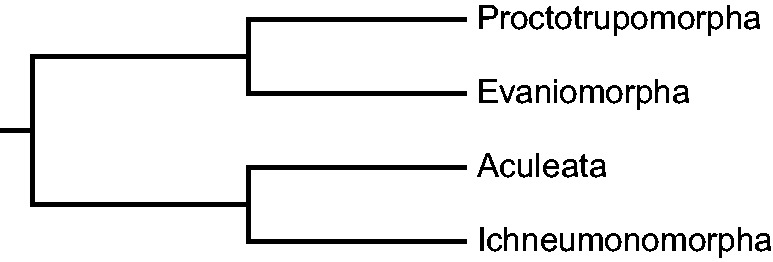
Simplified interpretation of the phylogenetic relationships among the major lineages within the Apocrita from Rasnitsyn (1998).
Prior to this study, there were only four evaniomorph mt genomes available in GenBank, representing three superfamilies (Evania appendigaster [Evanioidea: Evaniidae]; Pristaulacus compressus [Evanioidea: Aulacidae]; Schlettererius cinctipes [Stephanoidea: Stephanidae]; and Conostigmus sp. [Ceraphronoidea: Megaspilidae]) (Dowton, Cameron, Austin, et al. 2009; Wei, Tang, et al. 2010; Wei et al. 2013; Mao et al. 2014). Each of these mt genomes displayed a relatively small number of changes to their mt genome organization, when compared with the ancestral pancrustacean/hymenopteran organization, and these rearrangements involved both tRNA genes and (to a lesser extent) protein-coding genes. Schlettererius had evidence of two tRNA gene rearrangements (Dowton, Cameron, Dowavic, et al. 2009), Pristaulacus had evidence of one protein-coding gene (nad1) rearrangement and two tRNA gene rearrangements (Wei et al. 2013), Evania had evidence of four tRNA gene rearrangements (Wei, Tang, et al. 2010), whereas Conostigmus had 2 protein-coding genes rearranged (cox1, nad2) and 10 tRNA genes rearranged (Mao et al. 2014). Thus, this group represents a good candidate model system in which to investigate the dynamics of mt genome rearrangement, as individual rearrangements might be inferred through denser taxonomic sampling. For this reason, in this study we present four new mt genomes for representatives of four evaniomorph superfamilies, Megalyra sp. (Megalyroidea: Megalyridae), Orthogonalys pulchella (Trigonalyoidea: Trigonalyidae), Ceraphron sp. (Ceraphronoidea: Ceraphronidae) and Gasteruption sp. (Evanioidea: Gasteruptiidae). We also use these entire mt genomes to further investigate relationships among the Evaniomorpha and Aculeata. Because of the likely paraphyletic nature of the Evaniomorpha, we include the Aculeata to examine the phylogeny within a natural group.
Materials and Methods
DNA Extraction
The collection details for each study species are listed in table 1. Genomic DNA was extracted from 100% ethanol preserved specimens using the “salting out” protocol (Aljanabi and Martinez 1997). The DNA was resuspended in 100 μl of fresh TE solution (1 mM Tris–HCl, 0.1 mM ethylenediaminetetraacetic acid [pH 8]) and stored at 4° C.
Table 1.
Species Sequenced in This Study, Collection Data, and GenBank Accession Numbers
| Species | Library Reference | Family | Superfamily | Collection Locality | Accession Number |
|---|---|---|---|---|---|
| Ceraphron sp. | M247 | Ceraphronidae | Ceraphronoidea | CA | KJ570858 |
| Gasteruption sp. | M19 | Gasteruptionidae | Evanioidea | Auburn, South Australia | KJ619460 |
| Megalyra sp. | M272 | Megalyridae | Megalyroidea | Wistow, South Australia | KJ577600 |
| Orthogonalys pulchella | M16 | Trigonalyidae | Trigonalyoidea | Clarke County, WA | KJ619461 |
Mt Genome Amplification, Sequencing, and Annotation
Short gene fragments were amplified and sequenced using a range of universal insect mt primers (Simon et al. 1994, 2006) and primers that had been previously designed from consensus hymenopteran mt sequences. Using the sequence information obtained, taxon-specific primers were designed for each sample to amplify the remaining regions. Polymerase chain reaction and sequencing reactions were conducted as previously described (Mao et al. 2014). The sequences of both strands were determined for the entire mt genomes of Ceraphron sp., Gasteruption sp., and O. pulchella, In addition, we sequenced both strands for all coding regions and most of the A+T-rich region of Megalyra sp. but failed to get the full double-stranded sequence for the entire A+T-rich region due to an array of 50-bp tandem repeats. This array contained 20 repeats, and as a result, no unique internal sequencing primers could be designed.
Raw sequences were assembled into contigs in ChromasPro Ver 1.33 (Technelysium Ltd., Tewantin, Australia). tRNA genes were identified using tRNA-scan SE 1.21 (lowelab.ucsc.edu/tRNAscan-SE/, last accessed June 30, 2014) (Lowe and Eddy 1997) and ARWEN 1.2 (http://130.235.46.10/ARWEN/, last accessed June 30, 2014) (Laslett and Canbäck 2008). ORFinder (www.ncbi.nlm.nih.gov/gorf/gorf.html, last accessed June 30, 2014) was used to identify protein-coding genes, specifying the invertebrate mt genetic code. The start and stop codons of some genes were corrected according to the boundaries of tRNA genes and through alignment with other hymenopteran mt sequences. rRNA genes were identified using BLASTN, and the ends of genes were assumed to extend to the boundaries of the neighboring tRNA or protein-coding genes. However, the boundary between rrnL and rrnS was difficult to define in Megalyra sp., as there is no gene between them. In this case, we determined this boundary through sequence comparison with published hymenopteran mt rRNA sequences.
Nucleotide Composition, Gene Rearrangement, and Repeat Analyses
The A+T content was determined for the major strand of each of the four mt genomes by MEGA5 (Tamura et al. 2011). To investigate whether gene inversions influenced the nucleotide compositional biases, we also measured the AT and GC skews for some protein-coding genes (nad2, cox1, nad6, and cob) and two rRNA genes of 8 evaniomorph taxa and 12 aculeate taxa. These genes were inverted in at least one of the four new mt genomes (see below). The formulae used were AT-skew = (A − T)/(A + T) and GC-skew = (G − C)/(G + C) (Perna and Kocher, 1995).
Gene rearrangement analyses were conducted with CREx via the freely available CREx web server (http://pacosy.informatik.uni-leipzig.de/crex, last accessed June 30, 2014) (Bernt et al. 2007). We compared gene orders using a dissimilarity measurement—number of breakpoints and visually inspected the output diagram to identify shared, derived gene rearrangements.
Direct and inverted repeats longer than 12 bp were identified for the A+T-rich region and the entire mt genome of each evaniomorph taxa using UGENE v.1.13.1 (Okonechnikov et al. 2012). We calculated the number of different size repeats around the breakpoints and in the A+T-rich region to investigate whether any particular size of repeat facilitate gene rearrangement.
Sequence Alignment and Phylogenetic Analysis
A total of 24 taxa were analyzed in this study, including 8 evaniomorph taxa, 12 aculeate taxa, and 4 symphytan taxa. Nucleotide sequences for each of the 13 protein-coding genes and the 2 rRNA genes were imported into separate files using MEGA5 and aligned using Muscle (Edgar 2004) or MAFFT (Katoh et al. 2005). For the protein-coding genes (excluding the stop codons), an amino acid alignment was generated first for each gene in Muscle as implemented within MEGA5 or MAFFT at the freely available TranslatorX server (http://translatorx.co.uk/, last accessed June 30, 2014) (Abascal et al. 2010). A nucleotide alignment was then inferred from the amino acid alignment. The alignment parameters for all genes in Muscle were the default settings, which have been specified in a previous study (Mao et al. 2012). For the MAFFT alignment of the rRNA genes, we used the G-INS-i algorithm as implemented in the MAFFT web server (http://mafft.cbrc.jp/alignment/server/, last accessed June 30, 2014) (Katoh et al. 2005). MAFFT (G-INS-i) has been shown to be more accurate than other programs (Golubchik et al. 2007). No regions were excluded from the rRNA alignments. Individual gene alignments were concatenated prior to phylogenetic analysis.
The best partitioning schemes and corresponding nucleotide substitution models were determined with PartitionFinder version 1.0.1 (Lanfear et al. 2012) using the Bayesian Information Criterion and a heuristic search algorithm. A total of 41 data blocks were predefined (3 codon positions of 13 protein-coding genes + 2 rRNA genes). Maximum likelihood and Bayesian approaches were employed to infer phylogenetic trees. Maximum likelihood analyses were conducted with RAxML via the available RAxML BlackBox server (http://embnet.vital-it.ch/raxml-bb/, last accessed June 30, 2014) (Stamatakis et al. 2008). The GAMMA model of rate heterogeneity was employed for all partitions. The “Maximum likelihood search” and “Estimate proportion of invariable sites” boxes were selected, with a total of 100 bootstrap replicates performed. Bayesian analyses were conducted with MrBayes v. 3.2.2 (Ronquist et al. 2012) via the online CIPRES Science gateway portal (Miller et al. 2010). The Markov chain Monte Carlo process was set, so that four chains (three heated and one cold) ran simultaneously. Four separate runs using unlinked partitions (unlink statefreq = all; unlink revmat = all; unlink shape = all; unlink pinvar = all; prset applyto = all; ratepr = variable) for a total of 1,000,000 generations were performed, with sampling every 100 generations. Stationarity for each run was assessed by importing the parameter files into Tracer v. 1.5 (Rambaut and Drummond 2009).
Rogue Taxon Identification
“Rogue” taxa, defined as those that have an unstable position in topological trees (Wilkinson 1996), can obscure relationships that are consistently recovered during bootstrap and Bayesian analyses (Aberer et al. 2013). To reveal those relationships that were consistently recovered by our analyses, we conducted rogue taxon filtering using the approach implemented in RogueNaRok (http://rnr.h-its.org/rnr, last accessed June 30, 2014) (Aberer et al. 2013). The RAxML bootstrap tree sets and Bayesian tree sets (excluding burn-in) were employed to perform these analyses.
Results and Discussion
General Features of mt Genomes
Three complete and one nearly complete mt genomes were sequenced for this study: Ceraphron sp. (14,947 bp), Gasteruption sp. (17,884 bp), O. pulchella (17,277 bp), and Megalyra sp. (18,996 bp). For Megalyra sp., we were able to estimate the size of the genome with a reasonable degree of precision, because imperfect copies exist in the array of 50-bp tandem repeats, for which we failed to get the full double-stranded sequence. Each genome contains all of the 37 genes commonly found in animal mt genomes (Boore 1999). In addition, a second copy of trnE was identified in the O. pulchella mt genome. As the shortest hymenopteran complete mt genome to date, the Ceraphron sp. mt genome is extremely compact with a total of only 105 bp of short noncoding regions (excluding the A+T-rich region). The short noncoding regions in Megalyra sp., Gasteruption sp., and O. pulchella are 345-bp, 909-bp, and 919-bp long, respectively. The four mt genomes have a similar A+T content, ranging from 80% (Ceraphron sp.) to 83.8% (O. pulchella). The AT and GC skew analysis showed that gene inversions had no influence on nucleotide compositional biases (data not shown). Three of the conventional start codons (ATA, ATG, or ATT) could be assigned to all of the protein-coding genes. Most tRNA genes have a typical cloverleaf structure. The exceptions are trnS1 in all four species, trnS2 in Ceraphron sp. and trnR in Ceraphron sp. and Gasteruption sp. All of these tRNA genes lack the D-stem pairings in the dihydrouridine (DHU)-arm. The missing D-stem has been commonly noted for the trnS1 gene in insects (Sheffield et al. 2008; Mao et al. 2012; Yang et al. 2013), while to our knowledge, it has not been previously reported in trnS2 in insect mt genomes. The missing D-stem in trnR is likely a common feature of hymenopteran mt genomes. In addition to the two mt genomes reported here, it was also found in another two evaniomorph taxa (Conostigmus sp. and Schlettererius cinctipes) (Dowton, Cameron, Austin, et al. 2009; Mao et al. 2014) and in two sceliond mt genomes (Ceratobaeus sp. and Idris sp.) sequenced in our laboratory (Mao and Dowton 2014). Furthermore, the 3′-end of the aminoacyl acceptor stem of trnR overlaps with the downstream gene in Ceraphron sp., Megalyra sp., and O. pulchella. The truncated aminoacyl acceptor stem is probably restored to conventional structure by extensive tRNA editing (Dowton and Austin 1999; Lavrov et al. 2000; Masta and Boore 2004; Segovia et al. 2011). The considerable difference in genome size among the four evaniomorph taxa is mainly due to variation in the size of the A+T-rich region, which ranges from 692 bp (Ceraphron sp.) to 3,871 bp (Megalyra sp.). One or more series of tandem repeats could be found in the Megalyra sp., O. pulchella, and Gasteruption sp. mt genomes, whereas only three short nontandem repeats are present in the Ceraphron sp. mt genome (fig. 2 and supplementary fig. S1A, Supplementary Material online). Most notably, two copies of the A+T-rich region exist in the Gasteruption sp. mt genome, which are separated by trnL2. The second copy is 77.7% of the length of the first copy and the length difference is mainly due to the variation in repeat number of an 11-bp microsatellite sequence (supplementary fig. S1B, Supplementary Material online). Similarly, the major portion of the A+T-rich region in the O. pulchella mt genome is also duplicated (supplementary fig. S1C, Supplementary Material online). The presence of duplicated A+T-rich regions (control regions) has been reported in a diverse range of taxa, such as mantellid frogs (Kurabayashi et al. 2008), Amazona parrots (Eberhard et al. 2001), and Australasian Ixodes ticks (Shao et al. 2005). It is proposed that the presence of two control regions may be advantageous and be maintained either through stabilizing selection or through gene conversion (Eberhard et al. 2001).
Fig. 2.—

Structure of the A+T-rich region in the Ceraphron sp., Gasteruption sp., Megalyra sp., and Orthogonalys pulchella mt genomes. TR1-N and NTR1-N indicate the distinct tandem repeats and nontandem repeats in each genome, respectively. The 80-bp repeat in the O. pulchella mt genome, which was difficult to classify as either tandem or nontandem, is labeled as R1. The length and copy number of each repeat are indicated above each repeat. The small repeats that nest within the large repeats are not shown. The width of different boxes is not proportional but indicative of the size of a repeat within a particular genome. Different repeats in each mt genome are indicated with different colors. Boxes with asterisks underneath represent a partial copy of a repeat.
Genome Organizations
The four mt genome organizations are shown in figures 3–5. All of them possess dramatic gene rearrangements when compared with the ancestral pancrustacean mt genome organization (Cook 2005), which is also thought to represent the ancestral organization of the hymenopteran mt genome (Mao et al. 2012). Remarkably, each of the newly sequenced mt genomes has protein-coding or rRNA gene rearrangement(s), a feature which has not been commonly reported in the Hymenoptera (only 8 of the 38 available mt genomes [this number excludes congenerics] have protein-coding or rRNA gene rearrangements) (Castro et al. 2006; Oliveira et al. 2008; Dowton, Cameron, Dowavic, et al. 2009; Wei, Shi, et al. 2010; Xiao et al. 2011; Wei et al. 2013; Mao et al. 2014).
Fig. 3.—
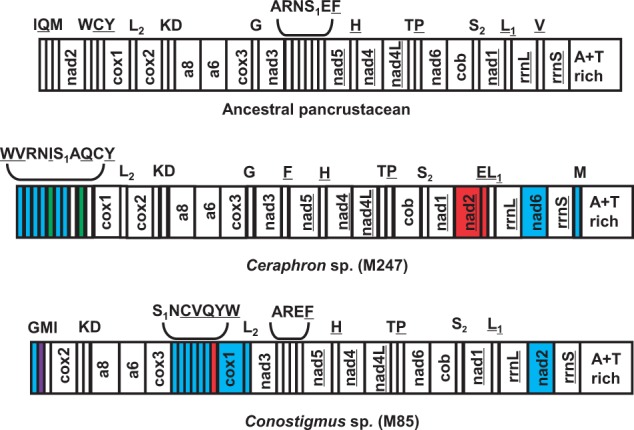
mt genome organization of two taxa from the Ceraphronoidea, Ceraphron sp. (present study) and Conostigmus sp. (Mao et al. 2014), compared with the ancestral pancrustacean/hymenopteran mt genome organization. tRNA genes are indicated by single-letter amino acid codes, L1, L2, S1, and S2 denote trnLCUN, trnLUUR, trnSAGN, and trnSUCN, respectively. Genes are transcribed from left to right except those indicated by underlining. Different types of gene movements, relative to the ancestral organization, are indicated with different colors: Blue represents long-distance translocation; red represents long-distance translocation and inversion; purple represents local translocation; green represents local inversion.
Fig. 4.—
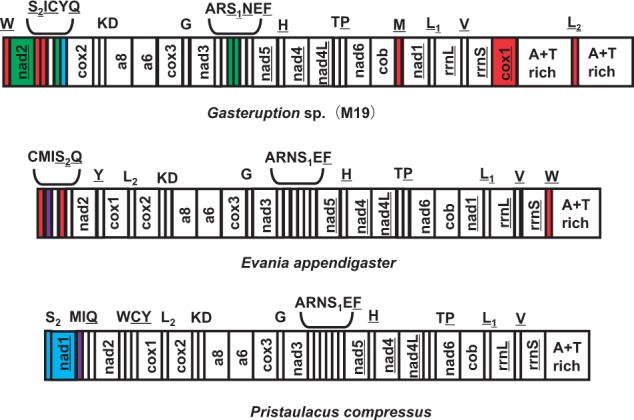
mt genome organization of three taxa from the Evanioidea: Gasteruption sp. (present study), Evania appendigaster (Wei, Tang, et al. 2010), and Pristaulacus compressus (Wei et al. 2013). Genes are transcribed from left to right except those indicated by underlining. Different types of gene movements, relative to the ancestral organization, are indicated with different colors: Blue represents long-distance translocation; red represents long-distance translocation and inversion; purple represents local translocation; green represents local inversion.
Fig. 5.—
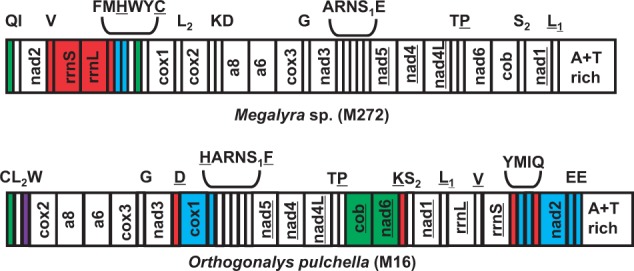
mt genome organization of Megalyra sp. (Megalyroidea) (present study) and Orthogonalys pulchella (Trigonalyoidea) (present study). The sister relationship of these two superfamilies is supported by present study and some other molecular studies. Genes are transcribed from left to right except those indicated by underlining. Different types of gene movements, relative to the ancestral organization, are indicated with different colors: Blue represents long-distance translocation; red represents long-distance translocation and inversion; purple represents local translocation; green represents local inversion.
The mt Genome of Ceraphron sp.
In Ceraphron sp., two protein coding genes (nad2 and nad6) and 10 tRNA genes have rearranged relative to their ancestral positions. The tRNA gene rearrangements mainly occur at the nad3-nad5 junction and around the A+T-rich region. Four tRNA genes (trnA, trnR, trnN, and trnS1) have moved from the nad3-nad5 junction to the tRNA gene cluster downstream of the A+T-rich region. trnE has also moved out of the nad3-nad5 junction, and we propose that it might be involved in two gene rearrangement events. First, it moved to the tRNA gene cluster downstream of the A+T-rich region together with the other four tRNA genes, then it inverted and moved to the nad1-trnL1 junction together with nad2. This is a more parsimonious scenario than independent movement with inversion of both genes (trnE and nad2) to the same gene junction. We also compared the mt genome organization of Ceraphron sp. with another ceraphronoid taxon, Conostigmus sp. (fig. 3), the sequence of which was reported in a previous study (Mao et al. 2014). In both ceraphronoids, the trnQ, trnN, trnS1, and trnV genes have moved to the tRNA gene cluster upstream of the cox1 gene, whereas the trnW gene has inverted. The nad2 gene has rearranged in both taxa but into different positions. The rearrangement of nad2 has also been reported in two chalcidoid taxa, Nasonia and Philotrypesis, in which nad2 has also moved into different positions (Oliveira et al. 2008; Xiao et al. 2011). The most striking gene rearrangement in both ceraphronoids is that the two rRNA genes are separated by protein-coding genes (nad6 in Ceraphron sp. and nad2 in Conostigmus sp.) instead of trnV lying between them, which has not been observed in other Hymenoptera. We consider that the existence of different protein-coding genes between the two rRNA genes might be useful markers to resolve the internal relationships of the Ceraphronoidea.
The mt Genome of Gasteruption sp.
The Gasteruption sp. mt genome possesses 11 gene rearrangements (two protein-coding genes and nine tRNA genes) and a duplication of the A+T-rich region (fig. 4) relative to the ancestral hymenopteran. The cox1 gene has inverted and moved into a position upstream of the A+T-rich region. The nad2 gene has also inverted but remains in its ancestral location. Among the rearranged tRNA genes, most of them have inverted and moved to positions remote from their ancestral locations, although trnY has inverted, but remains in its ancestral location (downstream of nad2). In addition, trnQ has moved to a position downstream of nad2. The existence of duplicate A+T-rich regions separated by trnL2 is consistent with the duplication/random loss model (Moritz et al. 1987). A parsimonious explanation is the cox1 and trnL2 genes inverted and moved to the position upstream of the A+T-rich region simultaneously (cox1-trnL2→trnL2-cox1), then a duplication of cox1, trnL2 and the A+T-rich region followed by random deletions of the redundant copy of the cox1 and trnL2 genes occurred. An important evidence for this explanation is that two copies of a 10-bp complementary repeat exist at the beginning of rrnS and at the end of trnL2 (AAAAGTATTT in rrnS and TTTTCATAAA in trnL2). We consider this short repeat may promote the cox1 and trnL2 inversion and long-range movement into the region between rrnS and A+T-rich region via recombination (Dowton and Campbell 2001; Mao et al. 2014). We then compared the mt genome organization of Gasteruption sp. with another two evanioid taxa, Evania appendigaster and Pristaulacus compressus (fig. 4). The gene positions are relatively conserved in Evania appendigaster and Pristaulacus compressus with only four and three gene rearrangements, respectively, when compared with the ancestral pancrustacean/hymenopteran. Although two tRNA genes (trnS2 and trnW) have inverted in both Gasteruption sp. and Evania appendigaster, no mt gene rearrangements are shared among the three taxa. Similarly, trnS2 is no longer adjacent to cob in all three taxa, but it has different, derived positions. Thus, among the three sequenced evanioid mt genomes, we identified 18 independent gene rearrangements, indicating that these could be extremely useful characters for assessing phylogeny in this superfamily.
The mt Genomes of Megalyra sp. and O. pulchella
Figure 5 shows the mt genomes of Megalyra sp. and O. pulchella sequenced in this study. Megalyra sp. is from the superfamily Megalyroidea, whereas O. pulchella is from the superfamily Trigonalyoidea. The sister relationship of these two superfamilies has been recovered by some molecular analyses (including this study, see below) (Dowton et al. 1997; Dowton and Austin 2001; Castro and Dowton 2006; Heraty et al. 2011; Klopfstein et al. 2013), so we illustrate them in one figure to facilitate comparisons. Both of them possess dramatic gene rearrangements when compared with the ancestral pancrustacean/hymenopteran. In Megalyra sp., there are eight gene rearrangements. The most striking rearrangement is the one in which the two rRNA genes have inverted and moved into the nad2-cox1 junction. Inverted rRNA genes have been reported in other insect orders, the Phthiraptera and Thysanoptera (Shao and Barker 2003; Covacin et al. 2006; Cameron et al. 2007, 2011). Interestingly, these four studies reported that both rRNA genes were inverted, similar to our observations in the Megalyra sp. mt genome. We suspect that there might be an advantage to having the two rRNA genes encoded on the same strand in insects. One candidate mechanism responsible for this phenomenon is the transcriptional feedback system regulating mtDNA replication. In this mechanism, the initiation of mtDNA replication is hypothesized to be positively correlated with the transcription rate, with the latter negatively regulated by the accumulation of gene products. Therefore, mtDNA replication is indirectly regulated by the amount of synthesized products (Axel and Thomas 2014). The rate of rRNA synthesis was found to be up to 60-fold higher than mRNA synthesis (i.e., of protein-coding genes) in humans due to the transcription termination factor binding mtDNA immediately downstream of the two rRNA genes (Martin et al. 2005). A significant difference between the transcription levels of rRNA and mRNA was also found in Drosophila (Torres et al. 2009). We suspect that the hymenopteran mt genome might also have a higher rate of rRNA synthesis. Therefore, the mechanism of transcriptional feedback system regulating mtDNA replication would confer an advantage to maintaining both rRNA genes on the same strand, as regulation of the synthesis of both rRNA genes would occur concomitantly. The retention of the rRNA genes on the minor strand in other available evaniomoph mt genomes indicates that the inversion occurred after the divergence of the Megalyroidea. In O. pulchella, 4 protein coding genes (nad2, cox1, nad6, and cob) and 10 tRNA genes have rearranged relative to the ancestral positions. nad2 and cox1 have moved into the position upstream of the A+T-rich region and the nad3-nad5 junction, respectively. nad6 and cob have inverted and swapped positions (nad6-cob→cob-nad6). The gene boundary cob-nad6 was also characterized in Venturia canescens (Ichneumonoidea) and Cotesia vestalis (Ichneumonoidea) (Dowton, Cameron, Dowavic, et al. 2009; Wei, Shi, et al. 2010), similar to our observations in O. pulchella. However, these two superfamilies (Trigonalyoidea and Ichneumonoidea) are not closely related, indicating that this arrangement has likely evolved on two or three occasions independently. Therefore, it is probably one of the few examples of convergent evolution of gene order involving protein-coding gene rearrangements. Two tRNA genes (trnQ and trnY) have inverted in both Megalyra sp. and O. pulchella, but they are in different, derived positions. In Megalyra sp., both trnQ and trnY remain in their ancestral locations, whereas in O. pulchella, they have moved to the position downstream of rrnS. Furthermore, trnH is no longer between nad5 and nad4, but the derived positions are different in the two taxa.
Evolutionary Dynamics of Evaniomorph mt Genomes
A total of 64 gene rearrangements were identified in the seven evaniomorph mt genomes, including 4 local translocations (within a tRNA gene cluster and without inversion), 32 gene inversions, and 49 long-range movements (some genes were involved in two rearrangement events). The local translocations are readily explained by the duplication/random loss model (Moritz et al. 1987), whereas the dominant rearrangement events (inversion and long-range movement) are more consistent with the intramolecular recombination mechanism (Dowton and Campbell 2001; Mao et al. 2014). In this mechanism, the repeat fragments located around the breakpoints may play an important role in facilitating the recombination (Mao et al. 2014). The A+T-rich region (control region) has been suggested as a “hot spot” of recombination (Kurabayashi et al. 2008). Among the 64 gene rearrangements, there are 43 rearrangements that occurred close to the A+T-rich region. A series of repeat sequences detected in each evaniomorph mt genome A+T-rich region may have played a role in the gene rearrangements, via recombination (table 2).
Table 2.
The Number of Repeats (Including Direct and Inverted Repeats) within the A+T-Rich Region and Gene Rearrangements Identified for Seven Evaniomorph Taxa
| Species | 12–20 bp | 21–50 bp | 51–100 bp | >100 bp | Closea | Not Closea | Inversion | Long Range | Local |
|---|---|---|---|---|---|---|---|---|---|
| Ceraphron sp. | 10 | 8 | 0 | 0 | 11 | 1 | 5 | 10 | 0 |
| Conostigmus sp. | 23 | 0 | 9 | 2 | 5 | 7 | 1 | 11 | 1 |
| Evania appendigaster | 35 | 31 | 0 | 0 | 4 | 0 | 3 | 3 | 1 |
| Gasteruption sp. | 50 | 0 | 0 | 2 | 7 | 4 | 10 | 7 | 0 |
| Megalyra sp. | 84 | 31 | 0 | 4 | 5 | 3 | 6 | 6 | 0 |
| Pristaulacus compressus | 37 | 0 | 0 | 0 | 3 | 0 | 0 | 2 | 1 |
| Orthogonalys pulchella | 21 | 2 | 5 | 2 | 8 | 6 | 7 | 10 | 1 |
Note.—aClose/not close refers to whether the gene rearrangements occurred close to or far from the A+T-rich region. The definition of “close to the A+T-rich region” is 1) when a tRNA gene moves to or moves out from a position between the rrnS-nad2 gene junction or 2) when a tRNA moves together with rrnS or nad2.
There have been few studies on the association of repeats with rearrangements in the mt genome. In a recent study on the plastid genome (Weng et al. 2014), repeats were strongly associated with the breakpoints in the rearranged genomes. In particular, large repeats (>20 bp and >60 bp) were significantly correlated with the degree of genome rearrangement. To better understand the relationship between repeats and gene rearrangements, we conducted a similar analysis for the A+T-rich region and the entire mt genome among the seven evaniomorph taxa. However, no significant correlations between the size of repeats and gene rearrangements could be detected (the number of different size of repeats in the A+T-rich region is listed in table 2). We suspect that this might be due to the high degree of divergence between the taxa studied here, with the number and type of repeats that are present at the time of rearrangement being obscured by subsequent evolution.
The Utility of Gene Rearrangements for Assessing Phylogeny in the Evaniomorpha
Gene rearrangement characters have been suggested as reliable markers for deducing phylogenetic relationships (Boore and Brown 1998; Dowton et al. 2002). To explore the potential phylogenetic signals in the evaniomorph mt gene orders, such as shared, derived gene rearrangements, we visually inspected each pairwise comparison of gene orders in the output diagrams from CREx. A large number of individual gene rearrangements were detected between the evaniomorph mt genomes. However, no shared, derived gene rearrangements could be identified. This result indicates that the frequency of gene rearrangements might still be too high to be useful for assessing higher-level relationships. We investigate this point further in the following section.
Rates of Gene Rearrangement in the Evaniomorpha
Does the Evaniomorpha have an intermediate rate of gene rearrangement? To answer this question, we calculated the breakpoint numbers of the evaniomorph, hemipteroid (hemipteran orders; Hemiptera, Phthiraptera, Psocodea, Thysanoptera), and nematoceran (Diptera: Nematocera; crane flies, gnats, and mosquitoes) mt genomes relative to the ancestral organization of insects. The hemipteroids (excluding the Hemiptera) have been identified as a group with a high rate of mt gene rearrangement (e.g., Shao et al. 2001, 2003), whereas most Nematocera have a low rate of mt gene rearrangement (e.g., Beckenbach 2012). As presented in table 3, the breakpoint numbers of the seven evaniomorph taxa range from 5 to 21, whereas the numbers of the three hemipteroid taxa (i.e., excluding Triatoma from the Hemiptera) are high (15–35), with most nematocerans low (0–5), although Paracladura is an obvious exception. These numbers confirm the intermediate rate of gene rearrangement of the Evaniomorpha. However, the inability to identify shared, derived gene rearrangements indicates that the rate of the gene rearrangement is still too high to be reliably used for reconstructing phylogenetic relationships. Therefore, increasing taxonomic sampling, particularly in those evaniomorph groups with a relatively lower rate of gene rearrangement might be more useful to establish phylogenetic relationships. Within the Evaniomorpha, the three evanioid taxa yield the lowest breakpoint numbers, which suggests the Evanioidea is the most promising candidate for further analyses. Although the monophyly of the Evanioidea was not recovered by some previous research (Dowton and Austin 2001; Castro and Dowton 2006), two most recently comprehensive studies robustly supported its monophyly (Heraty et al. 2011; Klopfstein et al. 2013).
Table 3.
Breakpoint Numbers of the Evaniomorph, Hemipteroid, and Nematoceran mt Genomes Relative to the Ancestral mt Genome Organization
| Species | Number of Breakpoints | Accession Number | References |
|---|---|---|---|
| Hemipteroids | |||
| Triatoma | 0 | NC_002609 | Dotson and Beard (2001) |
| Heterodoxus | 35 | NC_002651 | Shao et al. (2001) |
| Thrips | 30 | AF335993 | Shao and Barker (2003) |
| Lepidoscopid | 15 | AF335994 | Shao et al. (2003) |
| Diptera: Nematocera | |||
| Anopheles | 5 | NC_000875 | Mitchell et al. (1993) |
| Culicoides | 0 | NC_009809 | Matsumoto Y, Yanase T, Tshuda T, Noda H, unpublished |
| Cramptonomyia | 0 | JN861747 | Beckenbach (2012) |
| Tipula | 0 | JN861743 | Beckenbach (2012) |
| Sylvicola | 0 | JN861752 | Beckenbach (2012) |
| Paracladura | 21 | JN861751 | Beckenbach (2012) |
| Arachnocampa | 2 | JN861748 | Beckenbach (2012) |
| Mayetiola | 12 | GQ387648 | Beckenbach and Joy (2009) |
| Hymenoptera: Evaniomorpha | |||
| Megalyra | 16 | KJ577600 | Present study |
| Gasteruption | 14 | KJ619460 | Present study |
| Evania | 10 | FJ593187 | Wei, Tang, et al. (2010) |
| Pristaulacus | 5 | KF500406 | Wei et al. (2013) |
| Conostigmus | 16 | KF015227 | Mao et al. (2014) |
| Ceraphron | 18 | KJ570858 | Present study |
| Orthogonalys | 21 | KJ619461 | Present study |
Phylogenetic Analysis
Previous phylogenetic analyses of hymenopteran mt genome data by our group has established that uncontroversial relationships are only recovered with partitioned, model-based (Bayesian) analyses of nucleotide data (Dowton, Cameron, Austin, et al. 2009). Analysis of amino acid sequences using either maximum parsimony or Bayesian approaches failed to recover these uncontroversial relationships, as did analysis of nucleotide sequence data using the maximum parsimony criterion. Although our earlier studies also found that the exclusion of third codon positions markedly improved the recovery of these relationships, more recently we have found that the use of PartitionFinder (Lanfear et al. 2012) now facilitates the inclusion of third codon positions (Mao and Dowton 2014). PartitionFinder tends to group proteins into the strand on which they are encoded, an approach that we did not use in our earlier studies. For this reason, we analyzed the present data set using model-based analyses of nucleotide data, which was partitioned by PartitionFinder. Nevertheless, to assess how sensitive our phylogeny was to the analytical approach, we analyzed our data set using eight analytical approaches, which are outlined below.
We analyzed two taxon data sets (with and without rogue taxon deletion) using two alignment approaches (Muscle and MAFFT) and two phylogenetic approaches (maximum likelihood and Bayesian inference). Our initial data set contained all of the available evaniomorph and aculeate species. The trees yielded a number of expected relationships with high support (posterior probabilities 0.98–1.00 in Bayesian analyses and bootstrap proportions 57–100 in maximum likelihood analyses). For example, the Ceraphronoidea, Apoidea, Formicidae, and Vespidae were consistently recovered as monophyletic (fig. 6 and supplementary fig. S2, Supplementary Material online). However, some traditionally monophyletic groups failed to be recovered in all of the analyses. For example, the two chrysidoid genera (Cephalonomia and Primeuchroeus) failed to group together; Evania was placed within the Aculeata and grouped as a sister of Radoszkowskius. This latter relationship was also reported in a recent mt phylogeny of the Hymenoptera (Kaltenpoth et al. 2012). We then investigated whether some taxa were being placed in variable positions during the bootstrap and Bayesian analyses and as a result were obscuring other, more stable relationships (Wilkinson 1996). We prefer this objective approach to taxon pruning, rather than basing taxon exclusion upon, for example, inconsistency with previous phylogenetic hypotheses. Therefore, we conducted rogue taxon identification on all of the tree sets using RogueNaRok. For the RAxML bootstrap tree sets, Evania appendigaster and Primeuchroeus spp. were consistently identified as rogue taxa, whereas only Primeuchroeus spp. was identified in Bayesian tree sets. For consistency between analyses, we removed the concatenated sequences corresponding to the two rogue taxa (Evania and Primeuchroeus) and repeated the sequence alignment (as removal of rogue taxa might impact on the alignment) and phylogenetic analyses. The phylogenetic resolution was improved compared with the initial analyses. For example, a monophyletic Aculeata was consistently recovered in all analyses. Therefore, our discussion below focuses on the trees generated by the data set with these two taxa removed.
Fig. 6.—

Bayesian analysis of the initial taxon data set (without rogue taxon deletion) based on mitogenomic sequences including 13 protein-coding genes and two rRNA genes. All the Symphyta, Evaniomorpha, and Aculeata were recovered as paraphyletic grades. Posterior probabilities are shown at each node.
The general topologies were broadly congruent across the four analyses with similar levels of support for major clades (fig. 7 and supplementary fig. S3, Supplementary Material online). The two alignment approaches had no effect on topology and limited effect on nodal support (fig. 7 and supplementary fig. S3A, Supplementary Material online). When the two phylogenetic inference approaches are compared, there is only one topological difference, which was caused by the variable positions of Radoszkowskius (Vespoidea) and Cephalonomia (Chrysidoidea). In Bayesian analyses, the two taxa grouped together, and this clade was sister to Apoidea + the remaining Vespoidea (fig. 7 and supplementary fig. S3A, Supplementary Material online). However, in maximum likelihood analyses, Radoszkowskius was recovered as the sister group to the remaining Aculeata and Cephalonomia formed a clade with three formicid taxa (Camponotus, Pristomyrmex, and Solenopsis) (supplementary fig. S3B and C, Supplementary Material online). Nodal support was generally stronger for the Bayesian than the maximum likelihood analyses as has been commonly noted (Cameron et al. 2012; Nelson et al. 2012). The data matrix and the two Bayesian trees in figures 6 and 7 have been deposited in TreeBASE (accession number S16039, http://treebase.org/, last accessed June 30, 2014).
Fig. 7.—

Bayesian analysis of the reduced taxon data set (with rogue taxon deletion) based on mitogenomic sequences including 13 protein-coding genes and two rRNA genes. Both the Symphyta and Evaniomorpha were recovered as paraphyletic grades. Posterior probabilities are shown at each node.
In the pruned data set, there are 22 hymenopteran taxa, with four taxa coming from the Symphyta. Our analyses strongly supported the Cephoidea (Cephus) as sister to Orussoidea + Apocrita, which is congruent with previously morphological studies (Vilhelmsen 2001; Vilhelmsen et al. 2010). The Orussoidea (Orussus) was recovered as sister to the Apocrita with high nodal support.
Within the Apocrita, the Aculeata was consistently recovered as monophyletic. Ignoring the unstable positions of Radoszkowskius and Cephalonomia, the internal relationships are highly congruent with our previous study (Mao et al. 2012). Compared with the previous study, there are two new representatives: Camponotus from the Vespoidea and Philanthus from the Apoidea. As expected, Camponotus formed a clade with another two vespoid taxa: Pristomyrmex and Solenopsis. All these three taxa are from the same family Formicidae. Philanthus was recovered at the base of the Apoidea.
The Evaniomorpha was recovered as paraphyletic, with the Aculeata nested within them. Of the representatives from the Evaniomorpha, the Stephanoidea was placed as the sister group to the remaining Evaniomorpha + Aculeata. The sister relationship of the Ceraphronoidea and Evanioidea was also highly supported in each analysis. The Trigonalyoidea was recovered as sister to the Megalyroidea, and this clade was sister to the Aculeata. A paraphyletic Evaniomorpha with respect to the Aculeata has commonly been recovered in previous molecular studies (Dowton and Austin 2001; Castro and Dowton 2006; Heraty et al. 2011; Sharkey et al. 2011; Klopfstein et al. 2013). However, the likely sister group to the Aculeata remains controversial. In our previous studies, the Aculeata were generally recovered among a clade with the Stephanoidea, Trigonalyoidea, and Megalyroidea but with weak support (Dowton and Austin 2001; Castro and Dowton 2006). A recent analysis of combined morphological and molecular data supported a sister group of Aculeata + Evanioidea, but this clade was not supported by the morphology-only tree or individual gene trees (Sharkey et al. 2011). Additionally, some recent molecular analyses recovered the same sister group to the Aculeata as in this study, but the results were sensitive to the alignment approach (Heraty et al. 2011; Klopfstein et al. 2013). In contrast, in this study the Aculeata + (Trigonalyoidea + Megalyroidea) clade was insensitive to variations in alignment approach and phylogenetic inference approach. We consider this clade to be the best supported hypothesis, based on current evidence. The phylogenetic relationships among evaniomorph superfamilies are also far from settled. Since Rasnitsyn (1988) placed the Stephanoidea within the Evaniomorpha, there has been no robust morphological or molecular evidence to support this affiliation. Instead, most subsequent analyses placed the Stephanoidea as the most basal apocritan lineage (Whitfield, 1992; Dowton and Austin 1994; Vilhelmsen et al. 2010; Heraty et al. 2011; Peters et al. 2011; Sharkey et al. 2011). Our results supported the basal position of the Stephanoidea but failed to give a clear indication to the affiliation of this superfamily. The sister relationship between the Trigonalyoidea and Megalyroidea has been consistently proposed in molecular analyses (Dowton et al. 1997; Dowton and Austin 2001; Castro and Dowton 2006; Heraty et al. 2011; Klopfstein et al. 2013). However, recent morphological analyses favor the Ceraphronoidea as the sister to the Megalyroidea based on mesosomal characters (Vilhelmsen et al. 2010). Our results confirmed the Trigonalyoidea + Megalyroidea clade with robust molecular support and yielded a novel sister relationship of the Ceraphronoidea and Evanioidea. The inclusion of other taxa is required to further test this novel relationship.
To date, most molecular phylogenies on the internal relationships of the Apocrita have relied on relatively short mt and nuclear gene fragments. The few studies that have used the much larger mt genome to assess this group have done so with relatively few taxa—usually one representative from each superfamily. Our study is the first attempt to use the entire mt genome, and a denser taxonomic sampling, to reconstruct phylogeny. Nevertheless, we do so for just two of the four apocritan groups, the Evaniomorpha and Aculeata. The molecular phylogenetic hypothesis presented here provides a reliable basis for future analyses.
Supplementary Material
Supplementary figures S1–S3 are available at Genome Biology and Evolution online (http://www.gbe.oxfordjournals.org/).
Acknowledgments
The authors thank Andy Austin (Adelaide University) for the generous provision of biological samples and Xiushuai Yang for his kind assistance with preparing the figures. This work was supported by the Center for Medical and Molecular Bioscience, University of Wollongong.
Literature Cited
- Abascal F, Zardoya R, Telford MJ. TranslatorX: multiple alignment of nucleotide sequences guided by amino acid translations. Nucleic Acids Res. 2010;38:7–13. doi: 10.1093/nar/gkq291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aberer AJ, Krompass D, Stamatakis A. Pruning rogue taxa improves phylogenetic accuracy: an efficient algorithm and webservice. Syst Biol. 2013;62:162–166. doi: 10.1093/sysbio/sys078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aljanabi SM, Martinez I. Universal and rapid salt-extraction of high quality genomic DNA for PCR-based techniques. Nucleic Acids Res. 1997;25:4692–4693. doi: 10.1093/nar/25.22.4692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Axel K, Thomas BLK. Transcription could be the key to the selection advantage of mitochondrial deletion mutants in aging. Proc Natl Acad Sci U S A. 2014;111:2972–2977. doi: 10.1073/pnas.1314970111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beckenbach AT. Mitochondrial genome sequences of Nematocera (lower Diptera): evidence of rearrangement following a complete genome duplication in a winter crane fly. Genome Biol Evol. 2012;4:89–101. doi: 10.1093/gbe/evr131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beckenbach AT, Joy JB. Evolution of the mitochondrial genomes of gall midges (Diptera: Cecidomyiidae): rearrangement and severe truncation of tRNA genes. Genome Biol Evol. 2009;1:278–287. doi: 10.1093/gbe/evp027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernt M, et al. CREx: inferring genomic rearrangements based on common intervals. Bioinformatics. 2007;23:2957–2958. doi: 10.1093/bioinformatics/btm468. [DOI] [PubMed] [Google Scholar]
- Boore JL. Animal mitochondrial genomes. Nucleic Acids Res. 1999;27:1767–1780. doi: 10.1093/nar/27.8.1767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boore JL, Brown WM. Big trees from little genomes: mitochondrial gene order as a phylogenetic tool. Curr Opin Genet Dev. 1998;8:668–674. doi: 10.1016/s0959-437x(98)80035-x. [DOI] [PubMed] [Google Scholar]
- Cameron SL, Johnson KP, Whiting MF. The mitochondrial genome of the screamer louse Bothriometopus (Phthiraptera: Ischnocera): effects of extensive gene rearrangements on the evolution of the genome. J Mol Evol. 2007;65:589–604. doi: 10.1007/s00239-007-9042-8. [DOI] [PubMed] [Google Scholar]
- Cameron SL, Lo N, Bourguignon T, Svenson GJ, Evans TA. A mitochondrial genome phylogeny of termites (Blattodea: Termitoidae): robust support for interfamilial relationships and molecular synapomorphies define major clades. Mol Phylogenet Evol. 2012;65:163–173. doi: 10.1016/j.ympev.2012.05.034. [DOI] [PubMed] [Google Scholar]
- Cameron SL, Yoshizawa K, Mizukoshi A, Whiting MF, Johnson KP. Mitochondrial genome deletions and minicircles are common in lice. BMC Genomics. 2011;12:394. doi: 10.1186/1471-2164-12-394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castro LR, Dowton M. Molecular analyses of the Apocrita (Insecta: Hymenoptera) suggest that the Chalcidoidea are sister to the diaprioid complex. Invertebr Syst. 2006;20:603–614. [Google Scholar]
- Castro LR, Ruberu K, Dowton M. Mitochondrial genomes of Vanhornia eucnemidarum (Apocrita: Vanhorniidae) and Primeuchroeus spp. (Aculeata: Chrysididae): evidence of rearranged mitochondrial genomes within the Apocrita (Insecta: Hymenoptera) Genome. 2006;49:752–766. doi: 10.1139/g06-030. [DOI] [PubMed] [Google Scholar]
- Cook CE. The complete mitochondrial genome of the stomatopod crustacean Squilla mantis. BMC Genomics. 2005;6:105. doi: 10.1186/1471-2164-6-105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Covacin C, Shao R, Cameron S, Barker SC. Extraordinary number of gene rearrangements in the mitochondrial genomes of lice (Phthiraptera: Insecta) Insect Mol Biol. 2006;15:63–68. doi: 10.1111/j.1365-2583.2005.00608.x. [DOI] [PubMed] [Google Scholar]
- Dotson EM, Beard CB. Sequence and organization of the mitochondrial genome of the Chagas disease vector, Triatoma dimidiata. Insect Mol Biol. 2001;10:205–215. doi: 10.1046/j.1365-2583.2001.00258.x. [DOI] [PubMed] [Google Scholar]
- Dowton M, Austin AD. Molecular phylogeny of the insect order Hymenoptera: apocritan relationships. Proc Natl Acad Sci U S A. 1994;91:9911–9915. doi: 10.1073/pnas.91.21.9911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dowton M, Austin AD. Evolutionary dynamics of a mitochondrial rearrangement “hot spot” in the Hymenoptera. Mol Biol Evol. 1999;16:298–309. doi: 10.1093/oxfordjournals.molbev.a026111. [DOI] [PubMed] [Google Scholar]
- Dowton M, Austin AD. Simultaneous analysis of 16S, 28S, COI and morphology in the Hymenoptera: Apocrita—evolutionary transitions among parasitic wasps. Biol J Linn Soc. 2001;74:87–111. [Google Scholar]
- Dowton M, Austin AD, Dillon N, Bartowsky E. Molecular phylogeny of the apocritan wasps: the Proctotrupomorpha and Evaniomorpha. Syst Entomol. 1997;22:245–255. [Google Scholar]
- Dowton M, Cameron SL, Austin AD, Whiting MF. Phylogenetic approaches for the analysis of mitochondrial genome sequence datain the Hymenoptera—a lineage with both rapidly and slowly evolving mitochondrial genomes. Mol Phylogenet Evol. 2009;52:512–519. doi: 10.1016/j.ympev.2009.04.001. [DOI] [PubMed] [Google Scholar]
- Dowton M, Cameron SL, Dowavic JI, Austin AD, Whiting MF. Characterization of 67 mitochondrial tRNA gene rearrangements in the Hymenoptera suggests that mitochondrial tRNA gene position is selectively neutral. Mol Biol Evol. 2009;26:1607–1617. doi: 10.1093/molbev/msp072. [DOI] [PubMed] [Google Scholar]
- Dowton M, Campbell NJH. Intramitochondrial recombination—is it why some mitochondrial genes sleep around? Trends Ecol Evol. 2001;16:269–271. doi: 10.1016/s0169-5347(01)02182-6. [DOI] [PubMed] [Google Scholar]
- Dowton M, Castro LR, Austin AD. Mitochondrial gene rearrangements as phylogenetic characters in the invertebrates: theexamination of genome “morphology.”. Invertebr Syst. 2002;16:345–356. [Google Scholar]
- Eberhard JR, Wright TF, Bermingham E. Duplication and concerted evolution of the mitochondrial control region in the parrot genus Amazona. Mol Biol Evol. 2001;18:1330–1342. doi: 10.1093/oxfordjournals.molbev.a003917. [DOI] [PubMed] [Google Scholar]
- Edgar RC. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004;32:1792–1797. doi: 10.1093/nar/gkh340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibson GAP. Sister-group relationships of the Platygastroidea and Chalcidoidea (Hymenoptera)—an alternate hypothesis to Rasnitsyn (1988) Zool Scr. 1999;28:125–138. [Google Scholar]
- Golubchik T, Wise MJ, Easteal S, Jermiin LS. Mind the gaps: evidence of bias in estimates of multiple sequence alignments. Mol Biol Evol. 2007;24:2433–2442. doi: 10.1093/molbev/msm176. [DOI] [PubMed] [Google Scholar]
- Heraty J, et al. Evolution of the hymenopteran megaradiation. Mol Phylogenet Evol. 2011;60:73–88. doi: 10.1016/j.ympev.2011.04.003. [DOI] [PubMed] [Google Scholar]
- Huber JT. Biodiversity of Hymenoptera. In: Foottit RG, Adler PH, editors. Insect biodiversity: science and society. Oxford: Wiley-Blackwell; 2009. pp. 303–323. [Google Scholar]
- Kaltenpoth M, et al. Accelerated evolution of mitochondrial but not nuclear genomes of Hymenoptera: new evidence from crabronid wasps. PLoS One. 2012;7:e32826. doi: 10.1371/journal.pone.0032826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katoh K, Kuma K, Toh H, Miyata T. MAFFT version 5: improvement in accuracy of multiple sequence alignment. Nucleic Acids Res. 2005;33:511–518. doi: 10.1093/nar/gki198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klopfstein S, Vilhelmsen L, Heraty JM, Sharkey M, Ronquist F. The hymenopteran tree of life: evidence from protein-coding genes and objectively aligned ribosomal data. PLoS One. 2013;8:e69344. doi: 10.1371/journal.pone.0069344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurabayashi A, et al. Phylogeny, recombination, and mechanisms of stepwise mitochondrial genome reorganization in mantellid frogs from Madagascar. Mol Biol Evol. 2008;25:874–891. doi: 10.1093/molbev/msn031. [DOI] [PubMed] [Google Scholar]
- Lanfear R, Calcott B, Ho SYW, Guindon S. PartitionFinder: combined selection of partitioning schemes and substitution models for phylogenetic analyses. Mol Biol Evol. 2012;29:1695–1701. doi: 10.1093/molbev/mss020. [DOI] [PubMed] [Google Scholar]
- LaSalle J, Gauld ID. Hymenoptera: their diversity and their impact on the diversity of other organisms. In: LaSalle J, Gauld ID, editors. Hymenoptera and biodiversity. Wallingford (United Kingdom) C. A. B. International: 1993. pp. 1–26. [Google Scholar]
- Laslett D, Canbäck B. ARWEN: a program to detect tRNA genes in metazoan mitochondrial nucleotide sequences. Bioinformatics. 2008;24:172–175. doi: 10.1093/bioinformatics/btm573. [DOI] [PubMed] [Google Scholar]
- Lavrov DV, Brown WM, Boore JL. A novel type of RNA editing occurs in the mitochondrial tRNAs of the centipede Lithobius forficatus. Proc Natl Acad Sci U S A. 2000;97:13738–13742. doi: 10.1073/pnas.250402997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowe TM, Eddy SR. tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 1997;25:955–964. doi: 10.1093/nar/25.5.955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mao M, Austin AD, Johnson NF, Dowton M. Coexistence of minicircular and a highly rearranged mtDNA molecule suggests that recombination shapes mitochondrial genome organization. Mol Biol Evol. 2014;31:636–644. doi: 10.1093/molbev/mst255. [DOI] [PubMed] [Google Scholar]
- Mao M, Dowton M. Mol Biol Rep. 2014. Complete mitochondrial genomes of Ceratobaeus sp. and Idris sp. (Hymenoptera: Scelionidae): shared gene rearrangements as potential phylogenetic markers at the tribal level. Advance Access published July 3, 2014, doi: 10.1007/s11033-014-3522-x. [DOI] [PubMed] [Google Scholar]
- Mao M, Valerio A, Austin AD, Dowton M, Johnson NF. The first mitochondrial genome for the wasp superfamily Platygastroidea: the egg parasitoid Trissolcus basalis. Genome. 2012;55:194–204. doi: 10.1139/g2012-005. [DOI] [PubMed] [Google Scholar]
- Martin M, Cho J, Cesare AJ, Griffith JD, Attardi G. Terminationfactor-mediated DNA loop between termination and initiation sites drives mitochondrial rRNA synthesis. Cell. 2005;123:1227–1240. doi: 10.1016/j.cell.2005.09.040. [DOI] [PubMed] [Google Scholar]
- Masta SE, Boore JL. The complete mitochondrial genome sequence of the spider Habronattus oregonensis reveals rearranged and extremely truncated tRNAs. Mol Biol Evol. 2004;21:893–902. doi: 10.1093/molbev/msh096. [DOI] [PubMed] [Google Scholar]
- Miller MA, Pfeiffer W, Schwartz T. 2010. Creating the CIPRES Science Gateway for inference of large phylogenetic trees. Proceedings of the Gateway Computing Environments Workshop (GCE); 2010 Nov 14; New Orleans, LA. pp. 1–8. [Google Scholar]
- Mitchell SE, Cockburn AF, Seawright JA. The mitochondrial genome of Anopheles quadrimaculatus species A: complete nucleotide sequence and gene organization. Genome. 1993;36:1058–1073. doi: 10.1139/g93-141. [DOI] [PubMed] [Google Scholar]
- Moritz C, Dowling TE, Brown WM. Evolution of animal mitochondrial DNA: relevance for population biology and systematics. Annu Rev Ecol Syst. 1987;18:269–292. [Google Scholar]
- Nelson LA, et al. Beyond barcoding: a mitochondrial genomics approach to molecular phylogenetics and diagnostics of blowflies (Diptera: Calliphoridae) Gene. 2012;511:131–142. doi: 10.1016/j.gene.2012.09.103. [DOI] [PubMed] [Google Scholar]
- Okonechnikov K, Golosova O, Fursov M; UGENE team. 2012. Unipro UGENE: a unified bioinformatics toolkit. Bioinformatics 28:1166–1167. [DOI] [PubMed]
- Oliveira DCSG, Raychoudhury R, Lavrov DV, Werren JH. Rapidly evolving mitochondrial genome and directional selection in mitochondrial genes in the parasitic wasp Nasonia (Hymenoptera: Pteromalidae) Mol Biol Evol. 2008;25:2167–2180. doi: 10.1093/molbev/msn159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perna NT, Kocher TD. Patterns of nucleotide composition at fourfold degenerate sites of animal mitochondrial genomes. J Mol Evol. 1995;41:353–358. doi: 10.1007/BF00186547. [DOI] [PubMed] [Google Scholar]
- Peters RS, et al. The taming of an impossible child: a standardized all-in approach to the phylogeny of Hymenoptera using public database sequences. BMC Biol. 2011;9:55. doi: 10.1186/1741-7007-9-55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rambaut A, Drummond AJ. 2009. Tracer version 1.5. [Internet]. [cited 2014 Jun 30]. Available from: http://tree.bio.ed.ac.uk/software/tracer/ [Google Scholar]
- Rasnitsyn AP. An outline of the evolution of hymenopterous insects (order Vespida) Orient Insects. 1988;22:115–145. [Google Scholar]
- Rasnitsyn AP, Zhang HC. Early evolution of Apocrita (Insecta, Hymenoptera) as indicated by new findings in the middle Jurassic of Daohugou, Northeast China. Acta Geol Sin Engl. 2010;84:834–873. [Google Scholar]
- Ronquist F, Rasnitsyn AP, Roy A, Eriksson K, Lindgren M. Phylogeny of the Hymenoptera: a cladistic reanalysis of Rasnitsyn’s (1988) data. Zool Scr. 1999;28:13–50. [Google Scholar]
- Ronquist F, et al. MrBayes 3.2: efficient Bayesian phylogenetic inference and model choice across a large model space. Syst Biol. 2012;61:539–542. doi: 10.1093/sysbio/sys029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Segovia R, Pett W, Trewick S, Lavrov DV. Extensive and evolutionarily persistent mitochondrial tRNA editing in velvet worms (phylum Onychophora) Mol Biol Evol. 2011;28:2873–2881. doi: 10.1093/molbev/msr113. [DOI] [PubMed] [Google Scholar]
- Shao R, Barker SC. The highly rearranged mitochondrial genome of the plague thrips, Thrips imaginis (Insecta: Thysanoptera): convergence of two novel gene boundaries and an extraordinary arrangement of rRNA genes. Mol Biol Evol. 2003;20:362–370. doi: 10.1093/molbev/msg045. [DOI] [PubMed] [Google Scholar]
- Shao R, Barker SC, Mitani H, Aoki Y, Fukunaga M. Evolution of duplicate control regions in the mitochondrial genomes of metazoa: a case study with Australasian Ixodes ticks. Mol Biol Evol. 2005;22:620–629. doi: 10.1093/molbev/msi047. [DOI] [PubMed] [Google Scholar]
- Shao R, Campbell NJ, Barker SC. Numerous gene rearrangements in the mitochondrial genome of the wallaby louse, Heterodoxus macropus (Phthiraptera) Mol Biol Evol. 2001;18:858–865. doi: 10.1093/oxfordjournals.molbev.a003867. [DOI] [PubMed] [Google Scholar]
- Shao R, Dowton M, Murrell A, Barker SC. Rates of gene rearrangement and nucleotide substitution are correlated in the mitochondrial genomes of insects. Mol Biol Evol. 2003;20:1612–1619. doi: 10.1093/molbev/msg176. [DOI] [PubMed] [Google Scholar]
- Sharkey MJ. Phylogeny and classification of Hymenoptera. Zootaxa. 2007:521–548. [Google Scholar]
- Sharkey MJ, et al. Phylogenetic relationships among superfamilies of Hymenoptera. Cladistics. 2011;27:1–33. doi: 10.1111/j.1096-0031.2011.00366.x. [DOI] [PubMed] [Google Scholar]
- Sheffield NC, Song H, Cameron L, Whiting MF. A comparative analysis of mitochondrial genomes in Coleoptera (Arthropoda: Insecta) and genome descriptions of six new beetles. Mol Biol Evol. 2008;25:2499–2509. doi: 10.1093/molbev/msn198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simon C, Buckley TR, Frati F, Stewart JB, Beckenbach AT. Incorporating molecular evolution into phylogenetic analysis, and a new compilation of conserved polymerase chain reaction primersfor animal mitochondrial DNA. Annu Rev Ecol Evol Syst. 2006;37:545–579. [Google Scholar]
- Simon C, et al. Evolution, weighting, and phylogenetic utility of mitochondrial gene sequences and a compilation of conserved polymerase chain reaction primers. Ann Entomol Soc Am. 1994;87:651–701. [Google Scholar]
- Stamatakis A, Hoover P, Rougemont J. A rapid bootstrap algorithm for the RAxML web servers. Syst Biol. 2008;57:758–771. doi: 10.1080/10635150802429642. [DOI] [PubMed] [Google Scholar]
- Tamura K, et al. MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol Biol Evol. 2011;28:2731–2739. doi: 10.1093/molbev/msr121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torres TT, Dolezal M, Schlötterer C, Ottenwälder B. Expression profiling of Drosophila mitochondrial genes via deep mRNA sequencing. Nucleic Acids Res. 2009;37:7509–7518. doi: 10.1093/nar/gkp856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vilhelmsen L. Phylogeny and classification of the extant basal lineages of the Hymenoptera (Insecta) Zool J Linn Soc. 2001;131:393–442. [Google Scholar]
- Vilhelmsen L, Miko I, Krogmann L. Beyond the wasp-waist: structural diversity and phylogenetic significance of the mesosoma in apocritan wasps (Insecta: Hymenoptera) Zool J Linn Soc. 2010;159:22–194. [Google Scholar]
- Wei SJ, Shi M, Sharkey MJ, van Achterberg C, Chen XX. Comparative mitogenomics of Braconidae (Insecta: Hymenoptera) and the phylogenetic utility of mitochondrial genomes with special reference to Holometabolous insects. BMC Genomics. 2010;11:371. doi: 10.1186/1471-2164-11-371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei SJ, Tang P, Zheng LH, Shi M, Chen XX. The complete mitochondrial genome of Evania appendigaster (Hymenoptera: Evaniidae) has low A+T content and a long intergenic spacer between atp8 and atp6. Mol Biol Rep. 2010;37:1931–1942. doi: 10.1007/s11033-009-9640-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei SJ, Wu QL, van Achterberg K, Chen XX. Rearrangement of the nad1 gene in Pristaulacus compressus (Spinola) (Hymenoptera: Evanioidea: Aulacidae) mitochondrial genome. Mitochondrial DNA. 2013 doi: 10.3109/19401736.2013.834436. Advance Access published October 1, 2013, doi: 10.3109/19401736.2013.834436. [DOI] [PubMed] [Google Scholar]
- Weng ML, Blazier JC, Govindu M, Jansen RK. Reconstruction of the ancestral plastid genome in geraniaceae reveals a correlation between genome rearrangements, repeats, and nucleotide substitution rates. Mol Biol Evol. 2014;31:645–659. doi: 10.1093/molbev/mst257. [DOI] [PubMed] [Google Scholar]
- Whitfield JB. Phylogeny of the non-aculeate Apocrita and the evolution of parasitism in the Hymenoptera. J Hymenoptera Res. 1992;1:3–14. [Google Scholar]
- Wilkinson M. Majority-rule reduced consensus trees and their use in bootstrapping. Mol Biol Evol. 1996;13:437–444. doi: 10.1093/oxfordjournals.molbev.a025604. [DOI] [PubMed] [Google Scholar]
- Xiao JH, Jia JG, Murphy RW, Huang DW. Rapid evolution of the mitochondrial genome in chalcidoid wasps (Hymenoptera: Chalcidoidea) driven by parasitic lifestyles. PLoS One. 2011;6:e26645. doi: 10.1371/journal.pone.0026645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang X, Xue D, Han H. The complete mitochondrial genome of Biston panterinaria (Lepidoptera: Geometridae), with phylogenetic utility of mitochondrial genome in the Lepidoptera. Gene. 2013;515:349–358. doi: 10.1016/j.gene.2012.11.031. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.