

Abstract
Ligand binding to cell surface receptors activates signaling pathways in normal and pathologic conditions, and internalized ligand–receptor complexes may continue to signal from endosomes. Accessibility of cell surface receptors and the central function of ligand–receptor binding in signal transduction make ligand binding a prime target for therapeutic agents. We describe a Gaussia luciferase complementation method for imaging ligand–receptor binding in cell-based assays and living mice. While we illustrate this imaging method for chemokine ligand CXCL12 and its receptors CXCR4 and CXCR7, this imaging strategy can be generalized to a large number of ligand–receptor interactions.
Keywords: Molecular imaging, Optical imaging, Split luciferase, Bioluminescence, Protein complementation assay, PCA
1 Introduction
Protein fragment complementation assays based on luciferase enzymes, referred to as luciferase complementation or split luciferase assays, provide a powerful strategy to quantify protein–protein interactions in formats ranging from cell lysates to intact mice. Luciferase complementation entails fusing inactive amino (N)-terminal and carboxy (C)-terminal enzyme fragments to two different proteins of interest. Interactions between proteins of interest bring N- and C-terminal luciferase fragments together to reconstitute an active enzyme, producing bioluminescence as a quantitative measure of interactions between target proteins. Dissociation of proteins of interest also separates fused N-terminal and C-terminal luciferase fragments and reduces bioluminescence, allowing dynamic changes in protein–protein interactions to be quantified in real time. These assays may be used to quantify regulation of protein–protein complexes in response to signaling events, chemical probes, or drugs. When luciferase complementation reporters are expressed stably in mammalian cell lines, the same reporter cells can transition directly from intact cells to animal models, providing a facile approach for development and preclinical testing of candidate drugs.
Monitoring ligand–receptor binding places unique demands on a luciferase complementation assay system. These requirements include that the enzyme functions in the extracellular space and minimizes steric constraints imposed by fusing enzyme fragments to a ligand and receptor. Of available luciferase complementation assays, Gaussia luciferase (GLuc) best meets parameters for imaging ligand–receptor interactions [1]. GLuc does not require ATP, allowing enzyme activity in the extracellular space, and small size of N-terminal and C-terminal enzyme fragments (≈9–10 kDa) substantially reduces the potential to alter functions of fusion proteins. GLuc complementation is fully reversible, so ligand binding to a receptor and subsequent dissociation can be monitored in real time. We have used GLuc complementation to quantify binding of chemokine CXCL12 to its receptors CXCR4 and CXCR7 in intact cells and a mouse model of human breast cancer [2]. More generally, the GLuc complementation system is applicable to any ligand– receptor pair that can be modified to express N-terminal and C-terminal fragments of this enzyme.
2 Materials
2.1 Molecular Biology
DNA encoding open reading frames for desired interacting proteins.
Full-length Gaussia luciferase (GLuc) plasmid (New England Biolabs) or plasmids with NGLuc (amino acids 1–93) and CGLuc (amino acids 94–169) fragments.
Expression vectors with constitutive promoters for expression in mammalian cells.
Expression vector and packaging constructs for producing lentiviral vectors (optional).
Enzymes, buffers, and equipment for PCR, restriction digests of DNA, and ligations
2.2 Cell Culture
HEK-293 T cells or other cell line that can be transfected readily.
Desired cell line(s) for biologic question of interest.
General supplies for cell culture, including media, plasticware, and incubators.
2.3 Cell Imaging
96-Well plates with black sides, clear bottom, and lid for tissue culture.
Multichannel pipets for volumes from 1 to 200 μl.
Sterile pipette tips with low adherence coating adherence.
Sterile 1× phosphate-buffered saline (PBS) solution.
Stock solution of coelenterazine (Promega or other vendor) 1 mg/ml in methanol, stored in tightly closed container at −20 °C ( Gaussia luciferase substrate).
Acid wash solution: 0.2 M acetic acid, 0.5 M NaCl (optional) (see Note 1).
Bioluminescence imaging system with high sensitivity and software for data quantification and analysis (IVIS, Perkin-Elmer; or similar system).
2.4 Animal Imaging
Appropriate mouse strain for desired experimental system, such as immunocompromised mice (nude, SCID, or NSG) for human tumor xenografts.
Small animal shaver such as Wahl compact cordless trimmer (optional).
Depilatory solution such as Nair (optional).
10 mg/ml coelenterazine stock in acidified methanol, store in tightly sealed container at −20 °C (see Note 2).
Sterile solution 40 % DMSO in PBS for diluting coelenterazine immediately before injection and imaging.
28–30 gauge insulin syringe for intravenous tail vein injection in mice.
Restraint device for tail vein injections (Braintree Scientific Tail Vein Injection Platform or other similar device) (optional).
Bioluminescence imaging instrument with isoflurane anesthesia (IVIS or similar instrument as described in Subheading 2.3).
Methods
3.1 Construct Gaussia Luciferase Complementation Reporters
Select a ligand and corresponding receptor as interacting proteins and determine positions of NGLuc and CGLuc fusions to these proteins (Fig. 1) (see Note 3).
Design and optimize linkers between GLuc enzyme fragments and respective ligand and receptor pairs. While not required, linkers may limit steric constraints on ligand–receptor binding and folding of GLuc fragments (see Note 4).
Generate fusion proteins for ligand and receptor pairs using appropriate molecular biology procedures. We typically produce all logical orientations of fusions with NGLuc and CGLuc (see Note 5).
Generate relevant control constructs for nonspecific association of NGLuc and CGLuc (see Note 6).
Express complementation reporters in appropriate vectors for mammalian cells. Vectors should be selected with markers, such as co-expressed fluorescent proteins or antibiotic resistance genes, suitable for generating stable cell lines (see Note 7).
Confirm complementation constructs by DNA sequencing.
Fig. 1.
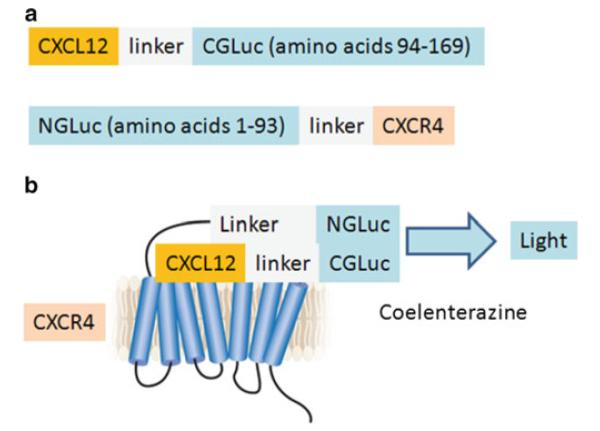
Schematic diagrams of GLuc complementation constructs and ligand– receptor interaction illustrated for CXCL12 and CXCR4. (a) Complementation reporter constructs with chemokine CXCL12 fused via a linker to CGLuc and NGLuc fused with an intervening linker to seven-transmembrane chemokine receptor CXCR4. These positions of NGLuc and CGLuc enzyme fragments allow complementation to occur in both extracellular and intracellular compartments. We also tested constructs with CXCL12 fused to NGLuc and CGLuc fused to CXCR4, but these reporters produced less bioluminescence upon ligand–receptor binding. (b) Binding of CXCL12-CGLuc to NGLuc-CXCR4 reconstitutes GLuc activity to oxidize the substrate coelenterazine and produce bioluminescence (fi gure adapted from ref. 10)
3.2 Cell-Based Bioluminescence Imaging
We initially test pairs of NGLuc and CGLuc fusions with ligand and receptor by transient transfection in 293T cells or another cell line that transfects readily. We include appropriate control NGLuc and CGLuc constructs in these tests. The purpose of these initial tests is to identify an optimal pair of NGLuc and CGLuc fusions for use in stable cell lines and subsequent cell-based assays and living mice. The format for cell-based assays is the same for transiently transfected cells or cells stably expressing reporter constructs (see Note 8).
Plate cells in black-walled, clear bottom 96-well plates for tissue culture. Cell density should be 1 × 10 4 −2 × 10 4 cells/per well in 100 μl complete growth medium with serum. Culture cells overnight under standard conditions in preparation for assays the following day (see Note 9).
Remove cell culture medium and replace with a minimum volume of fresh phenol red DMEM medium for assays (30–40 μl). For assays using inhibitors, we prepare 10× stocks of desired dilutions so that inhibitor or vehicle control can be added in a small volume, such as adding 4 μl of 10× inhibitor stock to 36 μl of medium per well. To perform time course studies, we remove standard culture medium and replace with phenol red free DMEM at staggered times so all wells in the plate are imaged at the end (see Note 10).
Measure Gaussia luciferase complementation signal by adding a 1:500 dilution of 1 mg/ml coelenterazine stock in phenol red free medium to each well using a multichannel pipette (see Note 11).
Image plate on the bioluminescence instrument as soon as possible after adding coelenterazine. A typical image requires 30–60 s with large binning (see Note 12).
Quantify bioluminescence by region-of-interest (ROI) analysis using software on the bioluminescence imaging instrument (Fig. 2).
Fig. 2.
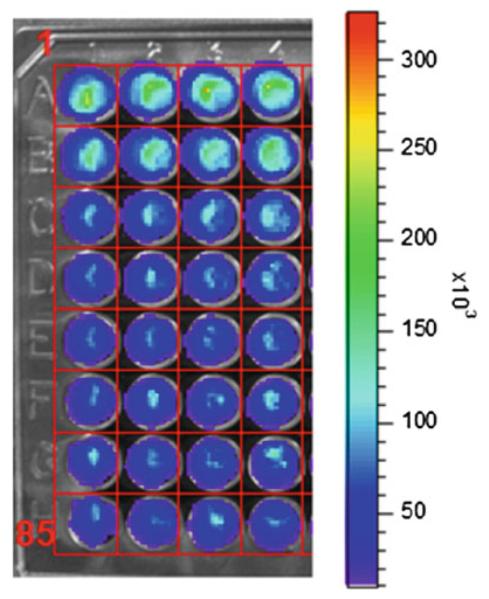
Cell-based GLuc complementation assay for inhibition of CXCL12-CGLuc binding to NGLuc-CXCR4. Equal numbers (1 × 10 4 cells each) of MDA-MB-231 cells secreting CXCL12-CGLuc (231-CXC12-CGLuc) or expressing NGLuc-CXCR4 (231-NGLuc-CXCR4) were cocultured in black wall 96 plates. Cells were incubated with vehicle control (row A) or increasing concentrations of the CXCL12-CXCR4 inhibitor AMD3100, which decreased bioluminescence from GLuc complementation. Grid overlay is used for ROI analysis. Scale bar shows range of photon fl ux values by pseudocolor display with red being highest and blue lowest numbers
3.3 Construction of Mouse Tumor Model
Implant mixture of stable complementation reporter cells for ligand and receptor (≈0.5–2 × 10 6 cells total) subcutaneously or orthotopically, such as in the mammary fat pad, in appropriate strain of mouse. We typically inject a 1:1 mixture of ligand and receptor complementation cells (see Note 13).
Begin imaging experiments when 4–5 mm diameter tumors form (see Note 14).
3.4 Mouse Imaging
Prepare coelenterazine for intravenous injection into one mouse by adding 44 μl of 10 mg/ml coelenterazine stock to 66 μl of 40 % DMSO/PBS solution. We use 100 μl of this solution for tail vein injection in each mouse (see Note 15).
Anesthetize mice with 1–2 % isoflurane and maintain mice under anesthesia during tail vein injection of coelenterazine (see Note 16).
Transfer mouse immediately to bioluminescence instrument and acquire image (see Note 17).
Remove mouse from imaging instrument and monitor for complete recovery from anesthesia.
Quantify imaging data by region-of-interest (ROI) analysis of bioluminescence produced by the tumor site, using units of photon flux (Fig. 3) (see Note 18).
Fig. 3.

Imaging ligand–receptor binding in living mice. Mice were implanted with equal numbers of 231-CXCL12-CGLuc and 231-NGLuc-CXCR4 cells as ortho-topic mammary tumor xenografts in NSG mice. Imaging began 20 s after tail vein injection of coelenterazine, using 2 min acquisition and large binning. Circles and values show photon fl ux measurements for ROIs around each tumor. Scale bar shows range of values depicted by pseudocolor display
Acknowledgement
This work was supported by NIH Grants R01CA136553, R01CA136829, R01CA142750, and P50CA093990.
4 Notes
Acid washing dissociates ligand–receptor complexes in the extracellular space, allowing quantification only of internalized receptors with bound ligand. This procedure is explained further in Note 11.
Acidified methanol is needed to keep higher concentrations of coelenterazine in solution. We dissolve 10 mg coelenterazine in 1 ml final volume of methanol and 3 N HCl (980 μl of methanol and 20 μl of 3 N HCl).
NGLuc and CGLuc must both localize to the extracellular space to detect ligand–receptor binding at the cell surface. To accomplish this objective, a GLuc fragment should be attached to the extracellular terminus of a transmembrane receptor and the terminus of the ligand that does not primarily determine receptor binding. We use the GLuc enzyme fragments identified by Remy et al. in which the enzyme is divided between amino acids G93 and E94 (omitting the 16 amino acid leader sequence) to form NGLuc and CGLuc with amino acids 1–93 and 94–169 [1].
We typically add a flexible linker, such as amino acids GGGSGGGS, between GLuc enzyme fragments and proteins of interest. Linkers allow placement of restriction enzyme sites for cloning fusion constructs and potentially reduce steric constraints on ligand–receptor pairs and GLuc enzyme fragments. A linker sequence is not required, and users should consider testing fusion constructs with shorter or no linking amino acids to determine optimum Gaussia luciferase complementation output for the selected ligand–receptor pair.
We test NGLuc and CGLuc fusions to both ligand and receptor in all orientations that allow complementation in the extra-cellular space with the goal of identifying a combination that optimizes ligand-dependent bioluminescence relative to background levels.
Control constructs could have mutations in key amino acids in either ligand or receptor that confer specific binding, secreted and/or membrane bound extracellular NGLuc or CGLuc fragments, or a mismatched pair of ligand and receptor.
We typically generate stable cell lines via lentiviral vectors with co-expressed fluorescent proteins, allowing us to use flow cytometry to sort for batch populations of transduced cells. Alternatively, investigators may transfect standard expression constructs into cells and select for cells with stable expression of the reporter transgene by drug resistance or fluorescence. We prefer batch populations of cells to avoid potential confounding effects of clonal cell lines.
We select the optimum reporter pair based on maximum induction of bioluminescence produced by matched ligand– receptor fusion proteins above background levels defined by control complementation reporters. After identifying the optimum orientations of fusion proteins, we generate stable cell lines expressing either the ligand or receptor complementation reporters, respectively.
We typically plate equal numbers of cells that either secrete a GLuc complementation ligand or express the cognate GLuc complementation receptor to make a total of 1 × 10 4 −2 × 10 4 cells/well. These cocultures reproduce chronic intercellular signaling, such as between two different cell types in a tumor. To test acute induction of complementation signal for a soluble ligand binding to its receptor, users can collect supernatants from ligand-secreting cells and add the supernatant to cells expressing the complementation receptor.
We use phenol red free medium to improve transmission of GLuc bioluminescence from cells. Since serum oxidizes coelenterazine, the substrate for GLuc, and generates substantial background signal, we routinely use serum-free medium for assays. We have performed complementation assays in serum-free medium for up to 24 h with no loss of signal. Users may need to tailor lengths of experiments and culture conditions based on specific cell types. If a low percentage of serum is needed to maintain cell viability for extended assays, users should make certain to have control cells with the same percentage of serum to determine background bioluminescence.
GLuc bioluminescence decreases by approximately 70 % within 1 min of adding coelenterazine [3], so we add coelenterazine to wells as rapidly as possible and begin imaging as soon as possible thereafter. Adding coelenterazine to medium already present in wells measures total ligand–receptor complexes in extracellular and intracellular compartments. To quantify only internalized, intact ligand–receptor complexes, users should use an acid wash protocol to dissociate extracellular ligand– receptor pairs. The acid wash procedure entails (a) removing all medium from wells; (b) incubating cells on ice with 150 μl per well ice-cold acid wash solution for 3–5 min; (c) removing acid wash; (d) washing wells once with 200 μl per well warm PBS; and (e) adding 1:500 coelenterazine diluted in PBS. Using PBS further decreases background bioluminescence as compared with phenol red free medium.
Imaging times need to be optimized for specific pairs of ligand and receptor to acquire detectable signal without saturating the detector system. If the bioluminescence signal is low, users may increase numbers of cells per well, use more concentrated coelenterazine (1:100 to 1:250 dilution of 1 mg/ml stock) or acquire images for longer periods of time.
Tumor xenografts provide a localized environment in which large numbers of ligand and receptor complementation pairs exist in a confined, relatively small volume. Adding Matrigel to implanted tumors may improve tumor take and generate more well-defined tumors. The configuration of an implanted tumor favors a large number of ligand–receptor binding events, enhancing detection of GLuc complementation. Since overlying tissue substantially attenuates blue light produced by GLuc, tumors in a superficial site (such as mammary fat pad or subcutaneous implant) produce a greater imaging signal than tumors in internal organs. Initial animal imaging studies should include a control group of mice with a control complementation pair that should not interact specifically, allowing quantification of background signal. Cancer cells have been shown to migrate from one tumor to another when two tumors are implanted in the same mouse, so we prefer to have separate cohorts of animals for ligand–receptor and control groups [4].
If the complementation signal is particularly weak, larger tumors may be needed to produce a detectable imaging signal. Tumor burden must comply with protocols approved by institutional committees for care and use of animals.
Coelenterazine oxidizes in aqueous solutions, so we prepare the diluted volume of coelenterazine in 40 % DMSO/PBS immediately before tail vein injection into each mouse. Larger amounts of coelenterazine typically are needed to detect GLuc complementation relative to full-length, intact enzyme since the complemented enzyme fragments generate only ≈25 % as much light. Adjust amounts of injected coelenterazine as needed to produce a detectable signal in mice.
We prefer isoflurane anesthesia because of its rapid induction, ease of adjusting dosage, and rapid recovery relative to injectable anesthetics. Anesthetizing mice prior to tail vein injection allows the mouse to be transferred immediately to the bioluminescence imaging instrument. Since GLuc has flash kinetics of bioluminescence, a delay between injection of coelenterazine and imaging results in loss of signal.
Image acquisition times vary depending on complementation signal intrinsic to the ligand–receptor pair, size of tumor, and amount of injected coelenterazine. Imaging times typically are 1–3 min per mouse, but times will need to be optimized for different ligand–receptor pairs and tumor sites.
IVIS and similar bioluminescence imaging instruments have dedicated software for defining ROIs and measuring photon flux. As tumors grow larger, investigators may need to acquire images for shorter periods of time to avoid saturating the detector system. Photon flux measurements correct for differences in imaging time used in different studies over the course of an experiment. When comparing bioluminescence for several mice in experimental and control groups, investigators should select the same minimum and maximum values for pseudocolor display prior to defining ROIs that encompass light emitted from a tumor or other site. This approach maintains consistency of data across different groups of mice, such as those treated with a specific inhibitor of ligand–receptor binding or vehicle control.
To account for effects of tumor size and relative numbers of cells on GLuc complementation signal in different mice, we typically co-express a second, constitutive reporter protein in each complementation cell line. For example, beetle lucifer-ases such as firefly or click beetle red can be used as a marker protein in one cell population. Bioluminescence from these enzymes can be distinguished readily from GLuc because beetle luciferases use a different substrate, luciferin. Beetle lucifer-ases can be imaged immediately following GLuc using previously described protocols for these enzymes in animal studies [5]. Typically, bioluminescence from beetle luciferases is substantially greater than GLuc complementation, particularly since the complementation signal decreases rapidly. If necessary, using an emission filter of 580–600 nm will further discriminate light from relatively red shifted beetle luciferases from GLuc bioluminescence. To mark a second population of complementation cells in a tumor, we have used a far red fluorescent protein such as eqFP650 [6], which can be detected readily in subcutaneous sites such as mammary fat pads. Since near-infrared light transmits through tissues even better than far red, the recently described IFP may allow even more sensitive detection of cells in vivo [7]. These independent markers allow changes in ligand–receptor binding and GLuc complementation to be normalized to changes in ligand-producing and receptor cell lines overtime.
Bioluminescence from GLuc peaks at 480 nm, so light from this enzyme is particularly sensitive to attenuation by overlying tissue, pigment, and fur [8]. To optimize detection of GLuc complementation in vivo, we typically shave fur overlying a tumor site and then use a depilatory agent to remove remaining hair. A depilatory agent should be applied briefly (<2 min), using a cotton swab or soft tissue to remove the lotion and fur. The skin site should be washed thoroughly with warm water to avoid chemical burns.
Coelenterazine is a substrate for the multidrug transporter ABCB1 (MDR1 P-glycoprotein), which limits access of this compound to the intact central nervous system because of ABCB1 in the blood–brain barrier [9]. ABCB1 also may limit intracellular accumulation of substrate and detection of internalized ligand–receptor complexes in cells that express this transporter. Investigators may need to test for expression of ABCB1 by flow cytometry or other assays and/or select an alternative cell line if needed to overcome this limitation.
References
- 1.Remy I, Michnick S. A highly sensitive protein-protein interaction assay based on Gaussia luciferase. Nat Methods. 2006;3:977–979. doi: 10.1038/nmeth979. [DOI] [PubMed] [Google Scholar]
- 2.Luker K, Mihalko L, Schmidt B, Lewin S, Ray P, et al. In vivo imaging of ligand receptor binding with Gaussia luciferase complementation. Nat Med. 2012;18:172–177. doi: 10.1038/nm.2590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Tannous B, Kim D, Fernandez J, Weissleder R, Breakefield X. Codon-optimized Gaussia luciferase cDNA for mammalian gene expression in culture and in vivo. Mol Ther. 2005;11:435–443. doi: 10.1016/j.ymthe.2004.10.016. [DOI] [PubMed] [Google Scholar]
- 4.Kim M, Oskarsson T, Acharyya S, Nguyen D, Zhang X, et al. Tumor self-seeding by circulating cancer cells. Cell. 2009;139:1315–1326. doi: 10.1016/j.cell.2009.11.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Luker G, Luker K. Luciferase protein complementation assays for bioluminescence imaging of cells and mice. Methods Mol Biol. 2011;680:29–43. doi: 10.1007/978-1-60761-901-7_2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Shcherbo D, Shemiakina I, Ryabova A, Luker K, Schmidt B, et al. Near infrared fluorescent proteins. Nat Methods. 2010;7:827–829. doi: 10.1038/nmeth.1501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Filonov G, Piatkevich K, Ting L, Zhang J, Kim K, et al. Bright and stable near-infrared fluorescent protein for in vivo imaging. Nat Biotechnol. 2011;29:757–761. doi: 10.1038/nbt.1918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Weissleder R, Ntziachristos V. Shedding light onto live molecular targets. Nat Med. 2003;9:123–128. doi: 10.1038/nm0103-123. [DOI] [PubMed] [Google Scholar]
- 9.Pichler A, Prior J, Piwnica-Worms D. Imaging reversal of multidrug resistance in living mice with bioluminescence: MDR1 P-glycoprotein transports coelenterazine. Proc Natl Acad Sci U S A. 2004;101:1702–1707. doi: 10.1073/pnas.0304326101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.McGarrigle D, Huang X-Y. GPCRs signaling directly through Src-family kinases. Sci STKE. 2007;2007:e35. doi: 10.1126/stke.3922007pe35. [DOI] [PubMed] [Google Scholar]