

Abstract
AL-Base, a curated database of human immunoglobulin (Ig) light chain (LC) sequences derived from patients with AL amyloidosis and controls, is described, along with a collection of analytical and graphic tools designed to facilitate their analysis. AL-Base is designed to compile and analyse amyloidogenic Ig LC sequences and to compare their predicted protein sequence and structure to non-amyloidogenic LC sequences. Currently, the database contains over 3000 de-identified LC nucleotide and amino acid sequences, of which 433 encode monoclonal proteins that were reported to form fibrillar deposits in AL patients. Each sequence is categorised according to germline gene usage, clinical status and sample source. Currently, tools are available to search for sequences by various criteria, to analyse the biochemical properties of the predicted amino acids at each position and to display the results in a graphical fashion. The likelihood that each sequence has evolved through somatic hypermutation can be predicted using an automated binomial or multinomial distribution model. AL-Base is available to the scientific community for research purposes.
Keywords: AL amyloidosis, online database, immunoglobulin light chain
Introduction
The pathology of amyloidosis in humans is due to the extra-cellular deposition of proteinaceous fibrils. Twenty-seven distinct proteins have been implicated in amyloid disease [1]. In many of these, a single amino acid substitution differentiates the amyloidogenic protein from its wild type counterpart. Thus, analysis of primary sequence is important in characterising the relationship between mutation and aggregation via changes in physicochemical properties [2,3]. AL amyloidosis presents a unique challenge in that the immunoglobulin (Ig) light chain (LC) proteins that comprise the deposited fibrils display inherent somatic sequence variability. For this reason, a large organised data set is needed to elucidate the links between protein sequence and aggregation. Conventional databases such as Gen-Bank (http://www.ncbi.nlm.nih.gov/Genbank/) and Uniprot (http://www.pir.uniprot.org/) store a wealth of nucleotide and protein sequence data. However, smaller, specialised databases can be useful for studying specific types of sequences, or sequences related to a particular disease such as amyloidosis. Previously, a database of Ig LC proteins (ftp://bioinformatics.anl.gov/VL-database) was established, containing the amino acid sequences of over 200 LCs derived from patients with a variety of diseases. However, the VL-database has not been updated since 2005 and has no integrated tools for searching or analysis. To address these issues, we have developed AL-Base, a visual platform analysis tool for the study of LC sequences associated with AL amyloidosis, which expands on the capabilities and the contents of the VL-database by adding sequences generated from mRNA, as well as including a searchable interface and visual tools for analysis. Currently, AL-Base contains 433 amyloidogenic LC sequences compiled from public sources, including 271 that we have derived from patients seen at the Boston University amyloid treatment and research program. New sequences are continually being added to the database as they become publicly available. AL-Base stores alignments for every LC sequence according to the international ImMunoGeneTics information system (IMGT) numbering for variable (VL) and constant (CL) domains [4,5]. Any set of Ig LC sequences can be pulled from the database and compared. All data, including alignments, are available to download in common formats such as FASTA [6] and ClustalW [7]. The database is currently available for public use at http://pulm.bumc.bu.edu/aldb/home/. We hope that with community input and involvement, the current feature set can be expanded.
Materials and methods
Software and hardware architecture
The database was developed on a Dell PowerEdge server with a 1.8 GHz Xeon processor and 2 GB of memory, using freely available and open source tools. The operating system is BU Linux 4.5 server edition (Zodiac). The main interface to the database is accessible through a web browser at http://pulm.bumc.bu.edu/aldb/home. The web server is Apache 2.0.1 (www.apache.org), and the web application framework is Ruby on Rails (www.rails.org). Ruby on Rails ties the back-end database to the front-end website, and simplifies the process of adding new features and functionality. Many features of the database make use of the BioRuby library (www.bioruby.org). The database is accessible via the internet. As all sequence processing is done on the back-end server, client side requirements are minimal; however, the large amount of data returning from some queries merits a recommendation of using at least a Pentium-4 PC with 512 MB of RAM, or a Macintosh of similar capabilities. The database was designed to be used with the Firefox web browser, but it can also be displayed in other browsers such as Internet Explorer, Safari or Opera.
Sequence collection
Sequences were culled from various publicly available sources, including GenBank, Uniprot and the PDB (http://www.rcsb.org/pdb). In addition to the amyloidogenic sequences, we have included all available LCs from other plasma cell disorders (PCD) such as multiple myeloma (MM), light chain deposition disease (LCDD) and Waldenstrom's macroglobulinemia (WM), as well as a heterogeneous collection of LCs from other disease states or sources. Sequence annotations were parsed to exclude non-functional sequences, such as those with out-of-frame insertions, or incorrect stop codons. When available, the pathologic nature of the sequence in vitro or in vivo (fibril, cast, amorphous deposit, non-pathologic) was noted. The original flat-file is also stored along with the sequence so that all available information is preserved.
Data storage
The relational database management system (RDBMS) used was MySQL 4.1.20 (http://www.mysql.org). The database schema includes tables for donor germline genes, nucleotide and protein LC sequences and alignments. The crucial first steps were creating an alignment standard, as well as automating the process of aligning new sequences as they were added to the database. The IMGT numbering for Ig VL and CL domains [4,5] was utilised based on several unique features, i.e. a current format and its easily parsed treatment of gaps in the VL domain complementarity determining regions (CDR). Germline gene information, all functional germline gene sequences and their respective alignments from the IMGT/GENE-DB (http://imgt.cines.fr) were downloaded in flat-file format, parsed and inserted into the database. Once this was complete, each nucleotide or protein LC sequence was assigned to its germline heritage using BLAST [8] in conjunction with the BioRuby BLAST module, parsed for CDR and framework regions (FR), aligned according to the IMGT numbering scheme and then inserted into the database.
Antigen selection
Binomial [9] and multinomial [10] antigen selection algorithms were implemented and applied to every LC sequence in the database. Briefly, these algorithms determine the probability of antigen selection by calculating the expected number of replacement (R) and silent (S) mutations in the FRs and CDRs of the LC VL region, and comparing these data to the total number of observed mutations. In a sequence selected by an antigen, there is an excess of R mutations in the CDR domains and S mutations in the FR domains. The results of the algorithms are stored in tables in the database for rapid access, and are available from the individual sequence entry page. The R statistical environment (www.r-project.org) was used to perform the algorithms.
Visual tools
A web-based interface was written for the database using the Ruby programming language (www.ruby-lang.org) and the Ruby on Rails web application framework (www.rubyonrails.org). Many features of the database make use of the BioRuby package (http://www.bioruby.org). All alignments and sequence data can be downloaded in common formats such as FASTA and ClustalW. For an alignment of any set of sequences, reports providing a summary of the germline gene usage, property scores, mutation rates and antigen selection results can be generated and downloaded.
Property-based statistical analysis
To highlight the utility of the database for statistical analysis of large data sets of sequences, we used the integrated tools to compare the mean property values for positions in alignments for the AL-PCD and other-PCD groups. First, we searched the database for all Vκ1 sequences for these two sets. For each group, we used the integrated tools to generate an alignment, and then downloaded reports in comma separated value (CSV) format for each property value currently available: hydrophobicity [11], flexibility [12], volume [13], accessible surface area [14], isoelectric point [15] and beta structure [16]. These reports were then imported into the R statistical environment, though the CSV format can also be read by common programs such as Microsoft Excel. Mean property values for each position of the alignment were compared between the AL-PCD and other-PCD group using a two-tailed T-test, and those positions and properties with significant differences were recorded based on the Type I error of 5%.
Results
As of February 2008, the database contained 433 AL LCs, 163 LCs from other PCDs and 3364 LCs from other sources. Additionally, 143 germline VL sequences, 19 germline joining (JL) region sequences and 15 germline CL region sequences have been entered; only functional, productive germline sequences were selected. Of 433 AL sequences, 26.3% were VLκ and 73.7% were VLλ. By subtype, the majority of LCs were in the κ1 (21.0%), λ3 (19.9%), λ1 (19.4%), λ6 (17.6%) and λ2 (15.7%) groups. The rest were divided among κ2–4 (5.3%) and λ4,7 and 10 (1.2%) subtypes. For those LCs from other PCDs, the distribution was 56.4% VLκ and 43.6% VLλ, with the majority in the κ1 (33.1%), λ3 (16.6%), λ2 (13.5%), κ4 (11.7%) and λ1 (10.4%) subtypes. There were very few λ6 sequences in both the other-PCD (1.2%) and non-PCD (1.6%) groups. The distribution (Table I) shows that AL is a disease primarily associated with VLλ LCs, as has been previously reported [17–21], whereas both the other-PCD and non-PCD groups contain predominantly VLκ LCs.
Table I.
Sequence counts by family and major category.
| VL LC family | Amyloidogenic sequences (n = 433) | Non-amyloidogenic sequences (n = 3527) | |
|---|---|---|---|
|
| |||
| Other-PCD (n = 163) | Non-PCD (n = 3364) | ||
| κ1 | 91 | 54 | 817 |
| κ2 | 4 | 9 | 425 |
| κ3 | 5 | 10 | 803 |
| κ4 | 14 | 19 | 148 |
| λ1 | 84 | 17 | 479 |
| λ2 | 68 | 22 | 217 |
| λ3 | 86 | 27 | 311 |
| λ4 | 3 | 1 | 50 |
| λ5 | 0 | 2 | 9 |
| λ6 | 76 | 2 | 53 |
| λ7 | 1 | 0 | 13 |
| λ8 | 0 | 0 | 16 |
| λ9 | 0 | 0 | 16 |
| λ10 | 1 | 0 | 7 |
LC sequences can be accessed from the database using the integrated search engine. Search identifiers include germline gene usage, clinical status and sample sources (molecule, tissue and cell types). Sequences that match criteria are then displayed on a sortable results page. Once a search has been performed and a set of LCs selected, the visual tools integrated into the database can be applied to simplify the analysis of individual or multiple sequences. Individual entries can be selected for more information, including a list of features and qualifiers, the sequence itself, a conceptual translation, germline gene usage, relevant hyperlinks and references. Sequences can be aligned and compared with their closest germline progenitors, for both the VL and CL regions. The number of nucleotide substitutions are counted, and a list of R and S nucleotide and protein substitutions can be generated (Figure 1).
Figure 1.
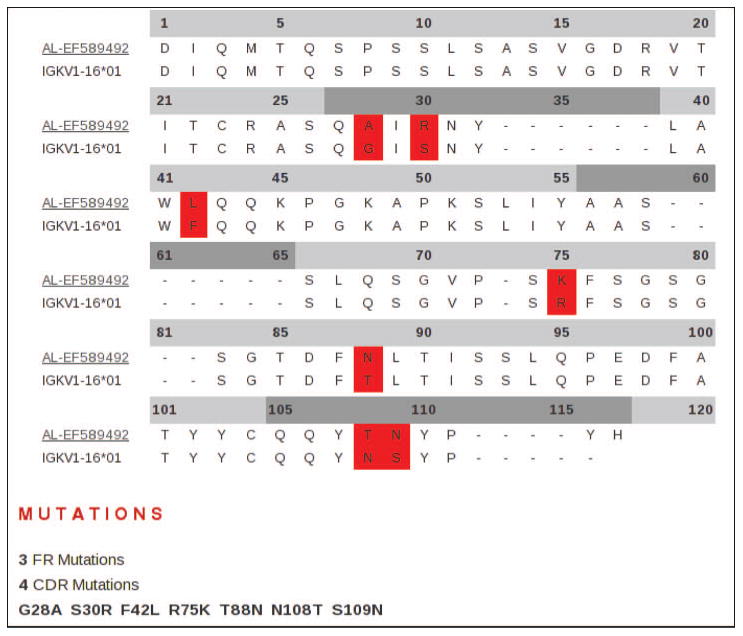
An example of an alignment of selected amyloidogenic VL LC to closest germline progenitor. Each LC in the database can be aligned to its most likely germline gene progenitor. A substitution list is generated and the comparison can be presented visually using a coloured alignment. FRs and CDRs are distinguished by light grey and dark grey header lines, respectively.
For multiple entries, alignments can be configured to show either nucleotide or protein sequences. Alignments are available for the VL region and, if available, the CL region of every LC; FR and CDR regions can be aligned individually if needed. All amino acid residues or only the substitutions can be characterised according to their physicochemical or biophysical properties using coloured alignments. Currently, residues can be coloured according to IMGT colour menus for a variety of amino acid classes based on hydropathy, volume and chemical characteristics (Figure 2) [22]. Furthermore, residues can be highlighted according to a scaled physicochemical or biophysical property (currently hydrophobicity, flexibility, volume, surface area, isoelectric point and beta structure), with shades of blue representing lower values and shades of red representing higher values. An ‘average’ value is coloured white. Values for residues are obtained from the AA index [23], a database of numerical indices representing various physicochemical and biochemical properties of amino acids. In many sequence to structure mapping problems, amino acid properties can be an extremely useful feature for prediction [24]. For large sets, a report can be generated for each position detailing the residue variability, along with a ‘score’ representing the average value for each selected property.
Figure 2.

An example of a multiple alignment of selected VL LC protein sequences. Once a set of LCs has been selected from a search, a multiple alignment can be performed. Either nucleotide or protein alignments can be shown. Alignments can be coloured according to the residue class or the property; in this case, by the 11 IMGT amino acid chemical characteristics classes are shown [22]. Different regions of the LC as defined by the IMGT can be individually aligned. Alignments can be downloaded in ClustalW format. Each sequence is identified by its clinical type, accession number, LC family subtype and germline gene. Sequences in a multiple alignment can be individually selected for more information. A report can be generated for each column, giving the counts of residues at that position.
Results for the antigen selection algorithms have been completed for 376 LCs in the AL category and 130 LCs in the other-PCD category. For any sequence, the results are publicly available from the antigen selection results page (Figure 3), which is accessed from the sequence features page. Tools to search for sequences based on these results are in development.
Figure 3.

Antigen selection algorithm results for LC EF589566. Top section shows nucleotide statistics for germline gene progenitor IGLV1–51★02. R, replacement sites; S, silent sites; RF, replacement frequency; LR, length of region compared with entire VL region. Bottom section shows results of antigen selection algorithm. R, observed replacement mutations; S, observed silent mutations; REXP, expected number of replacement mutations; PB, p-value as determined by binomial distribution model [9]; PM, p-value as determined by multinomial distribution model [10]. This LC shows evidence of antigen selection in both the FR and CDR regions.
The property-based statistical analysis identified 23 of 95 positions in the V-region of the Ig LC that had significant differences in mean property values between the AL-PCD and other-PCD groups. Of these positions, six were in the CDR and 17 were in the FR. These positions included some that have been previously identified, as well as several that are novel. A substitution list was generated for each recorded position for both groups (Table II).
Table II.
Results of property-based statistical analysis and substitution list for Vκ1 family.
| Region | Position (IMGT) | Property★ | Germline residue | Substitutions | |
|---|---|---|---|---|---|
|
| |||||
| AL-PCD (n = 91) | Other PCD (n = 53) | ||||
| FR1 | 3 | FX, HY, SA | Q/W | R, E(3), V(7) | R(2) |
| 17 | pI | D | A, N, G(2) | – | |
| 18 | BS, FX | R | K, G(2), N(2), T(2), S(4) | Y, T(3), V(5) | |
| 24 | SA, VM | R/Q/W | R, L(2), H(2) | R(3) | |
| 26 | FX | S | T, N(4) | R(2) | |
| CDR1 | 30 | BS | S | K, V, T, I, H(2), A(2), D(3), R(4), T(5), G(8), N(18) | V, K, S, R(2), T(2), L(3), N(3), G(5), I(5), A(5) |
| 31 | HY, FX, VM | S/N | E, A, R(2), Y(2), I(4), K(5), N(6), D(6), T(12) | R, Y, H, S(2), K(3), N(3), D(4), T(4), G(5) | |
| FR2 | 42 | FX, HY, SA | Y | W, L(3), F(4) | H, F(2) |
| 47 | pI, SA | G/E | A | – | |
| 52 | FX | L/S | I(2), F(3), V(5) | E, A, C, P, F(2), V(5) | |
| CDR2 | 56 | BS, FX | A/D/K | A, N, P, E(2), D(3), S(5), G(11) | V, K, N, T, S(2), A(3), G(3), E(4), D(4) |
| FR3 | 67 | BS | L | F, M, V(2) | – |
| 68 | FX | Q/E | K, L, A, R, E, I, D, Q(5) | A, R, Q(2), E(3), L(3), H(3) | |
| 69 | BS | S/T | K, T, N, P, R(2), A(3), I(3), G(4) | K, P, N(2), A(2), S(2), R(2), G(3), I(3) | |
| 72 | HY, pI, SA, VM | P | T, A(2), S(6) | T | |
| 75 | BS, pI, SA, VM | R | I, T, H, S(2), K(5), N(9) | M | |
| 86 | FX, pI | D/E | R, G(2), E(7), H(7), N(14) | N, Y, V(2), D(2), E(2) | |
| 88 | VM | T | Y, S(3), I(3), N(6) | A(2), S(4) | |
| 97 | VM | D/E | A, G(2), D(5) | V, D, E | |
| 103 | pI, VM | Y | S(2), F(3), H(4) | F(2) | |
| CDR3 | 107 | BS, FX, VM | S/Y/A | F, G, T | Y, N, D, E, I, H, R, G(2), T(2), S(3) |
| 108 | BS | Y/D/N | T, R, F, Q, A(2), D(3), K(3), N(3), E(3), H(3), Y(5), S(8) | Y, I, E, Q(2), T(2), S(3), N(3), H(4) | |
| 109 | FX | S/N | R, Y, I, H, K(2), F(2), N(4), S(5), D(10), T(14) | N, C, F, G, R(3), T(5), D(6), S(7) | |
HY, modified Kyte-Doolittle hydrophobicity scale; FX, average flexibility index; VM, average volume of residue; SA, average accessible surface area; pI, isoelectric point; BS, conformational parameter of beta-structure.
Property values identified to be significantly different between the AL-PCD and other-PCD groups.
Discussion
One of the challenges in creating the database was deciding which LCs should be included. There is a plethora of public LC sequence information available, including those generated from cDNA, genomic DNA and purified protein, which were obtained from a variety of sources such as bone marrow plasma cells, peripheral blood B cells, urine and tissue samples. AL-Base divides LCs into three major categories: AL, other-PCD and non-PCD. First and foremost, we searched for and included all available AL LCs. These included 271 sequences from patients seen in the amyloid treatment and research program at the Boston University Medical Centre (GenBank Accession Numbers EF589384-571, EU599319-361). Next, we searched for sequences that could be used as controls for comparison. This set is composed of LCs from other PCDs, not only from MM (n = 152) but also from LCDD (n = 7) and WM (n = 4). Although LCDD sequences by definition do not form fibrils [25], it is possible that some of the MM sequences could be amyloidogenic, as up to 10–20% of MM cases can develop fibrillar LC deposits [26–28]; thus, it is possible that some of the sequences classified as MM could actually be amyloidogenic. The final category, non-PCD, is a heterogeneous group that includes cDNA derived sequences from polyclonal healthy bone marrow, disease-related LCs and sequences of solved LC structures from the Protein Data Bank (www.rcsb.org/pdb). By including this category, many possibilities arise for studying AL LCs, such as finding structural models with similar sequence, or comparing the differences in family distribution for AL and another LC population.
The property-based statistical analysis identified 23 positions that had significant differences in mean property values between the AL-PCD and other-PCD sets (Table II). For each of the three invariant residues that form the folding core of the LC (C23, W41 and C104), significant differences were observed in adjacent residues (R/Q/W24, Y42 and Y103), as well as residues located nearby spatially (D/E86, T88 and L/S52). It may be that changes near folding core residues lead to decreased LC stability and amyloidosis, as has been previously suggested [29]. Additionally, Y42 and Y103 form part of the VL-VH interface, and substitutions in this region may prevent the LC from binding properly to the heavy chain or inhibit dimerisation [30]. Differences were also seen in certain clusters of residues that compose structurally important regions of the LC [31]. FR1 positions 17 and 18, which form the latter part of a turn between the A and B strands, had significant differences in isoelectric point, beta structure and flexibility between the two sets. P72, which interacts with F76 and forms a conserved loop, was replaced much more frequently in the AL group and had significant differences for 4 of 6 mean property indices. There were numerous replacements in the AL-PCD group at R75, the ‘smoking gun’ for LC pathogenicity [32] and salt bridge partner with D98. D/E97, which interacts with R75, was also replaced more frequently in the AL-PCD group. Of the CDR residues identified, positions 30 and 31 have been previously discussed for their role in amyloidogenicity [33]. Other AL sequence differences identified by this method included positions 3, 26, 47, 52, 56, 67–69, 86, 88 and 107–109.
The wide variety of substitutions seen at these positions illustrates the complexity in finding single point mutations that contribute to amyloidogenesis in AL. In understanding the pathogenesis of Ig LC deposition and fibril-forming diseases, one challenge lies in determining what combination of residues or substitutions can promote protein misfolding and fibril formation. To explore these changes, AL-Base provides an accessible and user-friendly way to work with large numbers of LC sequences and their alignments. Our aim was to create an open database usable by investigators interested in research involving AL Ig LC sequences. To that end, we have developed a fully featured, dynamic website that organises all publicly available AL LC sequences and associated data. Novel aspects of this database are the addition of searching and visual analysis tools, as well as its expandable architecture. Additions can be ‘plugged in’ to the current architecture as new algorithms or analysis tools are developed. This is illustrated by the antigen selection prediction capability. Each sequence entered into the database is analysed by the antigen selection algorithm, the results are stored and can be displayed on a results page. By using the IMGT numbering, whole LC sequences can be stored, and sequences from any LC family and source can be quickly analysed and compared. Colourful alignments aid comparative analysis and provide clues to the physicochemical and biochemical differences in the LC in different sets. We expect to maintain the database, continually adding new sequences when they become available, and adding new features as tools for analysis of nucleotide sequences and predicted proteins are developed. We hope that with community input and involvement, we can expand the feature set.
Acknowledgments
This project was supported by the NIH P01 HL68705 and by the Gerry Foundation. The authors thank David Ulrich of Gene Home Inc. (http://www.gene-home.com) and Dr. Avrum Spira of the BUSM Pulmonary Center for technical support and server hosting. They also thank Benjamin Forbes of the Gerry Laboratory for his assistance with sequence checking and categorisation.
Abbreviations
- AL
light chain amyloidosis
- CDR
complementarity determining region
- FR
framework region
- LC
light chain
- LCDD
light chain deposition disease
- MM
multiple myeloma
- PCD
plasma cell dyscrasia
- WM
Waldenstrom's macroglobulinemia
References
- 1.Westermark P, Benson MD, Buxbaum JN, Cohen AS, Frangione B, Ikeda S, Masters CL, Merlini G, Saraiva MJ, Sipe JD. A primer of amyloid nomenclature. Amyloid. 2007;14:179–183. doi: 10.1080/13506120701460923. [DOI] [PubMed] [Google Scholar]
- 2.Chiti F, Stefani M, Taddei N, Ramponi G, Dobson CM. Rationalization of the effects of mutations on peptide and protein aggregation rates. Nature. 2003;424:805–808. doi: 10.1038/nature01891. [DOI] [PubMed] [Google Scholar]
- 3.Dobson CM. Experimental investigation of protein folding and misfolding. Methods. 2004;34:4–14. doi: 10.1016/j.ymeth.2004.03.002. [DOI] [PubMed] [Google Scholar]
- 4.Lefranc MP, Pommie C, Kaas Q, Duprat E, Bosc N, Guiraudou D, Jean C, Ruiz M, Da Piedade I, Rouard M, Foulquier E, Thouvenin V, Lefranc G. IMGT unique numbering for immunoglobulin and T cell receptor constant domains and Ig superfamily C-like domains. Dev Comp Immunol. 2005;29:185–203. doi: 10.1016/j.dci.2004.07.003. [DOI] [PubMed] [Google Scholar]
- 5.Lefranc MP, Pommie C, Ruiz M, Giudicelli V, Foulquier E, Truong L, Thouvenin-Contet V, Lefranc G. IMGT unique numbering for immunoglobulin and T cell receptor variable domains and Ig superfamily V-like domains. Dev Comp Immunol. 2003;27:55–77. doi: 10.1016/s0145-305x(02)00039-3. [DOI] [PubMed] [Google Scholar]
- 6.Pearson WR, Lipman DJ. Improved tools for biological sequence comparison. Proc Natl Acad Sci USA. 1988;85:2444–2448. doi: 10.1073/pnas.85.8.2444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Higgins DG, Sharp PM. CLUSTAL: a package for performing multiple sequence alignment on a microcomputer. Gene. 1988;73:237–244. doi: 10.1016/0378-1119(88)90330-7. [DOI] [PubMed] [Google Scholar]
- 8.Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. J Mol Biol. 1990;215:403–410. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- 9.Chang B, Casali P. The CDR1 sequences of a major proportion of human germline Ig VH genes are inherently susceptible to amino acid replacement. Immunol Today. 1994;15:367–373. doi: 10.1016/0167-5699(94)90175-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lossos IS, Tibshirani R, Narasimhan B, Levy R. The inference of antigen selection on Ig genes. J Immunol. 2000;165:5122–5126. doi: 10.4049/jimmunol.165.9.5122. [DOI] [PubMed] [Google Scholar]
- 11.Juretic D, Lucic B, Zucic D, Trinajstic N. Protein transmembrane structure: recognition and prediction by using hydrophobicity scales through preference functions. Theor Comput Chem. 1998;5:405–445. [Google Scholar]
- 12.Bhaskaran R, Ponnuswamy PK. Positional flexibilities of amino acid residues in globular proteins. Int J Pept Protein Res. 1988;32:241–255. doi: 10.1111/j.1399-3011.1984.tb00944.x. [DOI] [PubMed] [Google Scholar]
- 13.Pontius J, Richelle J, Wodak SJ. Deviations from standard atomic volumes as a quality measure for protein crystal structures. J Mol Biol. 1996;264:121–136. doi: 10.1006/jmbi.1996.0628. [DOI] [PubMed] [Google Scholar]
- 14.Janin J, Wodak S, Levitt M, Maigret B. Conformation of amino acid side-chains in proteins. J Mol Biol. 1978;125:357–386. doi: 10.1016/0022-2836(78)90408-4. [DOI] [PubMed] [Google Scholar]
- 15.Zimmerman JM, Eliezer N, Simha R. The characterization of amino acid sequences in proteins by statistical methods. J Theor Biol. 1968;21:170–201. doi: 10.1016/0022-5193(68)90069-6. [DOI] [PubMed] [Google Scholar]
- 16.Beghin F, Dirkx J. Une methode statistique simple de prediction des conformations proteiques. Arch Int Physiol Biochim. 1975;83:167–168. [PubMed] [Google Scholar]
- 17.Comenzo RL, Wally J, Kica G, Murray J, Ericsson T, Skinner M, Zhang Y. Clonal immunoglobulin light chain variable region germline gene use in AL amyloidosis: association with dominant amyloid-related organ involvement and survival after stem cell transplantation. Br J Haematol. 1999;106:744–751. doi: 10.1046/j.1365-2141.1999.01591.x. [DOI] [PubMed] [Google Scholar]
- 18.Bellotti V, Merlini G, Bucciarelli E, Perfetti V, Quaglini S, Ascari E. Relevance of class, molecular weight and isoelectric point in predicting human light chain amyloidogenicity. Br J Haematol. 1990;74:65–69. doi: 10.1111/j.1365-2141.1990.tb02539.x. [DOI] [PubMed] [Google Scholar]
- 19.Alexanian R, Fraschini G, Smith L. Amyloidosis in multiple myeloma or without apparent cause. Arch Intern Med. 1984;144:2158–2160. [PubMed] [Google Scholar]
- 20.Solomon A, Frangione B, Franklin EC. Bence Jones proteins and light chains of immunoglobulins. Preferential association of the V lambda VI subgroup of human light chains with amyloidosis AL (lambda) J Clin Invest. 1982;70:453–460. doi: 10.1172/JCI110635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Perfetti V, Casarini S, Palladini G, Vignarelli MC, Klersy C, Diegoli M, Ascari E, Merlini G. Analysis of V(lambda)-J(lambda) expression in plasma cells from primary (AL) amyloidosis and normal bone marrow identifies 3r (lambdaIII) as a new amyloidassociated germline gene segment. Blood. 2002;100:948–953. doi: 10.1182/blood-2002-01-0114. [DOI] [PubMed] [Google Scholar]
- 22.Pommie C, Levadoux S, Sabatier R, Lefranc G, Lefranc MP. IMGT standardized criteria for statistical analysis of immunoglobulin V-REGION amino acid properties. J Mol Recognit. 2004;17:17–32. doi: 10.1002/jmr.647. [DOI] [PubMed] [Google Scholar]
- 23.Kawashima S, Ogata H, Kanehisa M. AAindex: amino acid index database. Nucleic Acids Res. 1999;27:368–369. doi: 10.1093/nar/27.1.368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ganapathiraju M, Manoharan V, Klein-Seetharaman J. BLMT: statistical sequence analysis using N-grams. Appl Bioinformatics. 2004;3:193–200. doi: 10.2165/00822942-200403020-00013. [DOI] [PubMed] [Google Scholar]
- 25.Gallo G, Picken M, Buxbaum J, Frangione B. The spectrum of monoclonal immunoglobulin deposition disease associated with immunocytic dyscrasias. Semin Hematol. 1989;26:234–245. [PubMed] [Google Scholar]
- 26.Stone MJ, Frendel EP. The clinical spectrum of light chain myeloma. A study of 35 patients with special reference to the occurrence of amyloidosis. Am J Med. 1975;58:601–619. doi: 10.1016/0002-9343(75)90496-9. [DOI] [PubMed] [Google Scholar]
- 27.Bahat G, Erten N, Saka B, Uzun S, Onur I, Kalayoglu-Besisik S, Buyukbabani N. Immunoglobulin D lambda multiple myeloma and amyloidosis with predominant soft tissue involvement. Amyloid. 2007;14:305–308. doi: 10.1080/13506120701614206. [DOI] [PubMed] [Google Scholar]
- 28.Gu M, Wilton R, Stevens FJ. Diversity and diversification of light chains in myeloma: the specter of amyloidogenesis by proxy. Contrib Nephrol. 2007;153:156–181. doi: 10.1159/000096766. [DOI] [PubMed] [Google Scholar]
- 29.Yu C, Zavaljevski N, Stevens FJ, Yackovich K, Reifman J. Classifying noisy protein sequence data: a case study of immunoglobulin light chains. Bioinformatics. 2005;21(Suppl 1):i495–i501. doi: 10.1093/bioinformatics/bti1024. [DOI] [PubMed] [Google Scholar]
- 30.Baden EM, Owen BA, Peterson FC, Volkman BF, Ramirez-Alvarado M, Thompson JR. Altered dimer interface decreases stability in an amyloidogenic protein. J Biol Chem. 2008;283:15853–15860. doi: 10.1074/jbc.M705347200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Stevens FJ, Argon Y. Pathogenic light chains and the B-cell repertoire. Immunol Today. 1999;20:451–457. doi: 10.1016/s0167-5699(99)01502-9. [DOI] [PubMed] [Google Scholar]
- 32.Stevens FJ. Four structural risk factors identify most fibril-forming kappa light chains. Amyloid. 2000;7:200–211. doi: 10.3109/13506120009146835. [DOI] [PubMed] [Google Scholar]
- 33.Raffen R, Dieckman LJ, Szpunar M, Wunschl C, Pokkuluri PR, Dave P, Wilkins Stevens P, Cai X, Schiffer M, Stevens FJ. Physicochemical consequences of amino acid variations that contribute to fibril formation by immunoglobulin light chains. Protein Sci. 1999;8:509–517. doi: 10.1110/ps.8.3.509. [DOI] [PMC free article] [PubMed] [Google Scholar]